")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Elucidating the in vivo fate of nanocrystals using a physiologically based pharmacokinetic model: a case study with the anticancer agent SNX-2112
Authors Dong D, Wang X, Wang H, Zhang X, Wang Y, Wu B
Received 22 December 2014
Accepted for publication 16 February 2015
Published 31 March 2015 Volume 2015:10(1) Pages 2521—2535
DOI https://doi.org/10.2147/IJN.S79734
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Dong Dong,1* Xiao Wang,1* Huailing Wang,1 Xingwang Zhang,2 Yifei Wang,1 Baojian Wu2
1Guangzhou Jinan Biomedicine Research and Development Center, 2Division of Pharmaceutics, College of Pharmacy, Jinan University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Introduction: SNX-2112 is a promising anticancer agent but has poor solubility in both water and oil. In the study reported here, we aimed to develop a nanocrystal formulation for SNX-2112 and to determine the pharmacokinetic behaviors of the prepared nanocrystals.
Methods: Nanocrystals of SNX-2112 were prepared using the wet-media milling technique and characterized by particle size, differential scanning calorimetry, drug release, etc. Physiologically based pharmacokinetic (PBPK) modeling was undertaken to evaluate the drug’s disposition in rats following administration of drug cosolvent or nanocrystals.
Results: The optimized SNX-2112 nanocrystals (with poloxamer 188 as the stabilizer) were 203 nm in size with a zeta potential of -11.6 mV. In addition, the nanocrystals showed a comparable release profile to the control (drug cosolvent). Further, the rat PBPK model incorporating the parameters of particulate uptake (into the liver and spleen) and of in vivo drug release was well fitted to the experimental data following administration of the drug nanocrystals. The results reveal that the nanocrystals rapidly released drug molecules in vivo, accounting for their cosolvent-like pharmacokinetic behaviors. Due to particulate uptake, drug accumulation in the liver and spleen was significant at the initial time points (within 1 hour).
Conclusion: The nanocrystals should be a good choice for the systemic delivery of the poorly soluble drug SNX-2112. Also, our study contributes to an improved understanding of the in vivo fate of nanocrystals.
Keywords: intravenous delivery, PBPK, tissue distribution, poloxamer 188
Introduction
Combinatorial chemistry and high-throughput screening are advancing drug discovery. However, many lead compounds have been categorized as Biopharmaceutics Classification System classes II or IV, with poor solubility.1 The insoluble nature causes significant challenges in drug delivery. Nano-drug systems are well-established approaches to meeting the formulation challenges of insoluble drugs.2 These promising carriers include liposomes,3,4 micro-/nanoemulsions,5 micelles,6,7 solid lipid nanoparticles,8 polymeric nanoparticles,9 and nanocrystals.10 Of these, liposomes and lipid emulsions have been successfully developed into injectable products such as Doxil® and Diprivan®. However, other nanocarriers have seen a low rate of commercial success due to physical instability, poor reproducibility, and potential toxicity. For those drugs that are insoluble both in water and in oil (or lipids), low drug load and significant drug leakage are the main obstacles to developing injectable nanocarriers. In this regard, nanocrystals are advantageous because they can greatly enhance drug solubility. It is noteworthy that a nanocrystal product of paliperidone for injection has been approved by the European Medicines Agency11 and five oral products of drug nanocrystals have been approved by the US Food and Drug Administration.10
Nanocrystals (or nanosuspensions) are a sub-micron colloidal dispersion system of pure drug particles in water.12–14 Nanocrystal systems are often stabilized by surfactants or polymers.12–14 The advantages of nanocrystals include: (1) they offer a high drug load (because the drug is suspended in a solid state); (2) there is no significant toxicity concern because a limited quantity of excipient (stabilizer) is used; (3) they are suitable for a diverse range of administration routes including oral, parenteral, pulmonary, and ocular pathways;15–17 and (4) they show potential in passive targeting and provide a depot effect.18–20 Drug nanocrystals are usually prepared using two types of methods, namely, anti-solvent precipitation of dissolved drugs (bottom-up)21,22 and comminution of bulk drugs with high-pressure homogenization or media milling (top-down).23,24 Although bottom-up technology is a straight-forward procedure to obtain drug nanocrystals, it has several limitations, such as the use of harmful organic solvents25,26 and there being great difficulty in controlling particle size.27 Top-down technology is more applicable to industrial production due to the facile and energy-saving processes involved.28
SNX-2112 is a novel Hsp90 inhibitor currently in clinical evaluation for cancer treatment (delivered by its prodrug SNX-5422; http://www.clinicaltrials.gov). The anticancer action of the compound is through abrogation of signaling via human epidermal growth factor receptor 2 (HER2), protein kinase B (Akt), and extracellular-signal-regulated kinase (ERK).29,30 SNX-2112 is hardly bioavailable after oral administration due to rather limited solubility and possible P-gp efflux.31 The prodrug SNX-5422 has been designed with an improved solubility profile, providing acceptable systemic exposure of SNX-2112. An effective alternative to circumvent the low bioavailability of SNX-2112 is to develop its parenteral formulations. Due to poor solubility both in water (8 μg/mL) and oil (eg, 5 μg/mL in soybean oil), nanocrystals are an excellent candidate for the intravenous delivery of the drug.
Physiologically based pharmacokinetic (PBPK) modeling has gained increasing attention in drug research and development.32,33 A PBPK model quantitatively describes drug absorption, distribution, metabolism, and elimination in the body, facilitating a deep understanding of the effects of these intricate processes on drug exposure and of how these processes interact with each other.34,35 An important feature of an PBPK model is that it allows data extrapolation from a preclinical animal species to humans, thereby providing excellent guidance on clinical trials.36 Although PBPK modeling has become a powerful tool in identifying the factors determining drug pharmacokinetics, its ability to evaluate the effects of the formulation factor on drug disposition has not been fully established.37
In the study reported here, we aimed to explore the possibility and feasibility of nanocrystals for the intravenous delivery of SNX-2112, and to determine the in vivo fate of the drug nanocrystals. Drug nanocrystals were prepared using the wet-media milling technique and characterized by particle size, differential scanning calorimetry (DSC), in vitro release, etc. PBPK modeling was performed to evaluate the drug disposition in rats following administration of the nanocrystals using drug cosolvent as a reference. A rat PBPK model incorporating the parameters of particulate uptake (into the liver and spleen) and of the in vivo drug release was used to describe and explain the experimental data.
Materials and methods
Materials
SNX-2112 and AT-533 (used as an internal standard) were synthesized in our laboratory as described previously.38 Poloxamer 188 (P188) was obtained from BASF SE (Ludwigshafen, Germany). Polysorbate 80 (Tween® 80) was obtained from Aladdin Reagent Co Ltd (Shanghai, People’s Republic of China). All other chemicals were of analytical grade and used as received.
Preparation of SNX-2112 nanocrystals
Drug nanocrystals were prepared using a top-down wet-milling technique.39 In brief, the stabilizer (0.2, 2.0, or 20.0 mg) was dissolved in 2 mL water, into which 20 mg of drug powder was dispersed. The resulting suspensions were subjected to homogenization in a homemade medium mill containing 2 g milling pearls (zirconium oxide, diameter of 1.2 and 0.8 mm at a ratio of 1:1) stirred at 1,100 rpm. After 24 hours’ homogenization, the nanocrystal suspensions were separated from the system by filtration against a nylon sieve.
Characterization of SNX-2112 nanocrystals
The particle size and zeta potential of the drug nanocrystals were determined using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at 25°C based on the dynamic light scattering principle. The sample (0.1 mL) was diluted with deionized water to 1 mL and measured after equilibrium to the given temperature in the cell. To calculate particle size, the data were recorded by and analyzed with the built-in software.
The morphology of the drug nanocrystals was observed by transmission electron microscopy (TEM). In brief, an aliquot of nanocrystals was dropped on a carbon-coated copper grid and then anchored, drying at ambient atmosphere, to the supporter. The supporter with fixed nanoparticles was immersed into 1% phosphotungstic acid solution for 100 seconds. The negatively stained particles were allowed to dry at ambient atmosphere and photographed at an acceleration voltage of 100 kV with TEM (Tecnai 10, Philips, Amsterdam, the Netherlands).
DSC
The samples of lyophilized nanocrystals, stabilizer (P188), pure drug, and physical mixtures of about 5 mg were weighed into a non-hermetically sealed aluminum pan, and proceeded to differential calorimetric scanning on a DSC 204A/G Phoenix® instrument (Netzsch GmbH & Co KG, Selb, Germany). The samples were heated from 25°C to 350°C at a stepping rate of 10°C/minute. The instrument was calibrated using indium. All the DSC measurements were carried out in a nitrogen atmosphere at a flow rate of 100 mL/minute.
