")
Back to Journals » International Journal of Nanomedicine » Volume 11
Efficient inhibition of ovarian cancer by degradable nanoparticle-delivered survivin T34A gene
Authors Luo L, Du T, Zhang J, Zhao W, Cheng H, Yang Y, Wu Y, Wang C, Men K , Gou M
Received 31 July 2015
Accepted for publication 18 November 2015
Published 2 February 2016 Volume 2016:11 Pages 501—513
DOI https://doi.org/10.2147/IJN.S93496
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Li Luo, 1,* Ting Du, 1,* Jiumeng Zhang, 1 Wei Zhao, 2 Hao Cheng, 1 Yuping Yang, 1 Yujiao Wu, 1 Chunmei Wang, 1 Ke Men, 1 Maling Gou 1
1State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center for Biotherapy, 2Department of Thoracic Oncology, Cancer Center, West China Hospital, Medical School, Sichuan University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Abstract: Gene therapy has promising applications in ovarian cancer therapy. Blocking the function of the survivin protein could lead to the growth inhibition of cancer cells. Herein, we used degradable heparin–polyethyleneimine (HPEI) nanoparticles to deliver a dominant-negative human survivin T34A (hs-T34A) gene to treat ovarian cancer. HPEI nanoparticles were characterized and were found to have a dynamic diameter of 66± 4.5 nm and a zeta potential of 27.1± 1.87 mV. The constructed hs-T34A gene expression plasmid could be effectively delivered into SKOV3 ovarian carcinoma cells by HPEI nanoparticles with low cytotoxicity. Intraperitoneal administration of HPEI/hs-T34A complexes could markedly inhibit tumor growth in a mouse xenograft model of SKOV3 human ovarian cancer. Moreover, according to our results, apparent apoptosis of cancer cells was observed both in vitro and in vivo. Taken together, the prepared HPEI/hs-T34A formulation showed potential applications in ovarian cancer gene therapy.
Keywords: human survivin T34A, heparin–polyethyleneimine nanoparticles, gene therapy, ovarian cancer
Corrigendum for this paper has been published
Introduction
Ovarian cancer is one of the most common cause of gynecologic cancer-related deaths, which is estimated as >220,000 new cases worldwide each year.1 The combination of surgical cytoreduction with platinum/taxane chemotherapy is the preferred traditional therapeutic strategy for ovarian cancer.2 Although much progress has been made by traditional therapeutic strategies, ovarian cancer still represents the worst type among gynecological malignancies with an overall 5-year survival rate of approximately 45%.3 Thus, developing novel therapeutic approaches for ovarian cancer is urgently required.
In the past 2 decades, gene therapy has been widely known as a potential therapeutic strategy for a wide range of diseases (such as cancer, neurodegenerative diseases, and cardiovascular diseases).4–7 Till now, ~64.2% of the 2,143 protocols of gene therapy has been approved for cancer clinical trials worldwide.8 Among these, efficient gene delivery carriers remain the major obstacles.9 For years, much effort has been devoted to the development of novel gene vectors, including viral-vectors and nonviral vectors. As efficient nucleic acid delivery vehicles, viral-vectors have a natural propensity to persist in human cells.10 However, their use is limited by broad tropism, difficulty in vector production, as well as cause of carcinogenesis and immunogenicity.9,11 On the other hand, nonviral vectors, including cationic lipids, lipid nanoemulsions, solid lipid nanoparticles, and polymer-based vectors, show advantages such as safety and production.12,13
In the past few years, synthetic cationic agents, such as polyethyleneimine (PEI) and 1,2-dioleoyl-3-trimethylammonim-propane (DOTAP), have been widely investigated and applied in gene delivery. However, the direct use of PEIs is limited because of its relatively high cytotoxicity and nonbiodegradable nature.14 To overcome these drawbacks, we have developed a novel nonviral gene vector that is based on nontoxic, low molecular weight polyethyleneimine (molecular weight 2,000 [PEI 2K]), which was crosslinked with biodegradable heparin, forming heparin–polyethyleneimine (HPEI) nanoparticles.15 In our previous study, the biodegradable cationic HPEI nanoparticles have been proved to be capable of delivering the gene into cells effectively and safely,16,17 demonstrating high potential in gene delivery.
