Back to Journals » Lung Cancer: Targets and Therapy » Volume 8
EGFR T790M: revealing the secrets of a gatekeeper
Authors Ko B, Paucar D, Halmos B
Received 16 May 2017
Accepted for publication 11 July 2017
Published 9 October 2017 Volume 2017:8 Pages 147—159
DOI https://doi.org/10.2147/LCTT.S117944
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pan-Chyr Yang
Brian Ko, Daniel Paucar, Balazs Halmos
Department of Oncology, Montefiore Medical Center, Albert Einstein College of Medicine, New York, NY, USA
Abstract: Non-small-cell lung cancers that harbor activating mutations in the EGFR gene represent an important molecularly defined subset of lung cancer. Despite dramatic initial responses with first- and second-generation EGFR-directed tyrosine-kinase inhibitors (TKIs) against these cancers, the development of a dominant and frequent resistance mechanism through a threonine–methionine amino acid substitution at position 790 (T790M) of EGFR has limited the long-term efficacy of these targeted therapies. This “gatekeeper” EGFR T790M alteration remains the only validated and relevant second-site resistance mutation for EGFR, allowing for focused research to understand and overcome EGFR T790M-mediated resistance. The current review focuses on EGFR T790M by discussing mechanisms of resistance mediated by EGFR T790M, reviewing development of novel third-generation EGFR TKIs targeting EGFR T790M, and highlighting current research on overcoming resistance to third-generation EGFR T790M TKIs.
Keywords: lung cancer, epidermal growth factor receptor, T790M, targeted therapy, resistance
Introduction
Numerous demonstrations of clinical efficacy with targeted therapies in a diverse range of cancer types have cemented molecularly targeted therapies as a major pillar in the treatment of many advanced solid-tumor and hematologic malignancies. For advanced non-small-cell lung cancers (NSCLCs) with certain activating mutations in the EGFR gene, first- and second-generation EGFR-directed tyrosine-kinase inhibitors (TKIs), including gefitinib, erlotinib, and afatinib1,2 have supplanted platinum-containing chemotherapy regimens as first-line therapy, marking a major paradigm shift in the treatment of NSCLC. Unfortunately, despite initial clinical and radiological responses, resistance to first- and second-generation EGFR TKIs invariably develops, with median progression-free survival of 9–16 months3–6 and approximately 60% of patients developing resistance mediated by drug-induced selection of tumor-cell clones harboring a threonine–methionine amino-acid substitution at position 790 (T790M) of EGFR, which is the putative gatekeeper residue.7
The current review focuses on EGFR T790M. We first outline the treatment of EGFR-mutant NSCLC with first- and second-generation EGFR TKIs and the discovery of EGFR T790M as a dominant resistance mechanism to first- and second-generation EGFR TKI treatment. We then discuss mechanisms of resistance mediated by EGFR T790M and the development of novel third-generation EGFR TKIs targeting EGFR T790M. Next, we introduce novel methods for detecting EGFR T790M, along with appropriate workflows for incorporating molecular testing into clinical management. Lastly, we briefly highlight current research on overcoming resistance to third-generation EGFR T790M TKIs.
Treatment of EGFR-mutant NSCLC with first- and second-generation EGFR TKIs
EGFR is a cell-surface receptor belonging to the ErbB family of receptor TKs that also includes ErbB2/Her2/Neu, ErbB3/Her3, and ErbB4/Her4. The binding of cognate ligands, such as EGF and TGFα, induces a conformational change in EGFR that facilitates receptor homo- or heterodimer formation, in which the C-terminal lobe of one kinase domain of the asymmetric dimer complex plays a role analogous to that of cyclin in activated cyclin-dependent kinase complexes.8,9 Subsequently, activated EGFR can phosphorylate a range of substrates that results in the activation of multiple important downstream-signaling pathways within the cell, including the RAS–RAF–MEK–ERK pathway and the PI3K–AKT–mTOR pathway, promoting cell proliferation and cell survival, respectively. With certain activating EGFR mutations, EGFR TK activity is permanently switched on, even in the absence of cognate ligands, through an intrinsic tendency to form asymmetric dimers, in which the C-terminal lobe of one kinase domain associates with the N-terminal lobe of a neighboring kinase domain, inducing an active conformation by orientating the regulatory C-helix to its inward position10 and resulting in inappropriate constitutive activation of oncogenic downstream-signaling pathways.
