Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Elucidating the Molecular Basis of Sorafenib Resistance in HCC: Current Findings and Future Directions
Authors Fornari F ![]() , Giovannini C, Piscaglia F
, Giovannini C, Piscaglia F ![]() , Gramantieri L
, Gramantieri L
Received 7 May 2021
Accepted for publication 11 June 2021
Published 2 July 2021 Volume 2021:8 Pages 741—757
DOI https://doi.org/10.2147/JHC.S285726
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Francesca Fornari,1,2 Catia Giovannini,1,3 Fabio Piscaglia,4,5 Laura Gramantieri4
1Centre for Applied Biomedical Research - CRBA, University of Bologna, St. Orsola Hospital, Bologna, Italy; 2Department for Life Quality Studies, University of Bologna, Rimini, Italy; 3Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Bologna, 40138, Italy; 4Division of Internal Medicine, Hepatobiliary and Immunoallergic Diseases, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy; 5Department of Medical and Surgical Sciences, University of Bologna, Bologna, Italy
Correspondence: Francesca Fornari; Laura Gramantieri Tel/Fax +390512143902
+390512142579
Email [email protected]; [email protected]
Abstract: Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related mortality worldwide. Sorafenib is the first multi-tyrosine kinase inhibitor approved for HCC and it has represented the standard of care for advanced HCC for almost 10 years, offering a survival benefit when compared to placebo. However, this benefit is limited, showing rare objective responses and a disease control rate approaching 50– 60%, with most patients experiencing disease progression at 6 months. These scant results dictate the urgent need for strategies to overcome both primary and acquired resistance. Herein we report several mechanisms supporting resistance to sorafenib in HCC patients, including activation of oncogenic pathways. Among these, the AKT/mTOR pathway plays a crucial role being at the crossroad of multiple driving events. Autophagy, multidrug-resistant phenotype, hypoxia-related mechanisms and endoplasmic reticulum stress are gaining more and more relevance as crucial events driving the response to anticancer drugs, including sorafenib. Several HCC-specific miRNAs take part to the regulation of these cellular processes. Remarkably, molecularly targeted strategies able to overcome resistance in these settings have also been reported. So far, the vast majority of data has been derived from laboratory studies, which means the need for an extensive validation. Indeed, most of the possible drug associations displaying promising effects in improving sorafenib efficacy herein described derive from preclinical explorations. Notably, data obtained in animal models can be inconsistent with regard to the human disease for efficacy, safety, side effects, best formulation and pharmacokinetics. However, they represent the necessary preliminary step to improve the management of advanced HCC.
Keywords: HCC, sorafenib, microRNA, autophagy
Plain Summary
Sorafenib has represented the standard of care for patients with advanced hepatocellular carcinoma for almost 10 years. Notwithstanding, the survival benefit of this treatment remains limited. Objective responses are rare and disease control can be obtained in approximately 50–60% of cases, with most patients experiencing progression at 6 months. Biomarkers predictive of primary and acquired resistance, together with novel strategies to increase sorafenib effectiveness are urgently needed. The understanding of molecular events leading to drug resistance and increasing treatment effectiveness represents the first step to improve hepatocellular carcinoma prognosis.
Introduction
Hepatocellular carcinoma (HCC) accounts for more than 80% of primary liver malignancies and is the second cause of cancer-related death worldwide.1 When diagnosed at advanced stages or with extrahepatic metastasis, systemic treatments are the only option.2 Sorafenib is a tyrosine kinase inhibitor (TKI) and targets multiple serine/threonine kinases, including c-RAF, wild type and mutant BRAF, transmembrane platelet-derived growth factor receptor beta (PDGFRB), vascular endothelial growth factor receptors (VEGFR1, VEGFR2, VEGFR3), hepatocyte factor receptor c-KIT (CD117), and Fms-like tyrosine kinase FLT3 (CD135).3,4 The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) and the insulin-like growth factor (IGF) signaling cascades play a major role among pathways that sorafenib fails to inhibit or that reactivate following treatment, gaining relevance in the context of resistance.5,6 In addition, following ligand-induced phosphorylation, insulin-like growth factor 1 receptor (IGF-1R) phosphorylates and activates both the PI3K/AKT and Ras/mitogen-activated protein kinase pathways further contributing to sorafenib resistance.7
Sorafenib is metabolized by hepatic CYP3A4 and UGT1A9, and the glucuronide conjugate is excreted via the biliary/fecal route.8 Sorafenib has represented the standard of care for advanced HCC for almost ten years, offering a survival benefit when compared to placebo.9 However, this benefit is limited, showing rare objective responses and a disease control rate approaching 50–60%, with most patients experiencing disease progression at 6 months.10 The poor effectiveness of sorafenib dictates the urgent need for strategies to overcome both the primary and the acquired resistance. The availability of novel treatments such as other TKIs and immune checkpoint inhibitors11–14 determines the urgent need of informative biomarkers to channel patients to the most promising first and second line treatments, according to personalized assessments. Informative biomarkers might only derive from a deep knowledge of molecular mechanisms driving primary and secondary resistance. The identification of these mechanisms might help to find biomarkers for treatment choice and its monitoring, and provide novel molecules for rational combined approaches to overcome sorafenib resistance. Indeed, beside primary resistance, sorafenib may also induce development of acquired resistance or intolerance. Several factors associated with sorafenib primary and acquired resistance were reported, including oncogenes and tumor suppressor genes, as well as cancer-related molecular pathways and non-coding RNAs.15 This last class of molecules includes microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) which role in liver carcinogenesis has been well established in the last two decades.16
Herein we will report some of the most representative studies dealing with molecular mechanisms involved in sorafenib resistance. Their knowledge is awaited to provide beneficial effects in terms of both prognostication and identification of novel treatments.
Driver genes and sorafenib resistance in HCC
High-throughput genome-wide screening tools have been employed for the identification of driver genes responsible for the drug resistant phenotype of cancer cells, leading to the discovery of new targets and possible treatment combinations. In a therapeutic perspective, it is crucial to distinguish between driver genes and bystander lesions that may result from genomic instability rather than from acquired mutations/deletions. Only the formers are responsible for clonal selection and increased aggressiveness. The advent of CRISPR/Cas9 technology17 further accelerated this process overcoming possible off-target effects and partial target silencing observed with the short hairpin RNA screening technology. Wei and coworkers performed a CRISPR/Cas9 knockout screening in sorafenib-treated HCC cells and identified phosphoglycerate dehydrogenase (PHGDH) as a crucial player in drug resistance. Indeed, PHGDH is the most negative-selected gene following sorafenib administration and, vice versa, it is highly upregulated in sorafenib-resistant cells. PHGDH is a rate-limiting enzyme of the serine synthesis pathway (SSP) whose activation during sorafenib treatment enables cancer cells to overcome reactive oxygen species (ROS) production and sorafenib-induced oxidative stress through the production of both antioxidant species and α-ketoglutarate. As a proof of concept, PHGDH knockdown induces apoptotic cell death and sensitizes tumor cells to sorafenib in xenograft mice. Metabolomics analysis revealed the accumulation of glycolytic metabolites that cannot be branched to the SSP in PHGDH silenced cells, and the decrease of the redox cell potential leading to mitochondrial ROS accumulation and sorafenib sensitization. The Authors also demonstrated the transcriptional regulation of PHGDH by nuclear factor erythroid 2-related factor 2 (NRF2) that indeed represents the principal regulator of the cell redox potential. Its induction by sorafenib might be responsible for acquired resistance. Interestingly, the association between the PHGDH inhibitor, NCT-503, and sorafenib triggered apoptosis in preclinical models overcoming sorafenib resistance. Notably, the activation of SSP is a common mechanism of resistance to TKIs as displayed following lenvatinib and regorafenib administration in HCC cells, suggesting PHGDH targeting as a promising combinational option for the treatment of HCC.18 A similar methodological approach applied on sorafenib-resistant clones identified the involvement of Keap1 downregulation in the acquired resistance to TKIs showing a reduction of ROS mainly due to NRF2 activation and upregulation of its transcriptional targets, NQO1, GPX2 and TXNRD1.19 Interestingly, a transposons-mediated delivery of miRNA-based short hairpin RNAs (shRNAs) against oncogenes located in amplification genomic regions allowed an in vivo negative selection of sorafenib-resistant genes. This approach led to the identification of Mapk14 as a therapeutic target whose blocking improves sorafenib efficacy and identified the activation of Mek-Erk and Atf2 downstream signaling cascades as responsible for acquired sorafenib resistance in mouse and human HCC. Intratumoral knockdown of Mapk14 did not affect tumor burden whereas it greatly increased overall survival of sorafenib-treated mice, suggesting its inhibition as a promising synergic strategy for HCC.20 The same shRNA screening also identified peptidase mitochondrial protein (PMCP) as a possible driver gene contributing to sorafenib resistance. A subsequent study demonstrated that silencing of PMCP subunit beta (PMPCB), but not subunit alpha (PMPCA), increases the survival of orthotopic tumor-bearing mice, as well as late stage HCCs, undergoing sorafenib treatment. Notably, a combination therapy with orally administered PMPCB shRNA-loaded nanoparticles plus sorafenib attenuated tumor burden and improved survival of HCC mice with respect to sorafenib monotherapy showing a comparable effectiveness with Mapk14 shRNA/sorafenib combination. The investigation of apoptosis-mediating PMPCB downstream signaling revealed the involvement of PTEN-induced putative kinase 1 (PINK1) and Parkin in the degradation of the anti-apoptotic gene MCL-1. Strikingly, MCL-1 staining correlates with poorer survival of HCC patients undergoing sorafenib treatment. The Authors proposed an elegant model where combinatorial strategies against PMPCB/MCL-1 pathway and Raf/Mek/Erk pathway are necessary to promote sorafenib sensitization.21 Preclinical tools can also be employed to determine novel mechanisms and gene signatures associated with sorafenib resistance. Tovar et al22 identified the enrichment of tumor-initiating cells (T-ICs) and the activation of fibroblast growth factor (FGF) and insulin growth factor (IGF) signaling cascades as crucial events leading to sorafenib resistance in a xenograft mouse model. A transcriptomic analysis revealed the upregulation of genes involved in cell cycle progression, proliferation, migration and progenitor cell-like features. Experimental settings confirmed the increase of sphere formation abilities and tumorigenic potential of cells from sorafenib-resistant tumors, proving the enrichment of T-ICs, which role in tumor relapse, drug resistance and disease progression is well known.23 Interestingly, the treatment of sorafenib-resistant HCCs with brivanib, an EGFR inhibitor, delayed tumor progression and increased survival by blocking the expression of IGFR1 and downstream effectors, ERK and phospho-RPS6. Therapeutic options directed against T-ICs and cancer stem cells (CSCs) might therefore overcome the development of tumor resistance and prevent tumor relapse, highlighting the combination or sequential employment of TKIs as a promising strategy in HCC.
