Back to Journals » International Journal of Nanomedicine » Volume 15
Electrosprayed Polymeric Nanospheres for Enhanced Solubility, Dissolution Rate, Oral Bioavailability and Antihyperlipidemic Activity of Bezafibrate
Authors Sun R, Shen C, Shafique S, Mustapha O ![]() , Hussain T, Khan IU
, Hussain T, Khan IU ![]() , Mehmood Y
, Mehmood Y ![]() , Anwer K, Shahzad Y
, Anwer K, Shahzad Y ![]() , Yousaf AM
, Yousaf AM ![]()
Received 18 October 2019
Accepted for publication 12 January 2020
Published 31 January 2020 Volume 2020:15 Pages 705—715
DOI https://doi.org/10.2147/IJN.S235146
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Ru Sun, 1 Chengwu Shen, 1 Shumaila Shafique, 2 Omer Mustapha, 2 Talib Hussain, 3 Ikram Ullah Khan, 4 Yasir Mehmood, 4 Khaleeq Anwer, 5 Yasser Shahzad, 3 Abid Mehmood Yousaf 3
1Department of Pharmacy, Shandong Provincial Hospital, Shandong University, Jinan, Shandong 250000, People’s Republic of China; 2Faculty of Pharmaceutical Sciences, Dow College of Pharmacy, Dow University of Health Sciences, Karachi 74200, Pakistan; 3Department of Pharmacy, COMSATS University Islamabad, Lahore 54000, Pakistan; 4Department of Pharmaceutics, Faculty of Pharmaceutical Sciences, Government College University Faisalabad, Faisalabad 38000, Pakistan; 5Office of Chief Executive Officer, District Health Authority, Pakpattan 57400, Pakistan
Correspondence: Abid Mehmood Yousaf; Talib Hussain
Department of Pharmacy, COMSATS University Islamabad, Lahore Campus, Lahore 54000, Pakistan
Tel +92-300-4774147;
+92-345-7220536
Email [email protected]; [email protected]
Background: Bezafibrate is a BCS class II drug as it presents very low solubility in water; therefore, its bioavailability after oral administration is very poor. The aim of this work was to enhance solubility and dissolution rate of bezafibrate in water in order to enhance its oral bioavailability.
Methods: Several formulations were prepared using PVP K30 and Cremophor ELP employing the solvent-evaporation method and the electrospraying technique. Solubility, release rate, bioavailability in male Sprague Dawley rats, and lipid profile attributes in Wistar rats were assessed in comparison with bezafibrate plain powder. Solid-state characterization was carried out using X-ray diffraction (XRD) analysis, differential scanning calorimetry (DSC), Fourier transform infrared (FTIR) spectroscopy and scanning electron microscopy (SEM).
Results: All the formulations exerted positive effect towards the desired goal. In particular, the optimized formulation furnished about 14-fold enhanced solubility and 85.48 ± 10.16% drug was released in 10 min as compared with bezafibrate alone (4.06 ± 2.59%). The drug existed in the amorphous state in the prepared sample as confirmed by XRD and DSC, whilst no drug-excipient interactions were observed through FTIR analysis. Moreover, SEM revealed smooth-surfaced spherical particles of the optimized formulation. A 5.5-fold higher oral bioavailability was achieved with the optimized formulation in comparison with bezafibrate plain powder. Also, TG, LDL and TC were decreased, and HDL was increased considerably in HFD-treated rats.
Conclusion: The optimized formulation consisting of bezafibrate, PVP K30 and cremophor ELP (1/12/1.5, w/w/w) might be a capable drug delivery system for orally administering poorly water-soluble bezafibrate with improved bioavailability and antihyperlipidemic effects.
Keywords: aqueous solubility, bezafibrate, electrospraying, lipid profile, oral bioavailability, solid dispersion
Introduction
Bezafibrate stimulates peroxisome proliferator-activated receptors (PPARs) such as PPARα, PPARϒ and PPARϭ.1 Stimulation of PPARα accelerates production of a high-density lipoprotein (HDL), and reduces triglycerides (TG), and low-density lipoprotein (LDL); therefore, bezafibrate is used in the treatment of hypercholesterolemia and hypertriglyceridemia.2 Activation of PPARϒ ameliorates insulin sensitivity and mitigates hyperglycaemia by enhancing glucose metabolism.1 Stimulation of PPARϭ speeds up biotransformation of fatty acids.1 Bezafibrate also lessens cardiovascular risks such as myocardial infarction in patients suffering from metabolic syndrome.3,4 The recommended daily dose of bezafibrate is 200 mg three times a day, or a dose of 400 mg daily in a sustained-release form can be administered alternatively.5 Bezafibrate is absolutely safe and effective in increasing HDL and decreasing TG.6
Bezafibrate is placed as a class II drug in Biopharmaceutics Classification System (BCS).7 The chemical entities included in BCS category II have low or no solubility in the aqueous media; however, they traverse cell membranes efficiently. The aqueous solubility of bezafibrate is about 1.55 µg/mL.8 The compounds having a solubility <100 µg/mL are practically insoluble in water.9 The gastrointestinal (GI) fluid mainly consists of water. A BCS category II drug is not adequately dissolved in the GI fluid. The drug molecules permeate across cell membranes only when they are in the dissolved state in GI fluid. Due to poor solubility in the GI fluid, a BCS category II drug is not absorbed adequately;10 accordingly, the drug reaching the systemic circulation is insufficient to stimulate the receptors to elicit the desired pharmacological effects. One option to get the optimum oral bioavailability is to increase the dose; however, this usually results in local toxicity, and not favourable economically. Another more appropriate way is to employ a suitable solubility enhancing technique to the drug before administration via the oral route.11