In vitro drug release study
The bulk-equilibrium reverse dialysis method was used for the release study of SNX-2112 nanocrystals.40 The release medium was a phosphate buffer (pH 7.4) with 0.2% Tween 80. After equilibrium with the bulk phase under magnetic agitation, 1 mL of nanocrystal (2 mg/mL) suspension was introduced and reversely dialyzed at 37°C against the dialysis bags. At predetermined intervals, the dialysis bags were withdrawn. SNX-2112 concentrations in the dialysate were determined and the percentage of drug release was calculated as mean ± standard deviation (n=3).
Pharmacokinetic study of SNX-2112
Pharmacokinetic study was performed with Sprague-Dawley rats (male; 193.0±8.9 g). The experimental procedures were approved by the Animal Ethics Committee of Jinan University (Guangzhou, China). The rats were randomly divided into two groups, the cosolvent (control) and nanocrystal groups. The control group of rats received SNX-2112 cosolvent (45% propylene glycerol) by intravenous injection (tail vein) at a dose of 2 mg/kg body weight, whereas the nanocrystal group received SNX-2112 nanocrystals intravenously at a same dose. At predetermined time points (10, 20, and 30 minutes; 1 hour; and 2, 4, 6, 8, 12, and 18 hours) after drug administration, the rats (n=5) were rendered unconscious for blood and tissue sampling by injecting phenobarbital. The blood was collected by cardiac puncture. After washout of blood with ice-cold saline, the heart, liver, spleen, lung, kidney, and intestine were rapidly removed, weighed, and stored at −80°C for further analysis.
Blood samples were processed as described previously.41 The concentrations of SNX-2112 in the samples were analyzed by the ultra-performance liquid chromatography quadrupole time-of flight mass spectrometry (UPLC-QTOF/MS) system. The heart, liver, spleen, lung, kidney, and intestine tissues were homogenized 1:2 (w/v) in saline solution. A 0.2 mL aliquot of tissue homogenate was mixed with 1 mL acetonitrile containing 2 μM internal standard (AT-533). The mixture was vortexed and centrifuged at 13,000× g for 15 minutes (4°C). The supernatant was collected and dried using a Concentrator plus (Eppendorf AG, Hamburg, Germany). The residue was reconstituted in 250 μL of water/methanol (50:50, v/v) and centrifuged at 13,000× g for 15 minutes (4°C). A 3 μL aliquot of the supernatant was injected into the UPLC-QTOF/MS system.
Quantification of SNX-2112 using UPLC-QTOF/MS
Quantification of SNX-2112 was performed using a Waters UPLC-QTOF/MS system consisting of an ACQUITY UPLC system and Xevo™ G2 QTof (Waters Corporation, Milford, MA, USA) as detailed in our previous publications.31,42 The analytical methods were rigorously validated with respect to linearity (4.88–5,000.00 nM), precision (>90%), and recovery (>80%).
Nonparticulate PBPK model
A nonparticulate PBPK model was constructed to describe disposition of nonparticulate SNX-2112 in rats. The whole-body PBPK model consisted of heart (ht), liver (li), spleen (sp), lung (lu), kidney (kd), and intestine (in) compartments. All other tissues were lumped into a remainder (rm) compartment (Figure 1A). Elimination of SNX-2112 primarily occurred in the liver,41 which was represented by the apparent hepatic clearance CLh. Drug concentrations in each tissue were determined by the tissue/plasma partition coefficient (Kp,ti), plasma flow (Qti), and organ volume (Vti). The Kp,ti values of SNX-2112 for the intestine, kidney, spleen, and heart were obtained experimentally from the tissue/plasma ratios toward the end of the study (close to 18 hours). Table 1 summarizes the plasma flow and organ volume values that were fixed during model fitting. Plasma cardiac output (Qco) for rats was set at 2.69 L/h.43 The mass balance equations (Equations 1–8) for the model follow. “C” denotes the concentration of SNX-2112 in each compartment. “V” denotes the volume in each compartment. The subscripts represent the compartment names.
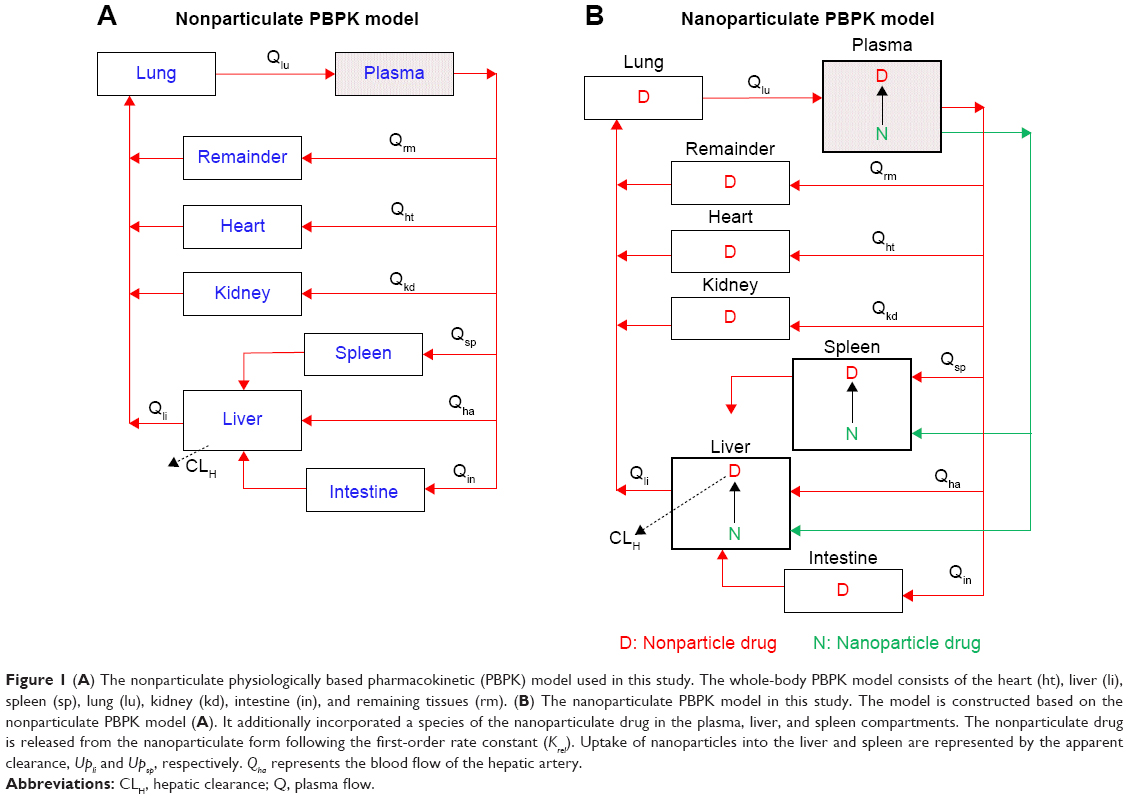 | Figure 1 (A) The nonparticulate physiologically based pharmacokinetic (PBPK) model used in this study. The whole-body PBPK model consists of the heart (ht), liver (li), spleen (sp), lung (lu), kidney (kd), intestine (in), and remaining tissues (rm). (B) The nanoparticulate PBPK model in this study. The model is constructed based on the nonparticulate PBPK model (A). It additionally incorporated a species of the nanoparticulate drug in the plasma, liver, and spleen compartments. The nonparticulate drug is released from the nanoparticulate form following the first-order rate constant (Krel). Uptake of nanoparticles into the liver and spleen are represented by the apparent clearance, Upli and Upsp, respectively. Qha represents the blood flow of the hepatic artery. |
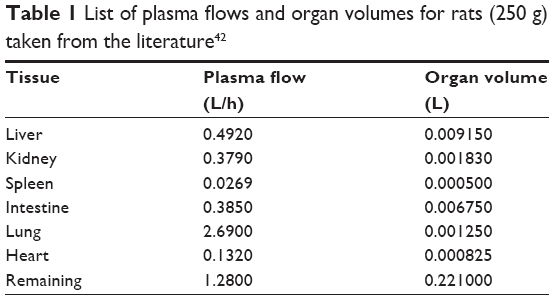 | Table 1 List of plasma flows and organ volumes for rats (250 g) taken from the literature42 |
|
|
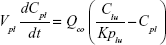
|
|
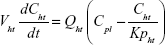
|
|
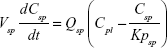
|
|
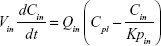
|
|
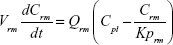
|
|
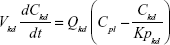
|
|
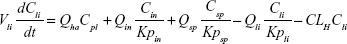
|
|
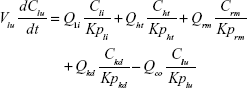
Nanoparticulate PBPK model
Following the administration of drug nanocrystals, the drug in the plasma (and tissues when uptake of nanoparticles occurred) had two forms, nonparticulate (D) and nanoparticulate (N) (Figure 1B). It was necessary to distinguish the two forms in pharmacokinetic modeling due to their differences in disposition processes, such as tissue partitioning. The nanoparticulate PBPK model was constructed based on the nonparticulate PBPK model (Figure 1A). The nanoparticulate model additionally incorporated a species of the nanoparticulate drug in the plasma, liver, and spleen compartments. The nonparticulate drug was released from the nanoparticulate form following the first-order rate constant (Krel). Uptake of nanoparticles into the liver and spleen is represented by the apparent clearance, Upli and Upsp, respectively. The mass balance equations (Equations 9–19) for the model follow. “C” and “CNano” denote the concentrations of the nonparticulate and particulate SNX-2112 in each compartment, respectively. “V” denotes the volume in each compartment. The subscripts represent the compartment names.
