Back to Journals » Vascular Health and Risk Management » Volume 17
Elevated Lipoprotein(a): Background, Current Insights and Future Potential Therapies
Authors Handhle A ![]() , Viljoen A, Wierzbicki AS
, Viljoen A, Wierzbicki AS ![]()
Received 17 June 2021
Accepted for publication 13 August 2021
Published 7 September 2021 Volume 2021:17 Pages 527—542
DOI https://doi.org/10.2147/VHRM.S266244
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Ahmed Handhle, 1 Adie Viljoen, 2 Anthony S Wierzbicki 3
1Department of Metabolic Medicine/Chemical Pathology, Addenbrookes Hospital, Cambridge, UK; 2Department of Metabolic Medicine/Chemical Pathology, North & East Hertfordshire Hospitals Trust, Lister Hospital, Hertfordshire, UK; 3Department of Metabolic Medicine/Chemical Pathology, Guy’s & St Thomas’, Hospitals, London, SE1 7EH, UK
Correspondence: Anthony S Wierzbicki
Metabolic Medicine/Chemical Pathology, St Thomas Hospital, Lambeth Palace Road, London, SE1 7EH, UK
Tel +44 207 188 1256
Email [email protected]
Abstract: Lipoprotein(a) forms a subfraction of the lipid profile and is characterized by the addition of apolipprotein(a) (apo(a)) to apoB100 derived particles. Its levels are mostly genetically determined inversely related to the number of protein domain (kringle) repeats in apo(a). In epidemiological studies, it shows consistent association with cardiovascular disease (CVD) and most recently with extent of aortic stenosis. Issues with standardizing the measurement of Lp(a) are being resolved and consensus statements favor its measurement in patients at high risk of, or with family histories of CVD events. Major lipid-lowering therapies such as statin, fibrates, and ezetimibe have little effect on Lp(a) levels. Therapies such as niacin or cholesterol ester transfer protein (CETP) inhibitors lower Lp(a) as well as reducing other lipid-related risk factors but have failed to clearly reduce CVD events. Proprotein convertase subtilisin kexin-9 (PCSK9) inhibitors reduce cholesterol and Lp(a) as well as reducing CVD events. New antisense therapies specifically targeting apo(a) and hence Lp(a) have greater and more specific effects and will help clarify the extent to which intervention in Lp(a) levels will reduce CVD events.
Keywords: lipoprotein (a), cardiovascular disease, aortic stenosis, apheresis, genetics, lipoprotein turnover, statin, PCSK9, antisense therapy
Plain Language Summary
Lipoprotein (a) (Lp(a)) forms a small fraction of cholesterol profile. It is related to the bad cholesterol (low-density-lipoprotein cholesterol; LDL-C) that drives artery narrowing (atherosclerosis) and hence heart attacks, strokes, and large artery disease. It differs from LDL in having an additional protein - apolipoprotein(a) bonded to the particle. This addition changes both the secretion rate and clearance of these particles. Lp(a) levels are mostly inherited with the variation driven by a number of repeats within the apo(a) molecule. The standard treatments for lowering LDL-C such as statins or ezetimibe have little effect on Lp(a) levels. Niacin and proprotein convertase subtilisin kexin-9 (PCSK9) inhibitors reduce Lp(a) but affect other fractions such as LDL-C as well. New specific inhibitors for Lp(a) have been developed and are in early trials.
 |
Figure 1 Distribution of Lp(a) within a density gradient profile compared to other lipid fractions showing the association of the distribution of apolipoprotein E with subfractions of Lp(a). |
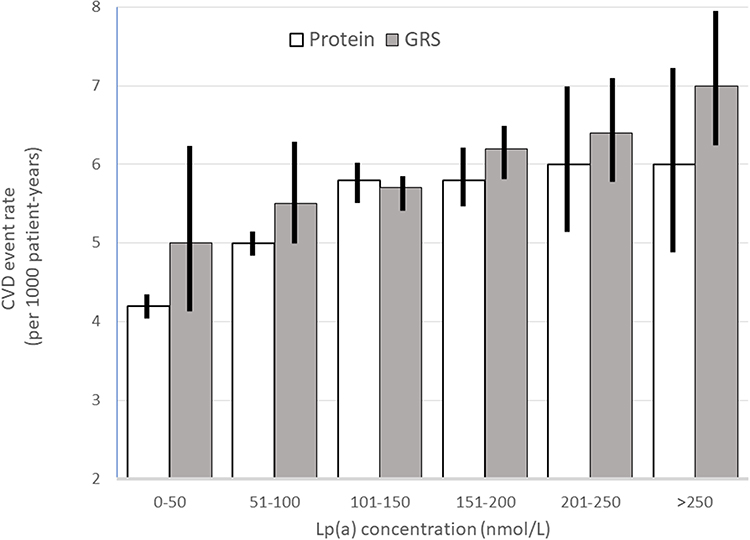 |
Figure 2 Relationship of measured Lp(a) levels and an equivalent polygenic risk score with CVD events in the UK Biobank study. Data from Trinder et al. 15 |
Introduction
A standard lipid profile consists of triglyceride-rich particles (especially post-prandially) and cholesterol-rich particles involved in transportation of lipids to body compartments (low-density lipoprotein; LDL) and those involved in transporting cholesterol back to the liver for disposal through the biliary system (high density lipoprotein; HDL). While these components are responsible for most of the plasma-related risk for cardiovascular disease (CVD) risk, other factors contribute including inflammation-related markers (eg, C-reactive protein), coagulation components (eg, fibrinogen or plasminogen), and lipid subfractions. These lipid subfractions include modifications to particle size distribution induced by insulin resistance (eg, small dense LDL) while a small proportion of both triglyceride-rich and LDL particles are distinguished by the covalent addition of apolipoprotein (a) (apo(a)) to apoB100 in an LDL-derived particle.
Structure and Genetics
Lp (a) is composed of a single apolipoprotein B100 (apoB100), covalently linked by a disulfide bond to a single apolipoprotein(a) (apo(a)) allied with associated cholesterol, triglyceride and phospholipid.1–3 Apo(a) is encoded by the LPA gene located on chromosome 6q26-27, bears homology to plasminogen and is expressed in hepatocytes. Whereas plasminogen contains 5 kringle (K-I to K-V) and a protease domain, apo(a) contains 10 kringle-IV subtypes (KIV1–10). Its molecular mass varies between 275–800 kDa due to the more than 40 allelic LPA variants4–6 with wide differences between ethnic groups.7 Whilst a single copy each of K-IV1 and K-IV3–10 are present, the number of K-IV2 copies can range from 1 to more than 40.8 Variable glycosylation of K-IV motifs and linker sequences that join individual kringles also contribute to this heterogeneity.9 Genetic studies have established that serum Lp(a) levels are predominantly genetically inherited in an autosomal co-dominant manner2,4 and that the allelic variation of the LPA gene is responsible for the large range (up to 1000-fold) seen in Lp(a) concentrations with concentrations inversely related to the number of KIV2 repeats.8 These repeats are too large and too varied to be clearly differentiated by next generation sequencing techniques with current reading depths. A pentanucleotide10 and 2 single nucleotide polymorphisms (rs10455872 and rs3798220) predict higher Lp(a) concentrations11,12 and the risk of premature CVD.13,14 An intra-genic risk score based on single nucleotide polymorphisms can be used to approximate Lp(a) concentrations and has similar predictive power to Lp(a) levels.15
Proteomic analysis has been conducted for Lp(a) particles and shows profound differences from LDL particles.16 In the first analysis 9 proteins were associated with LDL and 31 with Lp(a). 15 proteins were confirmed to be associated with Lp(a). These proteins were involved in negative regulation of peptidase activity, regulation or transport of insulin growth factors, extracellular structure organization, protein processing and binding.16 Proteins such as transthyretin, vitronectin, paraoxonase-1 and protease inhibitors might be preferentially transported by Lp(a). It is interesting that the genetic study suggesting an interaction of Lp(a) with apoH was not reproduced in this study.17
Assembly and Metabolism of Lp(a)
The site of Lp (a) assembly is unknown and may occur in hepatocytes, extracellularly in the space of Disse or in the plasma.18–20 Lp(a) is mostly assembled by addition of apo(a) to a newly synthesized LDL particle rather than added to a triglyceride-rich very low-density lipoprotein (VLDL) precursor particle. This assembly is accomplished by forming a covalent disulfide bond between K-IV9 of apo(a) and apoB100 of the LDL. In plasma, Lp(a) exists in 3 subfractions – a triglyceride-rich fraction in VLDL, an apoE-rich apoB-containing particle, and a predominant apoB-containing particle.21 (figure 1). The major ApoE polymorphism seems to affect the amount of Lp(a) with apoE4/E4 resulting in a 65% higher level than apoE2/E2.22
The mechanism of Lp(a) clearance from the plasma has not been fully elucidated.23 The attachment point for apo(a) is close to the LDL Receptor (LDLR) binding site on apo-B and thus interferes with binding to LDLR leading to reduced clearance and longer plasma half-life of Lp(a) compared to LDL-C. However, LDLR function may be relevant to the clearance of Lp(a) as patients with familial hypercholesterolemia (FH) have higher Lp(a) levels than their unaffected siblings with the effect being dependent on allele dose. Other undefined clearance mechanisms (eg, proteolytic cleavage of apo(a), scavenger receptors including B1 and plasminogen receptors) probably exist for Lp(a).23,24
Lp(a) is thought to promote atherosclerosis by two main mechanisms.25,26 The function of Lp(a) is unknown but these particles may be involved in clearance of oxidized phosphocholine phospholipids (OxPL) or their Schiff-based adducts to lysine resides in proteins.27 OxPL have multiple atherogenic and signaling properties.28 Lp(a) infiltrates into the arterial intima, space and binds to components of the extracellular matrix, enhancing macrophage infiltration and smooth muscle proliferation possibly through the effects of OxPL on macrophage function mediated by K-IV10 phospholipid-binding domain, interleukin-8, and the lipid scavenger receptor CD-36 and Toll-like-receptor-2 (TLR2).29,30 These mechanisms may be relevant to disease, as in the Dallas Heart Study (n=3381), OxPL, apoB, Lp(a) and OxPL/apoB levels differed between racial/ethnic subgroups, with blacks having the highest levels. OxPL/apoB levels correlated with Lp(a) (r=0.85, p<0.001) with the relationship being a “reverse L” shape for log-transformed values but not other CVD risk factors. The correlation was dependent on apo(a) isoform size; and became weaker with larger isoforms. The number of K-IV repeats negatively correlated with OxPL/apoB (r=−0.49, p<0.001) and Lp(a) (r=−0.61, p<0.001) but even after adjustment for apo(a) isoform size, the relationship between OxPL/apoB and Lp(a) remained (r=0.67, p<0.001).31