Solubility and oral bioavailability of poorly water-soluble substances can be ameliorated through fabrication of drug delivery systems such as nanoparticles, solid dispersions, inclusion complexes with cyclodextrins, encapsulation with hydrophilic polymers, adsorption on silica, self-emulsifying drug delivery systems (SEDDS), micro-emulsions, nano-emulsions, co-crystallization, co-precipitation and many more.12–14 Solid dispersion is a crystalline or amorphous drug-laden solid polymeric carrier system.15 Ternary type of solid dispersions are superior to binary type solid dispersions as far as amelioration in the aqueous solubility, dissolution rate and oral bioavailability is concerned.16 Solid dispersions can be prepared via the surface-attached method, solvent-wetting method and solvent-evaporation method. Investigators have demonstrated that solvent-evaporation method of solid dispersion preparation is superior to both the surface-attached and solvent-wetting methods.15 In this method, hydrophilic excipients and the drug are completely dissolved in a solvent system in order to obtain an absolutely transparent solution that ensures homogeneous intermingling of all the components at the molecular level and uniformly disseminated drug particles in the polymeric matrix upon drying. This results in high drug content and content uniformity as neither of the components is lost during the preparation process.17 Improvement in solubility and dissolution by the solvent-evaporated solid dispersion is usually a consequence of conversion of the crystalline form of the drug into its amorphous counterpart.18 Polymeric matrices present in this drug delivery system inhibit recrystallization of the drug during evaporation process.19 There are several sophisticated ways to evaporate the solution, for instance, tray-drying, spray-drying, electrospraying, lyophilisation, supercritical fluid technique and so on. Electrospraying is a promising technique that can produce drug-loaded spherical nanoparticles.18,20 Electrospraying has been elegantly performing in the fabrication of polymeric nanoparticles loaded with antibiotics,21–23 fibrates,24 anti-inflammatory drugs25,26 and hormones.27–29 A pharmaceutical nanoparticle is a drug-laden vehicle bearing a dimension of <1000 nm.30,31
In the present work, bezafibrate-loaded nanoparticulated ternary solid dispersions (nanospheres) were prepared with PVP K30 and cremophor ELP via the solvent-evaporation method and the electrospraying technique. Solubility and the release rate of bezafibrate in the solid dispersions were investigated. The crystalline physiognomies were determined using X-ray diffraction (XRD) and differential scanning calorimetric (DSC) methods. Fourier transform infrared (FTIR) spectroscopy was performed for investigating interactions between the drug and hydrophilic excipients. The morphological aspects were studied using scanning electron microscopy (SEM). Oral bioavailability was assessed in Sprague Dawley rats by determining area under the curve (AUC), peak plasma concentration (Cmax) and time to reach peak plasma level (Tmax). Also, concentrations of TG, LDL, HDL and TC were determined in High Fat Diet (HFD)-treated Wistar rats.
Materials and Methods
Materials
Bezafibrate was from Cayman Chemical Co. (Pittsfield Charter Township, MI, USA). Hydroxypropyl methylcellulose (HPMC), hydroxypropyl cellulose (HPC) and polyvinyl alcohol (PVA) were bought from Shin-Etsu Co. (Tokyo, Japan). Carboxymethylcellulose sodium (Na-CMC) and gelatin were purchased from Duksan Chemical Co. (Ansan, South Korea). Dextran, docusate sodium and hyaluronic acid were from Sigma-Aldrich (St. Louis, MO, USA), respectively. Polyvinylpyrrolidone (PVP K30), solutol HS15, poloxamer 188, poloxamer 407 and cremophor ELP were procured from BASF (Ludwigshafen, Germany). Polyethylene glycol 6000 and polyethylene glycol 1540 were acquired from Duksan Chemical Co. (Ansan, South Korea). Sodium lauryl sulphate (SLS), polysorbate 20 (Tween 20), polysorbate 80 (Tween 80), sorbitan monolaurate 20 (Span 20) and sorbitan monooleate 80 (Span 80) were obtained from Daejung Chemical Co. (Siheung, South Korea). Polysorbate 60 (Tween 60) (Croda, Singapore) was purchased from Masung and Co. (South Korea). All other solvents and chemical substances were of reagent grade.
Selection of Hydrophilic Excipients
For choosing the best performing hydrophilic excipients for bezafibrate-loaded solid dispersions, excess of plain drug powder was added to 0.5 mL of 1% (w/v) aqueous solution of each hydrophilic excipient in a 1.5 mL capacity Eppendorf microtube. The mixture was vortexed for a minute, secured on the agitator (100 rpm) in a water-bath (25°C), and left there for 5 days. Then, the tubes were vortexed again and centrifuged at 7000 ×g for 5 min. A 200 μL of supernatant was shifted to another 1.5 mL capacity Eppendorf microtube and diluted with 200 μL of acetonitrile. The diluent (20 μL) was analysed by HPLC (Model 1260 Infinity, Agilent Technologies, CA, USA), which was equipped with a C18 column (4.6 mm I.D. x 150 mm, 5 μm). The mobile phase was comprising of acetonitrile and 0.1% (v/v) aqueous trifluoroacetic acid at a ratio of 40:60 (v/v). The rate of mobile phase elution was set as 1 mL/min. The eluent was monitored at 232 nm for the determination of bezafibrate concentration.32
Preparation of Bezafibrate-Loaded Ternary Solid Dispersions
The solvent-evaporation method and the electrospraying technique were employed for the preparation of solid dispersions.24 The composition of trialed formulations is shown in Table 1. For each formulation, bezafibrate powder was completely dissolved in ethanol. Subsequently, cremophor ELP and PVP K30 were added under constant stirring. The final clear solution was subjected to electrospraying (ESR 100 NanoNC electrospraying assembly; Seoul, South Korea). The solution to be electrosprayed was taken in a glass syringe (Hamilton Co., Reno, NV, USA) whose plunger-tip was made of polytetrafluoroethylene (PTFE). The solution was allowed to flow through a single-lumen nozzle at a rate of 0.4 mL/h. The optimized voltage to split the jet to plume below the Taylor cone was 10.5 kV. The final formulation, nanosphere of solid dispersions, was delicately collected and stored in air-tight Eppendorf microtubes.
