")
Back to Journals » International Journal of Nanomedicine » Volume 19
EGFR Targeting of Liposomal Doxorubicin Improves Recognition and Suppression of Non-Small Cell Lung Cancer
Authors Moles E , Chang DW, Mansfeld FM, Duly A, Kimpton K, Logan A , Howard CB, Thurecht KJ , Kavallaris M
Received 18 November 2023
Accepted for publication 23 March 2024
Published 20 April 2024 Volume 2024:19 Pages 3623—3639
DOI https://doi.org/10.2147/IJN.S450534
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Eng San Thian
Ernest Moles,1– 4 David W Chang,1– 3 Friederike M Mansfeld,1– 3 Alastair Duly,1,2 Kathleen Kimpton,1 Amy Logan,1– 4 Christopher B Howard,5 Kristofer J Thurecht,5,6 Maria Kavallaris1– 4
1Children’s Cancer Institute, Lowy Cancer Research Centre, UNSW, Sydney, NSW, 2052, Australia; 2UNSW Australian Centre for Nanomedicine, Faculty of Engineering, UNSW, Sydney, NSW, 2052, Australia; 3School of Clinical Medicine, Faculty of Medicine & Health, UNSW, Sydney, NSW, 2052, Australia; 4UNSW RNA Institute, Faculty of Science, UNSW, Sydney, NSW, 2052, Australia; 5Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, St Lucia, QLD, 4072, Australia; 6Centre for Advanced Imaging, ARC Training Centre for Innovation in Biomedical Imaging Technologies, University of Queensland, St Lucia, QLD, 4072, Australia
Correspondence: Ernest Moles; Maria Kavallaris, Children’s Cancer Institute, Lowy Cancer Research Centre, UNSW, Sydney, NSW, 2052, Australia, Email [email protected]; [email protected]
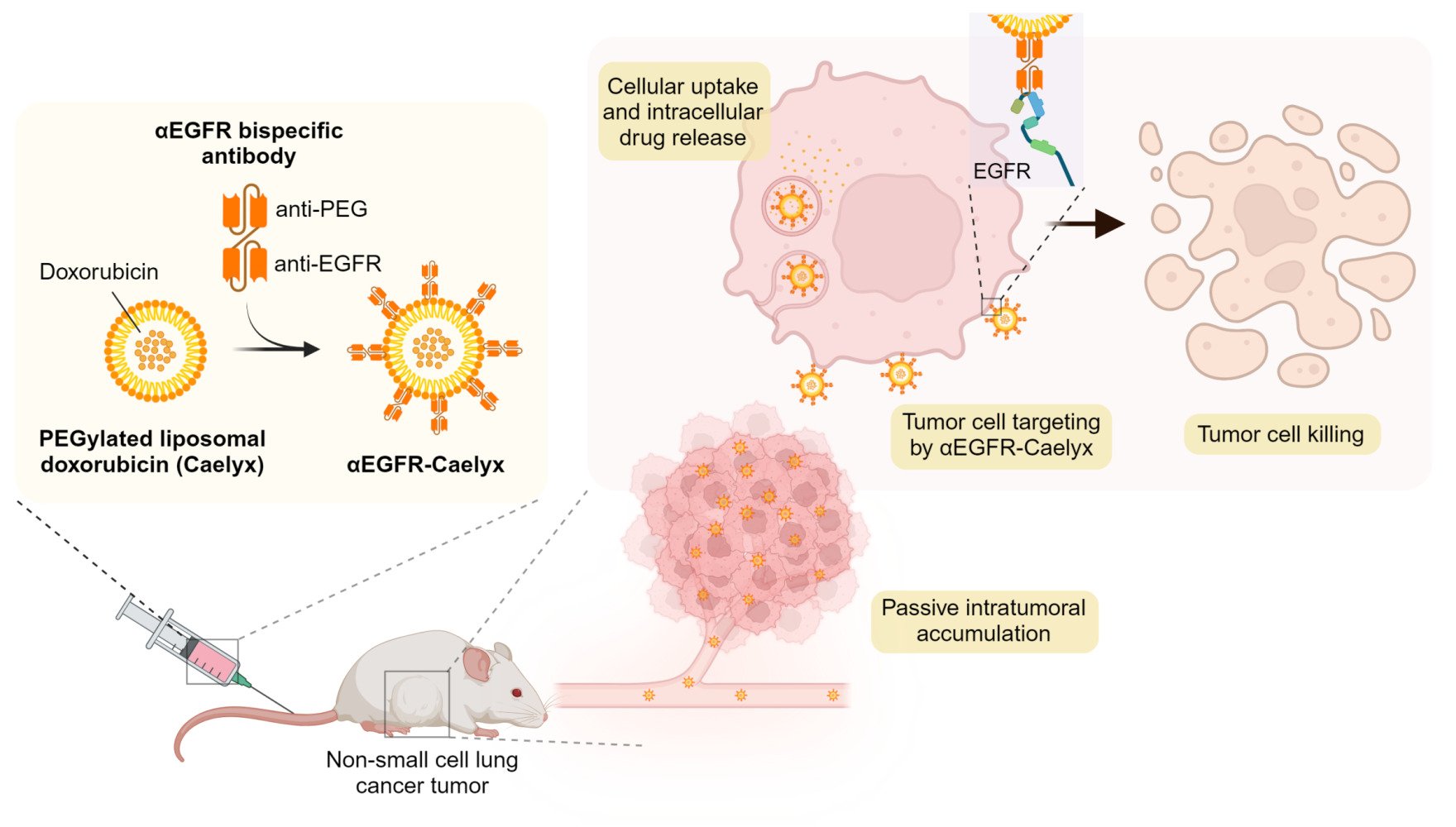
Introduction: Despite improvements in chemotherapy and molecularly targeted therapies, the life expectancy of patients with advanced non-small cell lung cancer (NSCLC) remains less than 1 year. There is thus a major global need to advance new treatment strategies that are more effective for NSCLC. Drug delivery using liposomal particles has shown success at improving the biodistribution and bioavailability of chemotherapy. Nevertheless, liposomal drugs lack selectivity for the cancer cells and have a limited ability to penetrate the tumor site, which severely limits their therapeutic potential. Epidermal growth factor receptor (EGFR) is overexpressed in NSCLC tumors in about 80% of patients, thus representing a promising NSCLC-specific target for redirecting liposome-embedded chemotherapy to the tumor site.
Methods: Herein, we investigated the targeting of PEGylated liposomal doxorubicin (Caelyx), a powerful off-the-shelf antitumoral liposomal drug, to EGFR as a therapeutic strategy to improve the specific delivery and intratumoral accumulation of chemotherapy in NSCLC. EGFR-targeting of Caelyx was enabled through its complexing with a polyethylene glycol (PEG)/EGFR bispecific antibody fragment. Tumor targeting and therapeutic potency of our treatment approach were investigated in vitro using a panel of NSCLC cell lines and 3D tumoroid models, and in vivo in a cell line-derived tumor xenograft model.
Results: Combining Caelyx with our bispecific antibody generated uniform EGFR-targeted particles with improved binding and cytotoxic efficacy toward NSCLC cells. Effects were exclusive to cancer cells expressing EGFR, and increments in efficacy positively correlated with EGFR density on the cancer cell surface. The approach demonstrated increased penetration within 3D spheroids and was effective at targeting and suppressing the growth of NSCLC tumors in vivo while reducing drug delivery to the heart.
Conclusion: EGFR targeting represents a successful approach to enhance the selectivity and therapeutic potency of liposomal chemotherapy toward NSCLC.
Keywords: targeted drug delivery, bispecific antibodies, PEGylated liposomal doxorubicin, EGFR targeting, non-small cell lung cancer
Graphical Abstract:
Introduction
Lung cancer is the leading cause of death from cancer worldwide. NSCLC is the most common form accounting for >80% of all cases. Over half of NSCLC patients have developed metastases at the time of diagnosis (stage IV cancer) and 5-year survival remains <26%. Systemic combination chemotherapy is the first-line treatment for NSCLC.1 However, this has shown limited effectiveness with most intense regimens having reached the limit of tolerability. Newer anticancer agents including tyrosine kinase inhibitors have led to high response rates and prolonged progression-free survival of advanced NSCLC patients in early phase clinical trials.2,3 Clinical studies in larger cohorts of patents are nevertheless needed to better delineate their potency and safety. There is thus an urgent need to develop new treatment strategies that are less toxic and more effective to improve outcomes for these patients.