The structural similarity of apo(a) to plasminogen could enhance anti-fibrinolytic effects and have been reported in biochemical studies.25 However, an analysis of Dal-Outcomes study failed to demonstrate any effect of Lp(a) in increasing thrombosis in patients with recent acute coronary syndromes where thrombosis was the main precipitant of events.32
Lipoprotein turnover studies of lipoprotein particles using labeled amino acids have been conducted for many years for VLDL and LDL. However, the lower concentration of Lp(a) limited the application of these techniques until recently. Leucine-label protein turnover studies of Lp(a) show that plasma levels are dependent on production rate and isoform size33 and have confirmed the far longer half-life of Lp(a) compared to LDL.34
A lipid turnover study in patients with FH (LDLR or PCSK9 gain-of-function mutations) those with PCSK9 loss-of-function mutations and controls.35 Subjects with PCSK9-loss-of-function mutations displayed reduced apoE concentrations associated with a reduced VLDL-apoE production rate. Lp(a) and VLDL-apoE absolute production rates were correlated (r=0.50; P<0.05) and apoE:apo(a) ratios in Lp(a) increased with plasma Lp(a) (r=0.96; P<0.001) but not with PCSK9 levels. Individuals with loss-of-function variants in PCSK9 (ie, increased LDL receptor expression) show lower concentrations of Lp(a) (63 vs 80nmol/L; p<0.001).36
Epidemiology of Lp(a) and Cardiovascular Disease
Numerous studies have described an association between elevated Lp(a) and CVD independent of LDL-C and other traditional CVD risk factors.11,12,37 These associations apply to mortality, CVD mortality, and individual vascular bed endpoints37 – myocardial infarction,38 stroke and peripheral arterial disease (PAD) in multiple populations mostly of European origin. Two studies of the Danish general population included Lp(a) concentration (n= 69,764), LPA K-IV2 repeats (n=98,810), and LPA rs10455872 genotype (n=119,094). An Lp(a)>93 mg/dL (199 nmol/L; 96th–100th centiles) compared with <10mg/dL (18 nmol/L; 1st–50th centiles) was associated with a hazard ratio (HR) of 1.50 (95% confidence interval 1.28–1.76) for CVD mortality and of 1.20 (1.10–1.30) for all-cause mortality. A 50mg/dL (105 nmol/L) increase in Lp(a) levels had a hazard ratio of 1.16 (1.09–1.23). For stroke, the multivariable-adjusted HR was 1.60 (1.24 to 2.05) and for a 50mg/dl (105 nmol/l) higher Lp(a) the HR was 1.20 (1.13 to 1.28).39 In the European Prospective Investigation of Cancer (EPIC)-Norfolk cohort with 212,981 person-years, Lp(a) levels were associated with PAD and CAD outcomes but not with ischemic stroke with HRs per 2.7-fold increase in Lp(a) of 1.37 (1.25–1.50), 1.13 (1.04–1.22) and 0.91 (0.79–1.03) respectively.40 In the Copenhagen General Population Study (CGPS), patients with familial hypercholesterolemia (FH) with Lp(a) levels >50mg/dl had a 1.4-fold HR of MI than those with FH and Lp(a) levels <50mg/dl.41
A study in UK Biobank in 370,049 individuals showed that a 120nmol/L increase in Lp(a) was associated with an 1.26 (1.23–1.28) excess risk of CVD while an intra-genic genetic risk score of >120 based on 43 single nucleotide polymorphisms gave similar results with an excess risk of 1.29 (1.26–1.33).15 (figure 2) Area under receiver operator curve (AUROC) was 0.64–0.642 for both methods but would be lower using a precision recall curve (AUPRC) which makes no assumption about balanced numbers in outcome groups.
Meta-analyses of epidemiological studies including the Emerging Risk Factors collaboration (36 studies; n=126,634). In the 24 cohort studies, the risk ratio for CHD (adjusted for age and sex) was 1.16 (1.11–1.22) per 3.5-fold higher Lp(a) concentration (ie, per 1 SD), and 1.13 (1.09–1.18) following further adjustment for lipids and other risk factors. The adjusted risk ratio for stroke was 1.10 (1.02–1.18).37 A plasma concentration of 20mg/dl (50nM) was associated with a 1.5-fold risk elevation while levels exceeding 50mg/dl (125nmol/L) were associated with a 2-fold risk elevation.37
As patients with CVD are now routinely treated with statins, there is increasing interest in the role of factors that drive recurrent events. Long-established data from epidemiological studies such as the Framingham Heart Study have identified age, standard lipid fractions, blood pressure, and diabetes as major predictors of recurrent events.42 In a study of 3359 patients from the Atherothrombosis Intervention in Metabolic Syndrome with low HDL/HIGH Triglycerides (AIM-HIGH) trial HRs for CVD events adjusted for age, gender, trial treatment, LDL-C, and other risk factors showed HRs increasing from 1.04 (0.82 to 1.32) to 1.51 (1.25 to 1.84) for Lp(a) 15–30mg/dl up to >70mg/dl (Figure 3.43 A continuous relation for total events was observed (HR=1.08 (1.04 to 1.12)) per 20mg/dL greater Lp(a).
 |
Figure 3 Relationship of increasing levels of Lp(a) within the AIM-HIGH study with CVD events. Lp(a) 0–15nM is taken as the reference group. Reproduced from Wong ND, Zhao Y, Sung J, Browne A. Relation of first and totalrecurrent atherosclerotic cardiovascular disease events toincreased lipoprotein(a) levels among statin treated adults withcardiovascular disease. Am J Cardiol. 2021;145:12–17. © 2021 Elsevier Inc. All rights reserved.43 |
Data on the relationship of Lp(a) levels with CVD risk in non-Caucasian populations are scarce and/or underpowered.44 The multi-ethnic INTERHEART study of 12,943 subjects reported data from 7 populations showing that Africans (n=775) have the highest Lp(a) concentrations (27mg/dl) and smallest isoform size (median=24 K-IV2 repeats) while Chinese (n=4443) ethnicity have the lowest concentrations (8mg/dl) and a median of 28 kringle repeats.45 Higher Lp(a) concentrations are associated with an increased risk of CVD in all populations but the exact thresholds for different risk levels vary by ethnic group.13,37 In West Africans, 3 polymorphisms and rare variants seem to account for the change in distribution.46
As most Caucasians have minimal Lp(a) concentrations through inheritance of high copy number K-IV alleles, high Lp(a) levels (low copy K-IV number) tend to show an autosomal dominant pattern of inheritance. Given the association of higher Lp(a) with CVD this suggests that Lp(a) should co-segregate with a family history of premature CVD. The Atherosclerosis Risk In Communities (ARIC) study included 12,149 subjects of average age 54 years, 56% women, 23% black, and 44% with a family history of CVD. Of these, 3114 had CVD events over 21 years of follow-up. Both family history (HR 1.17 (1.09–1.26)) and elevated Lp(a) (HR 1.25 (1.12 to 1.40)) were independently associated with CVD with no interaction (p=0.75).47 The highest risk was seen in those with a family history and high Lp(a) (HR: 1.43 (1.27 to 1.62)). Similar findings in ARIC were observed for coronary heart disease (CHD) risk in this study and in another for risk of stroke,48 and in analyses stratified by family history of premature CHD, as well as in the Dallas Heart Study cohort which had a higher proportion of African-Americans.47 In parallel with observational data from epidemiological studies, Mendelian randomization analyses provide strong evidence that the association between Lp(a) and risk of CVD is likely to be causal.12,13,38,49
A post hoc analysis of intravascular ultrasound regression trials (6 trials; 3943 patients) stratified into high (≥60 mg/dL; 17%) (17%) and low (<60 mg/dL; 83%) baseline serum Lp(a) showed that percent atheroma volume (PAV) was higher in the high Lp(a) group before adjustment for CVD risk factors (38 (33–44)% vs 37 (31–43), P=0.01) and more clearly after adjustment (39%±0.5 vs 38%±0.5, P<0.001). Risk-adjusted PAV increased across quintiles of Lp(a) (1–5; 37±0.5%, 37±0.5%, 37±0.5%, 38±0.5%, 39±0.5%, P=0.002).50
Relationships of Lp(a)51,52 with OxPL53,54 have also been shown in aortic stenosis and this association may explain part of the atheromatous appearance of degenerative aortic valve disease.55 Whether the association also occurs in mitral valve disease as opposed to mitral annulus calcification is unclear.56
Lp (a) Measurement
The standardized accurate measurement of Lp(a) concentrations remains a challenge. The mass measurement of Lp(a) includes all of the cholesterol, cholesterol esters, phospholipids, apoB100 and apo(a). The variability in K-IV2 repeats will thus influence the mass concentration whilst also rendering multiple epitopes available for immunoassays thereby making standardization with a single calibrant impossible.57,58 Furthermore, immunoassays must be specific for antigenic loci of apo (a) that are not present in plasminogen or apoB and are specific for K-IV2 which is a major contributor to apo(a) polymorphism. An example is an enzyme linked immunosorbent assay (ELISA) method employing monoclonal antibodies that are specific for a unique apo(a) epitope located in K-IV9 which shows excellent agreement with an ultraperformance liquid chromatography/mass spectrometry method.57 The selection of assay calibrators is difficult given the high degree of apo(a) size/KIV2 copy number variation but most kits now use 5-point calibration. Results should be expressed in nmol/l of Lp(a) particles, yet most of the previous literature reports Lp(a) concentrations in mass units and most papers do not specify the platform or traceability of reference materials. The use of a conversion factor of 2.4 to convert mass to molar units is not recommended given errors involved.57
A further complication is that most Lp(a) forms part of the small dense sub-fraction within the spectrum of LDL particle subspecies. It exists as 3 subspecies running in the VLDL fraction as a triglyceride-rich apoB-apo(a) particle, a subfraction of apoB-apo(a) with added apoE and an LDL apoB-apo(a) fraction. The density of Lp(a) means that it forms part of the calculated LDL-C (cLDL-C) level reported using the Friedewald equation by most laboratories. A number of formulae have been proposed to correct cLDL-C for Lp(a)-C but none have been widely adopted.59 Furthermore, the precipitation of Lp(a) by most direct LDL-C methods may vary.