|
|
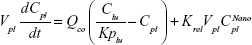
|
|
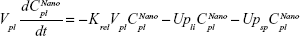
|
|
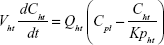
|
|
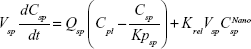
|
|
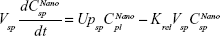
|
|
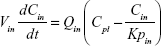
|
|
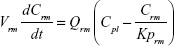
|
|
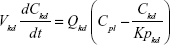
|
|
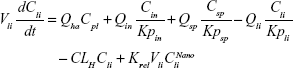
|
|
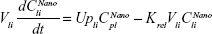
|
|
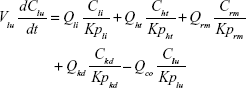
Data analysis
Modeling and simulations were performed with MATLAB® 8.0 software (The MathWorks, Inc, Natick, MA, USA). All parameters were estimated using a weighing scheme of 1/y2. Compartmental and non-compartmental analyses of plasma/tissue concentrations versus time were performed using Pharsight® Phoenix® WinNonlin® 6.3 software (Certara, LP, Princeton, NJ, USA). Data are expressed as mean ± standard deviation. The two-tailed Student’s t-test was used to compare the parameter difference between the cosolvent (control) and nanocrystal groups. The prior level of significance was set at 5% or P<0.05.
Results
Stabilizer selection for SNX-2112 nanocrystals
The stabilizer played an important role in determining the properties of the drug nanocrystals.24 We prepared SNX-2112 nanocrystals using different types of stabilizers including nonionic surfactants (Tween 80, P188, and P407), ionic surfactants (sodium dodecyl sulfate and sodium taurocholate hydrate), and polymers (hydroxypropyl methylcellulose and polyvinylpyrrolidone) (Figure 2). Also, the effects of the amount of stabilizer on the nanocrystals were evaluated (Figure 2). Clearly, the drug nanocrystals (with a particle <1 μm) can be formed by using any of tested stabilizers at a higher amount of 20 mg (Figure 2A). Although all other stabilizers showed potential in generating SNX-2112 nanocrystals, P188 was chosen as the optimal stabilizer for further evaluation because (1) the resulting nanocrystals showed acceptable particle size with narrow size distribution (Figure 2), and (2) P188 is a drug excipient for intravenous administration without safety concerns.44
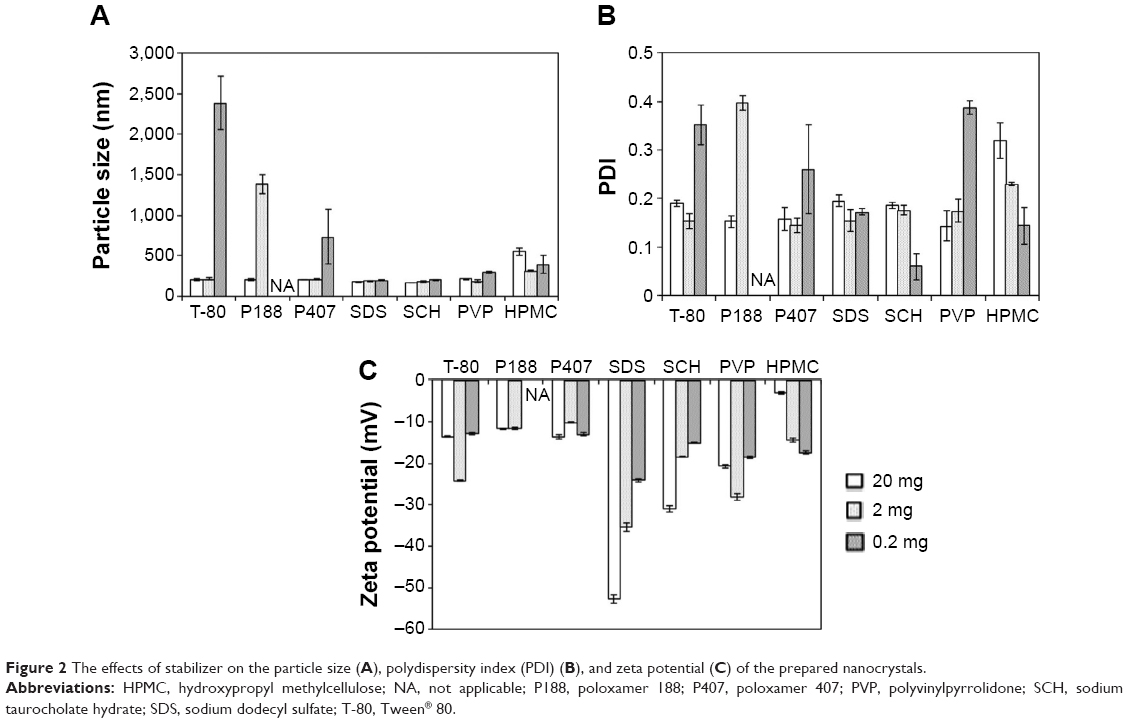 | Figure 2 The effects of stabilizer on the particle size (A), polydispersity index (PDI) (B), and zeta potential (C) of the prepared nanocrystals. |
Preparation and characterization of optimized SNX-2112 nanocrystals
The optimized SNX-2112 nanocrystals were prepared using P188 (20 mg) as the stabilizer. The obtained nanocrystals showed a particle size of 203 nm with a small polydispersity index value of 0.153 (Figure 3A). The zeta potential was determined to be −11.6 mV. This electric charge would help to stabilize the nanosuspensions, though the steric effects of P188 may play a major role in stabilization of the colloidal systems.45 In addition, SNX-2112 nanocrystals presented acicular architectures in their morphology as observed by TEM (Figure 3B), indicating that they were indeed nanocrystals rather than an amorphous substance. Preliminary stability studies also revealed that the prepared nanocrystals with P188 were stable for at least 1 month (Figure 4).