Beta-catenin is the second most frequently mutated gene in HCC and its activation associates with sorafenib resistance.24 Due to the undraggable nature of β-catenin itself, the identification of regulatory genes preventing its nuclear translocation might favor the design of novel strategies aimed at boosting sorafenib efficacy. Bioinformatics tools identified Nima-related kinase 2 (Nek2) as a β-catenin interacting protein and in vitro studies demonstrated its ability to stabilize β-catenin promoting its nuclear localization by interfering with its binding to the destruction complex. Nek2 is upregulated in HCC with respect to surrounding non-tumor tissue and is an independent predictor of HCC survival. Nek2 silencing in combination with sorafenib treatment abolished tumor progression improving sorafenib antitumor activity in xenograft mice.25
To sum up, several HCC drivers are implicated in sorafenib resistance (Figure 1). Their potential in combinatorial strategies able to circumvent the onset of resistant phenotypes was demonstrated in preclinical models and awaits further validation. Remarkably, before envisaging any possible translation, laboratory evidences need to be corroborated with safety and pharmacokinetic data.
 |
Figure 1 Driver genes involved in sorafenib resistance in HCC. Notes: Schematic representation of molecular pathways underneath the deregulation of driver genes and their effects in gene transcription, radical oxygen species (ROS) production and receptor pathway activation. |
Oncogenic Pathways and Sorafenib Resistance in HCC
HCC is a heterogeneous disease. In line, heterogeneous responses to sorafenib can be observed due to preexisting innate or acquired resistance. Innate resistance occurs when sorafenib is ineffective from the beginning of treatment and it is mainly related to reduced drug intake, activation/inhibition of signaling pathways and altered expression of molecular targets.26 The existence of an innate resistance is emphasized by in vitro studies showing that some HCC cell lines are more resistant to sorafenib compared to others.27,28 On the other end, acquired resistance occurs when sorafenib becomes ineffective during the course of treatment, after a clinical benefit has been observed. Acquired resistance most commonly results from the activation of signaling pathways that “bypass” the drug targets to maintain survival and proliferation. Indeed, sorafenib targets several cellular pathways and simultaneously it activates compensatory cascades responsible for acquired resistance. Most targets that are associated with drug resistance are also related with HCC development and progression. Among them, the Phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway plays a crucial role in different biological processes like proliferation and metastasis and it mediates acquired resistance to sorafenib. Resistant HCC cell lines obtained by prolonged exposition to sorafenib escalating doses are characterized by a strong activation of the PI3K/Akt signaling pathway and, accordingly, Akt overexpression in Huh7 cells resulted in sorafenib resistance.5 In sorafenib resistant cells PI3K/Akt activation upregulated the expression of the transcription factor Snail inducing the epithelial to mesenchymal transition (EMT), responsible for a more aggressive phenotype.29 Although phosphorylation of Akt is induced by protein phosphatase inactivation,30 it was shown that sorafenib-induced Akt phosphorylation was independent from protein phosphatase 2A (PP2A), one of the major serine/threonine phosphatase in eukaryotic cells. Thus, mechanisms responsible for Akt activation in resistance are still unclear.31 Once activated, Akt produces a number of downstream effects such as p27 inhibition, cytoplasmic Foxo localization and mTOR phosphorylation.32–34 Accordingly, by using a high density fluorescence reverse-phase protein array (RPPA) that is well suited for profiling of phosphorylated proteins, de novo activation of mTOR signaling was identified in sorafenib-resistant HCC cells.35 MTOR pathway activation is characterized by increased phosphorylation of ribosomal protein S6 (RPS6) that in turn regulates protein synthesis and allows progression from the G1 to the S phase of the cell cycle in HCC.36 Preclinical models confirmed the association between mTOR axis and HCC recurrence.37 In the clinical practice, mTOR activation is observed in up to 45% patients with HCC, and is associated to early recurrence and poor prognosis.38 Thus, it is not astounding that hyperactivation of PI3K/Akt/mTOR has been associated with attenuated sensitivity to sorafenib and it is therefore plausible that HCC patients overexpressing phospho-RPS6 (S235/236) could benefit from mTOR inhibition. The macrolide rapamycin is the most famous mTOR inhibitor, initially extensively used as an immunosuppressive drug in organ transplantation. Subsequently, multiple rapamycin analogs defined ‘rapalogs’ as well as other mTOR inhibitors have been developed. Among them, everolimus inhibits the mTOR pathway and reduces tumor growth in xenograft models of human HCC.39 The in vivo efficacy of everolimus is potentiated by combined treatment with sorafenib in a HCC orthotopic rat model, and this combination inhibits tumor angiogenesis.40 Unfortunately, a multicenter, randomized Phase 3 clinical trial (NCT01035229) evaluating everolimus in patients with HCC who progressed on sorafenib did not show any improvement in overall survival.41 Another multicenter randomized Phase 2 trial (NCT01005199) in patients with advanced HCC treated with sorafenib either alone or in association with everolimus did not show any survival benefit of the combination over sorafenib alone.42 To overcome the failings of rapalogs, other inhibitors have been developed. These include MLN0128 (sapanisertib), CC-233 or NVP-BEZ235 (dactolisib), and AZD-8055. Preclinical studies with second-generation mTOR inhibitors showed promising results and two clinical trials evaluating CC-233 and AZD-8055 in advanced HCC were completed. However, results have not been published to date.43 On the other hand, negative feedback loop exists between mTOR and PI3K axis that needs to be considered. Indeed, inhibition of mTOR can lead to Akt activation, thereby promoting cell proliferation and survival.44 This may explain why the effect of mTOR inhibition is not ideal to reverse sorafenib resistance. Negative results obtained so far with mTOR inhibitors suggest that biomarkers of mTOR or alternative pathways activation are necessary to select HCC patients who might benefit from mTOR suppression.
The Notch signaling cascade is a highly conserved pathway involved in cell fate, cell death and cell proliferation in several physiological and developmental processes.45 Four Notch receptors composed of a large extracellular domain for ligand binding and a cytoplasmic Notch intracellular domain (NICD) for signal transduction have been described in mammals. Notch receptors are activated by cell surface ligands present in neighboring cells that induce different cleavages by the ADAM/TACE family and subsequently by the γ-secretase complex.46 Although Notch signaling is a critical component in liver regeneration, there are conflicting data on its involvement in HCC. However, growing evidences indicate that most of Notch activity is associated with oncogenic activation.47 Accordingly, Villanueva et al showed that the conditional expression of NICD1 in a mouse model led to HCC in all test subjects during the first year.48 Aberrant expression of Notch receptors has been detected in HCC tissues when compared to the surrounding livers49 and several studies verified that targeting Notch pathway represents a potential therapeutic option for HCC.50 Notch3 silencing increased sorafenib-induced apoptosis via specific downregulation of p21 and upregulation of pGSK3β (Ser9) in HCC cells. Accordingly, in a mouse xenograft model, Notch3 depletion combined with sorafenib exerted an important antitumor effect preventing drug resistance.27
Different strategies inhibiting Notch have been developed including monoclonal antibodies preventing ligand receptor interaction and gamma-secretase inhibitors (GSIs) to block the proteolytic cleavage of the receptors. The GSI PF-03084014 showed antitumor activity in different human cancers including HCC51 and consequently its synergistic effect with sorafenib was evaluated in a HCC spheroid-derived orthotopic model. Both single agents failed to show an inhibitory effect, which was instead obtained by their combination, reaching a synergistic impact associated with inhibition of both angiogenesis and cell proliferation of HCC spheroid-derived orthotopic tumors.52 Again, the antitumor activity of the combined regimen was associated with reduced expression of PI3K/Akt signal. Recent studies have shown that the crosstalk between oncogenic pathways is involved in the development of resistance to cancer therapy and, in line, a crosstalk between Notch and Akt signaling in sorafenib‐resistant HCC cells was described. Knockdown of Akt in sorafenib-resistant HepG2 cells resulted in Notch1 reduction that overcomes sorafenib resistance. Accordingly, valproic acid (VPA), a histone deacetylase (HDAC) inhibitor that regulates the Notch signaling pathway in HCC53 combined with sorafenib enhanced the sensitivity of resistant cells reverting the increased levels of Notch1 and Akt observed in acquired resistance.54 This data suggests that the interplay between Notch and PI3K/Akt pathway needs to be further elucidated in order to explore possible combinations preventing the activation of both oncogenic pathways. Taken together, these findings provide a rationale to combine Notch inhibitors with sorafenib as a therapeutic strategy for HCC pointing out that targeting cancer through combined approaches might help overcome innate and acquired resistance.