Assays measuring Lp(a)-cholesterol content have been devised using lectin-based,60 ultracentifugation61,62 or immunofixation electrophoresis63 methods. The lectin Lp(a)-C assay was assessed in the Framingham Heart study (n=3121).60 The mean Lp(a)-C concentration in men with CHD (n=156) was 0.24±0. 20 mmol/L and 34% higher than in controls (P<0.001). The odds ratio for CHD risk in men with Lp(a)-C>0. 259mmol/L (>10mg/dL) was 2.29 (1.55–3.94; p<0.001). In this case, Lp(a)-C correlated highly with a mass immunoassay (ApotekTM Lp(a); r=0.83; P <0.0001). This finding was not reproduced in the Framingham Offspring study.64 For other methods Lp(a)-C shows a modest correlation with ELISA mass methods (r=0.56; P 0.01–0.001) accounting for 31% of the variance61 but results are often discordant.62 A new assay based on magnetic particle-based isolation of Lp(a) may be more practical.65 High Lp(a)-C levels may have an effect on calculated LDL-C and may affect classification in 38% of patients if an adjustment formula is used59 and even 3% of individuals for FH risk categories using the Dutch Lipid score.66 They might also affect the eligibility for therapies which are dependent on LDL-C levels such as proprotein convertase subtilisin kexin-9 (PCSK-9) inhibitors.
Lp (a) and Current Guidelines
Consensus groups and expert opinion suggest that Lp(a) should be measured at least once in high risk groups such as those with established CVD or with a family history of early onset CVD.24,67
In 2010, the European Atherosclerosis Society (EAS) Consensus Panel recommended screening for Lp (a) in a number of risk groups11 (Table 1). These categories were slightly amended in the 2019 European Society of Cardiology (ESC) guidelines for the management of dyslipidemia.68 Similar statements on measuring Lp(a) in patients at higher risk of CVD have been made by the US American Heart Association70 and in detail by the National Lipid Association.69 All these guidelines recommend that Lp(a) is measured once and used as an additional risk stratification tool especially in high-risk groups for CVD. In the UK, NICE guidelines have not reviewed the role of Lp(a) and neither has the National Screening committee reviewed whether it should be measured as part of CVD risk assessment. The HEART-UK consensus statement71 reproduced guidance from the ESC in the absence of assessment from NICE. These guidelines suggest a desirable level Lp(a) <50mg/dl for patients with established CVD if the primary goal of LDL-C lowering had been achieved, partly based on this level being the 80th centile of the general population distribution.11 Other authors have suggested an even lower target of 30mg/dl (90th centile).72,73
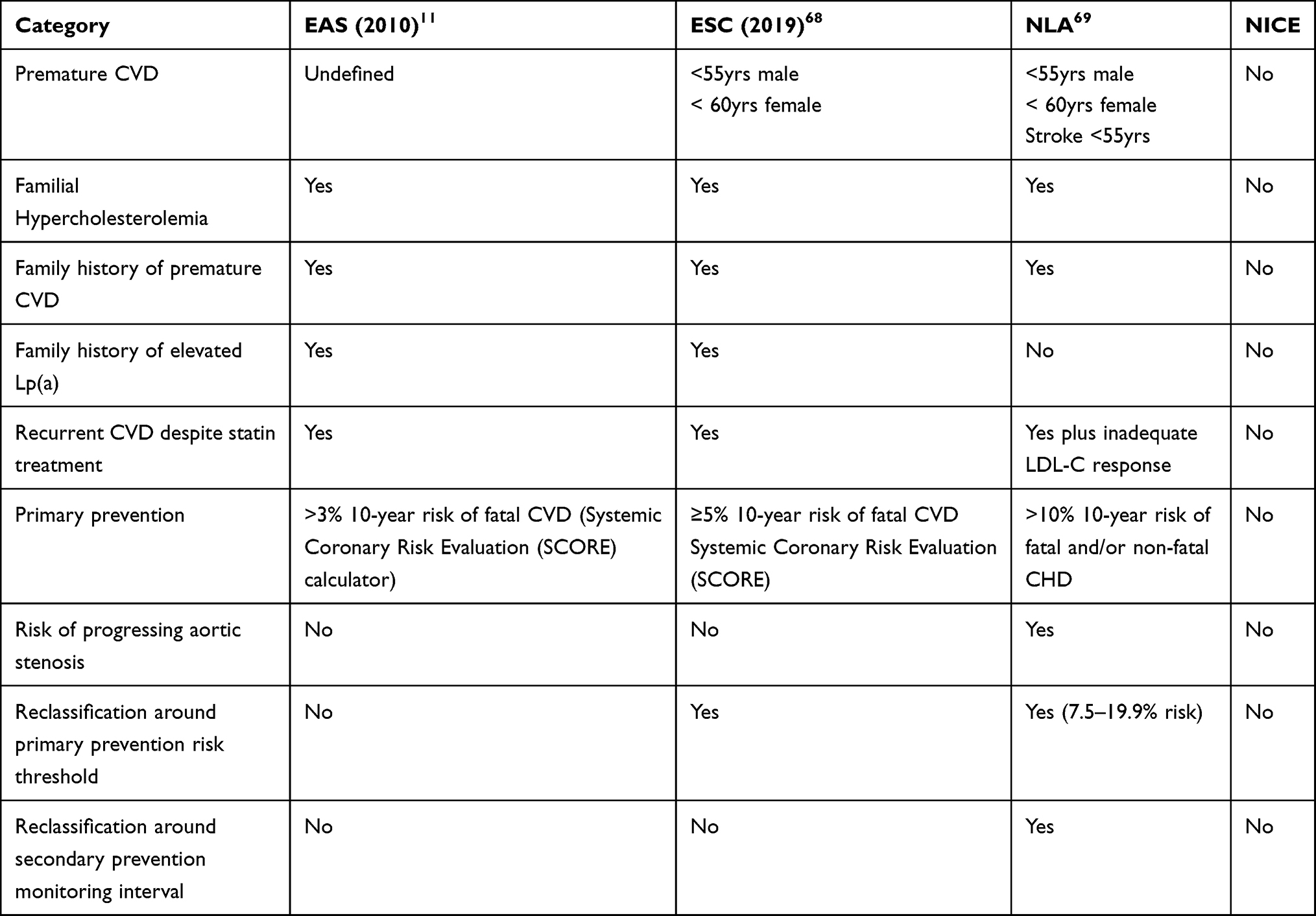 |
Table 1 Summary of Groups Recommended for Lipoprotein (a) Testing in Different International Guidelines |
With consensus guidelines recommending Lp(a) measurement in several patient groups the question on how a result will influence the clinician’s clinical decision making needs to be addressed. The use of Lp(a) levels to reclassify intermediate risk groups in primary prevention is feasible if risk modifiers can be agreed and added to calculator/web systems. Similarly, using Lp(a) as part of reclassification for recurrent disease and amending review intervals may be possible.
Lipid Lowering Therapies Indirectly Affecting Lp(a)
The effects of interventions on Lp(a) are very different to those on LDL particles which they partially resemble in structure apart from the addition of apo(a). Diet and lifestyle factors do not seem to influence Lp(a) concentrations74 though this has not been formally well tested in prospective intervention studies.72
Most studies that have investigated the effects of medications on Lp(a) have done this as a secondary endpoint or as a post hoc analysis. These studies (Table 2) generally do not select patients based on Lp(a) concentration, they are recruited from mostly Caucasian populations and report results either in the whole population or split at median values. Given the highly skewed distribution of Lp(a) this can lead to contrasting results depending on the population selected. The data on the effects of lipid-lowering drug therapies are presented based on meta-analyses of efficacy on Lp(a) levels in clinical trials; lipoprotein turnover studies suggesting the mechanism of any effect (production or catabolism) and any CVD outcome trial event data stratifying by Lp(a) concentration.