Survivin, a member of the inhibitor of apoptosis protein family, functions as a key regulator in mitosis and apoptosis. The survivin gene has been demonstrated to be overexpressed in various types of cancers (including ovarian cancer, colon cancer, brain cancer, breast cancer, and melanoma), but it is hardly detectable in most normal cells.18,19 It was reported that site mutation of survivin’s threonine 34 to alanine (survivin T34A) was able to induce cell apoptosis by abolishing cdc2–cyclin B1 complex-mediated phosphorylation to this gene and dissociation of caspase 9 with survivin.20 Recent studies have also found that survivin T34A could significantly induce apoptosis in ovarian cancer cells, enabling it a potential strategy for ovarian cancer therapy.21
In this study, HPEI/human survivin (hs)-T34A complexes were prepared and further characterized by size, zeta-potential, morphology, encapsulation efficiency, biodegradability, and tissue distribution; Furthermore, the anticancer ability of HPEI/hsT34A complexes against human ovarian cancer was evaluated both in vitro and in vivo. Our result suggested that the prepared HPEI/hs-T34A formulation has a potential application in ovarian cancer gene therapy.
Materials and methods
Materials
1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), 2-(N-morpholino) ethane-sulfonic acid, N-hydroxysuccinimide (NHS), PEI 2K, polyethyleneimine (molecular weight 25,000 [PEI 25K]), and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (St Louis, MO, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), penicillin, and streptomycin were purchased from HyClone™ (GE Healthcare UK Ltd, Little Chalfont, UK). DNA ladder and prestained protein ladder were provided by Fermentas (Thermo Fisher Scientific Inc., Waltham, MA, USA). All animals were obtained from the Beijing HFK Bioscience (Beijing, People’s Republic of China). Sprague Dawley rats (weighing 200±20 g) were used to perform the biodegradability study of HPEI nanoparticles. BALB/c mice (6–8 weeks old) were used to study the tissue distribution of HPEI nanoparticles. BALB/c nude mice (6–8 weeks old) were used to carry out the therapeutic study in vivo. Mice were humanely treated, and all animal procedures were approved and controlled by the Institutional Animal Care and Treatment Committee of Sichuan University.
Preparation of HPEI nanoparticles
HPEI nanoparticles were prepared as described previously.15 Briefly, 50 mg of heparin was dissolved in 2-(N-morpholino) ethane-sulfonic acid solution buffer (0.05 M, 100 mL), and then 30 mg of NHS and 20 mg of EDC were added to aforementioned solution for 2 hours to activate the carboxylic acid groups of heparin. The activated heparin solution was dropped into PEI 2K solution (7.5 mg/mL, 20 mL) under stirring constantly at room temperature overnight. The obtained HPEI nanoparticles were dialyzed (molecular weight cut off = 8,000–14,000) in distilled water for 3 days and filtered by using a syringe filter (pore size =0.22 μm) (Millex-LG; EMD Millipore, Billerica, MA, USA). The concentration was adjusted to 1 mg/mL, and the HPEI nanoparticles were stored at 4°C.
Characterization of HPEI nanoparticles
The morphology of the HPEI nanoparticles was observed under a transmission electron microscope (H-6009IV; Hitachi Ltd., Tokyo, Japan). The HPEI nanoparticles were diluted with distilled water and placed on a copper grid covered with nitrocellulose.
The size and zeta potential of the HPEI nanoparticles were determined by dynamic light scattering (Nano-ZS; Malvern Instruments, Malvern, UK) at 25°C. All results were taken as the mean of three test runs.