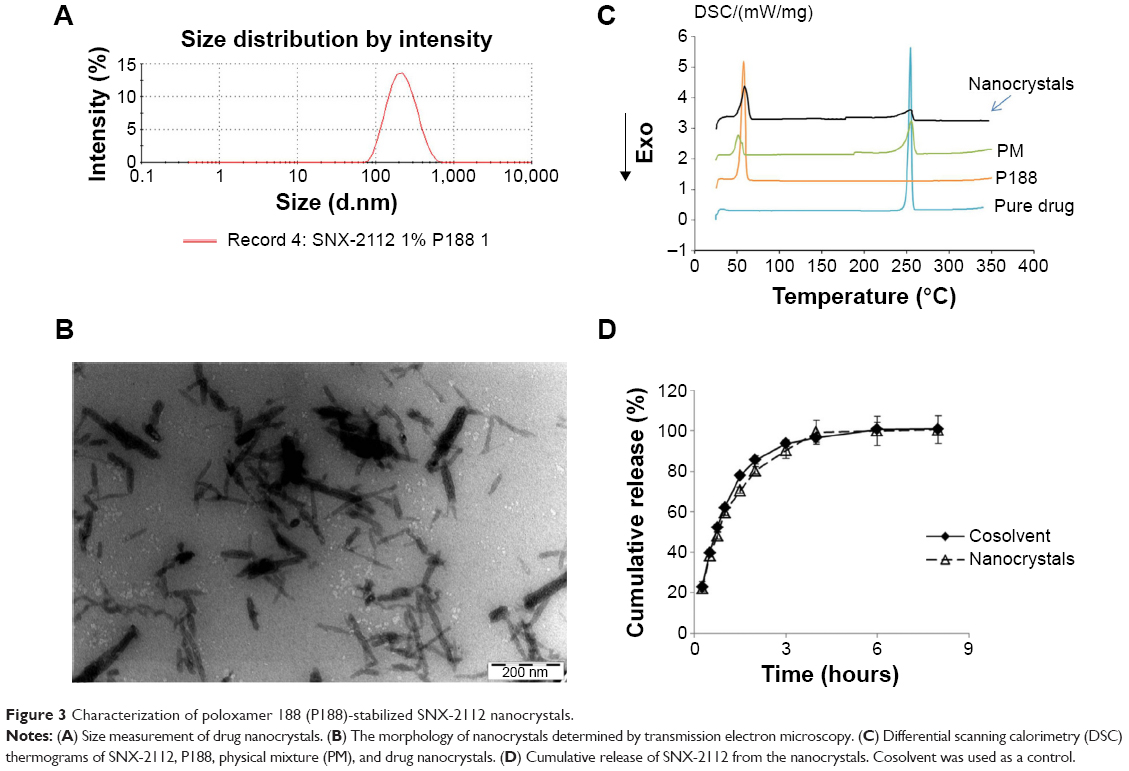 | Figure 3 Characterization of poloxamer 188 (P188)-stabilized SNX-2112 nanocrystals. |
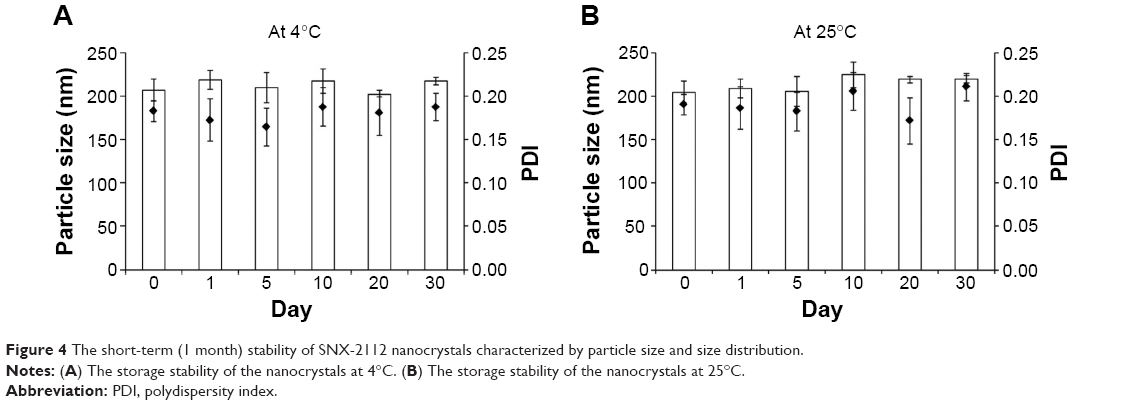 | Figure 4 The short-term (1 month) stability of SNX-2112 nanocrystals characterized by particle size and size distribution. |
DSC thermograms of the pure drug (SNX-2112), P188, physical mixture, and drug nanocrystals are shown in Figure 3C. The endothermic peaks at 57°C and 264°C corresponded to the melting points of P188 and SNX-2112, respectively (Figure 3C). The DSC profiles of the nanocrystals and physical mixture were superposition of those of individual SNX-2112 and P188. This lends strong support to the notion that the nanoscaled drug was crystalline. It has been reported in the literature that drug nanocrystals are microcrystalline rather than amorphous.46,47 The in vitro drug release profile of the nanocrystals was comparable to that of the control (drug cosolvent) (Figure 3D). The f2 value (similarity factor) between the two preparations was calculated to be 85. The release data indicate that release of the drug from the nanocrystals was rather rapid.
Pharmacokinetic and biodistribution profiles
The plasma concentration–time profiles displayed two distinct phases following intravenous administration of either the cosolvent (control) or nanocrystals (Figure 5A). These two phases appear to be the distribution (10 minutes–2 hours) and elimination (4–18 hours) phases. It was also noted that there were no significant differences in plasma concentrations (at the time points after 10 minutes) between the nanocrystal and cosolvent groups (P>0.05; Figure 5A). The data were analyzed using the compartmental approach and found to be well described by the two-compartment model (Figure 5A and Table 2). The derived parameters of the area under the concentration–time curve (AUC), elimination half-life (t1/2β), and systemic clearance (CL) were comparable between the nanocrystal and cosolvent groups (P>0.05; Table 2). This was consistent with the high similarity in the pharmacokinetic profile.
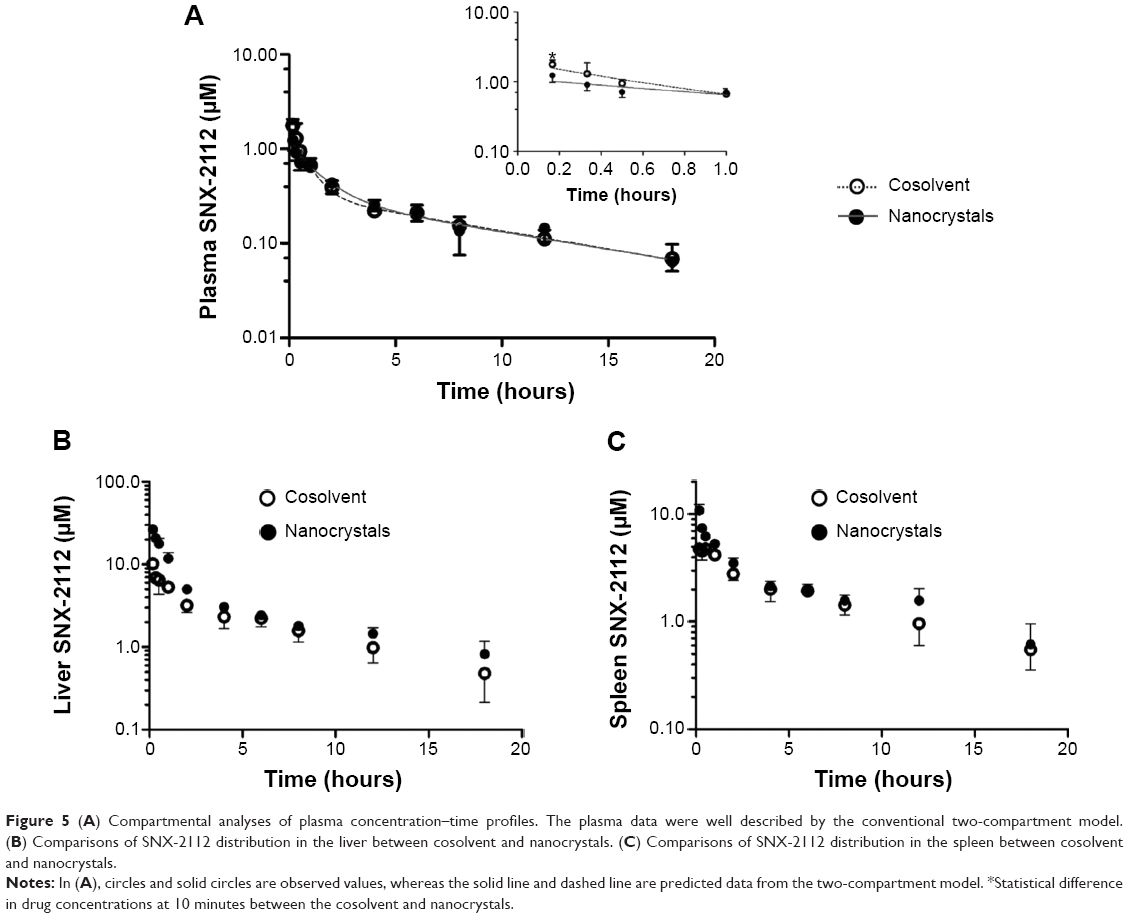 | Figure 5 (A) Compartmental analyses of plasma concentration–time profiles. The plasma data were well described by the conventional two-compartment model. (B) Comparisons of SNX-2112 distribution in the liver between cosolvent and nanocrystals. (C) Comparisons of SNX-2112 distribution in the spleen between cosolvent and nanocrystals. |
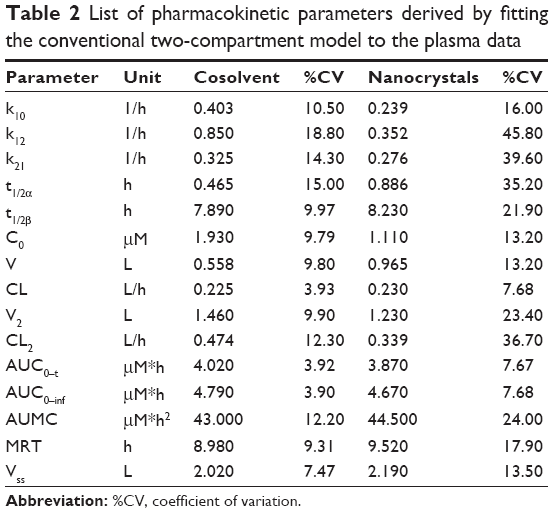 | Table 2 List of pharmacokinetic parameters derived by fitting the conventional two-compartment model to the plasma data |
The AUCs in the heart, liver, spleen, lung, kidney, and intestine were estimated by non-compartmental analysis (Table 3). The organs in which drug exposure was increased by greater than 20% were considered to exhibit a nanoparticle uptake effect. An apparently higher AUC(0–18 h) (≥27%) was found for the liver and spleen in the nanocrystal group (Table 3). This resulted largely from the significantly higher concentrations at the initial time points (within 1 hour) (Figure 5B and C). Therefore, uptake of nanoparticles into the liver and spleen had to be considered in pharmacokinetic modeling. By contrast, the AUC(0–18 h) for the heart, lung, kidney, and intestine remained relatively unchanged or slightly changed (Table 3). It was also noted that organ exposure was reduced in the intestine.