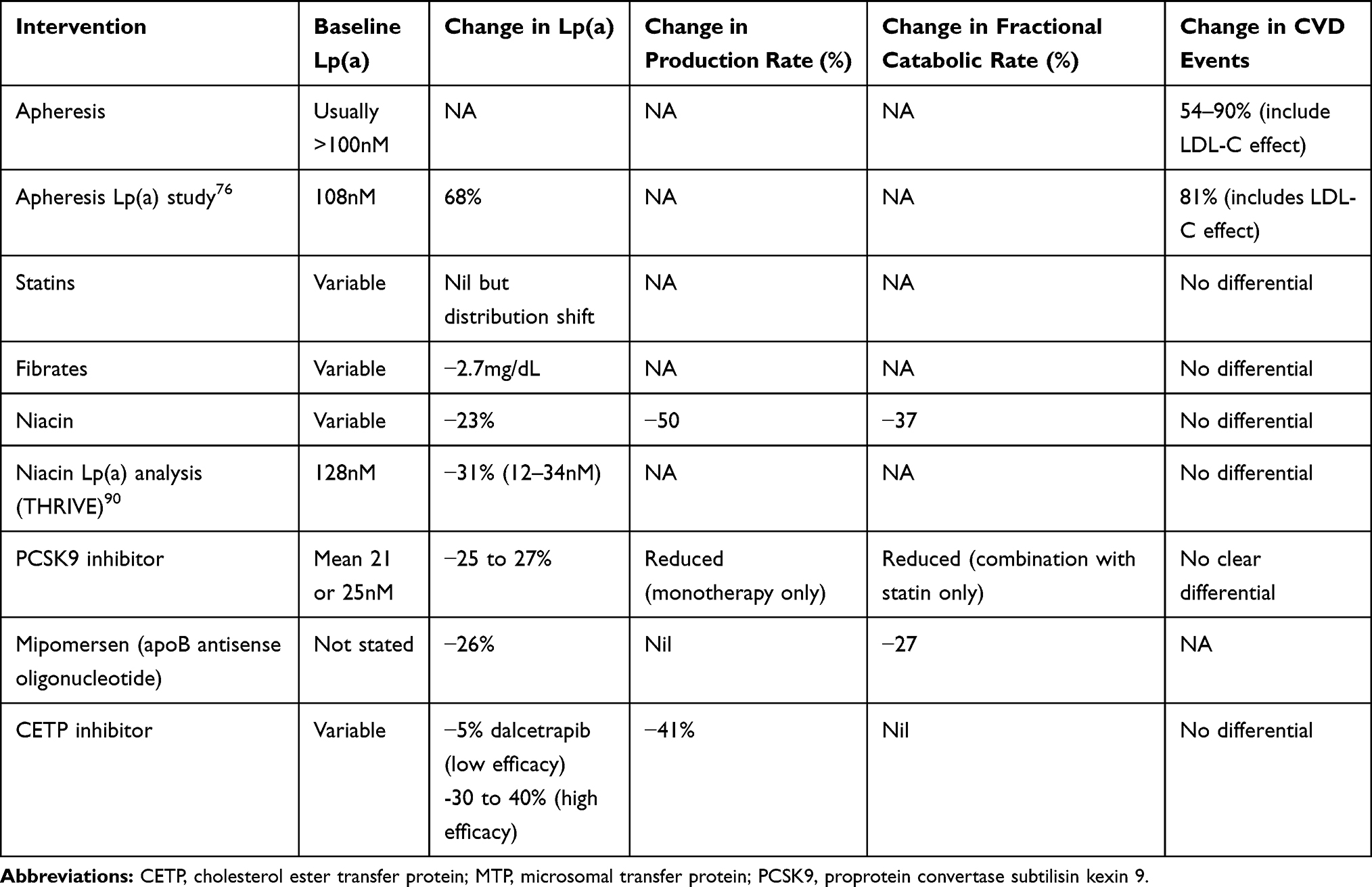 |
Table 2 Effects of Different Lipid-Lowering Drugs on Lp(a) Levels, Production and Catabolic Rates in Turnover Studies and Effects on CVD Outcomes Either Combined with LDL-C Changes or if Analyzed for Heterogeneity by Lp(a) Level Within Trials. Lp(a) Specific Studies are Quoted Separately |
Lipoprotein Apheresis
Apheresis methods including apheresis reduce Lp(a) levels about 65–75% immediately post-procedure and 40–50% on standard schedules. Most descriptions of the effects have involved apheresis techniques that remove LDL and Lp(a) particles with the majority of cases having concurrent homozygous or heterozygous FH but a smaller fraction with severe vascular disease, polygenic hypercholesterolemia, and high Lp(a) levels. A systematic review of early studies showed that apheresis reduced CVD events by 54–90%.75 A later study of 154 patients with baseline Lp(a) 108mg/dL showed apheresis reduced Lp(a) by 68% and reduced CVD events by 81%.76 One variant of apheresis (Lipopac) is specific for removing Lp(a) but data on this intervention are limited. In a study of 15 patients, Lp(a) specific apheresis reduced Lp(a) by 75% and showed angiographic benefit.77
Statins and Lp(a)
Most meta-analyses of the effects of statins (n=20; 23,605 patients) show minimal effect of these drugs on Lp(a) levels78 but selecting on uniform assays (6 studies; 5526 patients) gave different results.79 The only major study showing a different effect was JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Rosuvastatin therapy raised Lp(a) by 10% in a study of 9612 multi-ethnic patients (7746 Caucasian). Lp(a) concentrations (median (25–75th centile); nmol/L) were highest in blacks (60 (34–100)), then Asians (38 (18–60)), Hispanics (24 (11–46)), and whites (23 (10–50)).80 The median change in Lp(a) with rosuvastatin was zero, but statin therapy resulted in a positive shift in the Lp(a) distribution (P<0.0001). Rosuvastatin reduced CVD by 38% (HR 0.62 (0.43–0.90)) with Lp(a) above the median and 54% in those with Lp(a) below the median (HR 0.46 (0.30–0.72)), with no evidence of interaction.80 The effect of statins on Lp(a) is likely neutral but may be confounded by insulin resistance, or isotype distribution.
Other Established Lipid-Lowering Drugs
A meta-analysis (16 studies; 1388 patients) showed fibrates have a small effect in reducing Lp(a) (2.7 (0.8–4.5) mg/dl) which is increased in combination therapy with statins.81 Drugs with a primary site of action in the gut such as ezetimibe (an enterocyte-acting Niemann Pick-C1-like protein 1(NPC1L1) antagonist)82 have no effect on Lp(a) and the bile acid sequestrant cholestyramine showed similar effects in the Lipid Research Clinics study.83 There are little data on the effects of bempedoic acid (a hepatic acid-citrate lyase inhibitor) on Lp(a).84
Niacin and Lp(a)
A meta-analysis of studies (14 studies; n=14,375) with niacin have shown that this treatment reduces Lp(a) by 23 (19–27%).85 A crossover design lipoprotein turnover study in 8 patients treated with extended-release niacin showed it decreased triglycerides by 46%, raised HDL-C by 20%, and decreased apo(a) concentrations by 20%. It decreased apoB100 by 22% and PCSK9 levels by −29%. Apo(a) production rates were decreased by 50% and fractional catabolic rate by 37%.86 A further study of niacin treatment showed reductions in Lp(a) and VLDL-apoE absolute production rate were correlated (r=0.83; P=0.015). In contrast, PCSK9 reduction (−35%; P=0.008) was only correlated with that of VLDL-apoE absolute production rate (r=0.79; P=0.028).35
Coronary and carotid artery regression studies show the predicted effect of baseline Lp(a) on risk of progression but no effect of niacin therapy in post hoc median-split analyses where diabetes, metabolic syndrome and nonHDL-cholesterol predicted effects.87 In outcome studies, Lp(a) was not measured in the Coronary Drug Project but it was measured in the AIM-HIGH88 and HPS/THRIVE studies.89 The increased risk of events with baseline Lp(a) was seen in both studies. Niacin has multiple beneficial effects on lipid profiles including on Lp(a) but neither study showed any reduction in CVD events. A pre-specified analysis of HPS2/THRIVE investigated the effect of niacin-laropiprant on Lp(a) and CVD risk.90 Niacin therapy reduced mean Lp(a) by 12 (SE, 1) nmol/L overall and 34(6) nmol/L in the top quintile of Lp(a)(>128nmol/L). The mean reduction in Lp(a) with niacin was 31% but varied with predominant apolipoprotein(a) isoform size (PTrend=4×10−29) but was only 18% in the highest quintile (Lp(a)>128nM) with low isoform size.90
PCSK9 Inhibitors and Lp(a)
PCSK9 inhibitors, like statins, modulate expression of the LDL receptor. However, the 2 drug classes have different effects on Lp(a). Meta-analysis of the effects of PCSK9 inhibitors (41 studies; n=64,107) showed reduced Lp(a) by 28% in contrast to the lack of effect of statins.91 Lipoprotein turnover studies with PCSK9 inhibitors have shown confusing effects with a reduction in particle synthesis with monotherapy but changing to increased catabolism in combination with statins.92
Subgroup analyses of the FOURIER and ODYSSEY-Outcomes trials showed that higher baseline Lp(a) predicted CVD risk and thus was associated with a greater absolute risk of CVD reduction. In the FOURIER trial, in 27,564 patients with CVD, evolocumab reduced Lp(a) by a median 27 (6–47)%.93 The change in Lp(a) of median 25 (6–47)nmol/L correlated with change in LDL-C (r=0.37; p<0.001). Evolocumab reduced CVD events by 23% (HR 0.77 (0.67–0.88)) in patients with above median Lp(a) and by 7% (HR 0.93 (0.80–1.08); Pinteraction=0.07) in those below the median level. The higher baseline risk of 2.49% vs 0.95% and the greater absolute risk reductions translate to a number needed to treat over 3 years (NNT3) of 40 vs 105 individuals.93 The 25 nmol/L (12 mg/dL) reduction in Lp(a) corresponded to a 15% reduction in CVD events.94
Data from 4 Phase 3 trials with evolucumab comprising 895 patients showed heterogeneity between LDL-C and Lp(a) effects.95 Concordance was defined as LDL-C reduction >35% and Lp(a) reduction >10%. A discordant response was observed in 20% of patients with a higher prevalence in those with baseline Lp(a) concentrations >30mg/dL (26.5%) or >50 mg/dL (28.6%).95