The DNA–HPEI complexes with different ratios of nitrogen atoms of PEI to the phosphate group of DNA (N/P) were incubated at room temperature for 30 minutes and then electrophoresed on 1% (w/v) agarose gel (Thermo Fisher Scientific) for 30 minutes at 100 V. The agarose gel was dissolved in TAE buffer (40 mM Tris/HCl, 1% acetic acid, 1 mM ethylene diamine tetraacetic acid, pH 7.4) with Golden View™ as DNA stain. The electrophoresis gels were photographed by using a gel documentation system (Gel Doc 1000; Bio-Rad Laboratories Inc., Hercules, CA, USA).
Biodegradability study in vivo
Sprague Dawley rats were used for the biodegradability study of HPEI nanoparticles in vivo. Briefly, 5 mg of PEI 2K, HPEI, or PEI 25K were, respectively, injected intraperitoneally. The urine and feces of rats were collected by metabolism cages in the following 24 hours and were extracted by methanol. The resultant products were analyzed by using a Quattro Premier XE triple quadrupole mass spectrometer (Waters Corporation, Milford, MA, USA).
Tissue distribution of HPEI nanoparticles
For tissue imaging, FITC-labeled HPEI nanoparticles (1 mg) were administrated intraperitoneally into BALB/c mice. For the ex vivo imaging, the major organs of mice were then sacrificed and collected after 1, 2, 3, 4, or 6 hours (intraperitoneal [ip]) injection. Excised organs were imaged by using a Bio-Real in vivo imaging system (Bio-Real Quick View 3000; Geneway International, Salzburg, Austria).
Preparation of plasmid DNA
Oligonucleotide synthesis of the hs-T34A gene was performed by GENWIZ (Suzhou, Jiangsu, People’s Republic of China). The hs-T34A fragment incorporated a MluI site and a SalI site for cloning to the expression plasmid pVITRO2 (InvivoGen, San Diego, CA, USA). The recombinant plasmid pVITRO2–hs-T34A was confirmed by restriction endonuclease analysis, polymerase chain reaction (PCR), and DNA sequence analysis.
Plasmid pVITRO2 without survivin cDNA was used as an empty vector (named EP) control. The recombinant plasmid and empty vector were prepared using an Endofree Plasmid kit (Omega Bio-Tek, Norcross, GA, USA). Escherichia coli colonies containing hs-T34A and the null colony were cultured in Luria-Bertani broth with kanamycin (50 μg/mL). The DNA was then dissolved in sterile endotoxin-free water and stored at −20°C.
Cell culture and transfection experiments
Human ovarian carcinoma SKOV3 cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere containing 5% CO2 at 37°C.
The SKOV3 cells were seeded into six-well plates (Corning Incorporated, Corning, NY, USA) at 2×105 cells per well in 2 mL of complete medium (DMEM containing 10% FBS). After incubation for 24 hours, the medium in each well was replaced with 800 μL of fresh serum-free medium. pGFP as a report gene was placed at 2 μg/well, while the mass ratio of PEI 2K/pGFP, HPEI/pGFP, or PEI 25K/pGFP was 10:1, 10:1, or 1:1. The medium was replaced by complete medium after 6 hours. The expressed green fluorescent protein (GFP) was observed under an Olympus IX 71 inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan). The cell suspensions were harvested and analyzed by flow cytometer (BD Biosciences, San Jose, CA, USA) to determine the transfection efficiency of HPEI nanoparticles.
Cell viability assay
The cytotoxicity of HPEI nanoparticles on the SKOV3 cells was evaluated by cell viability assay. Briefly, SKOV3 cells were seeded in 96-well plates at a density of 5×103 cells per well in 100 μL of DMEM and treated with different concentrations of HPEI nanoparticles or PEI 25K for 48 hours. Subsequently, the cell viabilities were evaluated by MTT assays.