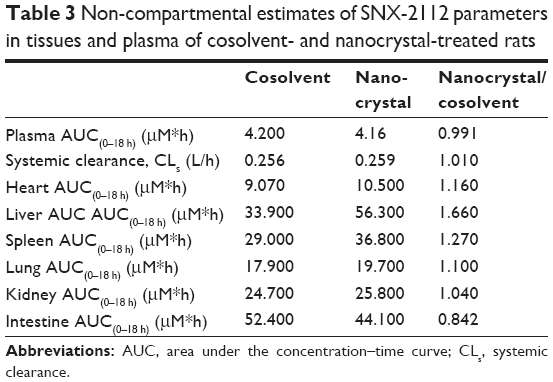 | Table 3 Non-compartmental estimates of SNX-2112 parameters in tissues and plasma of cosolvent- and nanocrystal-treated rats |
Nonparticulate PBPK modeling
The nonparticulate PBPK model (Figure 1A) was used to describe SNX-2112 disposition in rats following cosolvent administration. The obtained pharmacokinetic parameters associated with the nonparticulate drug will be fixed in later nanoparticulate PBPK modeling. The partitioning coefficient of SNX-2112 for those tissues to which the inlet drug concentrations equaled the plasma concentrations (ie, intestine, spleen, kidney, and heart) was determined experimentally as the ratio of AUC(0–18 h) of tissue normalized to that of plasma AUC(0–18 h).48 Tissue/Plasma AUC ratios of 12.2, 6.78, 5.92, and 2.24 were reached at 18 hours for the intestine, spleen, kidney, and heart, respectively (Figure 6 and Table 4). The partition coefficients for other tissues (liver, lung, and remaining tissues) and the hepatic clearance were estimated by simultaneous fitting of the model to the experimental data (Table 4). The observed values were largely close to the predicted ones from the model, indicating that the data were well described by the PBPK model (Figure 7).
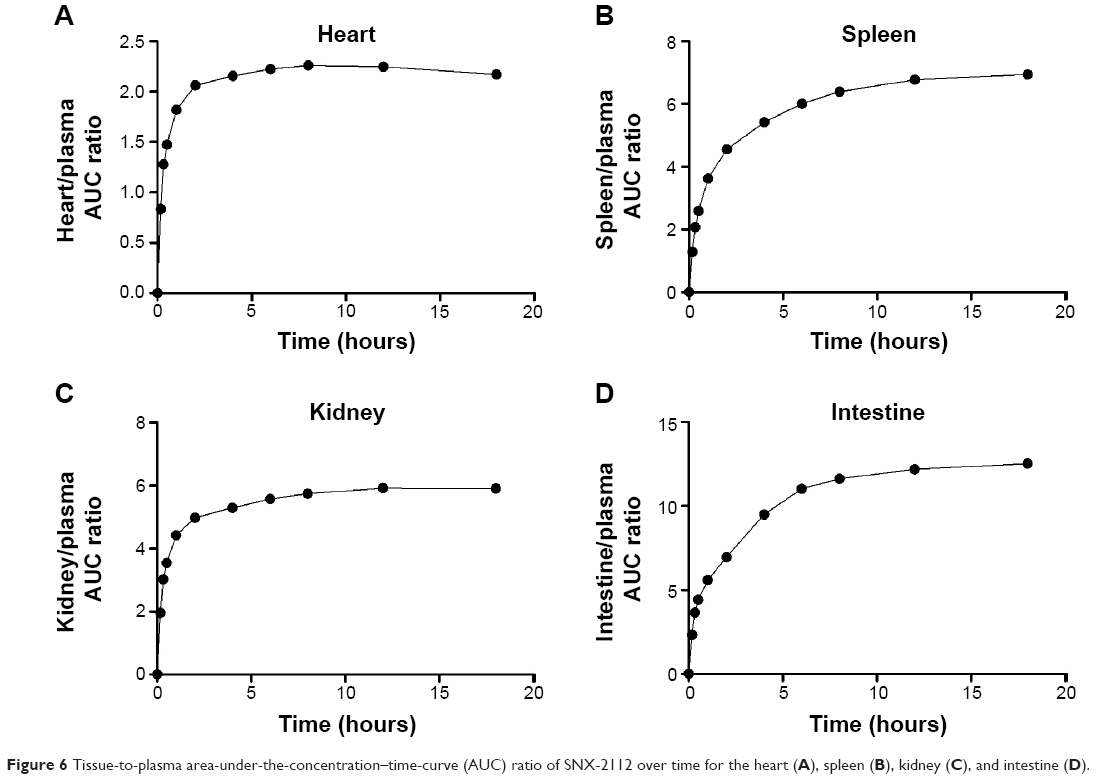 | Figure 6 Tissue-to-plasma area-under-the-concentration–time-curve (AUC) ratio of SNX-2112 over time for the heart (A), spleen (B), kidney (C), and intestine (D). |
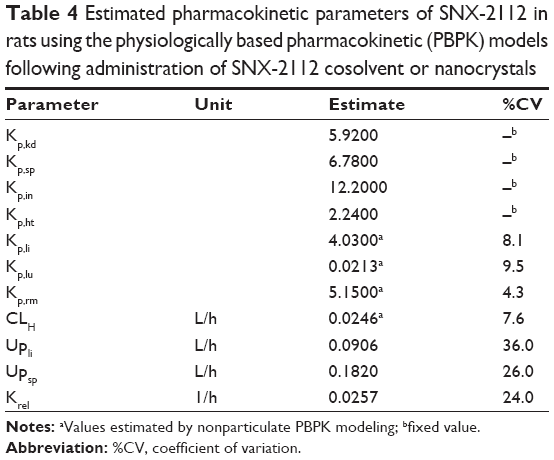 | Table 4 Estimated pharmacokinetic parameters of SNX-2112 in rats using the physiologically based pharmacokinetic (PBPK) models following administration of SNX-2112 cosolvent or nanocrystals |
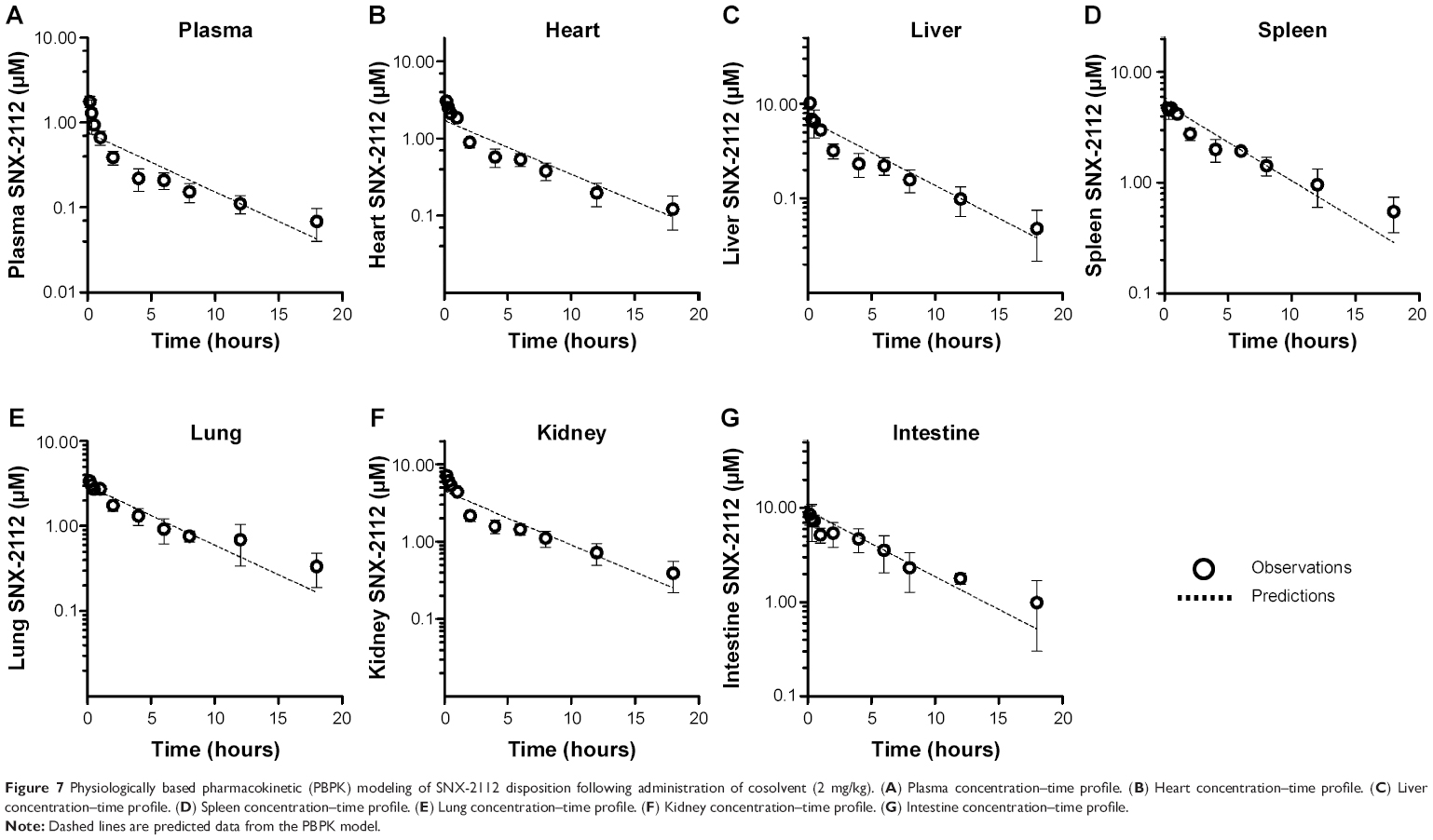 | Figure 7 Physiologically based pharmacokinetic (PBPK) modeling of SNX-2112 disposition following administration of cosolvent (2 mg/kg). (A) Plasma concentration–time profile. (B) Heart concentration–time profile. (C) Liver concentration–time profile. (D) Spleen concentration–time profile. (E) Lung concentration–time profile. (F) Kidney concentration–time profile. (G) Intestine concentration–time profile. |
Nanoparticulate PBPK modeling
The nanoparticulate PBPK model (Figure 1B) was specifically constructed to distinguish the nanoparticulate from the nonparticulate drug as nanocrystals were administered to the rats. Parameters associated the nonparticulate form were taken as the corresponding estimates from nonparticulate PBPK modeling and were fixed during model fitting. The nanoparticle-specific parameters (ie, Upsp, Upli, and Krel) were obtained by simultaneous fitting of the model to the experimental data (Table 4). The observed values versus the predicted ones from the model were plotted (Figure 8). It is clear the pharmacokinetic data were adequately modeled. In addition, the drug (in two forms) concentrations in the plasma, liver, and spleen were simulated (Figure 9). The results reveal that the nanocrystals rapidly released drug molecules in vivo, accounting for their cosolvent-like pharmacokinetic behaviors and biodistribution.