Tumor-associated EGFR mutations are present in approximately 10%–20% of NSCLC patients,1,2 with higher incidence in younger females, nonsmokers, and adenocarcinoma histology, occurring as frequently as 30%–50% in never-smokers with lung adenocarcinoma.11 These mutations most commonly occur within EGFR exons 18–21, which encode a critical portion of the EGFR-kinase domain, and usually do not occur concurrently with other NSCLC oncogenic mutations, such as KRAS mutations and ALK-gene rearrangements. Approximately 90% of these mutations are EGFR exon 19 deletions or exon 21 L858R point mutations,12 which increase the kinase activity of EGFR, leading to hyperactivation of downstream prosurvival signaling pathways, such as PI3K–AKT–mTOR and RAS–RAF–MEK–ERK,13 as well as induction of cyclin D1 expression.14 Through negative-feedback regulation, dual-specificity phosphatases, such as DUSP4 and DUSP6, can dampen excessive MAP-kinase signal transduction, while PHLDA1/2 can mitigate AKT activation.15–17 In addition, upregulation of the BH3-only polypeptide BIM correlates with gefitinib-induced apoptosis in gefitinib-sensitive EGFR-mutant lung cancer cells.18
Small-molecule adenosine triphosphate (ATP)-mimetic first-generation EGFR TKIs, such as gefitinib and erlotinib, inhibit EGFR TK activity by reversibly binding to the ATP-binding site of the receptor, while afatinib, a second-generation EGFR TKI, binds to the site irreversibly at amino-acid residue C797 at the cost of more severe epithelium-based toxicities (eg, rash, diarrhea) associated with increased binding to wild-type EGFR. In 2009, a pivotal Phase III study demonstrated superiority of gefitinib over carboplatin– paclitaxel as initial treatment for EGFR-mutated lung adenocarcinoma. The study was conducted among nonsmokers or former light smokers in East Asia with 12-month rates of progression-free survival of 24.9% with gefitinib and 6.7% with carboplatin–paclitaxel. In the subgroup of 261 patients with EGFR mutations, response rates were doubled and progression-free survival was significantly longer among those who received gefitinib than among those who received carboplatin–paclitaxel (HR for progression or death 0.48, 95% CI 0.36–0.64; P<0.001), establishing frontline EGFR TKI therapy as the optimal choice for this group of patients and highlighting the critical need for molecular testing in patients with advanced NSCLC.3 Since then, these results have been replicated multiple times with gefitinib, erlotinib, and afatinib with the LUX-Lung 319 and LUX-Lung 620 studies, demonstrating improvements in overall survival in patients with EGFR exon 19 deletion-positive tumors. Figure 1 summarizes the most clinically relevant EGFR mutations and pictorially demonstrates the distinct molecular targets of various EGFR inhibitors.
 | Figure 1 Molecular targets of EGFR inhibitors (A); exon map of EGFR mutations (B). Abbreviation: TKI, tyrosine-kinase inhibitor. |
EGFR T790M as a resistance mechanism to first- and second-generation EGFR TKIs
While first- and second-generation EGFR TKIs represent a prominent example of the clinical benefits targeted-therapy approaches can have on advanced oncogene-driven malignancies, the emergence of acquired resistance to these agents has prevented them from achieving long-term durable control of EGFR-mutant lung cancer. While approximately 60%–80% of patients with EGFR-mutant tumors initially respond to first- and second-generation EGFR TKIs with significant improvement in both objective response rates and progression-free survival compared to upfront platinum-containing chemotherapy regimens, resistance invariably develops, with a median progression-free survival of 9–16 months.3–6
First described in 2005, the most common mechanism of acquired resistance to first- and second-generation EGFR TKIs involves a threonine–methionine amino acid substitution at T790M of EGFR.7,21 Approximately 60% of patients treated with first- and second-generation EGFR TKIs harbor EGFR T790M mutations at the time of progression.22 In rare cases, EGFR T790M is reported as a germ-line alteration leading to a high lifetime risk of lung cancer,23 and recent research suggests that certain NSCLC tumors harbor genetically heterogeneous subclones with the EGFR T790M mutation at disease onset, instead of developing T790M de novo. Additionally, a subset of cells that are initially EGFR T790M-negative and survive EGFR TKI treatment can subsequently acquire EGFR T790M for a fitness advantage, representing a population of drug-tolerant cells that can evolve further acquired resistance mechanisms in the context of drug-related selective pressure.24 Several studies suggest that EGFR T790M-positive lung cancers tend to have more indolent progression and better prognosis compared to EGFR T790M-negative lung cancers,25,26 as the development of EGFR T790M may require less complex genetic and molecular changes compared to other resistance mechanisms.