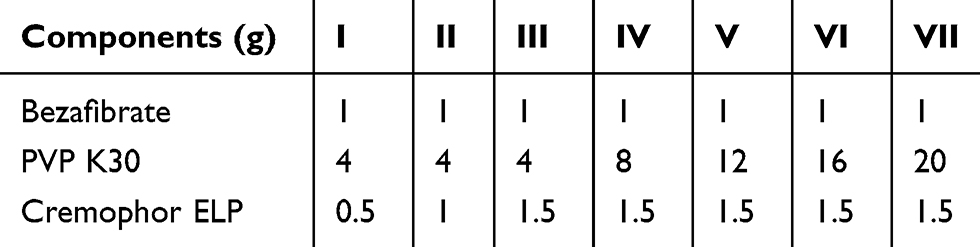 |
Table 1 Compositions of Electrosprayed Ternary Solid Dispersions |
Solubility of Bezafibrate in Electrosprayed Ternary Solid Dispersions
For each bezafibrate-loaded formulation, plenty of powder was added to 0.5 mL distilled water in a 1.5 mL capacity Eppendorf microtube and vortex-mixed. The test was carried out in triplicate for each formulation (n = 3). The microtubes were secured on the agitator in a water-bath (25°C) and agitated (100 rpm) for 5 days. Then, vortex-mixing was performed again and the samples were centrifuged at 7000 ×g for 5 min. A 200 μL of supernatant was shifted to another 1.5 mL capacity Eppendorf microtube and diluted with 200 μL of acetonitrile. Bezafibrate was quantified in each sample by the method described above.
Content of Bezafibrate in Ternary Solid Dispersions
Each bezafibrate-loaded formulation, equivalent to 25 mg drug, was completely dissolved in 250 mL of ethanol in a 250 mL capacity measuring flask resulting in a stock solution of 100 µg/mL concentration. This clear solution was filtered (0.22 μm), suitably diluted with acetonitrile and quantified. The content of bezafibrate in each ternary solid dispersion formulation was calculated using the following formula: BC = BA/BT × 100. Where BC stands for bezafibrate content in the formulation, BA stands for actual amount of bezafibrate in the formulation and BT stands for the theoretical concentration of the diluted sample. For each formulation, the test was performed in triplicate (n = 3).
Release Rate of Bezafibrate from the Electrosprayed Ternary Solid Dispersions
Release rate of bezafibrate from an electrosprayed polymeric formulation was determined using the basket apparatus (Model Vision Classic 6, Hanson Research Co., CA, USA). For each sample, ternary solid dispersion equivalent to 90 mg bezafibrate was sealed in a dialysis bag (12,000–14,000 Da) and enclosed in the basket. The rotating (100 rpm) basket was pulled down into 900 mL of 2% (w/v) aqueous solution of polysorbate 8033 which was pre-warmed to, and maintained at 37 ± 0.5°C. At each pre-decided time interval, 1 mL of release medium was withdrawn using a 1 mL syringe and passed through the syringe filter (0.22 μm). An aliquot of 20 μL was analyzed following the HPLC method as described above. For each formulation, the test was performed in sextuplicate (n = 6). The percent drug released at a specific time point was calculated and a graph was plotted against time.
X-Ray Diffraction
The intensity of crystallinity exhibited by a sample was perused using an XRD analysis setup (D-MAX 2500PC model, Rigaku Corporation, Tokyo, Japan). The instrument was equipped with a CuKα1 monochromatic emission source. The current flow and voltage settings were at 100 mA and 100 kV, respectively. Other scanning attributes such as scan speed, scanning mode and step-size were kept as 10°/min, 2θ and 0.02°/se, respectively. The patterns were recorded in the range of 10–50°.
Differential Scanning Calorimetry
The confirmation of crystalline intensity and other thermal attributes were determined by scanning the samples on a differential scanning calorimeter (DSC-Q20 model, TA Instruments, DE, USA). For each sample, about 5 mg powder was enclosed in an aluminum crucible and lid was snugly fitted to seal the sample. Then, it was placed in the calorimeter against an empty sealed pan of the same material and quality which served as a reference, and gradually heated at the rate of 10°C/min in the presence of 30 mL/min of nitrogen stream. The thermograms were recorded in the range of 30–300°C.
Scanning Electron Microscopy
Morphological aspects of particles such as shape, surface and size were studied using a scanning electron microscope (S 4800 model, Hitachi, Japan). The particles of bezafibrate plain powder and selected bezafibrate-loaded ternary solid dispersion were secured to the metallic stub using a double-side adhesive tape. Before inspecting through SEM, sputter coating (Model K-575-K, EMI-Teck Ion-Sputter Coater) of the samples was done in the presence of 8×10−3 mbar vacuum pressure, 20 mA current and turbo speed of 90%. The coating was performed with platinum in order to make the samples conductive for image production.
Fourier Transform Infrared Spectroscopy
FTIR spectrophotometer (Nicolet 6700 model, PA, USA) was employed for recording spectral features of the samples. For each sample, a little quantity of powder was introduced into the sampling disc below the scanning probe for direct scanning. The spectra were recorded in the range of 400–4000 cm−1. The resolution was set at 2 cm−1.
Bioavailability Assessment in Rats
Animal handling and care – For each sample, six male Sprague Dawley rats (230–280 g) were fasted for about 24 h prior to dose administration via the oral route. The rat-carrying cages were placed in a room with maintained environmental conditions of temperature (22–26°C) and relative humidity (55–60%). The animals had free access to drinking water. The animal care, handling, surgical procedure for cannulation, blood sampling and other experimental activities were accomplished in accordance with the “Guiding Principles in the Use of Animals in Toxicology” by the Society of Toxicology34 which were approved by Institutional Animal Care and Use Committee (IACUC) of Government College University Faisalabad (Ref. No. GCUF/ERC/2067) as well.
Cannulation and blood sampling procedure – A fasted rat was anesthetized and a polyethylene tube was surgically inserted into the right carotid artery. The tube was protected by the hollow flexible spring of the harness. Each rat, properly held with the infusion harness, was retained in a single rat-chamber where it was unrestricted to move and drink water. Bezafibrate plain powder or bezafibrate-loaded electrosprayed ternary solid dispersion, at a dose equivalent to 20 mg/kg body weight, was suspended in 500 μL water and immediately administered to the cannulated rat via the oral route using a gavage. At each of the following specified time points: 0.25, 0.50, 1.0, 2.0, 4.0, 6.0, 10, 14, 24 and 36 h, a 300 μL blood sample was collected and plasma was immediately separated by centrifugation (Hanil Science Industrial Co., Ltd; Smart 15, Korea) at 7000 ×g for 5 min. Thereafter, these plasma samples were stowed at −20°C.