Western blot analysis
Protein concentration was quantified by using a bicinchoninic acid assay protein assay kit (Bio-Rad Laboratories Inc.). The protein extracts were run on 12% sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred to polyvinylidenedifluoride membranes. The membranes were blocked with Tris-buffered saline and Tween 20-containing 5% nonfat dried milk at room temperature for 2 hours, washed, and incubated with antisurvivin (Cell Signaling Technology, Inc., Boston, MA, USA) or anti-β-actin (Santa Cruz Biotechnology Inc., Dallas, TX, USA) primary antibody at 4°C overnight. The membranes were then washed and incubated with corresponding secondary antibody conjugated with horseradish peroxidase (HRP) for 1 hour. Finally, the membranes were developed with enhanced chemiluminescence reagents (EMD Millipore, Billerica, MA, USA).
Real-time reverse transcription-polymerase chain reaction (real time RT-PCR)
Total RNA was isolated from cells or tissues by using an RNAsimple Total RNA Kit (TIANGEN, Haidian, Beijing, People’s Republic of China), and was then reverse-transcribed by using a PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Dalian, Liaoning, People’s Republic of China). The amount of cDNA used as templates was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The following sets of primers were used: survivin, forward: 5′-GCCCAGTGTTTCTTCTGCTT-3′, reverse: 5′-CCGGACGAATGCTTTTTATG-3′; GAPDH, forward: 5′-AGCCACATCGCTCAGACAC-3′, reverse: 5′-GCCCAATACGACCAAATCC-3′. The real time RT-PCR conditions comprised a 30-second step at 95°C, followed by 95°C for 5 seconds and 56°C for 20 seconds for 39 cycles with melting curve analysis. Relative quantification of each gene was calculated after normalization to GAPDH.
Flow cytometric analysis
Apoptosis assays were performed on SKOV3 cells. SKOV3 cells were treated with HPEI/hs-T34A complexes (50 μg HPEI/5 μg plasmid), HPEI/EP complexes, HPEI, or normal saline (NS) for 48 hours. Apoptosis was examined by flow cytometric analysis after propidium iodide staining.
In vivo therapy studies
Female BALB/c nude mice aged 6–8 weeks were injected intraperitoneally with 200 μL of SKOV3 cell suspension (~1×107 cells). The tumor-bearing mice were randomly divided into five groups based on their body weights. Treatment began on day 7 postinoculation. Mice received the following treatment by ip injection every 3 days: NS, HPEI nanoparticles (50 μg), HPEI/EP (50 μg HPEI combined with 5 μg EP), HPEI/hs-T34A complexes (50 μg HPEI combined with 5 μg hs-T34A), and PEI 25K/hs-T34A (5 μg PEI 25K combined with 5 μg hs-T34A). The mice were sacrificed when mice in the control group became very weak (typically 4 to 6 weeks). Tumors and vital organs were obtained and fixed in 4% paraformaldehyde or frozen in liquid nitrogen immediately. Ascites volume, tumor weight, and number of nodules were recorded.
Analysis of histology and apoptosis
For hematoxylin and eosin (HE) staining, tissues were fixed in 4% paraformaldehyde in phosphate buffered solution for at least 24 hours. Consecutive paraffin wax-embedded tissue sections (3–4 μm) were dewaxed and rehydrated. The hydrated sections were stained with Mayer’s HE.
For cluster of differentiation 31 (CD31) staining, tumor sections (3–4 μm) were made from tumor tissue specimens of each group, and then blocked with 3% H2O2 for 15 minutes at room temperature, incubated with goat serum for 1 hour at room temperature, and subsequently incubated with rabbit antimouse CD31 antibodies (AbcamPLC, Boston, MA, USA) at 4°C overnight. After washing with phosphate buffered solution for three times, the appropriate secondary antibody conjugated to HRP was added, and Histostain-Plus Kits (ZSGB-BIO, Beijing, People’s Republic of China) were used. HRP was detected with 3,3-diaminobenzidine substrate (DAB Kit; Maixin Bio Co., Fuzhou, Fujian, People’s Republic of China) for 1 minute. The slices were then washed and counterstained with hematoxylin (Beyotime Institute of Biotechnology, Shanghai, People’s Republic of China) for 45 seconds. The microvessel density was determined as the average number of CD31-positive small vessels in a field.