Systemic drug delivery using liposomal particles is an effective approach to optimize the biodistribution and pharmacokinetic profile of chemotherapy drugs for improved and less-toxic treatment of solid tumors.4,5 Liposomal particles display an optimal size of 100 to 150 nm for limited renal filtration and greater extravasation and infiltration into the tumor microenvironment.4 Coating of liposomal particles with methoxy polyethylene glycol (PEG) further minimizes their recognition by the reticuloendothelial system for extended plasma half-life and improved passive accumulation into solid tumors.6 Caelyx® or Doxil®, a PEGylated liposomal formulation of doxorubicin, became the first nanodrug to gain FDA approval in 1995 and has been the most investigated liposomal drug to treat solid tumors.7,8 Caelyx is currently indicated for treatment of Kaposi’s sarcoma, breast and ovarian cancer, and has shown clinical potency against lung cancer.9–12 Despite improved drug pharmacokinetics, Caelyx lacks selectivity for the cancer cells and largely accumulates within healthy tissues, inducing side effects whilst limiting the delivery of lethal drug payloads to the tumor for improved therapeutic action. Adverse events include hematological toxicity (neutropenia) with increased risk of infections, neuropathy, stomatitis, and cutaneous lesions.10,11
A strategy to improve recognition and delivery of chemotherapy drugs to the tumor site is to conjugate liposomal drugs with cancer cell-selective targeting ligands.13 Antibodies raised against aberrantly expressed surface receptors on the cancer cells, such as human epidermal growth factor receptor-2 (HER-2), epidermal growth factor receptor (EGFR), or EphrinA2 Receptor (EphA2), have been used effectively to target liposomal drugs to the tumor site with concomitant improvements in cancer remission and reduced toxicity. This strategy has shown success in the management of diverse cancers, safely suppressing the growth of tumors including breast, pancreas, colon, ovaries, esophagus, and glioblastoma in animal models.14–18 The therapeutic benefits of this approach have potential to be extended to treat NSCLC, for which targeted liposomal drugs have been poorly developed or have shown modest therapeutic efficacy.19–21
The development, testing and clinical translation of targeted nanodrugs is, however, hindered by technical hurdles in their de novo synthesis that result in a high batch-to-batch variability, extensive quality control testing, and poor reproducibility of their biological performance. Current methods for ligand-particle conjugation are difficult to reproduce, irreversibly alter nanoparticle and ligand structure or function, or rely on a precisely controlled environment to succeed.22,23 To circumvent these limitations, we have developed a targeting strategy featuring the non-covalent, one-step complexing of preformed PEGylated nanodrugs with dual-single chain variable fragment (scFv) bispecific antibodies binding PEG on the nanodrug and a receptor on the surface of the cancer cells.24–26 Importantly, this strategy further represents a significant opportunity to repurpose and enhance the selectivity towards solid tumors of clinically approved, yet toxic, off-the-shelf PEGylated nanodrugs, such as Caelyx.
Herein, we demonstrate how by complexing Caelyx with an EGFR/PEG scFv BsAb (named αEGFR) we can improve its specificity and therapeutic potency against EGFR+ NSCLC tumors. EGFR is widely overexpressed in patients with NSCLC27 and thus constitutes an ideal target to successfully deliver nanodrugs to the tumor site. Caelyx complexing with EGFR/PEG BsAb ameliorated recognition of EGFR+ NSCLC cells with concomitant improvements in biodistribution, on-site tumor accumulation, and improved suppression of tumor growth in a xenograft model of NSCLC.
Materials and Methods
Cell Culture
NSCLC cells H460 and H1299 were cultured in RPMI-1640 (Thermo Fisher Scientific, 11,835,030) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, A4766801) at 37 °C in 5% CO2. NSCLC cells A549, Calu-6, and breast cancer cells MCF-7 cells were cultured in Dulbecco’s modified eagle medium (DMEM, Thermo Fisher Scientific, 11,995,065) supplemented with 10% FBS at 37 °C in 5% CO2. All cells used in this study were sub-cultured using trypsin/EDTA when they reached 70 to 80% confluence and were all routinely screened and free of mycoplasma. Cell lines Calu-6, A549, H1299 and MCF-7 cells were obtained from ATCC (Invitro Technologies, Australia), and H460 were purchased from PerkinElmer (Australia). Detailed information about the cell lines can be found in Table S1. Human ethics was not required for established and commercially available human cell lines.
Synthesis of αEGFR and αCD19 BsAbs
BsAbs were synthesized as previously described.24,25 Briefly, the BsAbs generated comprised an EGFR-targeting or B cell-targeting scFv linked to a PEG-binding scFv by a glycine serine (G4S) peptide chain. ScFv sequences are indicated in Table S2. A 6 x Histidine motif at the N-terminus of the BsAb and a C-MYC epitope tag at the C-terminus were included to facilitate purification and detection of the BsAb following expression. The BsAb genes were cloned into pcDNA 3.1 (+) mammalian expression plasmid (Invitrogen) and transfected into CHO cells for transient production of BsAbs. Following culture, BsAbs were purified from CHO cells supernatant using a 5 mL Histrap excel column (GE Healthcare), and trapped BsAbs were eluted with a solution containing 20×10−3 M sodium phosphate, 500×10−3 M NaCl, and 500×10−3 M imidazole, pH 7.4. Eluted BsAb fractions were buffer exchanged into Phosphate Buffered Saline (PBS) containing 300 mM NaCl using a HiPrep 26/10 column (GE Healthcare), filter-sterilized through a 0.22 μm membrane and stored at −80 °C. After defrosting of aliquots, salinity of BsAb samples was adjusted to 290 mOsmg/Kg (isotonic salinity) by dilution in 10 mM phosphate buffer prior to their addition to Caelyx or cancer cell cultures.
Caelyx Complexing with BsAbs
Caelyx was provided by a healthcare supplier (Clifford Hallam Healthcare, 1,948,303) as 2 mg/mL PEGylated doxorubicin concentrate solution, 2.3×1014 Caelyx particles/mL, in histidine-sucrose buffer for injection and mixed with BsAbs at specified densities. Unless otherwise specified, BsAb-Caelyx mixtures were incubated for 2 hours at room temperature under orbital mixing and stored overnight at 4 °C prior to their addition to cell cultures or injection to animals. Size and PDI measurements of BsAb-Caelyx complexes were done after 1:30 v/v dilution in PBS by Dynamic and Electrophoretic Light Scattering (DLS) using a Malvern Zetasizer Ultra.
Cell Surface Expression Analysis of EGFR
Following trypsinization, 100,000 cells were dispersed in ice-cold PBS containing 1% (w/v) bovine serum albumin (PBS-BSA) solution. Cells were mixed in 100 µL of 1 µg/mL anti-EGFR mAb 111.6 (Thermo Fisher Scientific, MA5-13,269) or an IsoAb (Thermo Fisher Scientific, 10400C) and incubated for 1 hour on ice. Cells were then washed in ice-cold 1% BSA-PBS, incubated with a secondary AF 647-labelled IgG (1:1000 vol/vol) (Thermo Fisher Scientific, A21235) for 30 minutes on ice, and suspended in PBS-BSA. AF 647 fluorescence in cells was analyzed using a BD LSRFortessa flow cytometer (BD Biosciences).
Cell Targeting Assays
Cancer cells were plated in 48-well flat-bottom tissue culture plates and grown for 24 hours. Unless otherwise specified, NSCLC cells were seeded at 8750 cells/well. Breast cancer MCF-7 were plated at 12,000 cells/well. BsAb-Caelyx samples or Caelyx alone were then added to the cells at indicated concentrations and incubated for 3 hours at 37 °C. Following incubation, cells were washed with PBS, collected using trypsin, and finally dispersed in 200 µL ice-cold PBS. The percentage of cells testing positive for Caelyx fluorescence (% targeted cells) was measured at excitation/emission wavelengths of 488 nm/670 LP (long pass) using a FACS Canto II flow cytometer (BD Biosciences), or 488 nm/685–735 nm using a BD LSRFortessa flow cytometer (BD Biosciences).