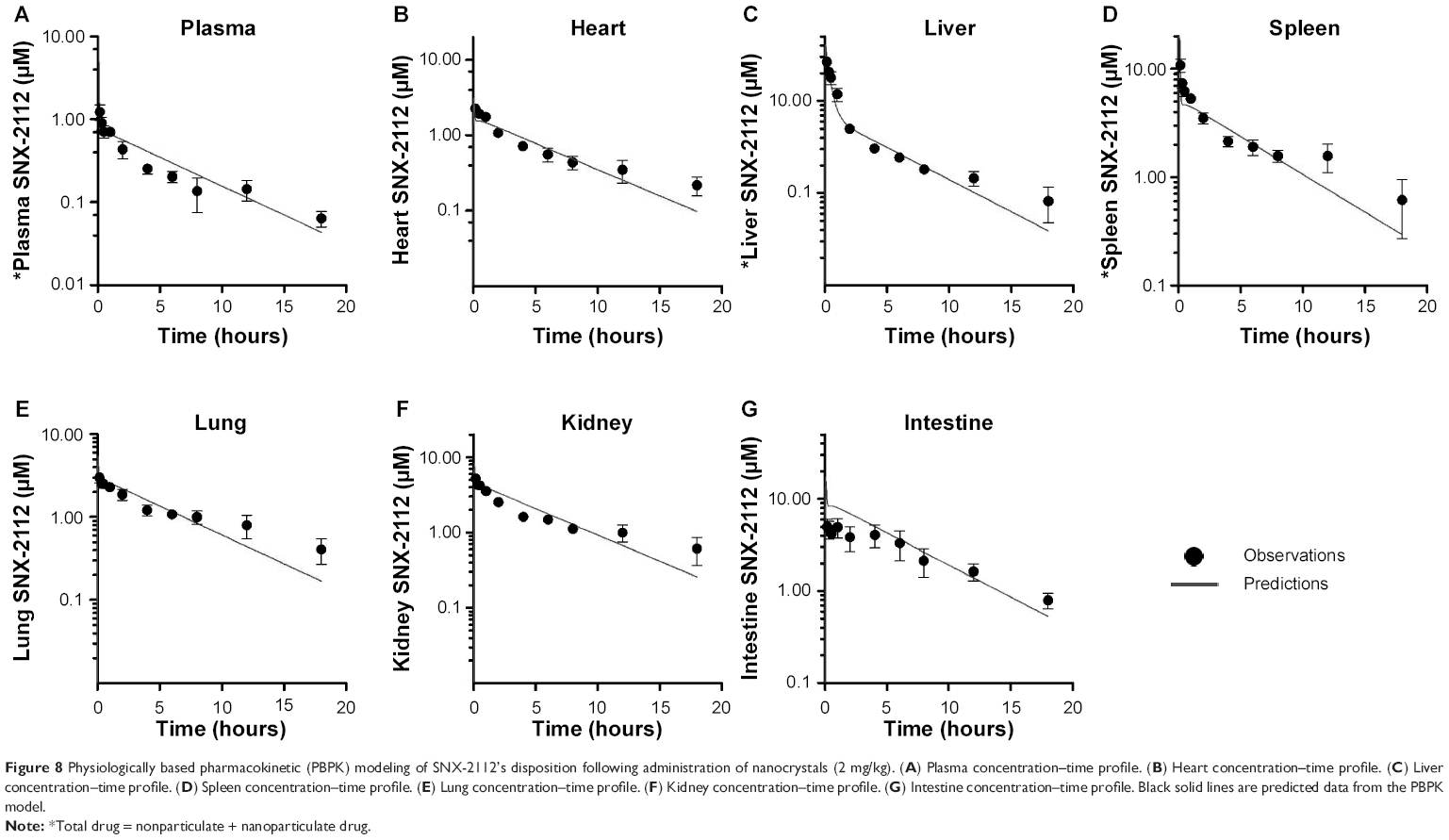 | Figure 8 Physiologically based pharmacokinetic (PBPK) modeling of SNX-2112’s disposition following administration of nanocrystals (2 mg/kg). (A) Plasma concentration–time profile. (B) Heart concentration–time profile. (C) Liver concentration–time profile. (D) Spleen concentration–time profile. (E) Lung concentration–time profile. (F) Kidney concentration–time profile. (G) Intestine concentration–time profile. Black solid lines are predicted data from the PBPK model. |
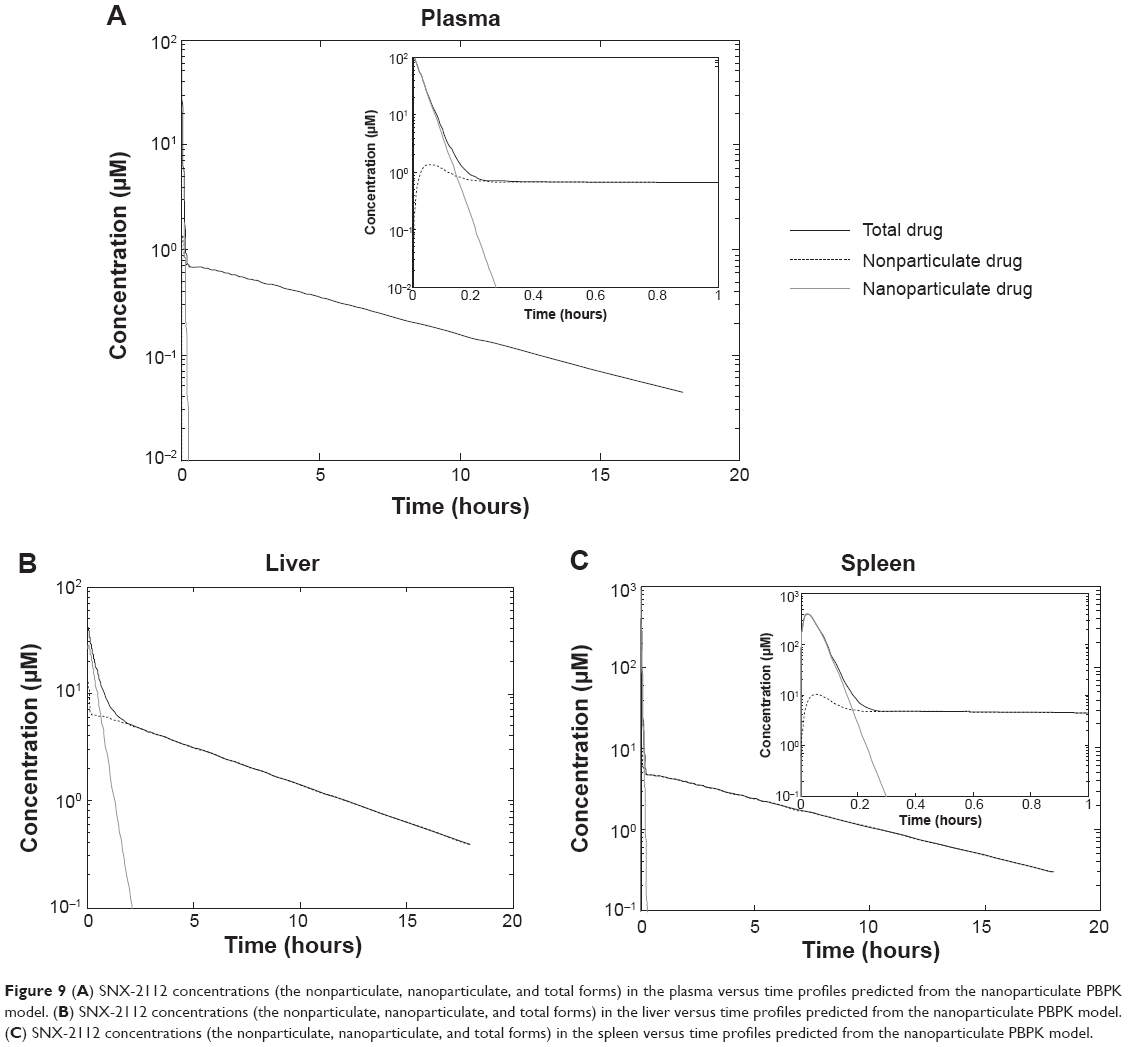 | Figure 9 (A) SNX-2112 concentrations (the nonparticulate, nanoparticulate, and total forms) in the plasma versus time profiles predicted from the nanoparticulate PBPK model. (B) SNX-2112 concentrations (the nonparticulate, nanoparticulate, and total forms) in the liver versus time profiles predicted from the nanoparticulate PBPK model. (C) SNX-2112 concentrations (the nonparticulate, nanoparticulate, and total forms) in the spleen versus time profiles predicted from the nanoparticulate PBPK model. |
Discussion
In this study, we, for the first time as far as we are aware, developed and characterized a nanocrystal system for systemic delivery of SNX-2112, a novel Hsp90 inhibitor currently in clinical evaluation for cancer treatment. Further, we elucidated the in vivo fate of SNX-2112 nanocrystals through pharmacokinetic determination and PBPK modeling. Our results reveal that the nanocrystals provided a rapid drug release in vivo. Also, significant uptake of nanocrystals into the spleen and liver occurred, accounting for the initial drug accumulation (within 1 hour) in these two tissues. The present study and another indicate that PBPK modeling is a powerful tool to describe body disposition of nanoformulations, and to facilitate a better understanding of their in vivo fate.37 In addition, our nanocrystals solve the solubility problem of SNX-2112 and overcome the difficulties in drug administration when it comes to clinical trials with the drug itself (the cosolvent is prohibited due to a large amount of organic solvent). Hence, the potential for nanocrystals to achieve the systemic delivery of SNX-2112 is worthy of further investigation in clinical trials.
Due to the insoluble nature of SNX-2112, a nanocrystal formulation is an excellent choice for its delivery. Attempts to develop other types of nanoformulations such as solid lipid nanoparticles and micro-/nanoemulsions for the compound were unsuccessful due to its low solubility in the oil phase. It is well known that the stabilizer plays an important role in determining the properties of drug nanocrystals. The authors of the present paper prepared SNX-2112 nanocrystals using different types of stabilizers including nonionic surfactants (Tween 80, P188, and P407), ionic surfactants (sodium dodecyl sulfate and sodium taurocholate hydrate), and polymers (hydroxypropyl methylcellulose and polyvinylpyrrolidone). It was found that P188-stabilized nanocrystals showed acceptable particle size with narrow size distribution. Preliminary stability studies also revealed that the prepared nanocrystals with P188 were stable for at least 1 month. Hence, P188-stabilized nanocrystals are a promising system to effectively deliver SNX-2112.
The SNX2112 nanocrystals showed a negative zeta potential (−11.6 mV) and extensive distribution in the liver and spleen. Hence, in addition to contributing to the physical stability of the formulation, the negative nature of the zeta potential may be a key determinant in the rate as well as the efficiency of monocyte/macrophage uptake (particularly in the liver and spleen). This is well supported by many other studies in the literature.49,50 For instance, the oridonin nanosuspensions, with a zeta potential value of −21.8 mV, developed by Gao et al demonstrated a high uptake by the liver and spleen.49 Rabinow et al evaluated the biodistribution of an intravenous itraconazole nanosuspension (with a zeta potential of −30.6 mV) and found that high accumulation of the drug occurred in the liver and spleen.50