To analyze apoptotic cells within abdominal cavity metastases of SKOV3 carcinoma tissue, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay of paraffin-embedded tumor sections (Promega Corporation, Fitchburg, WI, USA) was performed following the manufacturer’s instruction. TUNEL-positive cells were characterized by nuclei with bright green fluorescence staining.
Safety evaluation
To evaluate the potential side effects and toxicity, mice were continuously observed for relevant indices, including appearance, weight, independent activity, or toxic death. The body weights were measured and recorded. Sections of organs (including heart, liver, spleen, lung, and kidney) were stained with HE.
Statistical analysis
All experimental data are reported as mean ± standard deviation. Statistical analysis was performed with one-way analysis of variance using SPSS software (version 19.0.1; IBM Corporation, Armonk, NY, USA). P<0.05 was considered statistically significant.
Results
Preparation and characterization of HPEI nanoparticles
HPEI nanoparticles were prepared as schematically represented in Figure 1. In detail, heparin solution was activated by NHS and EDC, and PEI 2K solution was subsequently added to the activated heparin solution. As shown in Figure 2A, the dynamic diameter of HPEI nanoparticles was 66.0±4.5 nm with a polydispersity index of 0.163. The measured zeta potential was 27.1±1.87 mV (Figure 2B). The transmission electron microscopy analysis indicated that the mean size of these HPEI nanoparticles is 44 nm (Figure 2C). To evaluate the binding ability of HPEI with DNA, a gel retardation assay was performed. When the N/P ratio of HPEI to DNA was 4, no bright DNA band was observed, suggesting that the DNA was completely incorporated into HPEI (Figure 2D).
 | Figure 1 A brief workflow of the preparation of HPEI nanoparticles. |
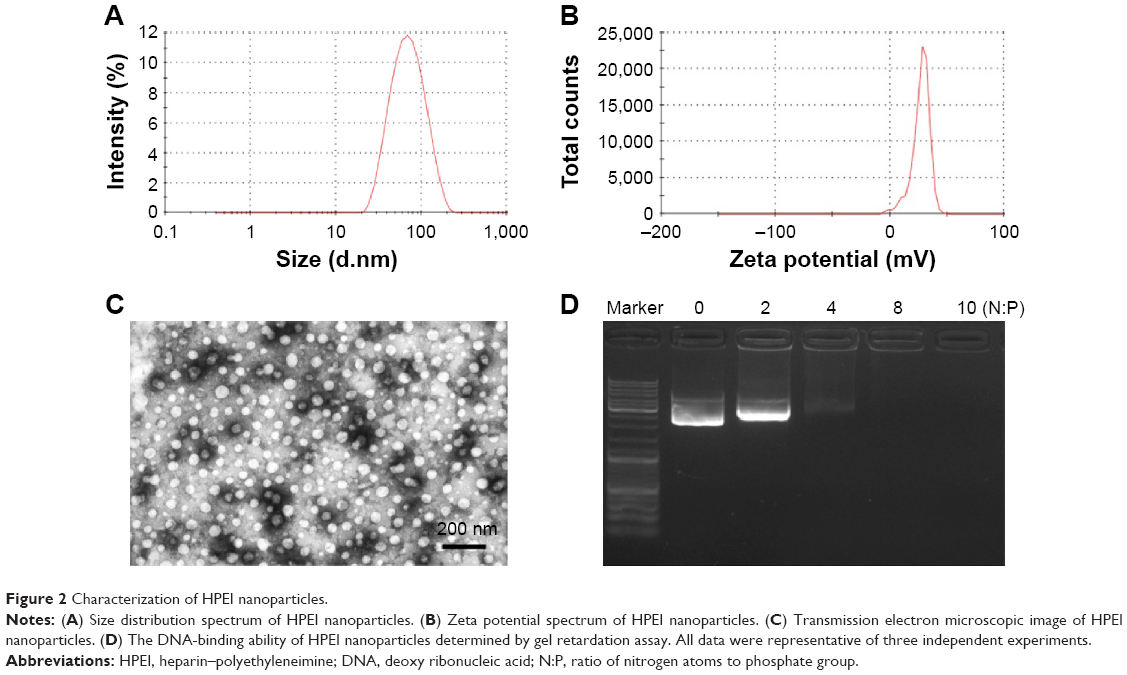 | Figure 2 Characterization of HPEI nanoparticles. |
We next evaluated the biodegradability of HPEI nanoparticles in rats in vivo. Five milligrams of HPEI, PEI 2K, or PEI 25K was injected intraperitoneally, respectively. The urine and feces were collected in the following 24 hours and then qualitatively determined by mass spectrometer. As shown in Figure 3A, the m/z range of PEI 2K was mainly composed of doubly or triply charged species. While in Figure 3B (HPEI-treated group) and Figure 3C (PEI 2K-treated group), doubly or triply charged species were found in the urine, implying the presence of low molecular weight PEI in urine. However, in Figure 3D (PEI 25K-treated group), only singly charged species was found, suggesting that there was no PEI in the urine. Additionally, HPEI, PEI 2K, or PEI 25K could not be detected in the PEI in feces, respectively (data not shown). Together, these results indicated that HPEI can be quickly degraded, and the degradation products are excreted by urine.