Competitive Cell Targeting Assays
H1299 cells were plated into 12-well flat-bottom tissue culture plates (65,000 cells/well) and grown for 24 hours. 2 µM Caelyx was mixed with αEGFR for 30 minutes to form αEGFR-Caelyx and then added to the cells in combination with a competing anti-EGFR mAb (Panitumumab, BioVision) or an isotype mAb control (anti-CD20 mAb clone 2H7; 14–0209-82, Thermo Fisher Scientific) at specified Ab-to-BsAb molar ratios and incubated for one hour at 37 °C. Following incubation, cells were washed with PBS, collected using trypsin, and suspended in 250 µL ice-cold PBS. Caelyx fluorescence in cancer cells was measured using a BD LSRFortessa flow cytometer (BD Biosciences; excitation/emission wavelengths of 488 nm/685–735 nm).
Cell Viability Assays
Cancer cells were plated in 48-well flat-bottom tissue culture plates and grown for 24 hours. Unless otherwise specified, cells were seeded at 8,750 cells/well. MCF-7 were plated at 12,000 cells/mL. BsAb-Caelyx samples or Caelyx alone were added to the cells at indicated concentrations and incubated for 3 hours at 37 °C. Following incubation, cells were washed with fresh medium and cultured for 48 hours at 37 °C. Cells were then collected using trypsin, mixed with cell counting beads (Beckman Coulter, 6,602,796), and viability of these cells was measured using Annexin V and Sytox markers of apoptosis and cell death, as described previously.24 Annexin V− Sytox−, Annexin V− Sytox+, Annexin V+ Sytox− and Annexin V+ Sytox+ are indicative of viable, necrotic, apoptotic, and dead cells, respectively. Stained cells were then suspended ice-cold PBS and analyzed using a BD LSRFortessa or a FACS Canto II flow cytometer (BD Biosciences). Cell count (cells/mL) in treated samples was determined relating the absolute number of counted cells to the total number of counted beads.
Confocal Microscopy
H460 and Calu-6 cells were seeded into 24-well glass bottom plates (10,000 cells/well; P24-1.5H-N 24, Cellvis) pretreated with 0.1 mg/mL of collagen I (A10483-01, ThermoFisher Scientific) for 40 minutes. Following 48 hours culture, cells were exposed to 20 µM αEGFR-Caelyx or Caelyx alone for 3 hours, and stained with cell culture medium containing 1:1000 v/v of CellMask Deep Red (C10046, ThermoFisher Scientific, USA) and 1:2000 v/v Hoechst 33,342 stain for 15 minutes at 37 °C. Cells were washed with fresh medium and imaged on a Zeiss LSM 880 confocal microscope (Carl Zeiss, Germany). Images were analyzed using ImageJ and Zeiss ZEN (Carl Zeiss, Germany).
Measuring Caelyx Penetration into 3D Tumor Spheroids by Confocal Microscopy
H460 cells were plated into Costar® ultra-low cluster round bottom 96 well plates (40 cells/well; Corning, USA) and grown for 4 days to form spheroids. These were then exposed to 20 µM αEGFR-Caelyx or Caelyx alone for 24 hours. Vehicle control spheroids were exposed to PBS for 24 hours. Following incubation, spheroids were washed with medium 5 times, transferred to 24-well glass bottom plates, and the equatorial plane of the spheroids (approximately 125 µm deep below the spheroid surface) was imaged using a Zeiss LSM 880 confocal microscope (Carl Zeiss, Germany). To measure αEGFR-Caelyx and Caelyx internalization, we drew 20 radii (0 to 100 µm below the spheroid edge; 0.83 µm/pixel) distributed across the imaged section, and measured absolute fluorescence intensity in them using Image J software. Internalization of particles in each spheroid was then plotted comparing median fluorescence intensity plus IQR of the 20 radii versus distance.
In vivo Studies
Animal studies were approved by the University of New South Wales Animal Ethics Committee (approval 21/98B) prior to commencement. SCID-beige mice (CB17.Cg-PrkdcscidLystbg-J/Crl) were bred in a colony at the Australian BioResources (ABR) facility and housed in groups of up to 6 mice with ad libitum food and water in rooms with 12-hour light/dark cycles. Animals were monitored for potential issues including appetite loss, diarrhea, dehydration, infection, hunching, abdominal guarding, inactivity, hypothermia, isolation from cage mates, non-responsiveness from environmental cues and weight loss. For in vivo biodistribution and efficacy studies, 0.5 million H460 cells in 50 µL PBS were mixed 1:1 v/v with growth factor-reduced MatrigelTM (BD Biosciences) and subcutaneously injected into the flank of 6- to 8-week old SCID-beige mice. Tumor size and animal weight were measured twice per week using calipers and scales. Tumor size was calculated as ½(length x width2). Treatment of xenograft mice with Caelyx samples (αEGFR-Caelyx, αCD19-Caelyx or Caelyx alone) or vehicle (PBS) began when tumor size exceeded 150 mm3 (approximately 10 days after inoculation of H460 cells). To measure biodistribution of Caelyx samples, the liver, heart and tumor were isolated at specified timepoints and processed for subsequent doxorubicin quantification (the drug component of Caelyx). Doxorubicin extraction was performed using acidified ethanol solution as reported previously,24 and quantified using a liquid chromatography system equipped with a BioSEC-5 column coupled to a fluorescence detector (Agilent 1290 UPLC; excitation/emission wavelengths of 480 nm/570 nm). Samples and standards (100 uL) were injected using a mobile phase of acetonitrile:water 30:70, pH2.8, with a flow rate of 1 mL per minute. Calibration equations were obtained plotting peak area versus nominal concentration of doxorubicin standards dispersed in organ extracts from PBS-injected mice. For therapeutic efficacy studies, tumor-bearing mice were intravenously injected with Caelyx samples or vector (PBS) weekly for four weeks, and tumor size and animal weight were followed over time. Once tumors size reached ≥ 1000 mm3 or 21 days after treatment initiation, mice were humanly killed by carbon dioxide asphyxiation followed by cervical dislocation.
Statistical Analysis
We used Mann–Whitney U-test to calculate statistical significance when comparing two groups with not-normally distributed data. Ratio paired t-test was used to study statistical significance between two related groups. For multiple group comparisons, statistical significance was determined using ANOVA followed by Dunnett’s multiple comparisons when data were normally distributed, or Kruskal–Wallis test followed by Dunn’s multiple comparisons test if data were not-normally distributed. Normality was tested using the Shapiro–Wilk test. Pearson’s correlation test was used to measure the statistical relationship between receptor expression and EC50 or IC50 fold change or to compare the relationship between changes in EC50 with IC50 values. A correlation coefficient of r > 0.5 was considered as positive correlation. A probability value of P < 0.05 was considered statistically significant. Prism version 8.4.2. software (GraphPad) was used for statistical analysis and graph production. FlowJo version 10.8.1 was used for flow cytometry analysis. Schematic figures were created using BioRender.com and CorelDRAW.
Results
αEGFR BsAb Redirects Caelyx to Target NSCLC Cells
Delivery of liposomal drugs to solid tumors remains a challenge. To improve the delivery and therapeutic selectivity of Caelyx toward NSCLC, we incorporated an EGFR/PEG-binding BsAb construct, αEGFR, as an active targeting agent. αEGFR was developed by fusing a methoxy PEG-targeting scFv with a scFv sequence targeting EGFR extracellular domain III (EGFRECDIII), a highly conserved domain in NSCLC cells where mutations are infrequent,28 derived from FDA-approved monoclonal antibody (mAb) panitumumab®. ScFv sequences (~50 kDa) are shown in Table S2 and bind EGFR and PEG with high affinity, KD = 1–10 × 10–9 M.25
We first studied whether complexing Caelyx with αEGFR can provide robust targeting of NSCLC cells. αEGFR and Caelyx were mixed for 1 hour and resulting complexes (defined as αEGFR-Caelyx hereafter, Figure 1A) were added to EGFR+ NSCLC cell cultures (Calu-6 and H460). Flow cytometry revealed a αEGFR dose-dependent association of Caelyx with NSCLC cells, with increasing BsAb densities leading to greater binding of Caelyx to the cancer cells. A cell-binding plateau with >90% Caelyx+ cells was obtained at 32 nM αEGFR combined with 4 µM Caelyx (~64 BsAb molecules per Caelyx particle) (Figure 1B) and was selected for further experiments. In addition, αEGFR-Caelyx complexes formed at 64 BsAb:Caelyx (mol:mol) ratio were similar in size to Caelyx alone (<20% increase in particle size) and monodisperse (~0.1 polydispersity units) (Figure 1C–1E).