The biochemical mechanisms of EGFR T790M-associated resistance are likely multifactorial, including the popular “gatekeeper” hypothesis, which stipulates that there is steric clash between the larger methionine moiety (compared to threonine) on the gatekeeper side chain of EGFR T790M and the aniline moiety of first-generation EGFR TKIs. Other mechanisms include drastically increased ATP-binding affinity for EGFR T790M, alterations in the catalytic domain, and changes in overall conformational dynamics.27 In contrast to other targetable kinases, such as ABL in chronic myeloid leukemia and ALK in ALK translocation-positive lung adenocarcinomas, in which a variety of second-site mutations can lead to resistance,28 EGFR T790M remains the only validated and relevant second-site resistance mutation for EGFR, as all other secondary mutations have been noted only in occasional case reports. This key dependence on T790M-mediated acquired resistance has allowed focused research into the development of T790M-targeting inhibitors.
Initial strategies to overcome EGFR T790M-mediated resistance
Concurrent with the original discovery of EGFR T790M, it was noted that while T790M leads to very high-level resistance (1,000-fold) against reversible EGFR inhibitors, such as gefitinib and erlotinib, partial sensitivity was maintained with irreversible EGFR inhibitors, such as neratinib, dacomitinib, and afatinib.29,30 Studies with afatinib have focused on patients with clinically defined acquired resistance to first-generation EGFR TKIs. In the pivotal LUX-Lung 1 study, a significant prolongation of progression-free survival was noted compared to placebo, along with a 7% response rate; however, this did not translate into improved overall survival.6 Similarly, other second-generation EGFR TKIs did not reach critical milestones in development for this space.31 The reason for the poor efficacy of afatinib and similar drugs in this setting is more related to excessive potency against wild-type EGFR leading to substantial epithelium-based toxicities, rather than lack of efficacy against the mutant form. Principally, the therapeutic window in this setting is not sufficiently wide for significant clinical impact.
Based on transgenic mouse studies with preclinical models of EGFR T790M-mutated lung adenocarcinomas,32 the combination of afatinib and cetuximab was developed to provide needed synergy for enhanced clinical benefit. While studies of this combination demonstrated a significant improvement in response rates to more than 30% without significant differences between cases with or without documented EGFR T790M,33 the excessive toxicity of the combination has limited overall enthusiasm for further development.
Targeting EGFR T790M with third-generation EGFR TKIs
Recognizing the limitations of second-generation EGFR TKIs, third-generation EGFR TKIs were developed specifically to target EGFR T790M and spare wild-type EGFR, thereby achieving much broader therapeutic windows compared to second-generation agents. Two compounds – osimertinib (AZD9291) and rociletinib (CO-1686) – were the first to be introduced to the clinic.34,35 Both are irreversible covalent inhibitors of EGFR via binding to C797, allowing them to achieve sustained and complete EGFR inhibition, even in the presence of high intracellular concentrations of ATP, unlike gefitinib and erlotinib. Both osimertinib and rociletinib bind more avidly to EGFR T790M mutants than wild-type EGFR, mitigating some of the off-target effects associated with afatinib.27 Table 1 summarizes the relative differences in activity of multiple EGFR-directed therapies against various EGFR mutations.