MicroRNAs and Sorafenib Resistance in HCC
MicroRNAs are a class of small (18–22 nucleotides long) non-coding RNAs with a regulatory function on target mRNA expression. They either impair translation, or promote mRNA degradation modulating the expression of hundreds of targets at once.55 Their functions are indeed more complex than single miRNA–mRNA interactions. They establish complex circuitry regulating key pathways at multiple levels interplaying with each other, ultimately leading to increased proliferation, invasion, and EMT, aberrant apoptosis and metabolic shift, accounting also for the response to extracellular challenges including drugs, hypoxia and nutrient deprivation.
Both tissue and circulating miRNAs were investigated in the setting of sorafenib treatment in HCC.56 Concerning tissue miRNAs predictive of response/resistance to treatments, the vast majority of data were generated in vitro or in animal models. This has to be ascribed to the fact that HCC diagnosis, especially at advanced stages, mostly relies upon imaging criteria, thus preventing the availability of tissue specimens to be investigated. Conversely, circulating miRNAs may provide non-invasive biomarkers to be evaluated over time during treatments. MiRNAs were suggested as potential diagnostic and prognostic markers and therapeutic targets in HCC. Several studies aimed to uncover the contribution of deregulated miRNAs to sorafenib resistance. Most of them focused on the molecular pathways primarily targeted by sorafenib while others explored miRNA-modulation of cancer-related processes such as apoptosis and multi-drug resistance (Figure 2).
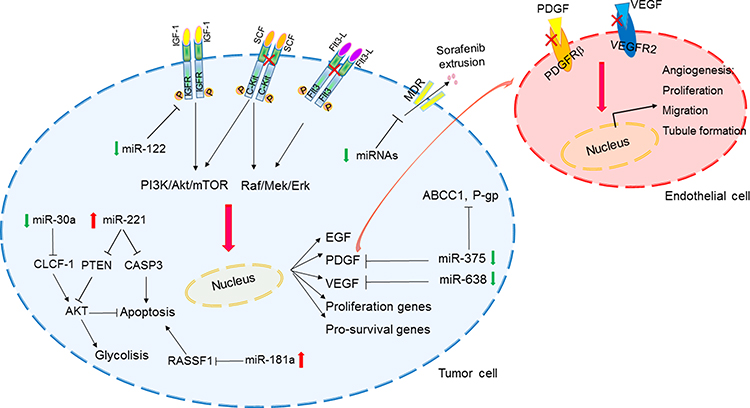 |
Figure 2 MiRNA-regulated pathways involved in sorafenib resistance in HCC. Abbreviations: P-gp, P-glycoprotein; SCF, stem cell factor; Red crosses, inhibition of sorafenib direct targets. Note: Schematic representation of HCC-deregulated miRNAs and downstream pathways influencing sorafenib response. Abbreviations: MDR: multi-drug-resistant transporters. |
VEGFR and PDGFR are the main transmembrane receptors whose function is directly impaired by sorafenib. VEGF is the main ligand triggering the activation of the VEGFR cascade stimulating endothelial cells proliferation and angiogenesis. MiR-638 directly targets VEGF. Remarkably, miR-638 downregulation in HCC cells and primary tumors inversely correlated with tumor size, portal vein invasion and poor prognosis. MiR-638 inhibited angiogenesis in in vitro models and tumor growth in a xenograft mouse model by blocking VEGF signaling. These findings were further corroborated by the presence of an inverse correlation between miR-638 and VEGF expression in HCC tissues.57 PDGF-C is a member of the PDGF/VEGF family of growth factors and it triggers the activation of PDGFR and plays an essential role in cell proliferation and blood vessel development. MiR-375 is downregulated in primary human HCCs and HCC-derived cell lines and it regulates PDGFC expression. MiR-375 antiangiogenic effects were demonstrated both in vitro and in vivo. In vitro, conditioned medium from miR-375 expressing cells impaired formation of tubular structures in HUVEC cells and it inhibited angiogenesis in rat aortic rings and chicken chorioallantoic membrane. Angiogenesis impairment was ascribed to the direct targeting of PDGFC, which effects were demonstrated both in vitro and in hepatoma xenograft mice. Interestingly, sorafenib induces the expression of miR-375, which, in turns, displays an antiangiogenic effect by targeting PDGFC. Among upstream regulators driving miR-375 overexpression under sorafenib-treatment, these authors identified the transcription factor achaete-scute homolog-1 (ASH1). Conversely, in the setting of sorafenib resistance elicited by long-term exposure, downregulation of miR-375 and an increase of MDR-related genes (MRP1 and P-glycoprotein) were reported. The enforced expression of miR-375 was able to restore sorafenib sensitiveness, at least in part by direct inhibition of the astrocyte elevated gene-1 (AEG1). Indeed, AEG1 is a multidrug resistance-related gene promoting MDR-1 expression; thus, its overexpression resulting from miR-375 downregulation participates to sorafenib resistance, while its downregulation resulting from miR-375 enforced expression contributes, at least in part, to sorafenib sensitivity.58
MiR-122 is downregulated in about 70% of primary HCC tissues59 and its downregulation associates with clonogenic survival, anchorage-independent growth, migration, invasion, EMT and sorafenib resistance in HCC cells. Among factors involved in sorafenib resistance resulting from miR-122 downregulation, Xu et al described the direct targeting of IGF-1R. Interestingly, this study showed that restoration of miR-122 in vitro elicited sorafenib sensitivity in resistant cells, mainly through apoptosis induction.6 Remarkably, sorafenib resistant HCCs displayed miR-122 downregulation and IGF-1R upregulation, which is indeed a common survival pathway to circumvent adverse conditions that gain reactivation during sorafenib resistance.60 MiR-181a is an oncogenic miRNA overexpressed in EpCAM+/AFP+ HCCs with stem cell features.61 MiR-181 was shown to contribute to sorafenib resistance through the direct targeting of Ras-association domain family 1 (RASSF1), a MAPK signaling factor. Indeed, besides its oncogenic effects, the Ras signaling is also involved in the activation of tumor suppression mechanisms. Among these, the RASSF1 family of proteins are activated by Ras to mediate tumor suppressive properties and apoptotic effects and their inhibition by miR-181a impairs sorafenib sensitivity.62
MiRNAs may modulate sorafenib effectiveness not only by regulating sorafenib canonical targets but also by hitting other cancer-related pathways. The adenosine triphosphate-binding cassette (ABC) transporter genes code for drug efflux pumps, responsible for the multidrug resistance phenotype. ABC transporters are highly expressed in normal and neoplastic hepatocytes. Twelve ABC transporter genes (ABCA2, ABCB1, ABCB6, ABCC1, ABCC2, ABCC3, ABCC4, ABCC5, ABCC10, ABCC11, ABCC12, and ABCE1) resulted upregulated in HCC tissue when compared to surrounding non-neoplastic liver and their upregulation was associated with the deregulated expression of miRNAs. In particular, 13 cellular miRNAs were confirmed to directly target and to upregulate the expression of five ABC genes, namely ABCA1, ABCC1, ABCC5, ABCC10, and ABCE1, with an inverse correlation between miRNAs and their corresponding target genes. Since these genes are directly involved in drug efflux, this study suggested evaluating miRNAs as non-conventional drugs in order to overcome multidrug resistance.63
Among tissue miRNAs predictive of sorafenib response, we have investigated the molecular mechanism underneath onco-miR-221 and tumor suppressor miR-30e deregulation in HCC. MiR-221 was assayed in two HCC animal models (DEN-induced HCC rats and xenograft mice), in a panel of HCC-derived cell lines and in human HCC tissues resected at surgery. Higher miR-221 levels turned out to be associated with sorafenib resistance at least in part through caspase 3 (CASP3) direct inhibition. Because of CASP3 role as a final effector of apoptotic cell death, its downregulation by miR-221 results in reduced apoptotic rate in HCC preclinical models.64 Remarkably, other targets of miR-221 such as PTEN may cooperate to induce sorafenib resistance. On the other hand, the tumor suppressor miR-30e-3p holds promise as a putative biomarker predictive of response to sorafenib in patients with HCC. MiR-30 family members downregulation was associated with proliferation, invasion and epithelial-to-mesenchymal transition, by targeting EMT-associated factors65,66 and cell adhesion molecules.67 MiR-30e-3p is downregulated in human and rat HCCs, and its downregulation associated with TP53 mutations. EpCAM, PTEN, and p27 are miR-30e-3p targets mediating its contribution to stemness and malignant features.68 Zhang et al provided a mechanistic insight explaining how miR-30a-5p, another member of the miR-30 family, participates to sorafenib resistance in HCC. More in detail, in sorafenib resistant settings, the downregulation of miR-30a-5p allows the overexpression of its direct target, cardiotrophin-like cytokine factor 1 (CLCF1). In turn, CLCF1 promotes aerobic glycolysis in HCC cells by activating PI3K/Akt signaling and its downstream genes, ultimately leading to a survival benefit. Thus, the miR-30a-5p/CLCF1/PI3K/AKT cascade links sorafenib resistance to metabolism in HCC cells, explaining the survival advantage of sorafenib resistant cells due to the increased aerobic glycolysis. Remarkably, the delivery of a cholesterol modified agomiR-30a-5p in a mouse model of sorafenib resistant HCC, displayed anticancer effects, suggesting the adoption of a combined approach in sorafenib resistant settings.69 To sum up, evidence supporting miRNAs involvement in sorafenib resistance in HCC are sound. Thus, miRNAs appear promising molecules to be validated as both biomarkers and therapeutic tools. Remarkably, their characteristics and the way they affect molecular mechanisms suggest to consider miRNA panels instead of single miRNAs.