Sample preparation and HPLC analysis – One hundred microliter plasma was taken in a 1.5 mL capacity Eppendorf microtube and 1 mL acetonitrile was added to it for liquid-liquid extraction. Then, a thorough vortex-mixing was carried out, and the sample was centrifuged at 7000 ×g for 5 min. The clear supernatant was carefully shifted to another clean microtube and placed at 40°C for evaporation. The residue was reconstituted with acetonitrile (100 μL) and shifted to a small volume HPLC vial. Then, bezafibrate in the sample was quantified by the HPLC method as described above.32
Pharmacokinetic parameters for bioavailability comparison – The area under the drug concentration–time curve (AUC, h.µg/mL), peak plasma-level of bezafibrate (Cmax, µg/mL) and time to reach Cmax (Tmax, h) were calculated using a non-compartmental analysis (WinNonlin, version 2.1, Pharsight Co., CA, USA). Student t-test was applied to estimate the levels of statistical significance (p-value) between two means for unpaired data at each time point. A p-value < 0.05 represented significant difference while a p-value > 0.05 suggested nonsignificant difference.
Lipid Panel Test
Twenty-four male Wistar rats (250–300 g) were used in this study. The animals were kept in a room with maintained environmental conditions of temperature (22–26°C) and relative humidity (55–60%). The rats were divided into four groups, each comprising of six rats. One group was given normal laboratory standard food and tap water. This group was used as a negative control. Other three groups were fed on a High Fat Diet (HFD), consisting of lamb’s tallow and coconut oil in 3:1 (w/w) ratio, for 21 days to develop model HFD. Then, HFD was stopped and all the rats were kept on normal laboratory standard diet. One HFD-treated group was used as a positive control. The second and third HFD-treated groups were used for testing the effects of bezafibrate plain powder and electrosprayed ternary solid dispersion formulation V at the daily drug dose of 10 mg/kg for 1 week, respectively. The blood samples were collected in heparinized microtubes via retro-orbital puncture, and sent to a medical laboratory for determination of TG, HDL and LDL titers. TC was calculated using the Friedewald formula: TC = LDL + HDL +. (TG/5)
Results and Discussion
PVP K30 and cremophor ELP were selected as the most apt excipients for bezafibrate-loaded ternary solid dispersions as they exhibited the best solubility results amongst the tested hydrophilic polymeric substances (Figure 1A) and solubilizers (Figure 1B), respectively. PVP K30 furnished a drug solubility of 18.57 ± 3.12 µg/mL while cremophor ELP exhibited 345.58 ± 24.43 µg/mL solubility of bezafibrate.
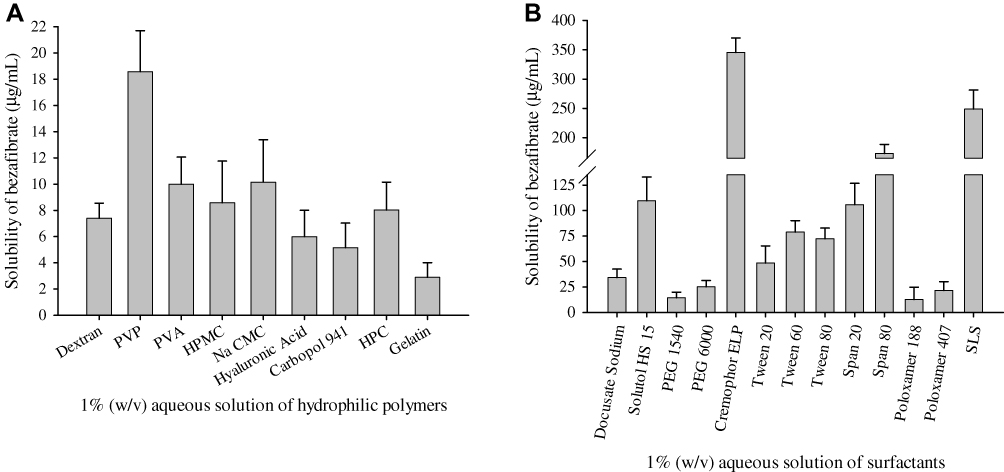 |
Figure 1 Saturation solubility of bezafibrate in 1% (w/v) aqueous solution of hydrophilic excipients: (A) hydrophilic polymeric matrices and (B) surfactants. Each value represents the Mean ± SD (n = 3). |
A drug-loaded entity having dimensions of less than 1 µm is declared as a pharmaceutical nanoparticle.30,31 Nano-sized solid dispersion is a magnificent drug delivery system to ameliorate dissolution rate and solubility of a poorly water-soluble substance in the aqueous media.18,24 Solvent-evaporation method in conjunction with electrospraying is a smart way to fabricate nanoparticulated solid dispersions;24,35 accordingly, in the present research work we adopted these techniques to obtain bezafibrate-loaded nanoparticulated solid dispersions (nanospheres). The recipe of each formulation is shown in Table 1.
The results of the aqueous solubility of bezafibrate in electrosprayed ternary solid dispersions are shown in Figure 2A. Solubility of the drug in formulations I-III was improved as the concentration of cremophor ELP increased. This was owing to solubilizing influence of the surfactant.36 Solubility of bezafibrate was improved further in formulations III-V due to gradually increased concentration of PVP K30. This enhancement in solubility might be ascribed to increased wetting of bezafibrate by the hydrophilic polymer.24 Moreover, as the quantity of PVP K30 increased further in formulations V-VII, an apparent decrease in solubility was observed. However, the solubility results were not significantly different (p > 0.05) from one another in these formulations. In essence, drug molecules are uniformly distributed in the polymeric matrix in the solid dispersions. The liberation of drug payload is dependent upon dissolution of polymer. The higher quantity of polymer in the formulation results in earlier supersaturation in the saturation solubility test; therefore, no further polymer is dissolved. The drug is embedded in the polymeric network; therefore, no further drug is dissolved as well.19,37 That is the why, the formulations VI and VII with higher polymer concentrations showed apparently decreased solubility of bezafibrate.37,38 Formulation V, consisting of bezafibrate, PVP K30 and cremophor ELP (1/12/1.5, w/w/w), showed apparently the highest solubility of bezafibrate (27.59 ± 4.90 mg/mL) which was about fivefold than that of the corresponding physical mixture. The physical mixture was obtained by simply triturating bezafibrate, PVP K30 and cremophor ELP (1/12/1.5, w/w/w) using a pestle and mortar.