 |
Figure 1 Bidirectional binding of αEGFR mediates the delivery of Caelyx to NSCLC cells in vitro. (A) Schematic illustration showing αEGFR BsAb construct, and Caelyx complexing step with αEGFR to form αEGFR-Caelyx. (B) Percentage of H460 or Calu-6 cells targeted by αEGFR-Caelyx complexes (Caelyx+ cells) formed with increasing densities of αEGFR. (C–E) Dynamic and Electrophoretic Light Scattering (DLS) measurement of Caelyx and αEGFR-Caelyx (prepared at 64:1 BsAb-to-Caelyx molar ratio), including Z-average size (C), polydispersity (D), and size distribution (E). (F) Percentage of H1299 cells targeted by αEGFR-Caelyx (Caelyx+ cells) mixed with anti-EGFR mAb panitumumab or IsoAb at increasing Ab-to-BsAb densities. (G) Representative flow cytometry plot (Caelyx fluorescence versus side scatter area or SCC indicating cell complexity) showing Caelyx+ H1299 cells after exposure to Caelyx alone, αEGFR-Caelyx, or αEGFR-Caelyx mixed with IsoAb or panibumumab (5:1 Ab-to-BsAb molar ratio). Histograms are shown for respective Caelyx and SSC channels. (H) Live cell confocal microscopy imaging of cancer cells after a 3-hour exposure to Caelyx alone or αEGFR-Caelyx. Nuclei and plasma membrane of cells were visualized using Hoechst 33,342 (blue), and CellMask Plasma Membrane Stain (green), respectively. Scale bars, 20 µm. In (B, F, and G), Caelyx+ cells were determined by flow cytometry analysis of Caelyx fluorescence in treated compared to untreated cells. Data in (B–F) are presented as means ± SDs of three (B–F) and five (C–E) independent experiments. |
Encouraged by these results, we proceeded to examine whether αEGFR-Caelyx binding to NSCLC cells is reliant on the specific recognition of cell-surface expressed EGFRECDIII. To do so, we exposed NSCLC cells to αEGFR-Caelyx in combination with EGFRECDIII-competing mAb, panitumumab, or a non-binder Ab isotype (IsoAb). Addition of panitumumab, but not the IsoAb control, induced a progressive decline in αEGFR-Caelyx binding to cancer cells and this was completely ablated at the highest density tested of 10:1 (mol:mol) panitumumab:αEGFR ratio, indicating that αEGFR interaction with cell-surface expressed EGFRECDIII is responsible for mediating the delivery of Caelyx to NSCLC cells (Figure1F and 1G). Live-cell confocal microscopy revealed that cell-bound αEGFR-Caelyx particles were effectively taken up by NSCLC cells, with all cells displaying a predominant intracellular drug fluorescent pattern, whilst minimal uptake was identified in the absence of αEGFR (Figure 1H). Overall, these results demonstrate that EGFR cell surface targeting using αEGFR effectively mediates the delivery of off-the-shelf PEGylated liposomal drugs to NSCLC cells.
αEGFR-Caelyx Demonstrates Improved Targeting to NSCLC in vitro
We next investigated the dose-dependent targeting efficacy of αEGFR-Caelyx versus Caelyx alone toward NSCLC cell monolayers (Figure 2A), and further determined the ability of αEGFR-Caelyx to recognize NSCLC cell lines with heterogeneous cell surface expression of EGFR, including cells expressing high (A549), intermediate (H1299), and low/intermediate (H460, Calu-6) densities of EGFR; MCF-7 breast cancer cells were used as low/negative EGFR expression control (Figure 2B and C). The cancer cell lines we used (Table S1) are derived from pleural effusions and metastatic sites of adults with NSCLC and represent the major types of this disease, including lung adenocarcinoma (Calu-6 and A549) and large cell carcinoma (H460 and H1299), a form of NSCLC that expands rapidly and is poorly responsive to chemotherapy.29 Dose–response curves and fold changes in the 50% cell targeting concentration (EC50) of Caelyx relative to αEGFR-Caelyx are indicated in Figure 2D and E. αEGFR-Caelyx demonstrated improved targeting efficacy toward all EGFR+ NSCLC cells, with 8.9- to 214.6-fold lower EC50 values compared to treatment with Caelyx alone (Figure 2E), and increments in EC50 fold-change showed a moderate positive correlation (r = 0.62) when compared against EGFR cell surface expression (Figure 2F). Calu-6 were the only cells to differ this tendency, which although expressing low/intermediate levels of EGFR, increments in targeting reached more than 200-fold. In contrast, EGFR− MCF-7 cells had no preferential binding of αEGFR-Caelyx or untargeted Caelyx (<2 EC50 fold change) (Figure 2D and E). Further, no differences in NSCLC cell targeting were obtained when complexing Caelyx with a BsAb binding PEG and CD19 (αCD19), a receptor with exclusive expression in B-lymphocyte lineage cells and therefore absent in NSCLC cells (Figure S1). Together, these results indicate that αEGFR-mediated improvements in NSCLC targeting ability of Caelyx are exclusive to EGFR expression in the cancer cells, and higher densities of EGFR in these cells may be predictive of increased binding by αEGFR-Caelyx.
 |
Figure 2 In vitro NSCLC cell targeting analysis of αEGFR-Caelyx. (A) Illustration showing cell targeting analysis of αEGFR-Caelyx and Caelyx alone. Percentage of cells targeted by αEGFR-Caelyx or Caelyx alone were determined by flow cytometry analysis, comparing Caelyx fluorescence in treated cells to cells receiving vector (autofluorescence). (B and C) Flow cytometry measurement of EGFR cell surface expression in cancer cells using a tandem of anti-human EGFR mAb or IsoAb plus a secondary Alexa Fluor (AF) 647-labelled IgG, including histograms (b) and AF 647 median fluorescence intensity (MFI) (C). (D) Dose–response curves showing percentage of targeted cancer cells (Caelyx+ cells) at increasing concentrations of αEGFR-Caelyx or untargeted Caelyx in a representative experiment. EC50 indicates drug concentration leading to 50% targeted cells. Data are means ± SDs of three experimental replicates. (E) Plots comparing EC50 of Caelyx alone versus αEGFR-Caelyx from three independent experiments. EC50 fold changes (Caelyx alone/αEGFR-Caelyx) were determined for individual experiments and plotted as means ± SDs. P values were computed using ratio paired t-test. (F) Correlation plot comparing EC50 fold change (Caelyx alone/αEGFR-Caelyx) against EGFR cell surface expression (AF 647 MFI). Pearson’s correlation coefficient, r. Error lines indicate 95% confidence bands of the best-fit line. Cutoff value (green) of twofold is defined to establish relevant changes in EC50. EGFR expression. In (C and F), data are presented as means (F) or means ± SDs (C) of three independent experiments. |
In vitro Treatment of NSCLC Cells with αEGFR-Caelyx is More Efficacious Than Caelyx Alone
We subsequently investigated whether the improved targeting ability of αEGFR-Caelyx toward NSCLC cell cultures correlates with potent cytotoxic action against these cells (Figure 3A). A 3-hour treatment of NSCLC cells with αEGFR-Caelyx caused more pronounced decrements in cell proliferation and increased percentage of apoptotic, dead, and necrotic cells, than treatment with Caelyx alone, resulting in marked reductions in the overall number of viable cancer cells (Figure 3B). Dose–response curves showing total counts of viable cancer cells post-treatment, half-maximal inhibitory concentration (IC50), and fold changes comparing IC50 of Caelyx relative to αEGFR-Caelyx (IC50 fold change) are shown in Figure 3C and D and Figure S2. IC50 fold changes were 3.5 to 9.7 in EGFR+ NSCLC cells, whilst no significant changes in IC50 were found in MCF-7 (EGFR−) cells (Figure 3D and Figure S3). Of note, αEGFR-induced increments in cytotoxicity of Caelyx (IC50 fold change) correlated with both higher EGFR cell surface expression on the cancer cells (r = 0.92) and improvements in cancer cell targeting (EC50 fold change; r = 0.61) (Figure 3E). Last, αCD19-complexed Caelyx (αCD19-Caelyx) displayed similar cytotoxicity to Caelyx alone (Figure S4), and αEGFR delivered alone to NSCLC cells had no significant effects in cancer cell viability or proliferation (Figure S5). These results show that complexing Caelyx with αEGFR effectively improves its cytotoxic potency to treat NSCLC, and greater anticancer effects can be obtained against NSCLC cells expressing higher densities of EGFR.