Autophagy and sorafenib resistance
Autophagy is an evolutionary conserved physiologic process through which damaged cell components, such as mitochondria, macromolecules, endoplasmic reticulum, are removed and degraded. It requires the formation of a transient double-membrane structure ‘autophagosome’ incorporating cytosolic proteins and organelles leading them to degradation following lysosomal fusion. Conversion of the soluble form of LC3 (LC3-I) to a lipidated and autophagosome-associated form (LC3-II) is a hallmark of autophagy, together with the overexpression of other molecules that are fundamentally involved in the autophagosome enucleation and maturation such Beclin-1 and autophagy-related genes (ATGs).
Autophagy occurs in basal conditions but it is further enhanced during stressful stimuli such as nutrient deprivation, growth factors withdrawal and hypoxia favoring the recycling of cellular components to obtain energetic fuel to sustain stressed cells. MTOR and AMPK (5’AMP-activated protein kinase) are the main pathways regulating autophagy process with opposite effects.70 On the one hand, mTOR inhibits autophagy in presence of growth factors and nutrient abundance. On the other hand, AMPK enhances autophagic flux when the cellular energetic potential is low. Autophagy enables cell survival in stressful conditions playing a key role in cancer promotion. Nevertheless, its effects are controversial leading to cytotoxicity and, in the long term, triggering cell death in specific contexts.71 Autophagy has been shown to play dual roles in tumor initiation and progression. Its deficiency is advantageous in tumor initiation but represses tumor progression, playing also a contradictory effect in cancer therapy including sorafenib treatment. Indeed, despite the well-known promoting activity of sorafenib on autophagy induction via Mcl-1-mediated Beclin-1 activation,72 conflicting studies report the pro-survival role of autophagy in the long-term sorafenib administration and, on the contrary, its lethal effect during acute treatment, highlighting the duplicity of autophagy depending on different cell-contexts.
Regarding autophagy role in sorafenib-mediated apoptosis, Zhai and coworkers showed that it is possible to convert protective autophagy to autophagic cell death by switching-off Akt pathway in HCC cells. Akt signaling re-activation is indeed a well-known hallmark of acquired resistance to sorafenib in HCC patients.5 The establishment of sorafenib-resistant (SR) cells following prolonged drug exposure represents a useful in vitro tool to investigate mechanisms involved in adaptive sorafenib response. These Authors reported a decrease of autophagy markers LC3B and Beclin-1 in SR cells. Notably, chronic exposure to sorafenib inhibits the mTOR pathway, resulting in the activation of Akt via the negative mTOR-dependent feedback loop in SR HCC cells. These molecular events might explain the lack of a synergic effect between sorafenib and rapamycin when treating SR cells. More importantly, they might be at the basis of clinical trials failure as in the case of the phase I study investigating everolimus and sorafenib combination in HCC patients.73 On the contrary, co-administration of the Akt inhibitor GDC0068 with sorafenib boosts autophagy process promoting cell death in both in vitro and in vivo models, representing an interesting strategy to overcome sorafenib-acquired resistance in HCC.31 Similarly, the study by Tang et al74 described the pro-survival role of large tumor suppressor 1 (LATS 1), a core kinase of the Hippo pathway, in HCC due to the inhibition of sorafenib-induced lethal autophagy. Specifically, LATS1 exerts a scaffolding function that stabilizes Beclin-1 through ubiquitination of its lysine residues leading to the formation of inactive dimers negatively regulating the autophagic flux in HCC cells. LATS1 is upregulated in SR Huh7 cells as well as in non-responder patients after sorafenib treatment, suggesting LATS1 as a possible biomarker for patients’ stratification to targeted therapy. In addition, LATS1 but not LATS2 is upregulated in HCC patients where its high expression correlates with poor overall and disease-free survival. Notably, LATS1 silencing in vivo does not alter tumor growth but significantly increases sorafenib antitumor activity in xenograft mice. Interestingly, LATS1 silencing exerts a similar effect on autophagy-mediated cell death in cabozantinib- and sunitinib-treated cells displaying a higher autophagic flux and a contemporaneous degradation of autophagy-adaptor protein p62/SQSTM1, suggesting LATS1-mediated autophagy inhibition as a general mechanism with therapeutic implications for the targeted treatment of HCC. This aspect acquires indeed a particular interest for HCC where several first and second-line targeted treatments granted approval in the last years in the absence of biomarkers for patients’ allocation. Consistently, several oncogenic miRNAs (miR-21, miR-153, miR-216a, miR-217, miR-494 and miR-10a-5p) targeting the principal inhibitor of the Akt pathway PTEN are upregulated in HepG2 and Huh7 SR cells leading to Akt signaling activation and consequent autophagy inhibition. Since miRNA regulatory networks are very complex, and redundant or compensatory events take part to the regulation of a single pathway, the simultaneous targeting of multiple miRNAs represents an interesting option as an anticancer strategy. This is the case of an artificial long non-coding RNA (AlncRNA) based adenoviral vector (Ad5-AlncRNA) containing antisense sequences able to specifically bind and sequester the above-mentioned sorafenib-regulated miRNAs. Ad5-AlncRNA infection reduces cell viability and promotes apoptosis in the presence of enhanced PTEN expression and autophagic rate as evidenced by the appearance of LC3B “puncta” in the cytoplasm of SR cells following sorafenib treatment.75 This study confirms the shutting down of Akt axis as a critical mechanism to prevent acquired resistance in HCC, so as to highlight the importance of oncogenic pathways that fall outside from direct sorafenib targets but that acquire an equal relevance in the multidrug-resistant (MDR) phenotype.
On the contrary, other studies suggest autophagy as a mechanism of drug-resistance, postulating its inhibition as a possible intervention to overcome sorafenib resistance in HCC. Importantly, sorafenib induces both autophagy and cell death in a mechanism involving endoplasmic reticulum (ER) stress with the upregulation of IRE-1 signal. Specifically, the disturbance of basal ER processes triggers an evolutionary-conserved cell stress response also known as the “unfolded protein response” (UPR) that restores ER homeostasis in case of a small damage or unleash apoptosis due to persistent stressful events. IRE-1 is a sensor of the UPR and its activation following sorafenib treatment reduces apoptosis via autophagy induction compensating for cellular ER stress loading. In agreement, combination of sorafenib with the autophagy inhibitor chloroquine (CLQ), blocking autophagosome–lysosome fusion, increases sorafenib effectiveness in preclinical models, suggesting autophagy targeting as a therapeutic strategy worthy of consideration.76 Of course, molecules with reduced toxicity with respect to CLQ should be investigated for a possible translational applicability in HCC. Lin et al demonstrated the influence of methyltransferase-like 3 (METTL3)/FOXO3 axis in sorafenib resistance via autophagy regulation in HCC. METTL3 is a key enzyme responsible for N6-methyladenosine (m6A) modification of target mRNAs regulating their stability and translation. METTL3 is downregulated in HCC patients developing sorafenib resistance following long-term treatment and its deregulation promotes the activation of angiogenesis-associated genes and autophagy. METTL3 silencing lead to decreased m6A modifications of FOXO3 mRNA reducing its expression in SR HCC cells. In turn, FOXO3 knockdown activates the transcription of autophagy-related genes, including ATG3, ATG5, ATG7, ATG12, ATG16L1, and MAP1LC3B, increasing autophagic activity in different SR cell lines. In vivo experiments assessed that FOXO3 overexpression in METTL3-knockdown xenografts rescues sorafenib sensitivity showing a significant reduction of tumor size, which is reflected at a cellular level by a decreased proliferation, autophagy and angiogenesis.77 MiR-541 represents another tile in the molecular regulation of autophagy in the context of sorafenib resistance in HCC. In particular, miR-541 is downregulated in HCC tissues and its low levels correlate with an aggressive phenotype, decreased recurrence-free survival and shorter overall survival. Remarkably, miR-541 belongs to the DLK1/DIO3 miRNA cluster, which plays a relevant role in HCC.78–80 At a molecular level, the interference with autophagy was demonstrated as acontribution of miR-541 to sorafenib resistance. In particular, miR-541 inhibits HCC proliferation, tumor growth and metastasis and enhances sensitivity to sorafenib by the direct targeting of two autophagy-associated genes, namely autophagy-related gene 2A (ATG2A) and Ras-related protein Rab-1B (RAB1B) oncogene. Notably, higher miR-541 tissue levels predict a better response to sorafenib treatment. A combination strategy with miR-541 potentiates sorafenib antitumor activity in both in vitro and in vivo models. The authors suggested miR-541 as a promising sorafenib sensitizer in HCC treatment due to autophagy suppression and proposed manipulation of miR-541/ATG2A/RAB1B axis as a strategy aimed to prevent tumor escape, especially in miR-541 low-expressing HCCs.81
Another interesting study reported the upregulation of Annexin A3 (ANXA3) in SR cells and patient-derived xenografts (PDX), and demonstrated that an in vivo strategy combining an anti-ANXA3 monoclonal antibody (mAb) with sorafenib/regorafenib treatment impaired tumor growth and increased survival. Mechanistically, ANXA3 overexpression in SR cells blocks protein kinase C (PKCδ)/p38-mediated apoptosis and activates autophagy, which represents a prosurvival event. In HCC tissues, ANXA3 is overexpressed and correlates with stemness features and autophagic marker LC3B and associates with poor survival in patients receiving sorafenib. Therefore, ANXA3 represents not only a promising biomarker to predict clinical response to sorafenib but also a candidate for the development of combinatorial trials testing anti-ANXA3 mAb with sorafenib or regorafenib. Regarding a possible clinical translation, these Authors reported consistent evidences in several preclinical models, ranging from SR cells, liver organoids, xenograft and orthotopically implanted PDXs to sorafenib resistant HCC mice, where anti-ANXA3 mAb alone or in combination were tested displaying not only a safety profile but also a strong antitumor effect via apoptosis activation and autophagy blockade.82 In summary, here we reported some examples (Table 1) illustrating how autophagy activation in response to therapy is context-dependent and we highlighted the importance to unravel mechanistic aspects regulating autophagy in HCC showing its targeting as a possible intervention to optimize therapeutic strategies. This field of investigation is gaining more and more relevance even though a translational application appears still far to be envisaged.