In vitro release study showed that drug release from the nanocrystals was rapid and was comparable to that from cosolvent (Figure 3D). Parallel release profiles for both the free drug (cosolvent) and the nanocrystals indicate that the nanocrystals rapidly dissolved in the medium upon dilution. However, drug release from the cosolvent was not as immediate as expected. The time for complete drug release was unusually long (more than 4 hours). This was because membrane resistance (the dialysis bag) had a retarding effect on the process of drug release. As a mimic of in vivo release, the dialysis technique is frequently used to study the release kinetics of drug nanocrystals.51,52 Although the release data obtained by the dialysis method did not fully represent the real release behavior of drug nanocrystals in the blood stream, the method was useful for the characterization and comparative analyses of drug release from different formulations.
In nonparticulate PBPK modeling, the tissue/plasma AUC ratios versus time were evaluated and steady-state in vivo conditions were verified (Figure 6). The Kp values were determined as the tissue/plasma AUC ratios under steady-state conditions, as described previously.46 The obtained values were used within the PBPK model without further optimization.46 It was noteworthy that the authors were unable to determine Kp values using such a method for the liver and lung because plasma did not directly flow to these two organs (ie, inlet drug concentrations to the two organs did not equal the plasma concentrations) (Figure 1A). However, the authors plotted the tissue/plasma AUC ratios versus time and also observed steady-state conditions for the liver and lung (data not shown). The AUC ratios toward 18 hours were 8.17 (for the liver) and 4.32 (for the lung), which markedly differed from the optimized Kp values (Table 4). Therefore, our PBPK modeling was carefully and appropriately performed.
Our results reveal that the nanocrystals were significantly taken up into the liver and spleen. This is consistent with the notion that nanoparticles possess the ability of targeting the macrophage system and can accumulate in the liver and spleen.53 The pharmacokinetic curves of the nanocrystals and cosolvent show a high similarity (Figure 5A). The fact that the nanocrystals did not markedly alter the pharmacokinetic profile of SNX-2112 was due to immediate drug release from the nanocrystals upon intravenous administration (Figure 9). It is noteworthy that such cosolvent-like behaviors of drug nanocrystals in the body have been observed in human subjects.54 Since the nanocrystals rapidly dissolved into the free drug, minimal systemic risk would be associated with particulate injection, highlighting the great potential of the nanocrystal formulation in practical uses.
Conclusion
By using the wet-media milling technique, we developed and characterized a nanocrystal system, allowing the systemic delivery of the insoluble drug SNX-2112. The obtained SNX-2112 nanocrystals were 203 nm in size with a zeta potential of −11.6 mv. In addition, the nanocrystals showed a comparable release profile to that of the control (drug cosolvent). Moreover, the nanoparticulate PBPK model incorporating the parameters of particulate uptake (into the liver and spleen) and of in vivo drug release was well fitted to the experimental data. The results reveal that the nanocrystals rapidly released drug molecules in vivo, accounting for their cosolvent-like pharmacokinetic behaviors. Due to particulate uptake, drug accumulation in the liver and spleen were significant at the initial time points (within 1 hour). This study is the first to report that the nanocrystal system is a good choice for the systemic delivery of SNX-2112. Also, our study contributes to an improved understanding of the in vivo fate of nanocrystals.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No 81373496), the Program for Pearl River New Stars of Science and Technology in Guangzhou (No 2014059), and the Doctoral Fund of Ministry of Education of China (20134401120014).
Disclosure
The authors declare no conflicts of interest in this work.