 |
Figure 3 Investigating cytotoxicity of αEGFR-Caelyx against NSCLC cells in vitro. (A) Illustration depicting cytotoxicity analysis αEGFR-Caelyx and Caelyx alone. Viable cells were defined by flow cytometry analysis as those double-negative for Annexin V-allophycocyanin (APC) and Sytox green markers of early apoptosis and cell death, respectively, and counted using flow cytometry counting beads. Flow cytometry plots indicate cell counts (cells/mL) in a representative experiment. (B) Percentage of viable, apoptotic, dead, and necrotic cells (clustered stacked chart; left), and total cell count (relative to vehicle; right) of 5 µM (Calu-6), 10 µM (H460) or 20 µM (A549 and H1299) Caelyx-treated cells compared to vehicle (PBS)-treated cells. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA followed by Dunnett’s multiple comparisons test) comparing percentage of viable cells or total cell count after treatment with αEGFR-Caelyx versus Caelyx alone. (C) Dose–response curves show viable cell counts (relative to vehicle) after treatment with increasing concentrations of EGFR-Caelyx or Caelyx alone (left) in the first of three repeated experiments; second and third replicates are shown in Figure S3. Data are means ± SDs of three experimental replicates. (D) Plots comparing IC50 of Caelyx alone versus αEGFR-Caelyx from three independent experiments. IC50 fold changes (Caelyx alone/αEGFR-Caelyx) were determined for individual experiments and plotted as means ± SDs. P values were computed using ratio paired t-test. (E) Correlation plot comparing IC50 fold change (Caelyx alone/αEGFR-Caelyx) against EGFR cell surface expression (AF 647 MFI). Pearson’s correlation coefficient, r. Error lines indicate 95% confidence bands of the best-fit line. Cutoff value (green) of twofold change is defined to establish relevant changes in EC50 and IC50. In (B and E), data are presented as means (E) or means ± SDs (B) of three independent experiments. |
αEGFR-Caelyx Shows Enhanced Internalization into NSCLC Spheroids in vitro
Motivated by the notable in vitro NSCLC targeting and anticancer effects of αEGFR-Caelyx, we next sought to investigate whether this targeted therapeutic would further display an improved efficacy to accumulate and penetrate into a three-dimensional (3D) NSCLC tumoroid model. 3D tumor models such as spheroids closely resemble the structural architecture and hypoxic conditions of the tumor environment in vivo,30 and consequently represent an advanced in vitro cell culture model to study the ability of targeted nanomaterials to bind and internalize into tumors. For this study, we used H460 cell spheroids grew to a diameter of approximately 250 µm. Spheroids were incubated with αEGFR-Caelyx or Caelyx alone and drug internalization was determined by live cell confocal microscopy, measuring Caelyx fluorescence from the surface to the center of the spheroids equatorial plane (Figure 4A). We found that αEGFR-Caelyx was more efficacious than Caelyx alone at accumulating and penetrating into the spheroids inner regions, resulting in significant increments in drug fluorescence of 32% (P = 0.043) and 73% (P = 0.016) at distances 0 to 50 µm (proliferative zone) and 50 to 100 µm (quiescent and necrotic zones) away from the spheroids surface, respectively (Figure 4B–D). In addition, we detected a fluorescence peak at the innermost region (90 to 100 µm below surface) of spheroids exposed to αEGFR-Caelyx, but not Caelyx alone (Figure 4C), indicating an improved ability of this targeted therapeutic to reach the necrotic zone of tumors to effectively suppress tumor growth.31
 |
Figure 4 Studying internalization of αEGFR-Caelyx into 3D spheroid models. (A) Illustration depicting the internalization analysis of αEGFR-Caelyx and Caelyx alone. We draw 20 radii (0 to 100 µm below spheroids surface) distributed across the spheroids equatorial plane and measured Caelyx fluorescence in them using live cell confocal microscopy. Plot shows grouped fluorescence intensity over distance of 20 radii in a representative spheroid; data are means +SDs. (B) Live confocal microscopy images of spheroids equatorial plane (scale bars, 125 µm); Caelyx fluorescence is shown in red. Images are representative of three independent experiments. An enlarged image (i) is also shown for spheroids exposed to αEGFR-Caelyx or Caelyx alone (scale bars, 50 µm). (C and D) Plots (C) and respective area under curves (AUCs) (D) show fluorescence intensity of Caelyx at different distances below the spheroids surface. Data represent means ± SDs of 6 spheroids per condition pooled from three independent experiments. P values comparing αEGFR-Caelyx versus Caelyx alone were computed using one-way ANOVA followed by Dunnett’s multiple comparisons test. |
Investigating the NSCLC Tumor Accumulation of αEGFR-Caelyx in vivo
Based on the demonstrated potency of αEGFR-Caelyx to accumulate and penetrate into NSCLC spheroids ex vivo, we next investigated its efficacy in targeting NSCLC tumors in vivo. We further measured in parallel whether αEGFR-Caelyx shows reduced off-target uptake by organs that are highly sensitive to its cytotoxic side effects (heart) or primarily responsible for the clearance of Caelyx in vivo (liver). Immunodeficient mice harboring H460 NSCLC tumors received a single intravenous injection of αEGFR-Caelyx (1 mg/kg) or Caelyx alone (1 mg/kg), and drug quantities in tumor, liver, and heart were measured 4, 24, or 48 hours post administration (p.a.) (Figure 5A). We found a 5-fold greater accumulation of αEGFR-Caelyx compared with Caelyx alone in the tumor at 4 and 24 hours p.a. (Figure 5B). Intratumoral amounts of αEGFR-Caelyx minimally decreased 48 hours p.a. (3-fold change versus Caelyx alone, Figure 5B). Of note, these results correlated with 3.2-fold and 5.5-fold reduced amounts of αEGFR-Caelyx compared to untargeted Caelyx in the heart at 4 and 24 hours p.a. (Figure 5C), and minor (<3-fold) differences in uptake were found in the liver across the 48 hours p.a. (Figure 5D).
 |
Figure 5 αEGFR-Caelyx accumulates in NSCLC tumors in vivo with minimal accumulation in the heart. (A) Timeline of biodistribution experiment. SCID-beige mice bearing subcutaneous H460 NSCLC tumors received an intravenous injection of αEGFR-Caelyx (1 mg/kg; n = 4 mice) or Caelyx alone (1 mg/kg; n = 4 mice) and drug amounts (nmol drug/g tissue) in tumor, liver, and heart were measured ex vivo at 4, 24 or 48 hours after injection. (B–D) Plots show drug amounts (median plus interquartile range (IQR); 4 animals per condition) in tumor (B), heart (C), and liver (D). P values were computed using Mann–Whitney U-test; *P < 0.05. |
αEGFR-Caelyx Suppresses Growth of NSCLC Tumors in a Xenograft Model
Anticancer efficacy of αEGFR-Caelyx compared to untargeted Caelyx was finally assessed in a xenograft model of NSCLC. Immunodeficient mice harboring H460 tumors received a treatment course of four intravenous injections, once weekly, with either phosphate-buffered saline (vehicle), Caelyx alone (1 mg/kg), or αEGFR-Caelyx (1 mg/kg), and tumor size was monitored until 24 hours after completing treatment (21 days from treatment initiation) (Figure 6A). A separate set of H460 tumor-bearing mice were injected with αCD19-Caelyx as non-NSCLC targeting control to demonstrate specificity of therapy. Treatment with αEGFR-Caelyx significantly ablated the expansion of tumors (Figure 6B–D and Table S3). Tumors of mice receiving αEGFR-Caelyx were 2.77-fold smaller in size relative to vehicle at the completion of the study (P < 0.001, Figure 6D). In contrast, effects of Caelyx alone or αCD19-Caelyx on tumor growth were negligible (<1.8-fold decrease in tumor size vs vehicle) and not significant (P > 0.59, Figure 6D). All treatments were well tolerated, with no signs of weight loss or other indicators of toxicity (Figure 6E).