 | Table 1 Relative qualitative activity of EGFR-directed therapies Notes: *Includes EGFR exon 19 deletions and insertions, exon 21 L858R and L861Q, and exon 18 G719X; **EGFR C797S can occur in either trans or cis with EGFR T790M EGFR. Abbreviation: TKIs, tyrosine-kinase inhibitors. |
Osimertinib (AZD9291) is a monoanilinopyrimidine compound that has little activity against wild-type EGFR, but irreversibly and very potently inhibits EGFR T790M and other common mutated forms of EGFR. It covalently binds cysteine 797 of EGFR, and has activity against some other kinases that harbor a cysteine residue in their analogous kinase domain, such as ErbB2, ErbB4, and BLK (formerly called BTK). Two circulating active metabolites, a desmethylindole analogue, AZ5104, and an N-demethylated analogue, AZ7550, have been observed in both preclinical and clinical settings, with significant dose-dependent tumor regression in NSCLC cell lines with activating mutations of EGFR and in tumor xenograft models.27
Despite tremendous initial excitement, clinical development of rociletinib was abruptly halted, due to high numbers of serious adverse events, including hyperglycemia and QT prolongation in clinical trials, which have been attributed to cross-reactivity with IGF1R.36,37 Osimertinib has a more favorable side-effect profile without cross-reactivity with IGF1R. Initial studies of osimertinib in an extended Phase I study of 253 patients with EGFR-mutated lung adenocarcinoma and resistance to prior EGFR TKI therapy demonstrated good tolerance across dose-escalation cohorts (20–240 mg) without identified dose-limiting toxicities.38 Five dose-expansion cohorts demonstrated an objective response rate of 51%, and in patients with documented EGFR T790M this was even higher, at 61%, with progression-free survival of 9.6 months (compared to 21% response rate for EGFR T790M-negative patients and progression-free survival of 2.8 months). Subsequently, based on similar activity in the AURA extension and AURA2 studies,39 osimertinib obtained an accelerated US Food and Drug Administration (FDA) indication for the treatment of metastatic EGFR T790M mutation-positive NSCLC in patients who have progressed on or after EGFR TKI therapy.40 In both single-arm studies, osimertinib was administered at 80 mg orally once daily, and objective response rates of 57% and 61% were noted in the AURA extension study and AURA2, respectively. Most observed adverse events were mild, and included diarrhea, rash, dry skin, and nail toxicities. Pneumonitis remains a concern like other EGFR TKIs, affecting a small percentage of patients (3.3% in these two studies). Osimertinib is now the mainstay of therapy for EGFR T790M mutation-positive NSCLC.
In 2016, the AURA3 Phase III trial demonstrated that in patients with EGFR T790M-positive advanced NSCLC who had disease progression after first-line EGFR TKI therapy, the median duration of progression-free survival was significantly longer with osimertinib than with platinum-containing chemotherapy (cisplatin or carboplatin) plus pemetrexed (10.1 months vs 4.4 months, HR 0.3, 95% CI 0.23– 0.41; P<0.001), with significantly longer median duration of progression-free survival in the subset of patients with central nervous system metastases (8.5 months vs 4.2 months, HR 0.32, 95% CI 0.21–0.49). The objective response rate was also significantly better with osimertinib (71%, 95% CI 65%–76%) than with platinum-containing chemotherapy plus pemetrexed (31%, 95% CI 24%–40%) (OR 5.39, 95% CI 3.47–8.48; P<0.001). In addition, the proportion of patients with adverse events of grade 3 or higher was lower with osimertinib (23%) than with platinum-containing chemotherapy plus pemetrexed (47%).41 These outstanding results led to full FDA approval of osimertinib in 2017.
Many ongoing studies are exploring combinations of osimertinib with other agents that have established synergy in the management of advanced NSCLC, including bevacizumab, ramucirumab, and necitumumab, as well as combinations with other agents affecting pathways of resistance, such as MET inhibition and immune-checkpoint inhibition. While the relatively atoxic nature of osimertinib makes it an ideal partner for drug combinations, combination studies of osimertinib and durvalumab have demonstrated an unexpectedly high rate of interstitial lung disease.42 A range of clinical studies featuring osimertinib in combination with other agents are highlighted in Table 2.