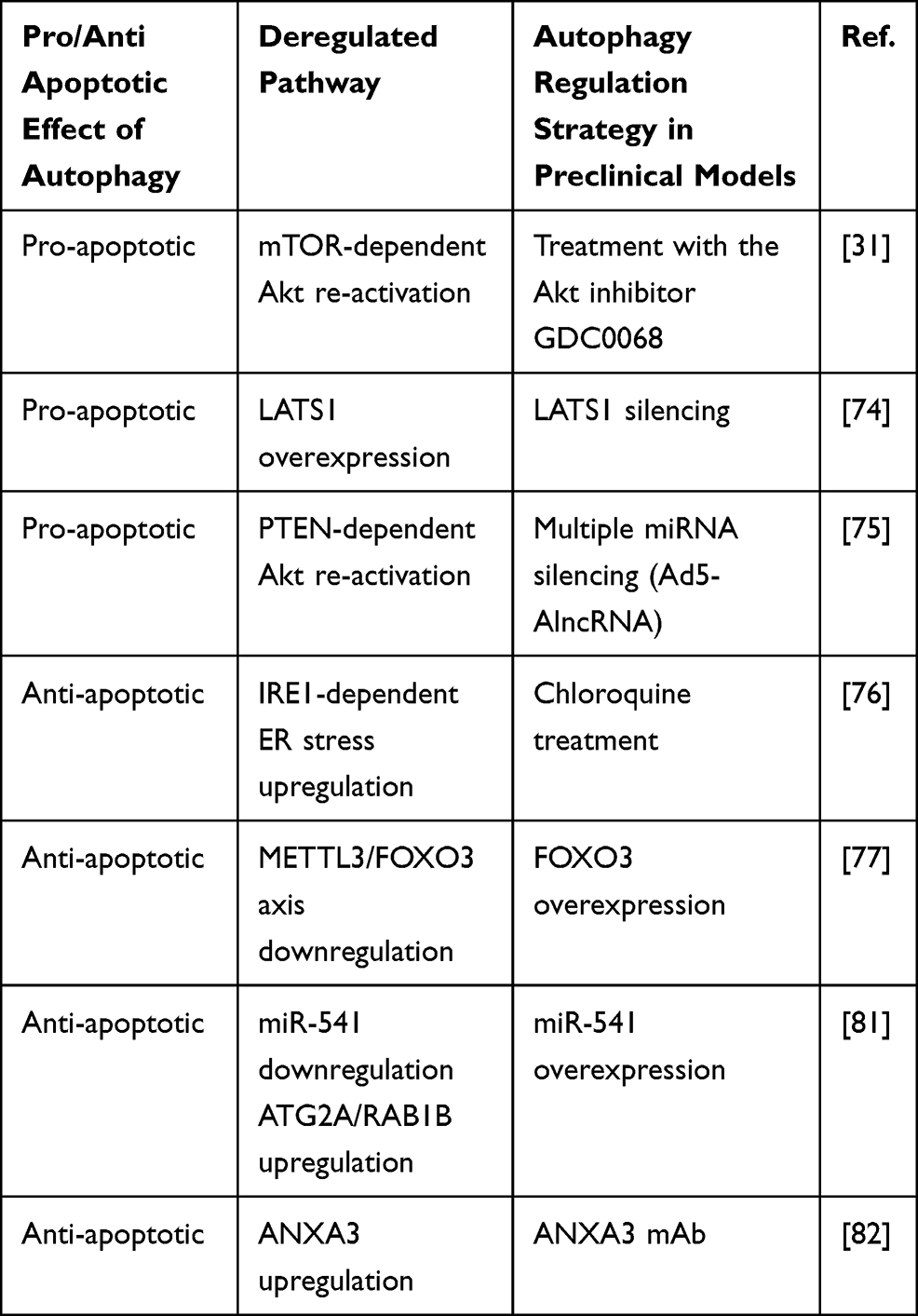 |
Table 1 Dual Role of Autophagy in Sorafenib Treatment |
Hypoxia and Sorafenib Resistance in HCC
The antitumor effect of the multi-TKI sorafenib is mediated by both autologous and non-autologous mechanisms. Specifically, VEGFR2 and PDGFβ are among sorafenib direct targets and important mediators of endothelial cell growth. Thus, several studies focused on the antitumor effect of sorafenib by investigating its interference with tumor angiogenesis impairing tumor vasculature and oxygen supply.83 The presence of a hypoxic microenvironment is a peculiar feature of many tumors, especially large-sized ones, promoting the activation of hypoxia-inducible factor 1 alpha (HIF-1α) and 2 alpha (HIF-2α) in cancer cells, which in turn regulate the expression of several proangiogenic factors to counteract the drug effectiveness.84 Indeed, the ubiquitous expressed HIF-1α and the cell specific expressed HIF-2α are well-known master regulators of cellular adaptation to hypoxic conditions and their role in tumor progression and therapy resistance is widely documented.85 Both HIF-1α and HIF-2α separately bind to HIF-1β (ARNT) to form a heterodimer (HIFs) that specifically recognizes consensus sequences in proximity of gene promoters termed as “hypoxia-response elements” (HREs) promoting transcriptional activation. Interestingly, HIFs expression is barely detectable in normoxic conditions, whereas its stability hugely increases in hypoxic contexts triggering the transcription of a vast repertoire of genes involved in angiogenesis, cell metabolism, apoptosis and metastasis. Oxygen-sensitive HIF-1α and HIF-2α subunits are both overexpressed in human HCCs but, despite a 48% sharing of gene homology, their activities are quite different and mediated by regulation of distinct targets, interaction with different molecules and activation at specific time points.86 Specifically, acute hypoxia mainly activates HIF-1α, whereas chronic hypoxia triggers HIF-2α, the latter regulating angiogenetic molecules in the liver and representing therefore an interesting candidate in HCC. Interestingly, compensatory mechanisms seem to take part to the mutual regulation of the two isoforms possibly contributing to sorafenib resistance. Indeed, sorafenib treatment was reported to block HIF-1α protein synthesis by downregulating AKT/mTOR and Raf/MEK/ERK pathways contributing to reduce tumor angiogenesis in vivo due to the inhibition of proangiogenic factor VEGF.87 On the other side, the auto-compensatory upregulation of HIF-2α following sorafenib-mediated HIF-1α inhibition was reported as a critical molecular event mediating sorafenib resistance in hypoxia-cultured cells. Specifically, increased HIF-2α protein levels trigger TGF-α transcriptional regulation activating EGFR phosphorylation and downstream signaling cascade concurring to sorafenib resistance. As a proof of principle, HIF-2α silencing or EGFR inhibition by gefitinib restores sorafenib sensitivity in hypoxic HepG2 and Huh7 cells and combination of sorafenib plus HIF-2α small interfering RNAs (siRNAs) synergistically reduces tumor growth in xenograft mice.88 Similarly, COX2/prostaglandin E2 (PGE2) axis promotes HIF-2α stabilization and nuclear translocation in hypoxic conditions via Von Hippel–Lindau protein (p-VHL) degradation and MAPK activation, respectively. Nuclear HIF carries out its prosurvival transcriptional program by activating genes such as VEGF, cyclin D1 and TGFα/EGFR influencing sorafenib resistance of hypoxic cells. High COX2/HIF-2α levels correlate with a worse overall and disease-free survival in HCC patients. In addition, COX2 selective inhibition synergistically enhances sorafenib efficacy in xenograft mice suggesting its therapeutic potential in combined strategies.89
Interestingly, Liang and coworkers proposed the induction of the hypoxia-based program due to antiangiogenic treatment as a cytoprotective adaptive response of cancer cells aiding the selection of aggressive neoplastic clones able to survive in a low-oxygen microenvironment. An increase of hypoxia is indeed observed in sorafenib-resistant tumors with respect to sensitive HCCs showing an upregulation of the P-glycoprotein (P-gp), encoded by the multidrug transporter (MDR) gene ABCB1, as well as a metabolic shift to glycolysis and the activation of the Nuclear Factor kappa B (NF-κB) signaling. EF24, a molecule with structural similarity to curcumin, was proved to overcome sorafenib resistance by sequestering HIF-1α into the cytoplasm favoring its degradation by p-VHL protein, which is part of the E3-ubiquitin ligase complex mediating protein ubiquitination and proteosomal degradation. Combinatorial treatment displayed a synergic effect in vivo hindering tumor growth and lung metastasis in immunocompromised mouse models.90 MiRNAs can also mediate sorafenib response by HIF-1α direct targeting, as in the case of the tumor suppressor miR-338-3p, which expression is downregulated not only in HCC tissues and cell lines but also in our well-characterized DEN-HCC rat model (ArrayExpress repository: accession number E-MTAB-7624) fully resembling the human pathology.91 In particular, miR-338-3p overexpression decreases HIF-1α levels and transcriptional potential, as evidenced by GLUT-1, P-gp and VEGF downregulation, compromising cell viability and potentiating sorafenib antitumor efficacy in HCC preclinical models. Notably, xenograft mice treated with miR-338-3p and sorafenib displayed smaller tumors than mice treated by sorafenib alone.