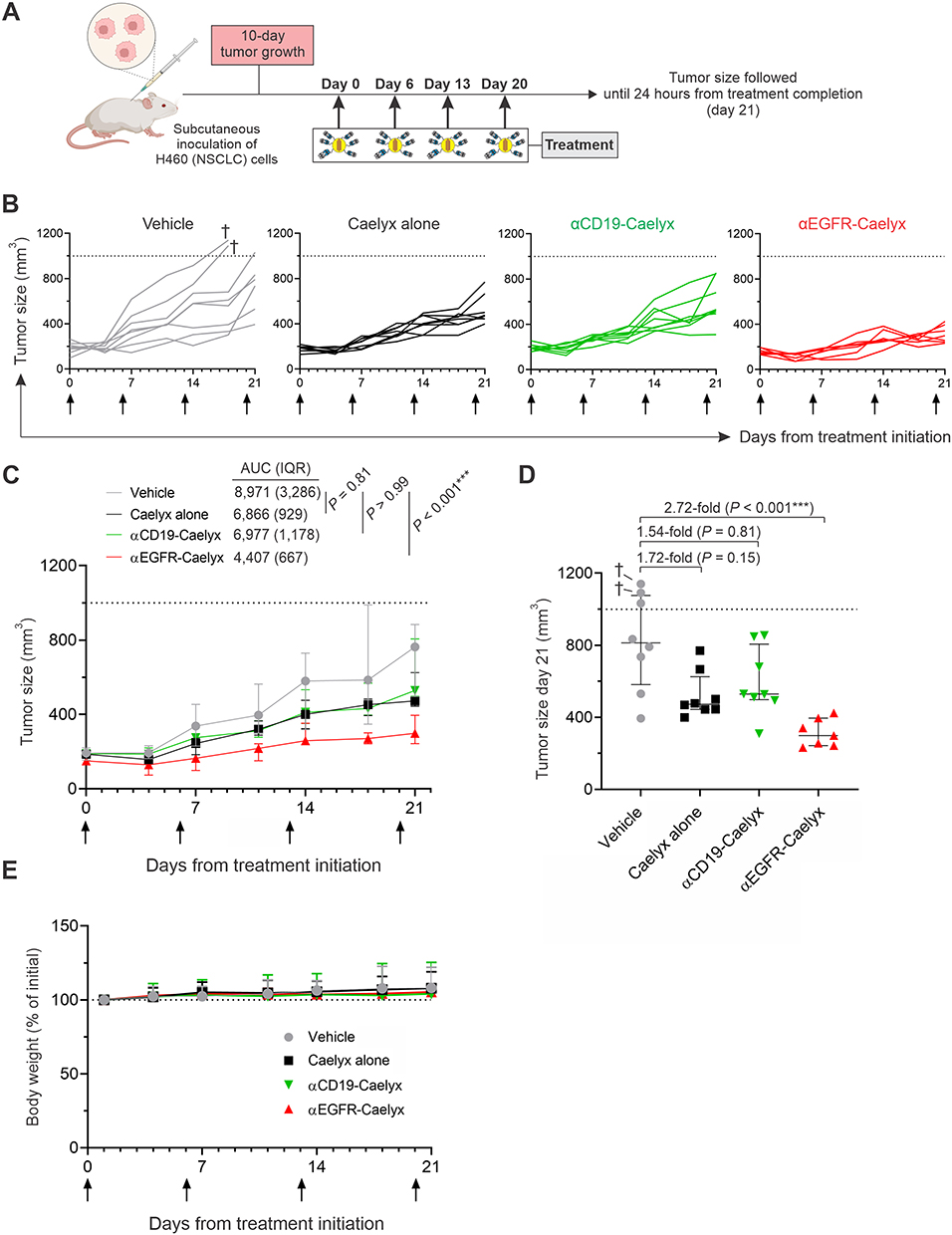 |
Figure 6 αEGFR-Caelyx significantly suppresses NSCLC progression in a tumor-xenograft model. (A) Timeline and treatment course of therapeutic experiments. SCID-beige mice engrafted with H460 cells were intravenously injected with vehicle (PBS; n = 8 mice), Caelyx alone (1 mg/kg; n = 8 mice), αCD19-Caelyx (1 mg/kg; n = 8 mice), or αEGFR-Caelyx (1 mg/kg; n = 7 mice). (B and C) Tumor size of treated mice over time; data are shown for individual (B) or grouped (C) mice. (D)Tumor size on day 21 (completion of the study). (E) Body weight of treated animals over time. Arrows in (A–C) and (E) indicate days of injection. Dash line in (B–D) indicates tumor size defined as event (1,000 mm3). In (B and D), (†) indicates individual mice that reached event on day 18. In (C and D), data are presented as medians plus IQR. P values comparing tumor size injection of Caelyx alone, αCD19-Caelyx, or αEGFR-Caelyx versus vehicle were computed using Kruskal–Wallis test followed by Dunn’s multiple comparisons test; ***P < 0.001. |
Discussion
NSCLC is the most aggressive and treatment-resistant form of lung cancer.32 High-dose chemotherapy is the gold standard against NSCLC and, although highly effective, its cytotoxic effects affect both cancerous and normal cells, causing severe dose-limiting toxicity.32 Sterically stabilized liposomal formulations of chemotherapy drugs provide extended circulation times in the blood and improve the passive delivery of packaged drugs to neovascularized tumors.33 However, the inability of liposomal drugs to discern between cancerous and healthy cells is a major roadblock that markedly limits their efficacy. Herein, we investigated the use of an anti-PEG/anti-EGFR BsAb (αEGFR) to provide FDA-approved PEGylated liposomal doxorubicin (Caelyx) with cancer selectivity and mediate its active targeted delivery to NSCLC. A single mixing step of Caelyx with αEGFR formed targeted complexes (αEGFR-Caelyx) with selective recognition and enhanced anticancer potency toward NSCLC cells. αEGFR further improved the ability of Caelyx to target solid tumors and suppress their growth in a H460 NSCLC xenograft model.
EGFR is detectable in approximately 80% of patients with NSCLC and its overexpression is a predictor of poor prognosis.27,34 EGFR binding to its multiple ligands induces its activation and transduction of pro-survival signaling cascades that promote tumor growth, angiogenesis, and stimulate metastasis.27,34 These findings have thus prompted the advancement of treatment strategies for NSCLC in the form of monoclonal antibodies blocking the binding of ligands with EGFR, or small-molecule inhibitors targeting its intracellular tyrosine kinase domain (TKD).35 However, these therapeutics are unable to eliminate the cancer cells on their own and thus need to be delivered in combination with high-dose chemotherapy to succeed, causing severe adverse effects.36,37 TKD inhibitors are also highly prone to fail due to the numerous mutations affecting this domain.38 In this work, we envisaged a differential therapeutic role for EGFR in NSCLC. Rather than blocking or inhibiting its oncogenic activity, we proposed to use EGFR as an anchoring point at the NSCLC cell surface to mediate the targeting, receptor-driven internalization, and intracellular release of liposomal-encapsulated doxorubicin, a potent DNA-damaging and cytotoxic drug, specifically into the cancer cells and not the healthy ones. This approach ameliorated the delivery and anticancer potency of Caelyx against EGFR+ NSCLC cells. Such effects were exclusive to cancer cells expressing EGFR, thus sparing cells that lacked this receptor (eg MCF-7 cells), and increments in efficacy were more pronounced in NSCLC cells expressing higher densities of EGFR (eg H460, H1299 and A549). In addition, the anticancer activity of our approach was linked to αEGFR-assisted delivery of Caelyx to the NSCLC cells rather than an inhibitory effect of αEGFR binding to EGFR on these cells. We and others provided proof-of-concept in previous works of targeting nanotherapeutics to EGFR as an efficacious approach to enhance their selectivity and delivery toward multiple cancers,25,35,39,40 yet this strategy has been poorly investigated in the management of NSCLC.41–43
BsAbs with bidirectional binding to PEG and EGFR constitute an advantageous technology for the facile functionalization of PEGylated nanomaterials and even smaller therapeutics to provide them with selectivity against EGFR-expressing cancers. Further, due to the low frequency of NSCLC patients presenting mutations in EGFRECDIII, BsAbs targeting this domain such as αEGFR would represent an efficacious approach to universally target NSCLC. Given the vast range of FDA-approved materials currently grafted with PEG, BsAbs targeting PEG and cancer cell-surface receptors can be of benefit for repurposing and improving the therapeutic index of numerous treatment strategies used in the management of cancer such as mRNA vaccines, RNA-interference therapy, enzymes, or chemotherapy-loaded nanotherapeutics.24,44–46 In addition, due to BsAb affinity for PEG, the targeted therapeutics can be formed in a time- and cost-effective manner (eg one-step 2-hour mixing prior to injection) without altering nanoparticle structure of function. Last, compared to BsAb targeting systems harboring anti-PEG/anti-receptor Fab/scFv fragments (91 kDa),47 our scFv/scFv BsAb format offers the advantages of smaller size (55 kDa) and lack of constant domains for reduced immunogenicity, easier production, and better tumor penetration whilst providing resistance to reducing conditions.