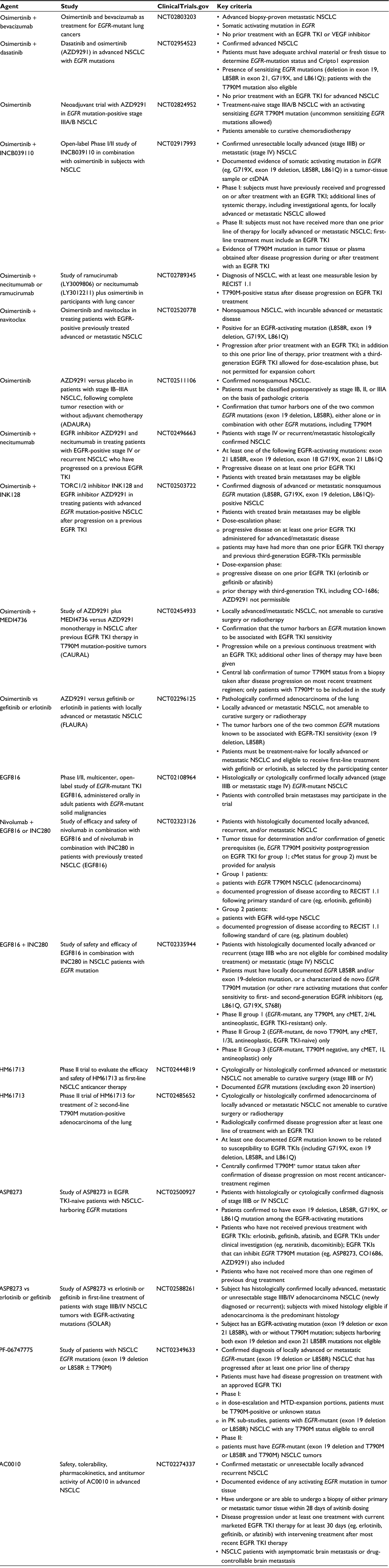 | Table 2 Sampling of clinical studies targeting EGFR-mutated lung cancer Abbreviations: NSCLC, non-small-cell lung cancer; TKI, tyrosine-kinase inhibitor; RECIST, Response Evaluation Criteria In Solid Tumors. |
Other third-generation EGFR inhibitors in development
Olmutinib (HM61713) is an irreversible EGFR TKI that (like osimertinib) binds EGFR C797 and has preferential activity against all mutant forms of EGFR, including EGFR T790M. In a Phase I/II study, 800 mg/day was defined to be the recommended dose for further studies; at this dose level, olmutinib appears generally well tolerated, with rare cases of interstitial lung disease, but no documented cases of hyperglycemia.43 A total of 76 EGFR T790M-positive patients received therapy at the 800 mg/day dose level, with an objective response rate of 54%. Central nervous system activity was also noted, and based on these promising results, olmutinib gained approval in South Korea in 2016. Current studies in the second-line (ELUXA 2) and frontline setting (ELUXA 1) are ongoing or recently completed. However, development of this drug in the US is currently on hold.
Nazartinib (EGF816) is another potent and selective EGFR-mutant inhibitor with irreversible binding to EGFR C797.44 Early-phase studies demonstrated promising activity, with an objective response rate of 46.9% and reported side effects of rash and diarrhea. Ongoing studies are exploring combination strategies with MET inhibitors and immune-checkpoint inhibitors.
ASP8273, which has mostly been developed in Asia, is another selective and potent third-generation EGFR inhibitor that has demonstrated good tolerance and excellent activity in early-phase studies, with an objective response rate of 64% in 70 EGFR T790M-positive patients treated at a dose of 300 mg/day. Current development focuses on first-line indication, with an ongoing pivotal Phase III study (SOLAR) comparing ASP8273 to gefitinib/erlotinib in treatment-naive patients with EGFR-mutation positive NSCLC.
Methods of detecting EGFR T790M
Given the high objective response rates of EGFR T790M mutation-positive lung cancers to osimertinib, it is crucial to accurately identify tumors that harbor EGFR T790M. Until recently, the standard of care for detecting EGFR T790M at the time of disease progression on EGFR TKI therapy was via tissue biopsy followed by sequencing using a range of testing platforms with differing sensitivities for the detection of subclonal presence of mutations. However, invasive biopsies cannot be performed in all patients (eg, tumors in anatomic locations that are difficult to access, very small metastatic lesions), and are associated with significant risks, such as bleeding, lung injury, and pneumothoraces. They can also inadequately sample genomically heterogeneous tumors, and might not accurately reflect tumor heterogeneity within a single patient. In addition, a single-biopsy procedure may provide insufficient material for the required battery of genetic and immunochemistry assays. With increasing recognition that serial molecular assessments are necessary to tailor treatments appropriately for lung cancer throughout the disease course,22 serial invasive biopsies represent a significant practical barrier in clinical care.