The ODYSSEY-Outcomes study randomized 18,924 patients to alirocumab or placebo and followed them for 2.8 years. Baseline Lp(a) levels were 21 (7–60)mg/dl and predicted CVD events.96 Alirocumab reduced Lp(a) by 5.0 (0–14) mg/dl, LDL-C by 51 (34–67)mg/dl, and reduced CVD events by 15% (HR 0.85 (0.78–0.93)). Alirocumab-induced reductions of Lp(a) and LDL-C independently predicted lower risk of CVD events, after adjustment for baseline concentrations, demographics and clinical characteristics. In a further analysis, ODYSSEY-Outcomes higher baseline Lp(a) levels were associated with a greater reduction in CVD events with alirocumab (HR Ptrend=0.045).97 A 1mg/dl reduction in Lp(a) with alirocumab was associated with a 0.6% reduction in CVD events96 while a 5mg/dL reduction in Lp(a) predicted a 2.5% reduction in CVD events.97
A post hoc analysis of the ODYSSEY program (10 trials) also showed discordance between Lp(a) and LDL-C responses. The total prevalence of discordant LDL-C/Lp(a) responses was 13% with LDL-C>35% reduction and Lp(a)<10% reduction; and 9% with LDL-C<35% reduction and Lp(a)>10% reduction.98
Little data are available, as yet, on Inclisiran -a modified siRNA targeting apo(a). In the ORION-1 study with 501 participants a single dose of inclisiran reduced apo B, non-HDL-C, and VLDL-cholesterol over 210 days. A second dose of inclisiran delivered additional lipid lowering. Inclisiran with reduced LDL-C and apoB similar to PCSK9 antibody therapies and Lp(a) reductions of 15–25% were obtained.99
Antisense Therapy to apoB
Mipomersen, a second-generation antisense oligonucleotide against apo-B100 was approved to treat homozygous FH (HoFH) but has now been withdrawn for commercial reasons. It has shown consistent effects in reducing Lp(a).100 Meta-analysis of all four phase 3 randomized trials including 382 patients found that mipomersen reduced Lp(a) by 26%.101 A lipoprotein turnover study in 14 healthy individuals using 150mg mipomersen showed a 21% reduction in Lp(a) driven by a 27% increase in the fractional catabolic rate, but no change in the production rate.102
Cholesterol Ester Transfer Protein Inhibitors
Cholesterol ester transfer protein (CETP) inhibitors reduce Lp(a) levels. Analysis of the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial showed that Lp(a) was dose-dependently increased with increasing atorvastatin doses during optimization. Torcetrapib therapy decreased Lp(a) by 11%.103 Evacetrapib decreased Lp(a) by up to −40% with evacetrapib 500 mg in dose ranging studies while evacetrapib combined with statins reduced Lp(a) by 31%.104 Dalcetrapib had lesser effects on lipids than torcetrapib, evacetrapib or anacetrapib and decreased Lp(a) by 5%.32
A lipoprotein turnover study investigated the effects of anacetrapib, statin or combined therapy. Anacetrapib treatment reduced Lp(a) by 34% (P<0.001). The decreases in Lp(a) levels were caused by a 41% reduction in the apo(a) production rate, with no effects on fractional catabolic rate.105
Specific Therapies for apo(a)
All the lipoprotein turnover data suggest that intervention on production rate of apo(a) is likely to reduce Lp(a). Antisense technology has been applied to apo(a) as a method of delivering a specific effect. The effect of ISIS 144367, a 2nd generation ASO to apo(a) was initially investigated in transgenic mouse models overexpressing human apo(a). It produced a decrease of 20, 30, and 86% in the three different models, respectively.106 ISIS 144367 was optimized and the new ASO called ISIS-APO(a)Rx was tested in cynomolgus monkeys achieving a reduction of 97% in hepatic apolipoprotein(a) mRNA of 90% in Lp(a) at the highest dose.107
The Phase 1 study of ISIS-APO(a)Rx in man (n=47) assigned 16 patients to single-dose treatment and 31 were assigned to a multi-dose cohort treated for 4 weeks (Table 3). After 36 days all 3 treated groups showed 100mg ISIS-APO(a)Rx reduced Lp(a) by 40%, 200mg 59%, and 300mg decreased Lp(a) by 79%. No serious adverse events were seen but one patient stopped due to injection site reaction and another stopped due to flu-like symptoms.108
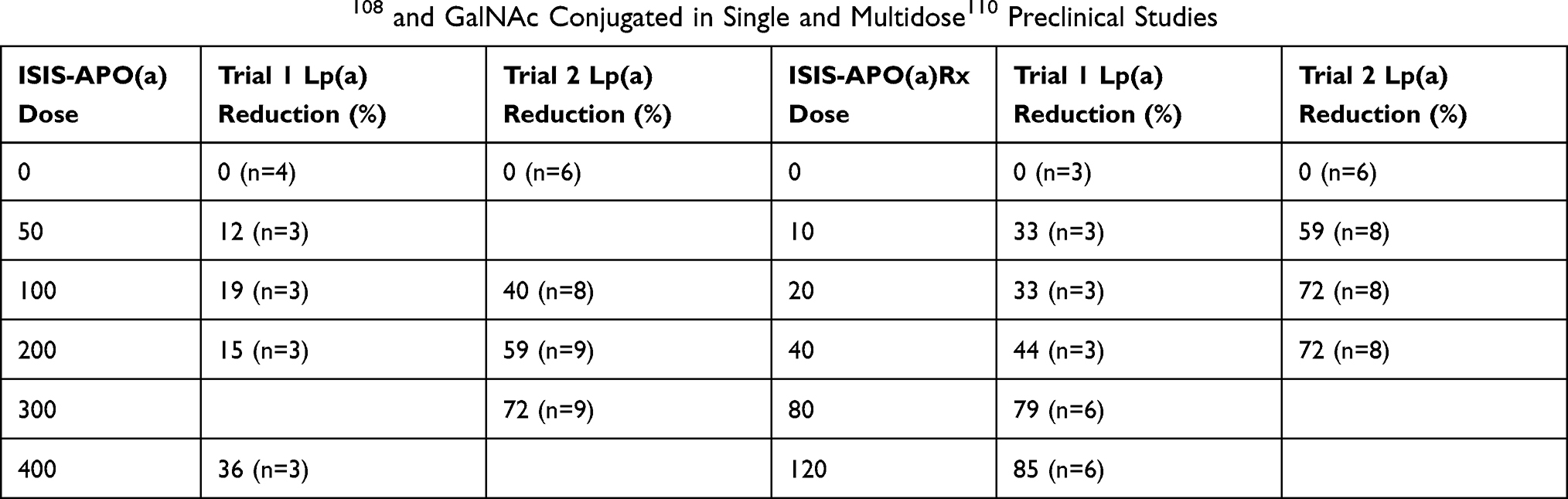 |
Table 3 Efficacy of Antisense108 and GalNAc Conjugated in Single and Multidose110 Preclinical Studies |
The Phase 2 study of ISIS-APO(a)Rx included 64 individuals with 51 assigned to cohort A with baseline Lp(a) levels from 50–175mg/dL (125–437 nmol/L) and 13 individuals (cohort B) had baseline Lp(a) levels >175 mg/dL (≥ 438 nmol/L).109 Both cohorts received 100 mg weekly for 4 weeks, then 200 mg for 4 weeks, and last 300 mg for 4 weeks.109 Levels of Lp(a) decreased by 67% for individuals in cohort A and 72% in cohort B, measured on day 85 or 99. No severe adverse events were recorded; but 2 individuals suffered a myocardial infarction, one in the placebo arm and one after a single dose of ISIS-APO(a)Rx. Regarding mild adverse events, 10% in cohort A and 19% in cohort B had injection site reactions.
Further modification of ISIS-APO(a)Rx (renamed IONIS/AKCEA-APO(a)-LRx; pelacarsen) involved adding a triantennary N-acetylgalactosamine (GalNAc) to induce high plasma clearance through the hepatocyte asialoglycoprotein (ASGP) receptor. A phase 1/2A dose-ranging study recruited 58 healthy volunteers assigning 28 to a single-dose cohort and 30 to a multiple-dose cohort (Table 1).110 Lp(a) was decreased by 59% in the 10mg group, 72% in the 20 mg group, and 72% in the 40 mg group at day 36. No serious adverse events were recorded, and no injection site reactions, bleeds, or changes in liver parameters were seen.
The phase 2 study of pelacarsen (ISIS 681257 now AKCEA-APO(a)-LRx; TQJ230) in 286 patients with CVD and Lp(a)>60 mg/dL (127nmol/L), divided them into 5 cohorts and a placebo group.111 At 6 months, the 20mg/4 weeks group showed a decrease in Lp(a) of 35%; 40mg/4 weeks achieved 56%; 60mg/4 weeks 72%; while 20mg/2 weeks reduced Lp(a) by 58%; and 20mg/week 80%. The most frequent adverse event was an injection site reaction that occurred in 26% of individuals.