Caelyx (also referred to as Doxil) was the first liposomal drug to provide clinical benefits in the management of solid tumors and since then, has been the most investigated and clinically reviewed nanomedicine in the management of cancer. However, despite its therapeutic potential, Caelyx vastly accumulates in healthy organs, reducing its bioavailability at the tumor site while causing numerous adverse events in humans that span stomatitis, high-grade cutaneous lesions, hematological toxicity, or cardiotoxicity, and which can lead to treatment discontinuation or even cause death in some patients.48 In this work, we proved that active tumor-targeting of Caelyx using our αEGFR is a successful approach to circumvent these limitations and improve its therapeutic efficacy. αEGFR-Caelyx demonstrated significantly better accumulation compared to Caelyx alone in H460 NSCLC tumors both in vitro using 3D spheroid cultures, and in vivo in tumor-bearing mice, showing five-fold increased intratumoral drug delivery in vivo at both 4 hours and 24 hours post administration, whilst leading to a three to five times lower amount of drug reaching the heart at equivalent timepoints. This correlated with an approximate three-fold suppression in H460 NSCLC tumor growth with no signs of toxicity in αEGFR-Caelyx treated animals compared to mice receiving vehicle, whereas no differences in cancer growth were obtained when treating animals with Caelyx alone or a non-NSCLC-specific BsAb-Caelyx conjugate (αCD19-Caelyx). Such reductions in tumor growth and differential biodistribution pattern may indicate an improved treatment potency along with a reduced risk for αEGFR-Caelyx to cause long-term cardiotoxic effects in humans.
Future studies with larger numbers of xenograft panels, including cell lines and patient-derived tumor isolates with heterogeneous EGFR expression, will illustrate the universality and clinical translatability of our treatment approach for the management of NSCLC. Investigation of immunocompetent mouse models is also required to elucidate any involvement of the immune system in boosting or reducing the anticancer effects and safety of our treatment approach.
Conclusion
In conclusion, our work demonstrates the power of using an anti-PEG/anti-EGFR BsAb for repurposing and improving the therapeutic potency of a historically effective FDA-approved liposomal drug, Caelyx, in the management of NSCLC. BsAb-assisted improvements in targeting and cytotoxic potency of Caelyx were demonstrated in vitro against a panel of NSCLC cell lines and a 3D tumoroid model, and in vivo in an animal model of the disease. The therapeutic advantages of this approach could be of relevance for NSCLC therapy by ameliorating its side effects whilst boosting efficacy, and may further translate to other PEGylated nanomedicines for applications in the management and diagnosis of the numerous solid tumors overexpressing EGFR (tumors in breast, ovary, lung, bladder, kidneys, pancreas, neck, and gliomas), thus facilitating the advancement and translation to clinical use of new targeted therapeutics with shorter development timelines.
Abbreviations
BsAb, bispecific antibody; EC50, 50% cell targeting concentration; Caelyx, PEGylated liposomal doxorubicin; EGFR, endothelial growth factor receptor; αEGFR, EGFR/PEG scFv BsAb; IC50, half-maximal inhibitory concentration; IsoAb, isotype antibody; mAb, monoclonal antibody; NSCLC, non-small cell lung cancer; PBS, phosphate buffered saline; PEG, polyethylene glycol; scFv, single chain variable fragment.
Data Sharing Statement
All data generated or analyzed during this study are included in the article and its supplementary materials.
Ethics Approval and Informed Consent
All animal experimentation studies were conducted with approval from the Animal Care and Ethics Committee of UNSW Sydney (Sydney, Australia) and in compliance with the Australian code for the care and use of animals for scientific purposes.
Acknowledgments
This work was supported by the Children’s Cancer Institute (CCI), which is affiliated with the University of New South Wales (UNSW Sydney), and the Sydney Children’s Hospital Network. This work was also supported by an Australian Government Research Training Program (RTP) Scholarship and Royal Australian and New Zealand College of Radiologists. We acknowledge that elements of this research utilized services provided by the Queensland node of the National Biologics Facility (NBF) and the Australian National Fabrication Facility (ANFF) – Queensland node. NBF is supported by Therapeutic Innovation Australia and both facilities are supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program. We would like to thank the Katharina Gaus Light Microscopy Facility at Mark Wainwright Analytical Centre (UNSW) and for their support and resources involved in this work; and UNSW Sydney for Research Infrastructure Support to establish the Liposomal Development lab at CCI.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded in part from the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (CE140100036, to M.K. and K.J.T.); ARC Training Centre for Innovation in Biomedical Imaging Technologies (IC170100035, to K.J.T.); National Health and Medical Research Council Investigator Grant (#2016464 to MK) and a Tour de Cure Grant (to MK). K.J.T. acknowledges the award of a Career Development Fellowship (APP1148582).
Disclosure
Professor Kristofer Thurecht reports the bispecific antibody technology has been awarded patent number WWO2016123675A1. The authors declare that they have no other competing interests in this work.
References
1. Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300. doi:10.21037/tlcr.2016.06.07
2. Li S, de Camargo Correia GS, Wang J, et al. Emerging targeted therapies in advanced Non-small-cell lung cancer. Cancers. 2023;15(11). doi:10.3390/cancers15112899
3. Sasaki A, Fujimoto Y, Inada T, et al. Efficacy of tyrosine kinase inhibitors in patients with non-small-cell lung cancer with performance status 4: a case series and review of the literature. J Med Case Rep. 2023;17(1):410. doi:10.1186/s13256-023-04145-z
4. Liu P, Chen G, Zhang J. A review of liposomes as a drug delivery system: current status of approved products, regulatory environments, and future perspectives. Molecules. 2022;27:4. doi:10.3390/molecules27041372
5. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi:10.1016/j.addr.2012.09.037
6. Suk JS, Xu Q, Kim N, et al. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. doi:10.1016/j.addr.2015.09.012
7. Barenholz Y. Doxil(R)- The first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi:10.1016/j.jconrel.2012.03.020
8. Gabizon AA. Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19(4):424–436.
9. Udhrain A, Skubitz KM, Northfelt DW. Pegylated liposomal doxorubicin in the treatment of AIDS-related Kaposi’s sarcoma. Int J Nanomed. 2007;2(3):345–352.