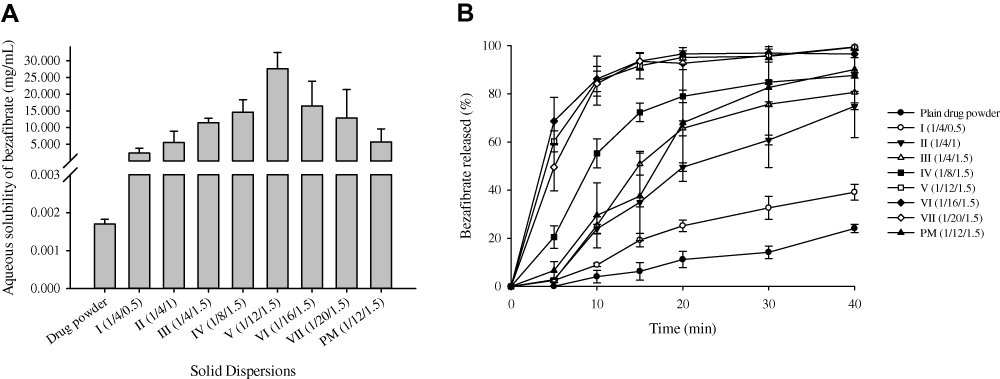 |
Figure 2 Effect of cremophor ELP (I-III) and PVP K30 (IV-VII) on the aqueous solubility (A) and dissolution rate (B) of bezafibrate in electrosprayed ternary solid dispersions. Each value denotes the Mean ± SD (n = 3 and n = 6, respectively). Solubility and dissolution of bezafibrate with physical mixture (PM) is also shown. |
The drug content in all the formulations was within ± 1%. The release rate of bezafibrate from the formulations is shown in Figure 2B. The release of the drug from formulations I-III was improved due to better solubilization of the drug by cremophor ELP. The further acceleration in release rate from formulations IV-VII might be attributed to conversion of the crystalline form of bezafibrate into the amorphous counterpart which increased the surface area of the drug. Hydrophilic polymer in solid dispersion hinders recrystallization of the drug.39,40 The intensity of crystallinity in the final solid dispersion is dependent on both the type41 and concentration42,43 of the hydrophilic polymer in the formulation. Formulation V showed about 85% release of bezafibrate in 10 min. The drug release behaviour in the physical mixture was erratic. Also, release profiles of formulations V-VII were not significantly different from one another. Hence, on the basis of the best apparent solubility and adequate release, formulation V was selected for further investigations.
The intensity of crystallinity was perused using XRD technique and further confirmed by DSC. The XRD pattern of plain drug powder suggested its typical crystalline nature (Figure 3A). The distinctive peaks of bezafibrate appeared at 11.3°, 11.9°, 14.4°, 16.1°, 16.5°, 17.9°, 18.1°, 20.8°, 24.6° and 25.2°. PVP K30 did not show any sharp spike as it was amorphous in nature (Figure 3B). Cremophor ELP produced couple of sharp spikes which suggested that it was semi-crystalline in nature (Figure 3C). The distinctive peaks of the drug and cremophor ELP were also seen in the pattern of physical mixture (Figure 3D). This suggested that both the drug and cremophor ELP retained their crystalline intensity in the physical mixture. The pattern of formulation V also exhibited peaks related to cremophor ELP; however, the spikes pertaining to the drug were absent (Figure 3E). Thus, the drug payload was in the amorphous state in the formulation. DSC results were in line with XRD results. A deep endotherm corresponding to the melting point of bezafibrate was witnessed at 180°C (Figure 4A). This confirmed the typical crystalline property of the drug. No endotherm was seen in case of PVP (Figure 4B); nevertheless, a broad descending curve appeared in the range of about 35–175°C which was because of vaporization of physically admixed moisture from the sample. A sharp endothermic conduit corresponding to the melting point of crystalline part of cremophor ELP was observed at about 65.8°C (Figure 4C) which confirmed the semi-crystalline property of the surfactant. The endotherms associated with the drug and cremophor ELP also appeared at their respective positions in the thermogram of physical mixture (Figure 4D). This confirmed that both the entities retained their crystalline properties in the physical mixture. In case of formulation V, the endotherm corresponding to the melting point of cremophor ELP appeared in the thermogram; however, the endotherm associated with the drug was absent (Figure 4E). This strongly advocated that the drug was completely changed into its amorphous counterpart in the formulation. During the preparation of formulations, the drug is likely to recrystallize during drying the transparent solution of components. Recrystallization of the drug is hindered, either completely or partially, by the polymeric component of the formulation.19
 |
Figure 3 XRD patterns: (A) bezafibrate, (B) PVP K30, (C) Cremophor ELP, (D) physical mixture and (E) electrosprayed ternary solid dispersion formulation V. |
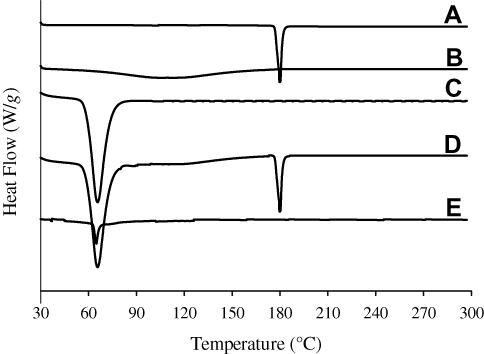 |
Figure 4 DSC thermograms: (A) bezafibrate, (B) PVP K30, (C) Cremophor ELP, (D) physical mixture and (E) electrosprayed ternary solid dispersion formulation V. |
Scanning electron micrographs of bezafibrate plain powder (Figure 5A) showed crystals with irregular shapes and surfaces. The particles of formulation V were smooth surfaced and round-shaped with a central depression on one side (Figure 5B). A pharmaceutical nanoparticle is a drug-loaded object possessing a diameter of not more than 1000 nm.24 From Figure 5B, it can be appraised that the average particle-size of the formulation was not more than 500 nm. Thus, particles of electrosprayed ternary solid dispersion formulation V were nano-sized particles.