Metabolic switch to aerobic glycolysis or Warburg effect is a crucial event in solid tumors accounting for cancer cells survival in critical oxygen and nutrient supply. The glycolytic activator 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase 3 (PFKFB3) is a well-known downstream target of HIF-1α. It is overexpressed in HCC where it regulates glycolysis and blood vessel formation enhancing tumor invasion and metastasis. PFKFB3 is also upregulated by sorafenib treatment in HCC cells where it mediates HIF-1α overexpression establishing a positive feedback loop that sustains sorafenib resistance.92 In contrast with the above-mentioned role of FOXO3 in autophagy regulation, Liang and coworkers described FOXO3a-mediated autophagy induction as a hypoxia-driven mechanism involved in long-term sorafenib resistance. FOXO3a transcriptionally activates Beclin-1 enhancing autophagosome enucleation and increasing the overall autophagic flux and its knockdown in vivo increased sorafenib efficacy.93 These findings raise controversial debates on the FOXO3 role in autophagy control under hypoxic conditions underlining once again the multifaceted aspects and effects of autophagy in cancer progression and sorafenib response. In summary, here we show the induction of hypoxia following sorafenib treatment as a double-edged sword that rather than prompting its antiangiogenic effect is especially implicated in long-term acquired resistance and tumor escape giving cancer cells a pro-survival advantage in low oxygen conditions (Figure 3). In this scenario, targeting hypoxia-mediators might represent a promising combinatorial strategy in HCC patients undergoing sorafenib treatment.
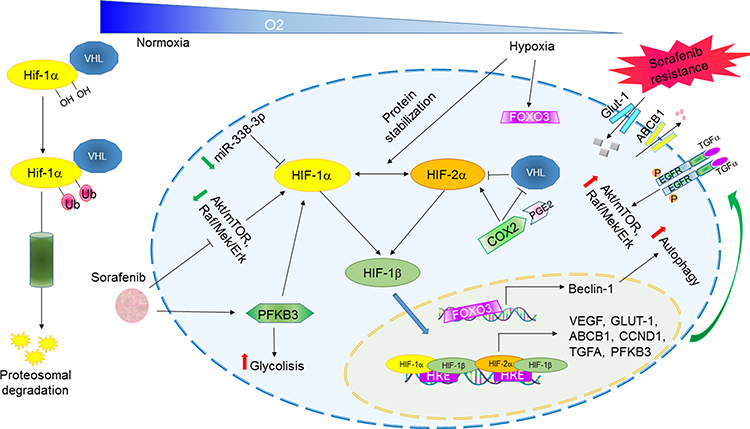 |
Figure 3 Hypoxia-regulated pathways involved in sorafenib resistance in HCC. Abbreviations: HRE, hypoxia-response element; Ub, ubiquitin. Note: Schematic representation of HIF-1α/HIF-1β activation in hypoxic conditions and downstream-mediated effects leading to sorafenib resistance. |
Combinational Therapies and Future Perspectives
Here, we have shown that several pathways and cellular processes take part to sorafenib resistance favoring tumor escape. Indeed, despite its clinical benefit in a proportion of HCCs, drug resistance and disease progression eventually occur during sorafenib treatment in the vast majority of patients. Given the central role of sorafenib in HCC therapy and its disappointing clinical outcome owing to drug resistance, the development of combined therapies is of utmost importance for the management of HCC. Indeed, the targeting of multiple intracellular signaling pathways might overcome single treatment failure. For instance, the compensatory activation of the PI3K/Akt/mTOR pathway in acquired resistance to sorafenib affects its antitumor activity and its inhibition is a promising autophagy-mediated strategy.31 Similarly, Ye et al described the synergic effect of the PI3K inhibitor copanlisib in combination with sorafenib in in vitro models of HCC. Copanlisib (BAY 80–6946), which received accelerated approval following a phase-II study (NCT01660451; part B) for relapsing follicular lymphoma, counteracted sorafenib-induced activation of the AKT signaling and potentiated its antineoplastic effect by causing cell cycle arrest and caspase-mediated apoptotic cell death in different SR cell lines.94 As above mentioned, sorafenib is an activator of autophagy-induced drug resistance. Sorafenib increases the number and stability of lysosomes in HCC cells and PDX models playing a critical role in drug sequestration and chemoresistance. Indeed, several chemotherapeutics accumulate within lysosomal lumen due to simple diffusion and, once inside they become protonated and compartmentalize into the lysosomal fraction. Sorafenib shows a pronounced lysosomotropism which concurs to impair its cellular availability and targets inhibition. Verteporfin is an FDA-approved photosensitizer that mediates lysosomes dysfunction by inducing pH alkalization, membrane permeability and tumor-selective proteotoxicity that is at the basis of sorafenib sensitization. Verteporfin exerts antitumor properties through autophagosome inhibition and interference with the autophagic flux progression blocking catabolic processes that mediate sorafenib adaptive response. Notably, verteporfin cytotoxic effect is specific to HCC cells whereas no sign of cell viability loss is observed in primary hepatocytes, possibly accounting for a safe use of this drug in advanced HCC patients whose liver function may be compromised.95 Strikingly, compensatory activation of growth factor-mediated signaling pathways contribute to sorafenib resistance but their single inhibition is often not sufficient to restore sorafenib sensitization due the presence of contemporaneous molecular alterations in drug-resistant cells. Ardelt and coworkers demonstrated by a multi-methodological approach that CDK5 inhibition perturbs the intracellular trafficking impairing cellular homeostasis through accumulation of enlarged vesicles that globally alters growth factor receptor signaling. This approach was more effective than single receptor inhibition. Notably, CDK5 is frequently overexpressed in HCCs and a clinically tested CDK5 small inhibitor, Dinaciclib, is currently available for CDK5 interference. Therefore, combination of sorafenib and Dinaciclib requires further evaluation to ameliorate the treatment of advanced HCC patients representing a promising approach for a clinical study.96 In conclusion, here we reported some examples of preclinical studies investigating the combination between small inhibitor molecules and sorafenib (Table 2), providing the molecular rationale for future evaluation in HCC. The understanding of molecular mechanisms is a fundamental step towards planning of efficient and safe drug combinations in selected patient subgroups. So far, the vast majority of data derive from laboratory studies, needing validation. Indeed, data obtained in animal models can be inconsistent because of heterogeneity of the human disease. However, they represent the necessary preliminary step to improve the management of advanced HCC.
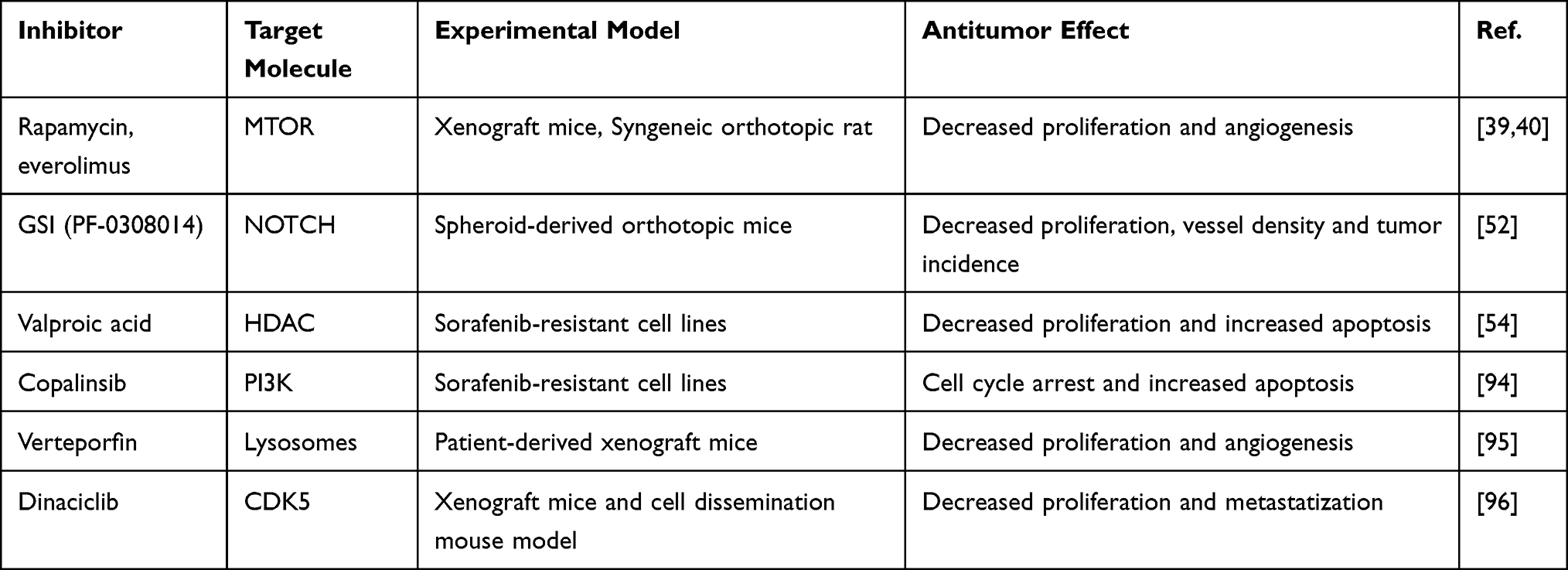 |
Table 2 Sorafenib Combined Treatments in Preclinical Models |
Remarkably, besides the molecular reasons, we should always consider the peculiar clinical features of HCC patients. HCC arises on a background of cirrhosis in more than 80% of cases, with associated liver functional impairment that may also alter drugs kinetics triggering side effects. However, effective modalities to selectively deliver drugs inside the tumor nodule are consolidated tools for HCC treatment and might be used for molecularly targeted treatments as they actually are for chemotherapeutical drugs and radioisotopes.