To address this problem directly, clinical development of noninvasive methods to detect genomic aberrations accurately is active in a wide range of tumor types, including isolation of circulating tumor cells (CTCs) and both plasma-based and urine-based ctDNA assays. While all cells shed short (usually 150–170 base pairs in length) double-stranded DNA fragments called cfDNA into the systemic circulation,45,46 malignant cells from primary and metastatic tumors shed CTCs and ctDNA, providing potential sources of systemic tumor-derived genetic material that carries mutations representative of tumor biology.47 In fact, ctDNA may more accurately reflect tumor heterogeneity within a given patient, and multiple studies have demonstrated that increasing solid-tumor burden is associated with higher detected levels of cfDNA and ctDNA. While capturing an adequate numbers of CTCs from blood samples has been a difficult technical challenge, more sensitive microfluidic-based platforms48 have further opened the possibility of noninvasive blood-based testing in combination with modern genotyping assays. In patients with advanced NSCLC, ctDNA-detection rates ranging from 80% to over 95% have been reported using current detection assays.49,50 With rapid progress in ctDNA-based technologies, multiple plasma-based, urine-based, and cerebrospinal fluid-based testing platforms have entered the clinical arena over the last few years. There are many platforms available or in active development based on droplet digital polymerase chain reaction and massively parallel DNA sequencing, otherwise known as next-generation sequencing, including cancer personalized profiling by deep sequencing, which can simultaneously detect insertions, deletions, rearrangements, single-nucleotide variants, and somatic copy-number alterations to examine a diverse array of potential resistance mechanisms.51 Key registration research utilized the Cobas EGFR-mutation test, which uses tumor tissue or plasma to detect 42 mutations in exons 18–21 of the EGFR gene, including T790M.52
In a prospective, multicenter exploratory analysis of 40 patients with EGFR-mutant tumors that had progressed on first- or second-generation EGFR TKIs, blood samples were drawn for CTC and ctDNA analyses at the same time as tumor biopsy to assess EGFR T790M test characteristics and concordance among the three diagnostic modalities. Interestingly, no clear “gold standard” emerged, and mutation data among tissue-based and blood-based analyses provided nonoverlapping and complementary information, as the noninvasive blood-based analyses identified the EGFR T790M mutation in 14 (35%) of patients who had negative or indeterminate invasive tumor biopsies.53 Further validation studies are clearly warranted, but it appears that the concurrent use of noninvasive blood-based analyses may help provide a more complete assessment of the biology of a given patient’s EGFR-mutant tumors. Indeed, FDA approval for the tissue and blood-based Cobas EGFR-mutation ctDNA assay has been secured in 2016, based on the aforementioned studies. Figure 2 provides a brief flowchart that demonstrates how ctDNA testing could be incorporated into the clinical management workflow for advanced NSCLC.
 | Figure 2 Flowchart for management of advanced NSCLC. Abbreviations: NSCLC, non-small-cell lung cancer; SCLC, small cell lung cancer; SC, small cell; TKI, tyrosine-kinase inhibitor. |
Acquired resistance to EGFR T790M-targeting TKIs
Unfortunately, novel acquired resistance mechanisms are emerging in specific response to EGFR T790M-directed therapy as well. For patients with EGFR T790M-positive lung cancer treated with osimertinib, progression-free survival usually ranges from 10 to 12 months. Tumor samples from patients treated with osimertinib and rociletinib during Phase I studies revealed several distinct acquired-resistance mechanisms, including the key EGFR C797S mutation, which drastically reduces the efficacy of all third-generation EGFR inhibitors.51,54 The detected frequency of C797S varied from study to study with different molecular monitoring methods, but it has been reported to be as high as 22%. Intriguingly, the allelic context of the occurrence of C797S might have treatment implications.55 If EGFR C797S occurs in trans with EGFR T790M on a different allele, the cells might retain sensitivity to a combination of first- and third-generation EGFR inhibitors. If the mutations are on the same allele in cis, high-level resistance is expected against all currently used EGFR TKIs; recent data suggest that most EGFR C797S mutations are occurring in cis. While tissue-based EGFR sequencing remains the standard, ctDNA approaches in this context are gaining clear importance. Since ctDNA exists typically as 150–170 base-pair fragments, the proximity of EGFR T790 and EGFR C797 allows these allelic relationships to be readily assessed. Interestingly, this EGFR C797S-resistance mutation is analogous to the recently described BTK-resistance mutation, C481S, which arises in the context of the BTK inhibitor ibrutinib in chronic lymphocytic leukemia.56
Besides EGFR C797S, multiple other resistance mechanisms to osimertinib therapy have been documented in case reports and case series, mirroring what has been reported with first-generation EGFR inhibitors.57 As anticipated, MET amplification, SC transformation, and downstream mutations in BRAF and PI3K have been noted. In addition to C797S, other EGFR mutations such as L792F might represent alternative resistance mechanisms. ctDNA platforms greatly accelerate the discovery of these mechanisms, and need to be integrated into ongoing studies.58,59 In addition, an interesting and recurrent observation has been that upon the development of resistance to EGFR T790M-directed therapy, the original T790M-mutated clone often becomes undetectable, highlighting some potential for retreatment with earlier-generation drugs for certain patients.