References
Williams HD, Trevaskis NL, Charman SA, et al. Strategies to address low drug solubility in discovery and development. Pharmacol Rev. 2013;65(1):315–499. | ||
Mattheolabakis G, Rigas B, Constantinides PP. Nanodelivery strategies in cancer chemotherapy: biological rationale and pharmaceutical perspectives. Nanomedicine (Lond). 2012;7(10):1577–1590. | ||
Liu XL, Lynn BC, Zhang JH, et al. A versatile prodrug approach for liposomal core-loading of water-insoluble camptothecin anticancer drugs. J Am Chem Soc. 2002;124(26):7650–7651. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Del Rev. 2013;65(1):36–48. | ||
Gupta S. Biocompatible microemulsion systems for drug encapsulation and delivery. Curr Sci. 2011;101(2):174–188. | ||
Zhang XJ, Zhang XG, Yu PE, Han Y, Li Y, Li C. Hydrotropic polymeric mixed micelles based on functional hyperbranched polyglycerol copolymers as hepatoma-targeting drug delivery system. J Pharm Sci. 2013;102(1):145–153. | ||
Lu Y, Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int J Pharm. 2013;453(1):198–214. | ||
Qi J, Lu Y, Wu W. Absorption, disposition and pharmacokinetics of solid lipid nanoparticles. Curr Drug Metab. 2012;13(4):418–428. | ||
Xu S, Olenyuk BZ, Okamoto CT, Hamm-Alvarez SF. Targeting receptor-mediated endocytotic pathways with nanoparticles: rationale and advances. Adv Drug Del Rev. 2013;65(1):121–138. | ||
Möschwitzer JP. Drug nanocrystals in the commercial pharmaceutical development process. Int J Pharm. 2013;453(1):142–156. | ||
Carter NJ. Extended-release intramuscular paliperidone palmitate: a review of its use in the treatment of schizophrenia. Drugs. 2012;72(8):1137–1160. | ||
Böhm BH, Müller RH. Lab-scale production unit design for nanosuspensions of sparingly soluble cytotoxic drugs. Pharm Sci Technolo Today. 1999;2(8):336–339. | ||
Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785–796. | ||
Sudhakar B, NagaJyothi K, Murthy KV. Nanosuspensions as a versatile carrier based drug delivery system – an overview. Curr Drug Deliv. 2014;11(3):299–305. | ||
Kesisoglou F, Panmai S, Wu Y. Nanosizing – oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59(7):631–644. | ||
Müller RH, Jacobs C. Buparvaquone mucoadhesive nanosuspension: preparation, optimisation and long-term stability. Int J Pharm. 2002;237(1–2):151–161. | ||
Pignatello R, Ricupero N, Bucolo C, Maugeri F, Maltese A, Puglisi G. Preparation and characterization of eudragit retard nanosuspensions for the ocular delivery of cloricromene. AAPS Pharm Sci Tech. 2006;24(1):E27. | ||
Xiong R, Lu W, Yue P, et al. Distribution of an intravenous injectable nimodipine nanosuspension in mice. J Pharm Pharmacol. 2008;60(9):1155–1159. | ||
Andes D. In vivo pharmacodynamics of antifungal drugs in treatment of candidiasis. Antimicrob Agents Chemother. 2003;47(4):1179–1186. | ||
Ganta S, Paxton JW, Baguley BC, Garg S. Formulation and pharmacokinetic evaluation of an asulacrine nanocrystalline suspension for intravenous delivery. Int J Pharm. 2009;367(1–2):179–186. | ||
Hollis CP1, Weiss HL, Leggas M, Evers BM, Gemeinhart RA, Li T. Biodistribution and bioimaging studies of hybrid paclitaxel nanocrystals: lessons learned of the EPR effect and image-guided drug delivery. J Control Release. 2003;172(1):12–21. | ||
Ali HS, York P, Blagden N. Preparation of hydrocortisone nanosuspension through a bottom-up nanoprecipitation technique using microfluidic reactors. Int J Pharm. 2009;375(1–2):107–113. | ||
Keck CM, Müller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm. 2006;62(1):3–16. | ||
Van Eerdenbrugh B, Van den Mooter G, Augustijns P. Top-down production of drug nanocrystals: nanosuspension stabilization, miniaturization and transformation into solid products. Int J Pharm. 2008;364(1):64–75. | ||
Chen H, Wan J, Wang Y, et al. A facile nanoaggregation strategy for oral delivery of hydrophobic drugs by utilizing acid-base neutralization reactions. Nanotechnology. 2008;19(37):375104. | ||
Gao L, Liu G, Wang X, Liu F, Xu Y, Ma J. Preparation of a chemically stable quercetin formulation using nanosuspension technology. Int J Pharm. 2011;404(1–2):231–237. | ||
Xia D, Gan Y, Cui F. Application of precipitation methods for the production of water-insoluble drug nanocrystals: production techniques and stability of nanocrystals. Curr Pharm Des. 2014;20(3):408–435. | ||
Müller RH, Gohla S, Keck CM. State of the art of nanocrystals – special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78(1):1–9. | ||
Chandarlapaty S, Sawai A, Ye Q, et al. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin Cancer Res. 2008;14(1):240–248. | ||
Okawa Y, Hideshima T, Steed P, et al. SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and ERK. Blood. 2009;113(4):846–855. | ||
Liu H, Sun H, Wu Z, Zhang X, Wu B. P-glycoprotein (P-gp)-mediated efflux limits intestinal absorption of the Hsp90 inhibitor SNX-2112 in rats. Xenobiotica. 2014;44(8):763–768. | ||
Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. | ||
Atkinson AJ Jr, Smith BP. Models of physiology and physiologically based models in clinical pharmacology. Clin Pharmacol Ther. 2012;92(1):3–6. | ||
Chow EC, Pang KS. Why we need proper PBPK models to examine intestine and liver oral drug absorption. Curr Drug Metab. 2013;14(1):57–79. | ||
Pang KS, Durk MR. Physiologically-based pharmacokinetic modeling for absorption, transport, metabolism and excretion. J Pharmacokinet Pharmacodyn. 2010;37(6):591–615. | ||
Rostami-Hodjegan A. Physiologically based pharmacokinetics joined with in vitro-in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther. 2012;92(1):50–61. | ||
Kagan L, Gershkovich P, Wasan KM, Mager DE. Dual physiologically based pharmacokinetic model of liposomal and nonliposomal amphotericin B disposition. Pharm Res. 2014;31(1):35–45. | ||
Barta TE, Veal JM, Rice JW, et al. Discovery of benzamide tetrahydro-4H-carbazol-4-ones as novel small molecule inhibitors of Hsp90. Bioorg Med Chem Lett. 2008;18(12):3517–3521. | ||
Tuomela A, Liu P, Puranen J, et al. Brinzolamide nanocrystal formulations for ophthalmic delivery: reduction of elevated intraocular pressure in vivo. Int J Pharm. 2014;467(1–2):34–41. | ||
Levy MY, Benita S. Drug release from submicronized o/w emulsion: a new in vitro kinetic evaluation model. Int J Pharm. 1990;66(1–3):29–37. | ||
Liu H, Sun H, Lu D, et al. Identification of glucuronidation and biliary excretion as the main mechanisms for gossypol clearance: in vivo and in vitro evidence. Xenobiotica. 2014;44(8):696–707. | ||
Liu W, Liu H, Sun H, et al. Metabolite elucidation of the Hsp90 inhibitor SNX-2112 using ultraperformance liquid chromatography/quadrupole time-of-flight mass spectrometry (UPLC-QTOF/MS). Xenobiotica. 2014;44(5):455–464. | ||
Kagan L, Gershkovich P, Wasan KM, Mager DE. Physiologically based pharmacokinetic model of amphotericin B disposition in rats following administration of deoxycholate formulation (Fungizone®): pooled analysis of published data. AAPS J. 2011;13(2):255–264. | ||
Wong J, Brugger A, Khare A, et al. Suspensions for intravenous (IV) injection: a review of development, preclinical and clinical aspects. Adv Drug Deliv Rev. 2008;60(8):939–954. | ||
Honary S, Zahir F. Effect of zeta potential on the properties of nano-drug delivery systems – a review (part 2). Trop J Pharm Res. 2013;12(2):265–273. | ||
Kenth S, Sylvestre JP, Fuhrmann K, Meunier M, Leroux JC. Fabrication of paclitaxel nanocrystals by femtosecond laser ablation and fragmentation. J Pharm Sci. 2011;100(3):1022–1030. | ||
Li Y, Sun S, Chang Q, et al. A strategy for the improvement of the bioavailability and antiosteoporosis activity of BCS IV flavonoid glycosides through the formulation of their lipophilic aglycone into nanocrystals. Mol Pharm. 2013;10(7):2534–2542. | ||
Chow EC, Durk MR, Cummins CL, Pang KS. 1Alpha,25-dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr(−/−) and fxr(+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J Pharmacol Exp Ther. 2011;337(3):846–859. | ||
Gao L, Zhang D, Chen M, et al. Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions. Int J Pharm. 2008;355(1–2):321–327. | ||
Rabinow B, Kipp J, Papadopoulos P, et al. Itraconazole IV nanosuspension enhances efficacy through altered pharmacokinetics in the rat. Int J Pharm. 2007;339(1–2):251–260. | ||
Liu Y, Huang L, Liu F. Paclitaxel nanocrystals for overcoming multidrug resistance in cancer. Mol Pharm. 2010;7(3):863–869. | ||
Zhang H, Hollis CP, Zhang Q, Li T. Preparation and antitumor study of camptothecin nanocrystals. Int J Pharm. 2011;415(1–2):293–300. | ||
Song H, Geng HQ, Ruan J, et al. Development of Polysorbate 80/Phospholipid mixed micellar formation for docetaxel and assessment of its in vivo distribution in animal models. Nanoscale Res Lett. 2011;6(1):354. | ||
Mouton JW, van Peer A, de Beule K, Van Vliet A, Donnelly JP, Soons PA. Pharmacokinetics of itraconazole and hydroxyitraconazole in healthy subjects after single and multiple doses of a novel formulation. Antimicrob Agents Chemother. 2006;50(12):4096–4102. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.