 |
Figure 5 SEM images: (A) bezafibrate (× 5000) and (B) electrosprayed ternary solid dispersion formulation V. (× 10,000). |
FTIR analysis revealed sharp distinctive peaks of bezafibrate, as shown in Figure 6A. FTIR spectrum of bezafibrate revealed p-substituted phenyl of Ar–Cl peaks appearing at 843.97 cm−1 and 1087.98 cm−1. Stretching vibrations of –CO of aryl alkyl ethers was visible at 1248.74 cm−1. Peaks appearing at 1502.79 cm−1 and 1598.96 cm−1 correspond to bending vibration of -NH and stretching vibration of –CO, respectively. Furthermore, carbonyl group (–CO) of carboxylic acid showed its stretching vibrational peak at 1728.14 cm−1. FTIR spectrum of bezafibrate also revealed –OH stretching vibration at 3457.72 cm−1.44 FTIR spectrum of PVP K30 and that of Cremophor ELP is shown in Figure 6B and C, respectively. The distinctive peaks of the drug were also seen at the same positions in both the spectra of physical mixture (Figure 6D) and electrosprayed ternary solid dispersion formulation V (Figure 6E). Furthermore, the spectrum of physical mixture was identical to that of the formulation; accordingly, this suggested absence of interactions between the drug and an excipient.
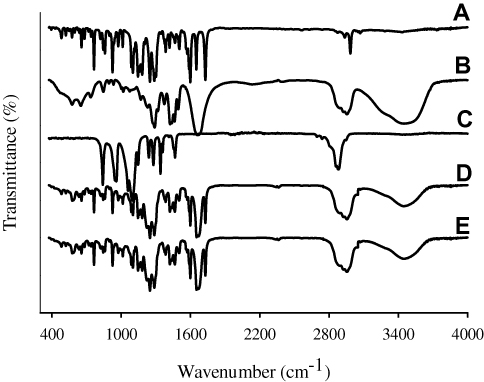 |
Figure 6 FTIR spectra: (A) bezafibrate, (B) PVP K30, (C) Cremophor ELP, (D) physical mixture and (E) electrosprayed ternary solid dispersion formulation V. |
Figure 7 shows the mean bezafibrate concentration-time curves produced after oral administration of bezafibrate alone (Figure 7A) and electrosprayed ternary solid dispersion formulation V (Figure 7B). The formulation furnished higher mean plasma concentrations of the drug at all time points between 1 and 24 h (t-test, p < 0.05) than did bezafibrate plain powder. The pharmacokinetic parameters, such as AUC, Cmax and Tmax, are shown in Table 2. The AUC and Cmax of the electrosprayed ternary solid dispersion formulation V were greatly enhanced as compared to those of bezafibrate plain powder (t-test, p < 0.05); however, Tmax did not change significantly (t-test, p > 0.05). As compared with bezafibrate plain powder, bioavailability was ~5.5-fold with electrosprayed ternary solid dispersion formulation V. The improved bioavailability might be ascribed to amelioration in the aqueous solubility and dissolution of bezafibrate resulting from improved wetting due to hydrophilic polymer, particle-size diminution and alteration of the crystalline form of the drug to its amorphous counterpart.18
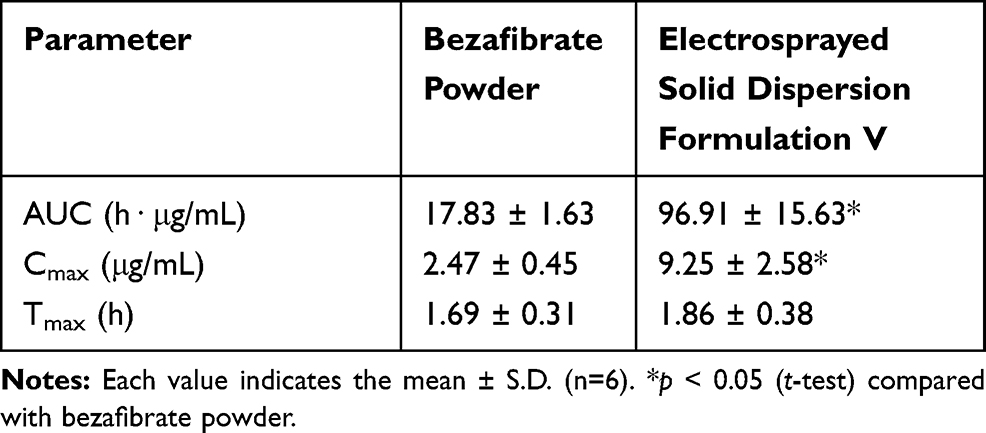 |
Table 2 Pharmacokinetic Parameters |
 |
Figure 7 Plasma level-time profiles: (A) bezafibrate and (B) electrosprayed ternary solid dispersion formulation V. *p < 0.05 as compared to bezafibrate plain powder. |
The mean values of TG, HDL, LDL and TC are shown in Figure 8. The negative control gave values 50.17 ± 12.25, 33.17 ± 7.98, 13.00 ± 3.85 and 55.87 ± 11.01 mg/dL, respectively (Figure 8A). The values appeared in positive control were 208.83 ± 27.97, 22.83 ± 6.43, 36.67 ± 6.53 and 101.27 ± 14.08 mg/dL, respectively (Figure 8B). The comparison shows that TG, LDL and TC were significantly higher (t-test, p < 0.05), and HDL was lower in HFD-treated rats than in rats fed on normal laboratory standard diet. As compared with the positive control, bezafibrate plain powder (Figure 8C) lowered TG (208.83 ± 27.97 vs 184.83 ± 22.43 mg/dL), LDL (36.67 ± 6.53 vs 31.66 ± 7.91 mg/dL) and TC (101.27 ± 14.08 vs 96.30 ± 11.96 mg/dL), and improved HDL (22.83 ± 6.43 vs 27.66 ± 5.16 mg/dL); however, the differences were not significant (t-test, p > 0.05). On the other hand, values given by the electrosprayed ternary solid dispersion formulation V (Figure 8D) were significantly different (p < 0.05) from those produced by the positive control and bezafibrate plain powder. As compared with bezafibrate plain powder, formulation V reduced TG (184.83 ± 22.43 vs 100.83 ± 11.53 mg/dL), LDL (31.66 ± 7.91 vs 21.17 ± 3.19 mg/dL) and TC (96.30 ± 11.96 vs 81.83 ± 13.25 mg/dL), and improved HDL (27.66 ± 5.16 vs 40.50 ± 10.11 mg/dL). This suggested that bezafibrate-loaded electrosprayed polymeric nanosphere formulation was more effective than bezafibrate plain powder when administered at the same drug dose. Thus, this formulation might be a promising nanoparticulated delivery system for bezafibrate with enhanced bioavailability and effectiveness.