Overcoming resistance to EGFR T790M-targeting TKIs
Since all currently FDA-approved EGFR TKIs target the ATP site of EGFR, a promising drug-discovery strategy involves the development of allosteric inhibitors that selectively target drug-resistant EGFR mutants while binding less avidly to wild-type EGFR. One such lead compound, EAI045, binds an allosteric site on the inactive conformation of EGFR and inhibits L858R/T790M-mutant EGFR with low-nanomolar potency (but not exon 19 deletion/T790M).60 However, as a single agent, EAI045 is ineffective in targeting EGFR asymmetric dimers and cannot block EGFR-driven cell proliferation. In combination with cetuximab, a recombinant human/mouse chimeric monoclonal antibody, which binds EGFR and prevents EGFR dimerization by competitively inhibiting the binding of cognate ligands, EAI045 effectively blocks EGFR-driven cell proliferation in mouse models of lung cancer driven by both L858R/T790M EGFR and L858R/T790M/C797S EGFR. Analogous to prior discussions with afatinib, resistant cells with T790M/C797S EGFR remain partially sensitive to cetuximab because of the disruption of EGFR dimerization. The combination of EAI045 and cetuximab demonstrated significant synergy in these elegant studies, and represents a possible approach to circumventing acquired resistance mediated by EGFR C797S.
Another promising approach utilizes the combination of the highly potent and selective ALK/EGFR T790M inhibitor brigatinib, along with cetuximab.61 While brigatinib has demonstrated modest efficacy as a single agent in early studies focused on EGFR T790M-positive patients, in an in vitro drug-screening Ba/F3 cell-line model it had the best efficacy among all screened compounds against triple-EGFR mutation-positive cells and structural prediction models suggest that this might be related to a better fit without steric crowding in the context of T790M and/or C797S compared to other EGFR inhibitors. While the in vitro efficacy of brigatinib monotherapy might not be clinically meaningful, given high required concentrations, concurrent treatment with cetuximab led to substantial synergy, yielding a potentially meaningful IC50 in the hundred-nanomolar range.
Conclusion
Since the original discovery of the pivotal EGFR T790M mutation driving acquired resistance in most EGFR-mutated lung adenocarcinomas exposed to first- or second-generation EGFR TKI therapy, a tremendous amount has been learned about the mechanism and biology of resistance. Tissue- and ctDNA-based testing for EGFR T790M has now become the standard of care. The high frequency of this key gatekeeper mutation permitted successful development of effective drugs specifically targeting this molecular aberration, yielding highly potent and safe drugs, such as osimertinib, that are extending the benefits of EGFR-directed therapy. Osimertinib will likely move into earlier lines of therapy, and rational drug combinations are now being developed to overcome and prevent such molecularly driven resistance. Lastly, the paradigm shift defined by acquired resistance via EGFR T790M has allowed rapid evolution of analogous knowledge for third-generation EGFR inhibitors. These lessons will continue to provide valuable insight into many other molecularly defined subsets of patients with a wide range of solid and hematological malignancies.
Disclosure
BH has received consulting fees from Foundation One, Genoptix, Boehringer-Ingelheim, Takeda, Novartis, AstraZeneca, Genentech, Pfizer, and Eli-Lilly, and research support from Merck, Bristol-Myers-Squibb, Pfizer, Mirati, Takeda, AstraZeneca, Boehringer-Ingelheim, Novartis, Eli-Lilly, and Genentech. The other authors report no conflicts of interest in this work.