Another relevant issue is the personalization of treatments based on molecular biomarkers to be used in either the first and second/third lines. Unfortunately, the validated molecular classifications of HCC have never entered the clinical practice so far. In the last years, a breakthrough has been experienced in terms of availability of novel therapeutic agents (TKIs and immune checkpoint inhibitors) for advanced-stage HCCs. In this scenario, the identification of biomarkers of treatment response represents an urgent clinical need in HCC, favoring patient allocation to the best treatment. In this regard, we showed that miRNAs might represent ideal candidates for the selection of sorafenib-responder patients68 or for the identification of early tumor escape64 aiding subgroups stratification as well as a prompt shift to second-line treatments. Despite their promising characteristics and easy technical assessment, circulating miRNAs have not yet routinely entered in the diagnostics because of lack of consistent validation studies due to methodological issues (eg internal control) and different control groups (eg healthy volunteers versus cirrhotic patients). Therefore, several efforts are still needed to translate both preclinical and preliminry clinica data into the clinical practice leading to the identification of multi-biomarker classifiers taking into account disease complexity. Finally, the understanding of sorafenib resistance mechanisms will have a great significance for the development of new therapeutic targets to be evaluated as combined or sequential therapeutic regimens in well-defined molecular subclasses of patients preventing or delaying the onset of acquired resistance phenotypes.
Funding
This work was supported by funds from the Italian Association for Cancer Research (AIRC IG-25187, “Identification of circulating biomarkers for patient allocation to the best treatment in hepatocellular carcinoma”) to FF and Programma di Ricerca Regione-Università, Regione Emilia Romagna, Bando “Ricerca Innovativa” (Innovative approaches to the diagnosis and pharmacogenetics-based therapies of primary hepatic tumors, peripheral B and T-cell lymphomas and lymphoblastic leukaemias) to LG.
Disclosure
Fabio Piscaglia reports a research contract with Esaote (Genova, Italy); personal fees from AstraZeneca and Roche; advisory board for Alkermes; speaker bureau for Bayer, Bracco, EISAI, IPSEN and MSD; speaker bureau and advisory board for GE and Tiziana Life Sciences; consultant for BMS; and speaker bureau for Samsung. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi:10.3322/caac.21332
2. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
3. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi:10.1158/0008-5472.CAN-04-1443
4. Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs. 2009;69(2):223–240. doi:10.2165/00003495-200969020-00006
5. Chen KF, Chen HL, Tai WT, et al. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther. 2011;337(1):155–161. doi:10.1124/jpet.110.175786
6. Xu Y, Huang J, Ma L, et al. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016;371(2):171–181. doi:10.1016/j.canlet.2015.11.034
7. Denduluri SK, Idowu O, Wang Z, et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015;2(1):13–25. doi:10.1016/j.gendis.2014.10.004
8. Edginton AN, Zimmerman EI, Vasilyeva A, Baker SD, Panetta JC. Sorafenib metabolism, transport, and enterohepatic recycling: physiologically based modeling and simulation in mice. Cancer Chemother Pharmacol. 2016;77(5):1039–1052. doi:10.1007/s00280-016-3018-6
9. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. New Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
10. Zhai B, Sun XY. Mechanisms of resistance to sorafenib and the corresponding strategies in hepatocellular carcinoma. World J Hepatol. 2013;5(7):345–352. doi:10.4254/wjh.v5.i7.345
11. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–1173. doi:10.1016/S0140-6736(18)30207-1
12. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. doi:10.1016/S0140-6736(16)32453-9
13. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, Phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi:10.1016/S0140-6736(17)31046-2
14. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. New Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
15. Cabral LKD, Tiribelli C, Sukowati CHC. Sorafenib Resistance in Hepatocellular Carcinoma: the Relevance of Genetic Heterogeneity. Cancers. 2020;12:6. doi:10.3390/cancers12061576
16. Wong CM, Tsang FH, Ng IO. Non-coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol. 2018;15(3):137–151. doi:10.1038/nrgastro.2017.169
17. Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. doi:10.1126/science.1138140
18. Wei L, Lee D, Law CT, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun. 2019;10(1):4681. doi:10.1038/s41467-019-12606-7
19. Zheng A, Chevalier N, Calderoni M, et al. CRISPR/Cas9 genome-wide screening identifies KEAP1 as a sorafenib, lenvatinib, and regorafenib sensitivity gene in hepatocellular carcinoma. Oncotarget. 2019;10(66):7058–7070. doi:10.18632/oncotarget.27361
20. Rudalska R, Dauch D, Longerich T, et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat Med. 2014;20(10):1138–1146. doi:10.1038/nm.3679
21. Zheng JF, He S, Zeng Z, Gu X, Cai L, Qi G. PMPCB silencing sensitizes HCC tumor cells to sorafenib therapy. Mol Ther. 2019;27(10):1784–1795. doi:10.1016/j.ymthe.2019.06.014
22. Tovar V, Cornella H, Moeini A, et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut. 2017;66(3):530–540. doi:10.1136/gutjnl-2015-309501
23. Visvader JE. Cells of origin in cancer. Nature. 2011;469(7330):314–322. doi:10.1038/nature09781
24. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi:10.1038/nrdp.2016.18
25. Deng L, Sun J, Chen X, Liu L, Wu D. Nek2 augments sorafenib resistance by regulating the ubiquitination and localization of beta-catenin in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):316. doi:10.1186/s13046-019-1311-z
26. Marin JJ, Romero MR, Briz O. Molecular bases of liver cancer refractoriness to pharmacological treatment. Curr Med Chem. 2010;17(8):709–740. doi:10.2174/092986710790514462
27. Giovannini C, Baglioni M, Baron Toaldo M, et al. Notch3 inhibition enhances sorafenib cytotoxic efficacy by promoting GSK3b phosphorylation and p21 down-regulation in hepatocellular carcinoma. Oncotarget. 2013;4(10):1618–1631. doi:10.18632/oncotarget.1221
28. Xin H, Wang K, Hu G, et al. Establishment and characterization of 7 novel hepatocellular carcinoma cell lines from patient-derived tumor xenografts. PLoS One. 2014;9(1):e85308. doi:10.1371/journal.pone.0085308
29. Wang H, Xu L, Zhu X, Wang P, Chi H, Meng Z. Activation of phosphatidylinositol 3-kinase/Akt signaling mediates sorafenib-induced invasion and metastasis in hepatocellular carcinoma. Oncol Rep. 2014;32(4):1465–1472. doi:10.3892/or.2014.3352
30. Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13(3):234–244. doi:10.2174/1568009611313030002
31. Zhai B, Hu F, Jiang X, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014;13(6):1589–1598. doi:10.1158/1535-7163.MCT-13-1043
32. Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280(37):32081–32089. doi:10.1074/jbc.M502876200
33. Shanmugasundaram K, Block K, Nayak BK, Livi CB, Venkatachalam MA, Sudarshan S. PI3K regulation of the SKP-2/p27 axis through mTORC2. Oncogene. 2013;32(16):2027–2036. doi:10.1038/onc.2012.226
34. Tzivion G, Hay N. PI3K-AKT-FoxO axis in cancer and aging. Biochim Biophys Acta. 2011;1813(11):1925. doi:10.1016/j.bbamcr.2011.08.014
35. Masuda M, Chen WY, Miyanaga A, et al. Alternative mammalian target of rapamycin (mTOR) signal activation in sorafenib-resistant hepatocellular carcinoma cells revealed by array-based pathway profiling. Mol Cell Proteomics. 2014;13(6):1429–1438. doi:10.1074/mcp.M113.033845
36. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4(5):335–348. doi:10.1038/nrc1362
37. Villanueva A, Chiang DY, Newell P, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135(6):
38. Zhou L, Huang Y, Li J, Wang Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol. 2010;27(2):255–261. doi:10.1007/s12032-009-9201-4
39. Wang Z, Zhou J, Fan J, et al. Effect of rapamycin alone and in combination with sorafenib in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res. 2008;14(16):5124–5130.