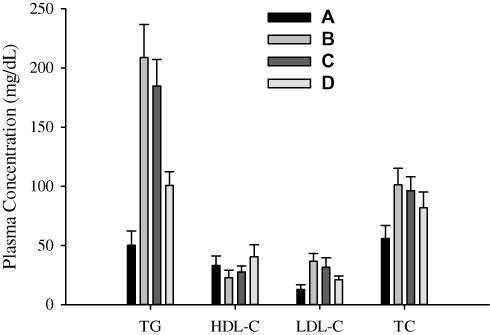 |
Figure 8 Lipid profile characteristics: (A) negative control, (B) positive control, (C) bezafibrate and (D) electrosprayed ternary solid dispersion formulation V. Abbreviations: TG, triglycerides; HDL-C, high-density lipoprotein cholesterol, LDL-C, low-density lipoprotein cholesterol; TC, total cholesterol. |
Conclusion
Electrosprayed polymeric nanosphere formulation V, consisting of bezafibrate, PVP K30 and Cremophor ELP at the ratio of 1/12/1.5 (w/w/w), demonstrated the most enhanced solubility (27.59 ± 4.90 mg/mL) and an excellent dissolution (85.48 ± 10.16% in 10 min). The drug existed in the amorphous state in the nanospheres, and had no strong bonding with the polymeric matrix. The amelioration in solubility and dissolution of bezafibrate in the aqueous media might be attributed to improved wetting of the drug due to the presence of PVP K30, enhanced solubilization because of cremophor ELP and conversion of bezafibrate from its crystalline form to the amorphous counterpart in electrosprayed polymeric nanospheres. The improvement in solubility and dissolution of bezafibrate in nanospheres resulted in greater bioavailability and effectiveness of the drug. As compared with bezafibrate plain powder, formulation V lowered titers of TG (184.83 ± 22.43 vs 100.83 ± 11.53 mg/dL, respectively), LDL (31.66 ± 7.91 vs 21.17 ± 3.19 mg/dL, respectively) and TC (96.30 ± 11.96 vs 81.83 ± 13.25 mg/dL, respectively), and improved HDL (27.66 ± 5.16 vs 40.50 ± 10.11 mg/dL, respectively) in HFD-treated rats. Thus, this formulation exhibited more antihyperlipidemic activity than that shown by bezafibrate plain powder when administered at the same drug dose.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Aronson JK. 44 - Drugs that affect lipid metabolism. In: Aronson JK, editor. Side Effects of Drugs Annual. Vol. 32. Elsevier; 2010:803–826.
2. Tenenbaum A, Motro M, Fisman EZ. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: the bezafibrate lessons. Cardiovasc Diabetol. 2005;4(1):14. doi:10.1186/1475-2840-4-14
3. Bezafibrate IPB. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation. 2000;102(1):21.
4. Tenenbaum A, Motro M, Fisman EZ, Tanne D, Boyko V, Behar S. Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch Intern Med. 2005;165(10):1154–1160. doi:10.1001/archinte.165.10.1154
5. Monk JP, Todd PA. Bezafibrate. Drugs. 1987;33(6):539–576. doi:10.2165/00003495-198733060-00002
6. Bezafibrate IP. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation. 2000;102(1):21–27. doi:10.1161/01.CIR.102.1.21
7. Oliveira M, Silva G, Campos MST. Chemical degradation kinetics of fibrates: bezafibrate, ciprofibrate and fenofibrate. Braz J Pharm Sci. 2016;52(3):545–553. doi:10.1590/s1984-82502016000300019
8. Das S, Ray NM, Wan J, Khan A, Chakraborty T, Ray MB. Micropollutants in wastewater: fate and removal processes. Physi Chem Wastewater Treat Res Recovery. 2017;75 InTech.