10. Gibson JM, Alzghari S, Ahn C, et al. The role of pegylated liposomal doxorubicin in ovarian cancer: a meta-analysis of randomized clinical trials. Oncologist. 2013;18(9):1022–1031. doi:10.1634/theoncologist.2013-0126
11. Leighl NB, Burkes RL, Dancey JE, et al. A Phase I study of pegylated liposomal doxorubicin hydrochloride (Caelyx) in combination with cyclophosphamide and vincristine as second-line treatment of patients with small-cell lung cancer. Clin Lung Cancer. 2003;5(2):107–112. doi:10.3816/CLC.2003.n.024
12. Mlineritsch B, Mayer P, Rass C, et al. Phase II study of single-agent pegylated liposomal doxorubicin HCl (PLD) in metastatic breast cancer after first-line treatment failure. Onkologie. 2004;27(5):441–446. doi:10.1159/000080363
13. Makwana V, Karanjia J, Haselhorst T, et al. Liposomal doxorubicin as targeted delivery platform: current trends in surface functionalization. Int J Pharm. 2021;593:120117. doi:10.1016/j.ijpharm.2020.120117
14. Jahan S, Karim ME, Chowdhury EH. Nanoparticles targeting receptors on breast cancer for efficient delivery of chemotherapeutics. Biomedicines. 2021;9(2). doi:10.3390/biomedicines9020114
15. Kim B, Shin J, Wu J, et al. Engineering peptide-targeted liposomal nanoparticles optimized for improved selectivity for HER2-positive breast cancer cells to achieve enhanced in vivo efficacy. J Control Release. 2020;322:530–541. doi:10.1016/j.jconrel.2020.04.010
16. Kamoun WS, Kirpotin DB, Huang ZR, et al. Antitumour activity and tolerability of an EphA2-targeted nanotherapeutic in multiple mouse models. Nat Biomed Eng. 2019;3(4):264–280. doi:10.1038/s41551-019-0385-4
17. Sandoval MA, Sloat BR, Lansakara PD, et al. EGFR-targeted stearoyl gemcitabine nanoparticles show enhanced anti-tumor activity. J Control Release. 2012;157(2):287–296. doi:10.1016/j.jconrel.2011.08.015
18. Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65(24):11631–11638. doi:10.1158/0008-5472.CAN-05-1093
19. Chang DK, Li PC, Lu RM, et al. Peptide-mediated liposomal Doxorubicin enhances drug delivery efficiency and therapeutic efficacy in animal models. PLoS One. 2013;8(12):e83239. doi:10.1371/journal.pone.0083239
20. Cheng L, Huang FZ, Cheng LF, et al. GE11-modified liposomes for non-small cell lung cancer targeting: preparation, ex vitro and in vivo evaluation. Int J Nanomed. 2014;9:921–935. doi:10.2147/IJN.S53310
21. Chi YH, Hsiao JK, Lin MH, et al. Lung cancer-targeting peptides with multi-subtype indication for combinational drug delivery and molecular imaging. Theranostics. 2017;7(6):1612–1632. doi:10.7150/thno.17573
22. Eras A, Castillo D, Suarez M, et al. Chemical conjugation in drug delivery systems. Front Chem. 2022;10:889083. doi:10.3389/fchem.2022.889083
23. Sivaram AJ, Wardiana A, Howard CB, et al. Recent advances in the generation of antibody-nanomaterial conjugates. Adv Healthc Mater. 2018;7(1). doi:10.1002/adhm.201700607
24. Moles E, Howard CB, Huda P, et al. Delivery of PEGylated liposomal doxorubicin by bispecific antibodies improves treatment in models of high-risk childhood leukemia. Sci Transl Med. 2023;15(696):eabm1262. doi:10.1126/scitranslmed.abm1262
25. Howard CB, Fletcher N, Houston ZH, et al. Overcoming instability of antibody-nanomaterial conjugates: next generation targeted nanomedicines using bispecific antibodies. Adv. Healthc Mater. 2016;5(16):2055–2068. doi:10.1002/adhm.201600263
26. Logan A, Howard CB, Huda P, et al. Targeted delivery of polo-like kinase 1 siRNA nanoparticles using an EGFR-PEG bispecific antibody inhibits proliferation of high-risk neuroblastoma. J Control Release. 2024;367:806–820. doi:10.1016/j.jconrel.2024.02.007
27. Karlsen EA, Kahler S, Tefay J, et al. Epidermal growth factor receptor expression and resistance patterns to targeted therapy in non-small cell lung cancer: a review. Cells. 2021;10(5). doi:10.3390/cells10051206
28. Wang Q, Wang Y, Zhang X, et al. Efficacy of erlotinib in NSCLC harboring rare EGFR extracellular domain mutation (T263P) and common mutations: case report and literature review. Front Oncol. 2022;12:954026. doi:10.3389/fonc.2022.954026
29. Tai Q, Zhang L, Hu X. Clinical characteristics and treatments of large cell lung carcinoma: a retrospective study using SEER data. Transl Cancer Res. 2020;9(3):1455–1464. doi:10.21037/tcr.2020.01.40
30. Pinto B, Henriques AC, Silva PMA, Bousbaa H. Three-dimensional spheroids as in vitro preclinical models for cancer research. Pharmaceutics. 2020;12(12). doi:10.3390/pharmaceutics12121186
31. Yamamoto A, Huang Y, Krajina BA, et al. Metastasis from the tumor interior and necrotic core formation are regulated by breast cancer-derived angiopoietin-like 7. Proc Natl Acad Sci U S A. 2023;120(10):e2214888120. doi:10.1073/pnas.2214888120
32. Chen Z, Fillmore CM, Hammerman PS, et al. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. doi:10.1038/nrc3775
33. Wang S, Chen Y, Guo J, Huang Q. Liposomes for tumor targeted therapy: a review. Int J Mol Sci. 2023;24(3). doi:10.3390/ijms24032643
34. Bethune G, Bethune D, Ridgway N, Xu Z. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2010;2(1):48–51.
35. Santos EDS, Nogueira KAB, Fernandes LCC, et al. EGFR targeting for cancer therapy: pharmacology and immunoconjugates with drugs and nanoparticles. Int J Pharm. 2021;592:120082. doi:10.1016/j.ijpharm.2020.120082
36. Cai WQ, Zeng LS, Wang LF, et al. The latest battles between EGFR monoclonal antibodies and resistant tumor cells. Front Oncol. 2020;10:1249. doi:10.3389/fonc.2020.01249
37. Zubair T, Bandyopadhyay D. Small molecule EGFR inhibitors as anti-cancer agents: discovery, mechanisms of action, and opportunities. Int J Mol Sci. 2023;24(3). doi:10.3390/ijms24032651
38. Hsu WH, Yang JC, Mok TS, Loong HH. Overview of current systemic management of EGFR-mutant NSCLC. Ann Oncol. 2018;29(suppl_1):i3–i9. doi:10.1093/annonc/mdx702
39. Y-C S, Burnouf P-A, Chuang K-H, et al. Conditional internalization of PEGylated nanomedicines by PEG engagers for triple negative breast cancer therapy. Nat Commun. 2017;8(1):15507. doi:10.1038/ncomms15507
40. Cui J, Ju Y, Houston ZH, et al. Modulating targeting of poly(ethylene glycol) particles to tumor cells using bispecific antibodies. Adv Healthc Mater. 2019;8(9):e1801607. doi:10.1002/adhm.201801607
41. Lu X, Liu S, Han M, et al. Afatinib-loaded immunoliposomes functionalized with cetuximab: a novel strategy targeting the epidermal growth factor receptor for treatment of non-small-cell lung cancer. Int J Pharm. 2019;560:126–135. doi:10.1016/j.ijpharm.2019.02.001
42. Patel J, Amrutiya J, Bhatt P, et al. Targeted delivery of monoclonal antibody conjugated docetaxel loaded PLGA nanoparticles into EGFR overexpressed lung tumour cells. J Microencapsul. 2018;35(2):204–217. doi:10.1080/02652048.2018.1453560
43. Master AM, Sen Gupta A. EGF receptor-targeted nanocarriers for enhanced cancer treatment. Nanomedicine. 2012;7(12):1895–1906. doi:10.2217/nnm.12.160
44. Dinndorf PA, Gootenberg J, Cohen MH, et al. FDA drug approval summary: pegaspargase (oncaspar) for the first-line treatment of children with acute lymphoblastic leukemia (ALL). Oncologist. 2007;12(8):991–998. doi:10.1634/theoncologist.12-8-991
45. Urits I, Swanson D, Swett MC, et al. A review of Patisiran (ONPATTRO(R)) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther. 2020;9(2):301–315. doi:10.1007/s40120-020-00208-1
46. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6(12):1078–1094. doi:10.1038/s41578-021-00358-0
47. Kao CH, Wang JY, Chuang KH, et al. One-step mixing with humanized anti-mPEG bispecific antibody enhances tumor accumulation and therapeutic efficacy of mPEGylated nanoparticles. Biomaterials. 2014;35(37):9930–9940. doi:10.1016/j.biomaterials.2014.08.032
48. Li XR, Cheng XH, Zhang GN, et al. Cardiac safety analysis of first-line chemotherapy drug pegylated liposomal doxorubicin in ovarian cancer. J Ovarian Res. 2022;15(1):96. doi:10.1186/s13048-022-01029-6
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.