Other siRNA-based therapies (ARC-LPa as known as AMG890 and SLN360) targeting LPA RNA are also in early development. These have just started phase 1 and 2 studies in man.112 Structural work on the binding properties of Kringle domains has led to the development of AZ-005 which inhibits the function of K-IV10 domain and this small molecule inhibitor may proceed to human trials.113
Cardiovascular Outcome Studies for Lp(a)
All therapies licensed to date either have minimal effects on Lp(a) or have multiple actions on other parts of the lipid profile including LDL-C (PCSK-9 inhibitors); triglycerides and HDL-C (niacin). The development of specific therapies for Lp(a) means it is now possible to investigate whether intervention specifically on this risk factor will translate into clinical benefits. The first question that needs to be addressed is how to deliver sufficient statistical power to answer the question. Data from HPS-THRIVE suggested that Lp(a) reductions were predicted to reduce CAD risk by ≈2% overall and 6% in the top quintile by Lp(a) levels, so new therapies needed to reduce Lp(a) levels by >80nmol/L to produce worthwhile benefits.90 A Mendelian randomization study showed that a 102mg/dl (≈260nmol/L) reduction would reproduce the effects seen with a 1mmol/L reduction in LDL-C based on a 10mg/dl (≈25nmol/L) translating to a 6% reduction in CVD events using a genetic risk score.49 Patients with Lp(a) levels>100nmol/L account for 5.7% of CVD events in the UK Biobank cohort, so recruiting patients with CVD and Lp(a) >175nmol/L may reduce CVD risk by 20%, assuming causality, if the intervention reduces Lp(a) by 80%.114
The second question is whether the effects of LDL-C and Lp(a) on CVD outcomes are independent. Data from observational studies suggest that Lp(a) may not have a significant effect in driving CVD risk if LDL-C<2mmol/L.115 Data from a study of 2769 patients with possible CVD who had coronary angiography with Lp(a) 16mg/dl (>30 mg/dl in 38%) showed elevated Lp(a) was associated with a 2.3 (1.7–3.2) fold likelihood of significant angiographic stenosis (P<0.001) and 1.5 (1.3–1.7) fold chance of 3-vessel disease. Lp(a) levels were related to CVD outcomes in patients with LDL-C 70–100 mg/dl (1.8–2.4mmol/L) (P=0.05) and >100mg/dl (2.4mmol/L) (P=0.02), but not in those with LDL-C<70mg/dl (1.8mmol/L) (p=0.77).115 Current guidelines all suggest that patients with established CVD should attain LDL-C<2mmol/L70 (or be on highest dose of potent statin likely to have similar effects).116 Yet recent studies in patients with acute coronary syndromes with ezetimibe and PCSK9 inhibitors showed that LDL-C 1.6mmol/L was attained with ezetimibe117 and 0.9mmol/L with PCSK9 inhibitors118,119 with further benefits in CVD outcomes. This has led the ESC to suggest a target LDL-C of 1.4 mmol/L.68 Whether and to what extent Lp(a) will remain a significant risk factor in many patients after attainment of such low LDL-C remains unclear.
A CVD outcomes trial is underway with an Lp(a) reducing therapy (https://clinicaltrials.gov/ct2/show/NCT04023552). The trial aims to recruit 7680 patients with established CVD, likely a treated LDL-C <2mmol/L, and an Lp(a)>70mg/dl (≈175nmol/L). Patients had to have had a CHD or CVA event within 0.25–10yrs or have PAD. The trial is designed for a follow-up of 4 yrs. A pre-specified analysis will also be conducted in patients with Lp(a)>90mg/dL.
Conclusion
Assessment of Lp(a) levels may be useful, yet standardized measurement of Lp(a) concentrations remains a challenge which restricts and complicates inferences made across studies, intervention trials, and the usefulness of measurement in routine clinical practice. There is a clear consensus that elevated Lp(a) levels are associated with increased risk of CVD events and may aid reclassification or review intervals. However, detailed health economic based policy recommendations for identifying exact threshold levels and which high-risk subgroups should be targeted or screened, remain to be developed.44 Recent CVD epidemiology previously showed enthusiasm for homocysteine measurement which had similar evidence for reclassification, but clinical trials failed to show any benefit on intervention. Modern lipid management of CVD is becoming more aggressive and targeting lower LDL-C levels. Whether reducing Lp(a) has any role once LDL-C has been optimally controlled, remains unclear. The results of ongoing intervention trials which target Lp(a) reduction with concomitant effects on other lipid sub-fractions, are eagerly awaited. These trials will help clarify the role of Lp(a) in atherosclerotic CVD.
Disclosure
Dr Adie Viljoen reports personal fees, non-financial support from Novo Nordisk, personal fees from Novartis, non-financial support from Lilly, personal fees from Boehringer Ingelheim, grants, personal fees from Sanofi, personal fees from Napp, personal fees from Novartis, personal fees from Pfizer, personal fees, non-financial support from Astra Zeneca, outside the submitted work. Professor Anthony S Wierzbicki is: Site clinical trial investigator: volanesorsen for Akcea, Site clinical trial investigator: Evinacumab for Regeneron, Site clinical trial investigator: Evolocumab for Amgen, outside the submitted work. The authors report no other conflicts in this work.
References
1. Scanu AM, Nakajima K, Edelstein C. Apolipoprotein(a): structure and biology. Front Biosci. 2001;6:D546–D554.
2. Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57(8):1339–1359. doi:10.1194/jlr.R067314
3. Utermann G, Weber W. Protein composition of Lp(a) lipoprotein from human plasma. FEBS Lett. 1983;154(2):357–361. doi:10.1016/0014-5793(83)80182-3
4. Utermann G, Menzel HJ, Kraft HG, Duba HC, Kemmler HG, Seitz C. Lp(a) glycoprotein phenotypes. Inheritance and relation to Lp(a)-lipoprotein concentrations in plasma. JClinInvest. 1987;80(2):458–465. doi:10.1172/JCI113093
5. Utermann G, Kraft HG, Menzel HJ, Hopferwieser T, Seitz C. Genetics of the quantitative Lp(a) lipoprotein trait. I. Relation of LP(a) glycoprotein phenotypes to Lp(a) lipoprotein concentrations in plasma. Hum Genet. 1988;78(1):41–46.
6. Utermann G, Duba C, Menzel HJ. Genetics of the quantitative Lp(a) lipoprotein trait. II. Inheritance of Lp(a) glycoprotein phenotypes. Hum Genet. 1988;78(1):47–50.
7. Sandholzer C, Hallman DM, Saha N, et al. Effects of the apolipoprotein(a) size polymorphism on the lipoprotein(a) concentration in 7 ethnic groups. Hum Genet. 1991;86(6):607–614.
8. Lackner C, Boerwinkle E, Leffert CC, Rahmig T, Hobbs HH. Molecular basis of apolipoprotein (a) isoform size heterogeneity as revealed by pulsed-field gel electrophoresis. J Clin Invest. 1991;87(6):2153–2161. doi:10.1172/JCI115248
9. Scanu AM. Lipoprotein(a). Link between structure and pathology. Ann Epidemiol. 1992;2(4):407–412.
10. Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Pentanucleotide repeat polymorphism, lipoprotein(a) levels, and risk of ischemic heart disease. J Clin Endocrinol Metab. 2008;93(10):3769–3776. doi:10.1210/jc.2008-0830
11. Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–2853. doi:10.1093/eurheartj/ehq386
12. Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res. 2016;57(11):1953–1975. doi:10.1194/jlr.R071233
13. Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–2528. doi:10.1056/NEJMoa0902604
14. Paquette M, Bernard S, Baass A. SLC22A3 is associated with lipoprotein (a) concentration and cardiovascular disease in familial hypercholesterolemia. Clin Biochem. 2019;66:44–48. doi:10.1016/j.clinbiochem.2019.02.008
15. Trinder M, Uddin MM, Finneran P, Aragam KG, Natarajan P. Clinical utility of Lipoprotein(a) and LPA genetic risk score in risk prediction of incident atherosclerotic cardiovascular disease. JAMA Cardiol. 2020;6(3):287–295. doi:10.1001/jamacardio.2020.5398
16. Bourgeois R, Girard A, Perrot N, et al. A comparative analysis of the Lipoprotein(a) and low-density lipoprotein proteomic profiles combining mass spectrometry and Mendelian randomization. CJC Open. 2021;3(4):450–459. doi:10.1016/j.cjco.2020.11.019
17. Hoekstra M, Chen HY, Rong J, et al. Genome-wide association study highlights APOH as a novel locus for Lipoprotein(a) levels-brief report. Arterioscler Thromb Vasc Biol. 2021;41(1):458–464. doi:10.1161/ATVBAHA.120.314965
18. Frischmann ME, Ikewaki K, Trenkwalder E, et al. In vivo stable-isotope kinetic study suggests intracellular assembly of lipoprotein(a). Atherosclerosis. 2012;225(2):322–327. doi:10.1016/j.atherosclerosis.2012.09.031
19. Becker L, Nesheim ME, Koschinsky ML. Catalysis of covalent Lp(a) assembly: evidence for an extracellular enzyme activity that enhances disulfide bond formation. Biochemistry. 2006;45(32):9919–9928. doi:10.1021/bi060283t