References
Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | ||
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | ||
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. | ||
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. | ||
Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. | ||
Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2B/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. | ||
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. | ||
Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. | ||
Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol. 2008;26(10):1742–1751. | ||
Park JH, Liu Y, Lemmon MA, Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem J. 2012;448(3):417–423. | ||
D’Angelo SP, Pietanza MC, Johnson ML, et al. Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol. 2011;29(15):2066–2070. | ||
Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol. 2008;21 Suppl 2:S16–S22. | ||
Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–1167. | ||
Kobayashi S, Shimamura T, Monti S, et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 2006;66(23):11389–11398. | ||
Zhang Z, Kobayashi S, Borczuk AC, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis. 2010;31(4):577–586. | ||
Chitale D, Gong Y, Taylor BS, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28(31):2773–2783. | ||
Li G, Wang X, Hibshoosh H, Jin C, Halmos B. Modulation of ErbB2 blockade in ErbB2-positive cancers: the role of ErbB2 mutations and PHLDA1. PLoS One. 2014;9(9):e106349. | ||
Costa DB, Halmos B, Kumar A, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4(10):1669–1680. | ||
Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. | ||
Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–222. | ||
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Bell DW, Gore I, Okimoto RA, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005;37(12):1315–1316. | ||
Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–269. | ||
Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17(6):1616–1622. | ||
Hata A, Katakami N, Yoshioka H, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: comparison between T790M mutation-positive and mutation-negative populations. Cancer. 2013;119(24):4325–4332. | ||
Cheng H, Nair SK, Murray BW. Recent progress on third generation covalent EGFR inhibitors. Bioorg Med Chem Lett. 2016;26(8):1861–1868. | ||
Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–1133. | ||
Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102(21):7665–7670. | ||
Kobayashi S, Ji H, Yuza Y, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65(16):7096–7101. | ||
Ou SH, Soo RA. Dacomitinib in lung cancer: a “lost generation” EGFR tyrosine-kinase inhibitor from a bygone era? Drug Des Devel Ther. 2015;9:5641–5653. | ||
Regales L, Gong Y, Shen R, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119(10):3000–3010. | ||
Janjigian YY, Smit EF, Groen HJ, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4(9):1036–1045. | ||
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | ||
Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404-15. | ||
Sequist LV, Goldman JW, Wakelee HA, et al. Efficacy of rociletinib (CO-1686) in plasma-genotyped T790M-positive non-small cell lung cancer (NSCLC) patients (pts). J Clin Oncol. 2015;33 Suppl:8001. | ||
Van Der Steen N, Caparello C, Rolfo C, Pauwels P, Peters GJ, Giovannetti E. New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong? Onco Targets Ther. 2016;9:6065–6074. | ||
Jänne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. | ||
Goss G, Tsai CM, Shepherd FA, et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016;17(12):1643–1652. | ||
Khozin S, Weinstock C, Blumenthal GM, et al. Osimertinib for the treatment of metastatic epidermal growth factor T970M positive non-small cell lung cancer. Clin Cancer Res. Epub 2016 Dec 6. | ||
Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376(7):629–640. | ||
Ahn MJ, Sun JM, Lee SH, Ahn JS, Park K. EGFR TKI combination with immunotherapy in non-small cell lung cancer. Expert Opin Drug Saf. 2017;16(4):465–469. | ||
Tan CS, Cho BC, Soo RA. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor-mutant non-small cell lung cancer. Lung Cancer. 2016;93:59–68. | ||
Jia Y, Juarez J, Li J, et al. EGF816 exerts anticancer effects in non-small cell lung cancer by irreversibly and selectively targeting primary and acquired activating mutations in the EGF receptor. Cancer Res. 2016;76(6):1591–1602. | ||
Levy B, Hu ZI, Cordova KN, Close S, Lee K, Becker D. Clinical utility of liquid diagnostic platforms in non-small cell lung cancer. Oncologist. 2016;21(9):1121–1130. | ||
Chandrananda D, Thorne NP, Bahlo M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med Genomics. 2015;8:29. | ||
Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–586. | ||
Stott SL, Hsu CH, Tsukrov DI, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A. 2010;107(43):18392–18397. | ||
Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–554. | ||
Thompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin Cancer Res. 2016;22(23):5772–5782. | ||
Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. | ||
Thress KS, Brant R, Carr TH, et al. EGFR mutation detection in ctDNA from NSCLC patient plasma: a cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 2015;90(3):509–515. | ||
Sundaresan TK, Sequist LV, Heymach JV, et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res. 2016;22(5):1103–1110. | ||
Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–562. | ||
Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21(17):3924–3933. | ||
Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. | ||
Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front Med. 2016;10(4):383–388. | ||
Kobayashi Y, Azuma K, Nagai H, et al. Characterization of EGFR T790M, L792F, and C797S mutations as mechanisms of acquired resistance to afatinib in lung cancer. Mol Cancer Ther. 2017;16(2):357–364. | ||
Lin JJ, Fairclough SR, Nagly RJ, et al. Identification of on-target mechanisms of resistance to EGFR inhibitors using cfDNA next-generation sequencing. Poster presented at: 17th World Conference on Lung Cancer; December 4–7, 2016; Vienna, Austria. | ||
Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–132. | ||
Uchibori K, Inase N, Araki M, et al. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat Commun. 2017;8:14768. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.