9. Dressman J, Butler J, Hempenstall J, Reppas C. The BCS: where do we go from here? Pharm Technol. 2001;25(7):68–77.
10. Adkins J, Faulds D. Micronised fenofibrate. Drugs. 1997;54(4):615–633. doi:10.2165/00003495-199754040-00007
11. Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. doi:10.1016/j.ejps.2006.04.016
12. Goddeeris C, Coacci J, Van den Mooter G. Correlation between digestion of the lipid phase of smedds and release of the anti-HIV drug UC 781 and the anti-mycotic drug enilconazole from smedds. Eur J Pharm Biopharm. 2007;66(2):173–181. doi:10.1016/j.ejpb.2006.10.005
13. Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. doi:10.1016/S0939-6411(00)00076-X
14. Perrut M, Jung J, Leboeuf F. Enhancement of dissolution rate of poorly-soluble active ingredients by supercritical fluid processes: part I: micronization of neat particles. Int J Pharm. 2005;288(1):3–10. doi:10.1016/j.ijpharm.2004.09.007
15. Joe JH, Lee WM, Park Y-J, et al. Effect of the solid-dispersion method on the solubility and crystalline property of tacrolimus. Int J Pharm. 2010;395(1–2):161–166. doi:10.1016/j.ijpharm.2010.05.023
16. Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discovery Today. 2007;12(23–24):1068–1075. doi:10.1016/j.drudis.2007.09.005
17. Shahzad Y, Saeed S, Ghori MU, et al. Influence of polymer ratio and surfactants on controlled drug release from cellulosic microsponges. Int J Biol Macromol. 2018;109:963–970. doi:10.1016/j.ijbiomac.2017.11.089
18. Yousaf AM, Kim DW, Oh Y-K, Yong CS, Kim JO, Choi H-G. Enhanced oral bioavailability of fenofibrate using polymeric nanoparticulated systems: physicochemical characterization and in vivo investigation. Int J Nanomedicine. 2015;10:1819–1830. doi:10.2147/IJN.S78895
19. Yousaf AM, Ramzan M, Shahzad Y, Mahmood T, Jamshaid M. Fabrication and in vitro characterization of fenofibric acid-loaded hyaluronic acid–polyethylene glycol polymeric composites with enhanced drug solubility and dissolution rate. Int J Polym Mater Polym Biomater. 2019;68:510–515. doi:10.1080/00914037.2018.1466137
20. Luo C-F, Yuan M, Chen M-S, et al. Pharmacokinetics, tissue distribution and relative bioavailability of puerarin solid lipid nanoparticles following oral administration. Int J Pharm. 2011;410(1–2):138–144. doi:10.1016/j.ijpharm.2011.02.064
21. Arya N, Chakraborty S, Dube N, Katti DS. Electrospraying: a facile technique for synthesis of chitosan‐based micro/nanospheres for drug delivery applications. J Biomed Mater Res Part B. 2009;88(1):17–31. doi:10.1002/jbm.b.31085
22. Hong Y, Li Y, Yin Y, Li D, Zou G. Electrohydrodynamic atomization of quasi-monodisperse drug-loaded spherical/wrinkled microparticles. J Aerosol Sci. 2008;39(6):525–536. doi:10.1016/j.jaerosci.2008.02.004
23. Ding L, Lee T, Wang C-H. Fabrication of monodispersed Taxol-loaded particles using electrohydrodynamic atomization. J Control Release. 2005;102(2):395–413. doi:10.1016/j.jconrel.2004.10.011
24. Yousaf AM, Mustapha O, Kim DW, et al. Novel electrosprayed nanospherules for enhanced aqueous solubility and oral bioavailability of poorly water-soluble fenofibrate. Int J Nanomedicine. 2016;11:213–221. doi:10.2147/IJN.S97496
25. Bohr A, Kristensen J, Stride E, Dyas M, Edirisinghe M. Preparation of microspheres containing low solubility drug compound by electrohydrodynamic spraying. Int J Pharm. 2011;412(1):59–67. doi:10.1016/j.ijpharm.2011.04.005
26. Yu D-G, Williams GR, Yang J-H, Wang X, Yang J-M, Li X-Y. Solid lipid nanoparticles self-assembled from electrosprayed polymer-based microparticles. J Mater Chem. 2011;21(40):15957–15961. doi:10.1039/c1jm12720a
27. Enayati M, Ahmad Z, Stride E, Edirisinghe M. Size mapping of electric field-assisted production of polycaprolactone particles. J R Soc Interface. 2010;7. doi:10.1098/rsif.2010.0099.focus.
28. Trotta M, Cavalli R, Trotta C, Bussano R, Costa L. Electrospray technique for solid lipid-based particle production. Drug Dev Ind Pharm. 2010;36(4):431–438. doi:10.3109/03639040903241817
29. Cavalli R, Bisazza A, Bussano R, et al. Poly (amidoamine)-cholesterol conjugate nanoparticles obtained by electrospraying as novel tamoxifen delivery system. J Drug Deliv. 2011;2011:1–9. doi:10.1155/2011/587604
30. Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine. 2006;2(1):8–21. doi:10.1016/j.nano.2005.12.003
31. Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70(1):1–20. doi:10.1016/S0168-3659(00)00339-4
32. Chen D, Zhang J, Peng X, et al. Pharmacokinetic study of bezafibrate in rat by high performance liquid chromatography. Lat Am J Pharm. 2016;35(10):2279–2283.
33. Shah V, Konecny J, Everett R, McCullough B, Noorizadeh AC, Skelly J. In vitro dissolution profile of water-insoluble drug dosage forms in the presence of surfactants. Pharm Res. 1989;6(7):612–618. doi:10.1023/A:1015909716312
34. Toxicology So. Guiding Principles in the Use of Animals in Toxicology. Virginia: Society of Toxicology Reston; 1999. https://www.toxicology.org/pubs/statements/statements.asp.
35. Mustapha O, Din F, Kim DW, et al. Novel piroxicam-loaded nanospheres generated by the electrospraying technique: physicochemical characterisation and oral bioavailability evaluation. J Microencapsul. 2016;33(4):323–330. doi:10.1080/02652048.2016.1185475
36. Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21(2):201–230. doi:10.1023/B:PHAM.0000016235.32639.23
37. Yousaf AM, Zulfiqar S, Shahzad Y, Hussain T, Mahmood T, Jamshaid M. The preparation and physicochemical characterization of eprosartan mesylate-laden polymeric ternary solid dispersions for enhanced solubility and dissolution rate of the drug. Polim Med. 2018;48(2):69–75. doi:10.17219/pim/102976
38. Yousaf AM, Malik UR, Shahzad Y, Mahmood T, Silymarin-laden HT. PVP-PEG polymeric composite for enhanced aqueous solubility and dissolution rate: preparation and in vitro characterization. J Pharm Anal. 2019;9(1):34–39. doi:10.1016/j.jpha.2018.09.003
39. Taylor L, Zografi G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm Res. 1997;14(12):1691–1698. doi:10.1023/A:1012167410376
40. Doherty C, York P. Accelerated stability of an X-ray amorphous frusemide-polyvinylpyrrolidone solid dispersion. Drug Dev Ind Pharm. 1989;15(12):1969–1987. doi:10.3109/03639048909052513
41. Konno H, Handa T, Alonzo DE, Taylor LS. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur J Pharm Biopharm. 2008;70(2):493–499. doi:10.1016/j.ejpb.2008.05.023
42. Gupta P, Kakumanu VK, Bansal AK. Stability and solubility of celecoxib-PVP amorphous dispersions: a molecular perspective. Pharm Res. 2004;21(10):1762–1769. doi:10.1023/B:PHAM.0000045226.42859.b8
43. Tanno F, Nishiyama Y, Kokubo H, Obara S. Evaluation of hypromellose acetate succinate (HPMCAS) as a carrier in solid dispersions. Drug Dev Ind Pharm. 2004;30(1):9–17. doi:10.1081/DDC-120027506
44. Shen C-S, Zhou C-R. Investigation of the thermal decomposition kinetics of bezafibrate. J Therm Anal Calorim. 2016;126(2):959–967. doi:10.1007/s10973-016-5565-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.