20. Koschinsky ML, Marcovina SM. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol. 2004;15(2):167–174.
21. Hoppichler F, Kraft HG, Sandholzer C, Lechleitner M, Patsch JR, Utermann G. Lipoprotein(a) is increased in triglyceride-rich lipoproteins in men with coronary heart disease, but does not change acutely following oral fat ingestion. Atherosclerosis. 1996;122(1):127–134. doi:10.1016/0021-9150(96)05803-0
22. Moriarty PM, Varvel SA, Gordts PL, McConnell JP, Tsimikas S. Lipoprotein(a) mass levels increase significantly according to APOE genotype: an analysis of 431,239 patients. Arterioscler Thromb Vasc Biol. 2017;37(3):580–588. doi:10.1161/ATVBAHA.116.308704
23. McCormick SPA, Schneider WJ. Lipoprotein(a) catabolism: a case of multiple receptors. Pathology. 2019;51(2):155–164. doi:10.1016/j.pathol.2018.11.003
24. Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692–711. doi:10.1016/j.jacc.2016.11.042
25. Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57(5):745–757. doi:10.1194/jlr.R060582
26. Tsimikas S. Lipoprotein(a): novel target and emergence of novel therapies to lower cardiovascular disease risk. Curr Opin Endocrinol Diabetes Obes. 2016;23(2):157–164. doi:10.1097/MED.0000000000000237
27. Edelstein C, Pfaffinger D, Hinman J, et al. Lysine-phosphatidylcholine adducts in kringle V impart unique immunological and potential pro-inflammatory properties to human apolipoprotein(a). J Biol Chem. 2003;278(52):52841–52847. doi:10.1074/jbc.M310425200
28. Berliner JA, Leitinger N, Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009;50(Suppl):S207–12. doi:10.1194/jlr.R800074-JLR200
29. Scipione CA, Sayegh SE, Romagnuolo R, et al. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a). J Lipid Res. 2015;56(12):2273–2285. doi:10.1194/jlr.M060210
30. van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611–624. doi:10.1161/CIRCULATIONAHA.116.020838
31. Tsimikas S, Clopton P, Brilakis ES, et al. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: results from the Dallas Heart Study. Circulation. 2009;119(13):1711–1719. doi:10.1161/CIRCULATIONAHA.108.836940
32. Schwartz GG, Ballantyne CM, Barter PJ, et al. Association of lipoprotein(a) with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-outcomes randomized clinical trial. JAMA Cardiol. 2018;3(2):164–168. doi:10.1001/jamacardio.2017.3833
33. Chan DC, Watts GF, Coll B, Wasserman SM, Marcovina SM, Barrett PHR. Lipoprotein(a) particle production as a determinant of plasma lipoprotein(a) concentration across varying apolipoprotein(a) isoform sizes and background cholesterol-lowering therapy. J Am Heart Assoc. 2019;8(7):e011781. doi:10.1161/JAHA.118.011781
34. Ma L, Waldmann E, Ooi EMM, et al. Lipoprotein (a) and Low-density lipoprotein apolipoprotein B metabolism following apheresis in patients with elevated lipoprotein(a) and coronary artery disease. Eur J Clin Invest. 2019;49(2):e13053. doi:10.1111/eci.13053
35. Croyal M, Blanchard V, Ouguerram K, et al. VLDL (Very-Low-Density Lipoprotein)-Apo E (Apolipoprotein E) may influence Lp(a) (lipoprotein [a]) synthesis or assembly. Arterioscler Thromb Vasc Biol. 2020;40(3):819–829. doi:10.1161/ATVBAHA.119.313877
36. Mefford MT, Marcovina SM, Bittner V, et al. PCSK9 loss-of-function variants and Lp(a) phenotypes among black US adults. J Lipid Res. 2019;60(11):1946–1952. doi:10.1194/jlr.P119000173
37. Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–423. doi:10.1001/jama.2009.1063.
38. Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–2339. doi:10.1001/jama.2009.801
39. Langsted A, Nordestgaard BG, Kamstrup PR. Elevated lipoprotein(a) and risk of ischemic stroke. J Am Coll Cardiol. 2019;74(1):54–66. doi:10.1016/j.jacc.2019.03.524
40. Gurdasani D, Sjouke B, Tsimikas S, et al. Lipoprotein(a) and risk of coronary, cerebrovascular, and peripheral artery disease: the EPIC-Norfolk prospective population study. Arterioscler Thromb Vasc Biol. 2012;32(12):3058–3065. doi:10.1161/ATVBAHA.112.255521
41. Langsted A, Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4(7):577–587. doi:10.1016/S2213-8587(16)30042-0
42. Wong ND, Cupples LA, Ostfeld AM, Levy D, Kannel WB. Risk factors for long-term coronary prognosis after initial myocardial infarction: the Framingham Study. Am J Epidemiol. 1989;130(3):469–480. doi:10.1093/oxfordjournals.aje.a115360
43. Wong ND, Zhao Y, Sung J, Browne A. Relation of first and total recurrent atherosclerotic cardiovascular disease events to increased lipoprotein(a) levels among statin treated adults with cardiovascular disease. Am J Cardiol. 2021;145:12–17. doi:10.1016/j.amjcard.2020.12.075
44. Kostner KM, Kostner GM, Wierzbicki AS. Is Lp(a) ready for prime time use in the clinic? A pros-and-cons debate. Atherosclerosis. 2018;274:16–22. doi:10.1016/j.atherosclerosis.2018.04.032
45. Pare G, Caku A, McQueen M, et al. Lipoprotein(a) levels and the risk of myocardial infarction among 7 ethnic groups. Circulation. 2019;139(12):1472–1482. doi:10.1161/CIRCULATIONAHA.118.034311
46. Chretien JP, Coresh J, Berthier-Schaad Y, et al. Three single-nucleotide polymorphisms in LPA account for most of the increase in lipoprotein(a) level elevation in African Americans compared with European Americans. J Med Genet. 2006;43(12):917–923. doi:10.1136/jmg.2006.042119
47. Mehta A, Virani SS, Ayers CR, et al. Lipoprotein(a) and family history predict cardiovascular disease risk. J Am Coll Cardiol. 2020;76(7):781–793. doi:10.1016/j.jacc.2020.06.040
48. Ohira T, Schreiner PJ, Morrisett JD, Chambless LE, Rosamond WD, Folsom AR. Lipoprotein(a) and incident ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) study. Stroke. 2006;37(6):1407–1412. doi:10.1161/01.STR.0000222666.21482.b6
49. Burgess S, Ference BA, Staley JR, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a Mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619–627. doi:10.1001/jamacardio.2018.1470
50. Huded CP, Shah NP, Puri R, et al. Association of serum lipoprotein (a) levels and coronary atheroma volume by intravascular ultrasound. J Am Heart Assoc. 2020;9(23):e018023. doi:10.1161/JAHA.120.018023
51. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470–477. doi:10.1016/j.jacc.2013.09.038
52. Capoulade R, Chan KL, Yeang C, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. 2015;66(11):1236–1246. doi:10.1016/j.jacc.2015.07.020
53. Zheng KH, Tsimikas S, Pawade T, et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73(17):2150–2162. doi:10.1016/j.jacc.2019.01.070
54. Tsimikas S. Potential causality and emerging medical therapies for lipoprotein(a) and its associated oxidized phospholipids in calcific aortic valve stenosis. Circ Res. 2019;124(3):405–415. doi:10.1161/CIRCRESAHA.118.313864
55. Wierzbicki AS, Viljoen A, Chambers JB. Aortic stenosis and lipids: does intervention work? Curr Opin Cardiol. 2010;25(4):379–384. doi:10.1097/HCO.0b013e3283393c9b
56. Garg PK, Guan W, Karger AB, Steffen BT, Budoff M, Tsai MY. Lipoprotein (a) and risk for calcification of the coronary arteries, mitral valve, and thoracic aorta: the multi-ethnic study of atherosclerosis. J Cardiovasc Comput Tomogr. 2021;15(2):154–160. doi:10.1016/j.jcct.2020.06.002
57. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526–537. doi:10.1194/jlr.R061648
58. Tsimikas S, Fazio S, Ferdinand KC, et al. NHLBI working group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71(2):177–192. doi:10.1016/j.jacc.2017.11.014
59. Moriarty PM, Yeang C, Varvel SA, McConnell JP, Tsimikas S. Removing the lipoprotein (a) from the LDL-C measurement results in a 38% reduction in prevalence of elevated LDL-C at any threshold: implications for the prevalence and diagnosis of LDL-mediated risk. J Am Coll Cardiol. 2018;71(11):A1783. doi:10.1016/S0735-1097(18)32324-6
60. Seman LJ, DeLuca C, Jenner JL, et al. Lipoprotein(a)-cholesterol and coronary heart disease in the Framingham Heart Study. Clin Chem. 1999;45(7):1039–1046. doi:10.1093/clinchem/45.7.1039
61. Yeang C, Clopton PC, Tsimikas S. Lipoprotein(a)-cholesterol levels estimated by vertical auto profile correlate poorly with Lp(a) mass in hyperlipidemic subjects: implications for clinical practice interpretation of Lp(a)-mediated risk. J Clin Lipidol. 2016;10(6):1389–1396. doi:10.1016/j.jacl.2016.09.012
62. Konerman M, Kulkarni K, Toth PP, Jones SR. Lipoprotein(a) particle concentration and lipoprotein(a) cholesterol assays yield discordant classification of patients into four physiologically discrete groups. J Clin Lipidol. 2012;6(4):368–373. doi:10.1016/j.jacl.2012.01.004
63. Guadagno PA, Summers Bellin EG, Harris WS, et al. Validation of a lipoprotein(a) particle concentration assay by quantitative lipoprotein immunofixation electrophoresis. Clin Chim Acta. 2015;439:219–224. doi:10.1016/j.cca.2014.10.013
64. Lamon-Fava S, Marcovina SM, Albers JJ, et al. Lipoprotein(a) levels, apo(a) isoform size, and coronary heart disease risk in the Framingham Offspring Study. J Lipid Res. 2011;52(6):1181–1187. doi:10.1194/jlr.M012526
65. Yeang C, Witztum JL, Tsimikas S. Novel method for quantification of lipoprotein(a)-cholesterol: implications for improving accuracy of LDL-C measurements. J Lipid Res. 2021;62:100053. doi:10.1016/j.jlr.2021.100053
66. Fatica EM, Meeusen JW, Vasile VC, Jaffe AS, Donato LJ. Measuring the contribution of Lp(a) cholesterol towards LDL-C interpretation. Clin Biochem. 2020;86:45–51. doi:10.1016/j.clinbiochem.2020.09.007
67. Nordestgaard BG, Langlois MR, Langsted A, et al. Quantifying atherogenic lipoproteins for lipid-lowering strategies: consensus-based recommendations from EAS and EFLM. Atherosclerosis. 2020;294:46–61. doi:10.1016/j.atherosclerosis.2019.12.005
68. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2019;41(1):111–188. doi:10.1093/eurheartj/ehz455
69. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374–392. doi:10.1016/j.jacl.2019.04.010
70. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. 2019;139(25):e1082–e1143. doi:10.1161/CIR.0000000000000625
71. Cegla J, Neely RDG, France M, et al. HEART UK consensus statement on Lipoprotein(a): a call to action. Atherosclerosis. 2019;291:62–70. doi:10.1016/j.atherosclerosis.2019.10.011
72. Gencer B, Kronenberg F, Stroes ES, Mach F. Lipoprotein(a): the revenant. Eur Heart J. 2017;38(20):1553–1560. doi:10.1093/eurheartj/ehx033
73. Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36(11):2239–2245. doi:10.1161/ATVBAHA.116.308011
74. Kronenberg F. Human genetics and the causal role of lipoprotein(a) for various diseases. Cardiovasc Drugs Ther. 2016;30(1):87–100. doi:10.1007/s10557-016-6648-3
75. Franchini M, Capuzzo E, Liumbruno GM. Lipoprotein apheresis for the treatment of elevated circulating levels of lipoprotein(a): a critical literature review. Blood Transfus. 2016;14(5):413–418. doi:10.2450/2015.0163-15
76. Roeseler E, Julius U, Heigl F, et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36(9):2019–2027. doi:10.1161/ATVBAHA.116.307983
77. Safarova MS, Ezhov MV, Afanasieva OI, et al. Effect of specific lipoprotein(a) apheresis on coronary atherosclerosis regression assessed by quantitative coronary angiography. Atheroscler Suppl. 2013;14(1):93–99. doi:10.1016/j.atherosclerosissup.2012.10.015
78. Wang X, Li J, Ju J, Fan Y, Xu H. Effect of different types and dosages of statins on plasma lipoprotein(a) levels: a network meta-analysis. Pharmacol Res. 2021;163:105275. doi:10.1016/j.phrs.2020.105275
79. Tsimikas S, Gordts P, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41(24):2275–2284. doi:10.1093/eurheartj/ehz310
80. Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129(6):635–642. doi:10.1161/CIRCULATIONAHA.113.004406
81. Sahebkar A, Simental-Mendia LE, Watts GF, et al. Comparison of the effects of fibrates versus statins on plasma lipoprotein(a) concentrations: a systematic review and meta-analysis of head-to-head randomized controlled trials. BMC Med. 2017;15(1):22. doi:10.1186/s12916-017-0787-7
82. Sahebkar A, Simental-Mendia LE, Pirro M, et al. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: a systematic review and meta-analysis of randomized controlled trials. Sci Rep. 2018;8(1):17887. doi:10.1038/s41598-018-36204-7
83. Schaefer EJ, Lamon-Fava S, Jenner JL, et al. Lipoprotein(a) levels and risk of coronary heart disease in men. The lipid research clinics coronary primary prevention trial. JAMA. 1994;271(13):999–1003. doi:10.1001/jama.1994.03510370051031
84. Wang X, Luo S, Gan X, He C, Huang R. Safety and efficacy of ETC-1002 in hypercholesterolaemic patients: a meta-analysis of randomised controlled trials. Kardiol Pol. 2019;77(2):207–216. doi:10.5603/KP.a2019.0013
85. Sahebkar A, Reiner Z, Simental-Mendia LE, Ferretti G, Cicero AF. Effect of extended-release niacin on plasma lipoprotein(a) levels: a systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism. 2016;65(11):1664–1678. doi:10.1016/j.metabol.2016.08.007
86. Croyal M, Ouguerram K, Passard M, et al. Effects of extended-release nicotinic acid on apolipoprotein (a) kinetics in hypertriglyceridemic patients. Arterioscler Thromb Vasc Biol. 2015;35(9):2042–2047. doi:10.1161/ATVBAHA.115.305835
87. Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110(23):3512–3517. doi:10.1161/01.CIR.0000148955.19792.8D
88. Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. doi:10.1056/NEJMoa1107579
89. HPS THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–212. doi:10.1056/NEJMoa1300955.
90. Parish S, Hopewell JC, Hill MR, et al. Impact of apolipoprotein(a) isoform size on lipoprotein(a) lowering in the HPS2-THRIVE study. Circ Genom Precis Med. 2018;11(2):e001696. doi:10.1161/CIRCGEN.117.001696
91. Farmakis I, Doundoulakis I, Pagiantza A, et al. Lipoprotein(a) reduction with proprotein convertase subtilisin/kexin type 9 inhibitors: a systematic review and meta-analysis. J Cardiovasc Pharmacol. 2021;77(3):397–407. doi:10.1097/FJC.0000000000000963
92. Watts GF, Chan DC, Somaratne R, et al. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur Heart J. 2018;39:2577–2585. doi:10.1093/eurheartj/ehy122
93. O’Donoghue ML, Fazio S, Giugliano RP, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139(12):1483–1492. doi:10.1161/CIRCULATIONAHA.118.037184
94. Ruscica M, Greco MF, Ferri N, Corsini A. Lipoprotein(a) and PCSK9 inhibition: clinical evidence. Eur Heart J. 2020;22(SupplL):L53–L56. doi:10.1093/eurheartj/suaa135
95. Shapiro MD, Minnier J, Tavori H, et al. Relationship between low-density lipoprotein cholesterol and lipoprotein(a) lowering in response to PCSK9 inhibition with evolocumab. J Am Heart Assoc. 2019;8(4):e010932. doi:10.1161/JAHA.118.010932
96. Bittner VA, Szarek M, Aylward PE, et al. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75(2):133–144. doi:10.1016/j.jacc.2019.10.057
97. Szarek M, Bittner VA, Aylward P, et al. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur Heart J. 2020;41(44):4245–4255. doi:10.1093/eurheartj/ehaa649
98. Mahmood T, Minnier J, Ito MK, et al. Discordant responses of plasma low-density lipoprotein cholesterol and lipoprotein(a) to alirocumab: a pooled analysis from 10 ODYSSEY phase 3 studies. Eur J Prev Cardiol. 2020;28(8):816–822. . doi:10.1177/2047487320915803.
99. Ray KK, Stoekenbroek RM, Kallend D, et al. Effect of an siRNA therapeutic targeting PCSK9 on atherogenic lipoproteins: prespecified secondary end points in ORION 1. Circulation. 2018;138(13):1304–1316. doi:10.1161/CIRCULATIONAHA.118.034710
100. Fogacci F, Ferri N, Toth PP, Ruscica M, Corsini A, Cicero AFG. Efficacy and safety of mipomersen: a systematic review and meta-analysis of randomized clinical trials. Drugs. 2019;79(7):751–766. doi:10.1007/s40265-019-01114-z
101. Santos RD, Raal FJ, Catapano AL, Witztum JL, Steinhagen-Thiessen E, Tsimikas S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arterioscler Thromb Vasc Biol. 2015;35(3):689–699. doi:10.1161/ATVBAHA.114.304549
102. Nandakumar R, Matveyenko A, Thomas T, et al. Effects of mipomersen, an apolipoprotein B100 antisense, on lipoprotein (a) metabolism in healthy subjects. J Lipid Res. 2018;59(12):2397–2402. doi:10.1194/jlr.P082834
103. Arsenault BJ, Petrides F, Tabet F, et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J Clin Lipidol. 2018;12(1):130–136. doi:10.1016/j.jacl.2017.10.001
104. Nicholls SJ, Ruotolo G, Brewer HB, et al. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J Clin Lipidol. 2016;10(3):519–527 e4. doi:10.1016/j.jacl.2015.11.014
105. Thomas T, Zhou H, Karmally W, et al. CETP (Cholesteryl Ester Transfer Protein) inhibition with anacetrapib decreases production of lipoprotein(a) in mildly hypercholesterolemic subjects. Arterioscler Thromb Vasc Biol. 2017;37(9):1770–1775. doi:10.1161/ATVBAHA.117.309549
106. Merki E, Graham MJ, Mullick AE, et al. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118(7):743–753. doi:10.1161/CIRCULATIONAHA.108.786822
107. Graham MJ, Viney N, Crooke RM, Tsimikas S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J Lipid Res. 2016;57(3):340–351. doi:10.1194/jlr.R052258
108. Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386(10002):1472–1483. doi:10.1016/S0140-6736(15)61252-1
109. Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239–2253. doi:10.1016/S0140-6736(16)31009-1
110. Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Safety and efficacy of AKCEA-APO(a)-LRx to lower lipoprotein(a) levels in patients with established cardiovascular disease: a phase 2 dose-ranging trial American Heart Association: scientific sessions 2018; 2021. Available from: https://www.abstractsonline.com/pp8/#!/4682/presentation/59761.
111. Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382(3):244–255. doi:10.1056/NEJMoa1905239
112. Swerdlow DI, Rider DA, Yavari A, Lindholm MW, Campion GV, Nissen SE. Treatment and prevention of lipoprotein(a)-mediated cardiovascular disease: the emerging potential of RNA interference therapeutics. Cardiovasc Res. 2021;cvab100. doi:10.1093/cvr/cvab100
113. Sandmark J, Tigerstrom A, Akerud T, et al. Identification and analyses of inhibitors targeting apolipoprotein(a) kringle domains KIV-7, KIV-10, and KV provide insight into kringle domain function. J Biol Chem. 2020;295(15):5136–5151. doi:10.1074/jbc.RA119.011251
114. Welsh P, Welsh C, Celis-Morales CA, et al. Lipoprotein(a) and cardiovascular disease: prediction, attributable risk fraction, and estimating benefits from novel interventions. Eur J Prev Cardiol. 2020;zwaa063. doi:10.1093/eurjpc/zwaa063
115. Nicholls SJ, Tang WH, Scoffone H, et al. Lipoprotein(a) levels and long-term cardiovascular risk in the contemporary era of statin therapy. J Lipid Res. 2010;51(10):3055–3061. doi:10.1194/jlr.M008961
116. Rabar S, Harker M, O’Flynn N, Wierzbicki AS; Guideline Development Group. Lipid modification and cardiovascular risk assessment for the primary and secondary prevention of cardiovascular disease: summary of updated NICE guidance. BMJ. 2014;349:g4356. doi:10.1136/bmj.g4356.
117. Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372(25):2387–2397. doi:10.1056/NEJMoa1410489
118. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–1722. doi:10.1056/NEJMoa1615664
119. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097–2107. doi:10.1056/NEJMoa1801174
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.