40. Piguet AC, Saar B, Hlushchuk R, et al. Everolimus augments the effects of sorafenib in a syngeneic orthotopic model of hepatocellular carcinoma. Mol Cancer Ther. 2011;10(6):1007–1017. doi:10.1158/1535-7163.MCT-10-0666
41. Zhu AX, Kudo M, Assenat E, et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA. 2014;312(1):57–67. doi:10.1001/jama.2014.7189
42. Koeberle D, Dufour JF, Demeter G, et al. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): a randomized multicenter, multinational Phase II trial (SAKK 77/08 and SASL 29). Ann Oncol. 2016;27(5):856–861. doi:10.1093/annonc/mdw054
43. Lu X, Paliogiannis P, Calvisi DF, Chen X. Role of the Mammalian Target of Rapamycin Pathway in Liver Cancer: from Molecular Genetics to Targeted Therapies. Hepatology. 2021;73(Suppl 1):49–61. doi:10.1002/hep.31310
44. Chandarlapaty S, Sawai A, Scaltriti M, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. doi:10.1016/j.ccr.2010.10.031
45. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7(9):678–689. doi:10.1038/nrm2009
46. Henrique D, Schweisguth F. Mechanisms of Notch signaling: a simple logic deployed in time and space. Development. 2019;146:3. doi:10.1242/dev.172148
47. Sokolowski KM, Balamurugan M, Kunnimalaiyaan T, Gamblin C, Kunnimalaiyaan M. Notch signaling in hepatocellular carcinoma: molecular targeting in an advanced disease. Hepatoma Res. 2015;1:11–18. doi:10.4103/2394-5079.154132
48. Villanueva A, Alsinet C, Yanger K, et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143(6):1660–9 e7. doi:10.1053/j.gastro.2012.09.002
49. Gramantieri L, Giovannini C, Lanzi A, et al. Aberrant Notch3 and Notch4 expression in human hepatocellular carcinoma. Liver Int. 2007;27(7):997–1007. doi:10.1111/j.1478-3231.2007.01544.x
50. Jia H, Wang Z, Zhang J, Feng F. gamma-Secretase inhibitors for breast cancer and hepatocellular carcinoma: from mechanism to treatment. Life Sci. 2021;268:119007. doi:10.1016/j.lfs.2020.119007
51. Wu CX, Xu A, Zhang CC, et al. Notch inhibitor PF-03084014 inhibits hepatocellular carcinoma growth and metastasis via suppression of cancer stemness due to reduced activation of Notch1-Stat3. Mol Cancer Ther. 2017;16(8):1531–1543. doi:10.1158/1535-7163.MCT-17-0001
52. Yang X, Xia W, Chen L, et al. Synergistic antitumor effect of a gamma-secretase inhibitor PF-03084014 and sorafenib in hepatocellular carcinoma. Oncotarget. 2018;9(79):34996–35007. doi:10.18632/oncotarget.26209
53. Sun G, Mackey LV, Coy DH, Yu CY, Sun L. The histone deacetylase inhibitor vaproic acid induces cell growth arrest in hepatocellular carcinoma cells via suppressing notch signaling. J Cancer. 2015;6(10):996–1004. doi:10.7150/jca.12135
54. Yang X, Liu J, Liang Q, Sun G. Valproic acid reverses sorafenib resistance through inhibiting activated Notch/Akt signaling pathway in hepatocellular carcinoma. Fundam Clin Pharmacol. 2020. doi:10.1111/fcp.12608
55. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi:10.1016/S0092-8674(04)00045-5
56. Wei L, Wang X, Lv L, et al. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol Cancer. 2019;18(1):147. doi:10.1186/s12943-019-1086-z
57. Cheng J, Chen Y, Zhao P, et al. Downregulation of miRNA-638 promotes angiogenesis and growth of hepatocellular carcinoma by targeting VEGF. Oncotarget. 2016;7(21):30702–30711. doi:10.18632/oncotarget.8930
58. Li D, Wang T, Sun FF, et al. MicroRNA-375 represses tumor angiogenesis and reverses resistance to sorafenib in hepatocarcinoma. Cancer Gene Ther. 2021;28(1–2):126–140. doi:10.1038/s41417-020-0191-x
59. Gramantieri L, Ferracin M, Fornari F, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67(13):6092–6099. doi:10.1158/0008-5472.CAN-06-4607
60. Wang F, Bank T, Malnassy G, et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol Commun. 2018;2(6):732–746. doi:10.1002/hep4.1181
61. Ji J, Yamashita T, Budhu A, et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50(2):472–480. doi:10.1002/hep.22989
62. Azumi J, Tsubota T, Sakabe T, Shiota G. miR-181a induces sorafenib resistance of hepatocellular carcinoma cells through downregulation of RASSF1 expression. Cancer Sci. 2016;107(9):1256–1262. doi:10.1111/cas.13006
63. Borel F, Han R, Visser A, et al. Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology. 2012;55(3):821–832. doi:10.1002/hep.24682
64. Gramantieri L, Pollutri D, Gagliardi M, et al. MiR-30e-3p influences tumor phenotype through MDM2/TP53 axis and predicts sorafenib resistance in hepatocellular carcinoma. Cancer Res. 2020;80(8):1720–1734. doi:10.1158/0008-5472.CAN-19-0472
65. Chen Z, Zhang J, Zhang Z, et al. The putative tumor suppressor microRNA-30a-5p modulates clear cell renal cell carcinoma aggressiveness through repression of ZEB2. Cell Death Dis. 2017;8(6):e2859. doi:10.1038/cddis.2017.252
66. Kumarswamy R, Mudduluru G, Ceppi P, et al. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012;130(9):2044–2053. doi:10.1002/ijc.26218
67. Zhang Q, Yu L, Qin D, et al. Role of microRNA-30c targeting ADAM19 in colorectal cancer. PLoS One. 2015;10(3):e0120698. doi:10.1371/journal.pone.0120698
68. Fornari F, Pollutri D, Patrizi C, et al. In Hepatocellular Carcinoma miR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3-Mediated Apoptosis. Clin Cancer Res. 2017;23(14):3953–3965. doi:10.1158/1078-0432.CCR-16-1464
69. Zhang Z, Tan X, Luo J, Yao H, Si Z, Tong JS. The miR-30a-5p/CLCF1 axis regulates sorafenib resistance and aerobic glycolysis in hepatocellular carcinoma. Cell Death Dis. 2020;11(10):902. doi:10.1038/s41419-020-03123-3
70. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
71. Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14(4):376–391. doi:10.1007/s10495-008-0307-5
72. Tai WT, Shiau CW, Chen HL, et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013;4:e485. doi:10.1038/cddis.2013.18
73. Finn RS, Poon RT, Yau T, et al. Phase I study investigating everolimus combined with sorafenib in patients with advanced hepatocellular carcinoma. J Hepatol. 2013;59(6):1271–1277. doi:10.1016/j.jhep.2013.07.029
74. Tang F, Gao R, Jeevan-Raj B, et al. LATS1 but not LATS2 represses autophagy by a kinase-independent scaffold function. Nat Commun. 2019;10(1):5755. doi:10.1038/s41467-019-13591-7
75. Tang S, Tan G, Jiang X, et al. An artificial lncRNA targeting multiple miRNAs overcomes sorafenib resistance in hepatocellular carcinoma cells. Oncotarget. 2016;7(45):73257–73269. doi:10.18632/oncotarget.12304
76. Shi YH, Ding ZB, Zhou J, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011;7(10):1159–1172. doi:10.4161/auto.7.10.16818
77. Lin Z, Niu Y, Wan A, et al. RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. THE EMBO J. 2020;39(12):e103181. doi:10.15252/embj.2019103181
78. Lim L, Balakrishnan A, Huskey N, et al. MicroRNA-494 within an oncogenic microRNA megacluster regulates G1/S transition in liver tumorigenesis through suppression of mutated in colorectal cancer. Hepatology. 2014;59(1):202–215. doi:10.1002/hep.26662
79. Luk JM, Burchard J, Zhang C, et al. DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J Biol Chem. 2011;286(35):30706–30713.
80. Pollutri D, Patrizi C, Marinelli S, et al. The epigenetically regulated miR-494 associates with stem-cell phenotype and induces sorafenib resistance in hepatocellular carcinoma. Cell Death Dis. 2018;9(1):4. doi:10.1038/s41419-017-0076-6
81. Xu WP, Liu JP, Feng JF, et al. miR-541 potentiates the response of human hepatocellular carcinoma to sorafenib treatment by inhibiting autophagy. Gut. 2020;69(7):1309–1321. doi:10.1136/gutjnl-2019-318830
82. Tong M, Che N, Zhou L, et al. Efficacy of annexin A3 blockade in sensitizing hepatocellular carcinoma to sorafenib and regorafenib. J Hepatol. 2018;69(4):826–839. doi:10.1016/j.jhep.2018.05.034
83. Chang YS, Adnane J, Trail PA, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chem Pharmacol. 2007;59(5):561–574. doi:10.1007/s00280-006-0393-4
84. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Nat Cancer Inst. 2007;99(19):1441–1454. doi:10.1093/jnci/djm135
85. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11(6):393–410. doi:10.1038/nrc3064
86. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. doi:10.1038/nrc3183
87. Liu LP, Ho RL, Chen GG, Lai PB. Sorafenib inhibits hypoxia-inducible factor-1alpha synthesis: implications for antiangiogenic activity in hepatocellular carcinoma. Clin Cancer Res. 2012;18(20):5662–5671. doi:10.1158/1078-0432.CCR-12-0552
88. Zhao D, Zhai B, He C, et al. Upregulation of HIF-2alpha induced by sorafenib contributes to the resistance by activating the TGF-alpha/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014;26(5):1030–1039. doi:10.1016/j.cellsig.2014.01.026
89. Dong XF, Liu TQ, Zhi XT, et al. COX-2/PGE2 Axis Regulates HIF2alpha Activity to Promote Hepatocellular Carcinoma Hypoxic Response and Reduce the Sensitivity of Sorafenib Treatment. Clin Cancer Res. 2018;24(13):3204–3216. doi:10.1158/1078-0432.CCR-17-2725
90. Liang Y, Zheng T, Song R, et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1alpha inhibition in hepatocellular carcinoma. Hepatology. 2013;57(5):1847–1857. doi:10.1002/hep.26224
91. Fornari F, Gramantieri L, Callegari E, et al. MicroRNAs in Animal Models of HCC. Cancers. 2019;11:12. doi:10.3390/cancers11121906
92. Long Q, Zou X, Song Y, Duan Z, Liu L. PFKFB3/HIF-1alpha feedback loop modulates sorafenib resistance in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2019;513(3):642–650. doi:10.1016/j.bbrc.2019.03.109
93. Liang C, Dong Z, Cai X, et al. Hypoxia induces sorafenib resistance mediated by autophagy via activating FOXO3a in hepatocellular carcinoma. Cell Death Dis. 2020;11(11):1017. doi:10.1038/s41419-020-03233-y
94. Ye L, Mayerle J, Ziesch A, Reiter FP, Gerbes AL, De Toni EN. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019;5:86. doi:10.1038/s41420-019-0165-7
95. Gavini J, Dommann N, Jakob MO, et al. Verteporfin-induced lysosomal compartment dysregulation potentiates the effect of sorafenib in hepatocellular carcinoma. Cell Death Dis. 2019;10(10):749. doi:10.1038/s41419-019-1989-z
96. Ardelt MA, Frohlich T, Martini E, et al. Inhibition of Cyclin-Dependent Kinase 5: a Strategy to Improve Sorafenib Response in Hepatocellular Carcinoma Therapy. Hepatology. 2019;69(1):376–393. doi:10.1002/hep.30190
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.