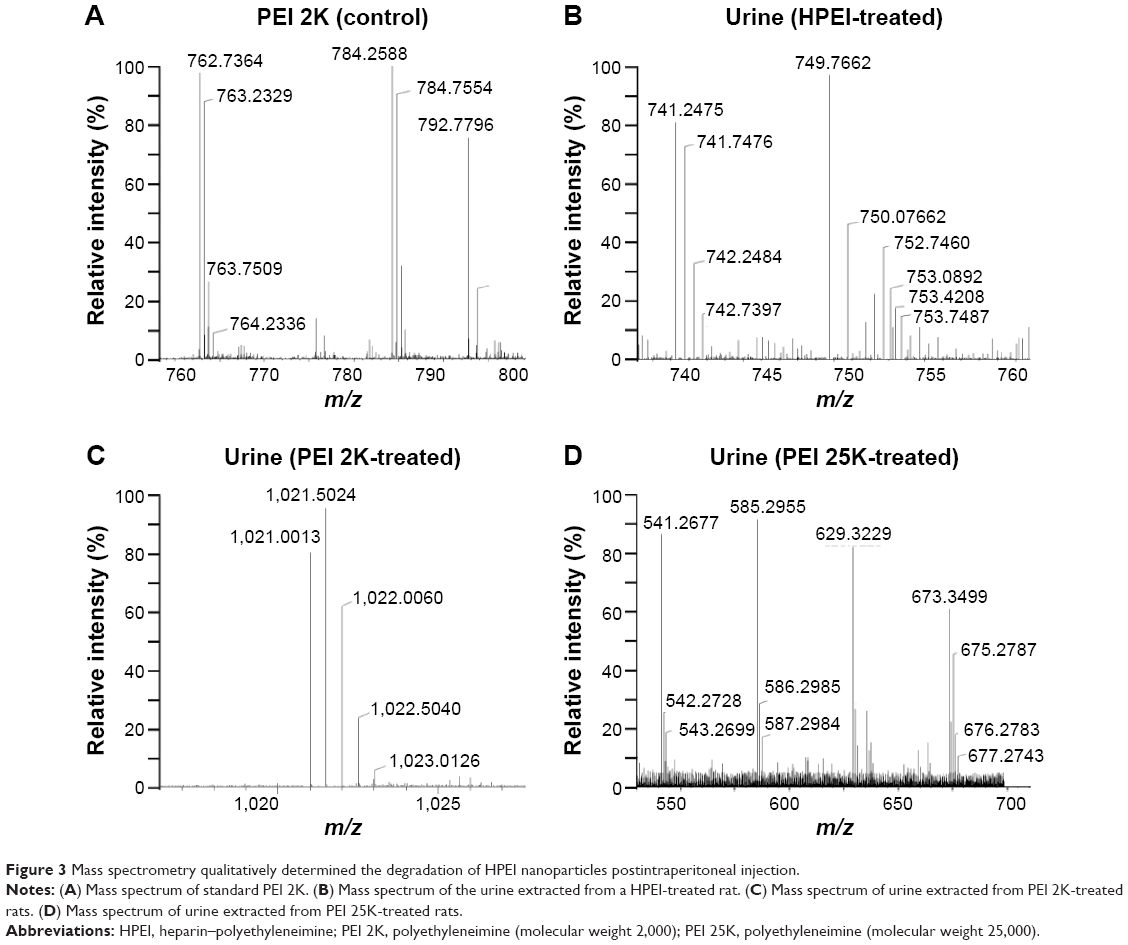 | Figure 3 Mass spectrometry qualitatively determined the degradation of HPEI nanoparticles postintraperitoneal injection. |
The tissue distribution of HPEI nanoparticles in nude mice was further investigated. Mice were sacrificed at different time points (0, 1, 2, 4, 6, 8, 24, and 30 hours) postintraperitoneal injection. Heart, liver, spleen, lung, and kidney were harvested, respectively. As shown in Figure 4, green fluorescence was observed in liver and kidney (but not in the spleen, lung, or heart) at 1 hour postinjection of HPEI. These results are consistent with previous reports that heparin is liver targeting.22 In addition, the existence of HPEI in kidney further suggested that HPEI nanoparticles are cleared by urine.
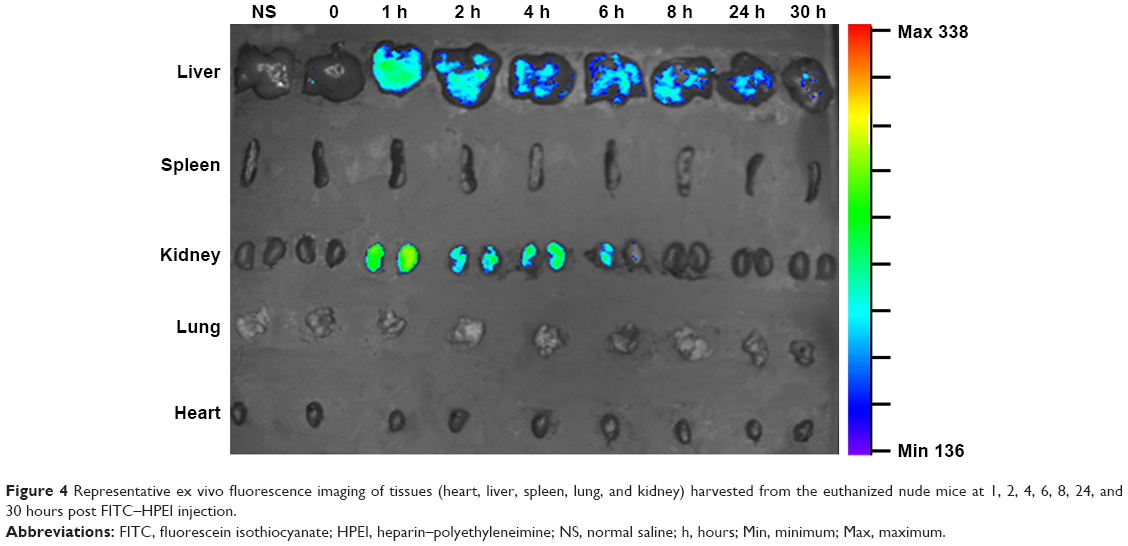 | Figure 4 Representative ex vivo fluorescence imaging of tissues (heart, liver, spleen, lung, and kidney) harvested from the euthanized nude mice at 1, 2, 4, 6, 8, 24, and 30 hours post FITC–HPEI injection. |
In addition, we examined the cytotoxicity and transfection efficiency of HPEI nanoparticles. MTT assay revealed that, at a concentration of 50 μg/mL, HPEI nanoparticles barely inhibited cell viability of SKOV3 cells, while PEI 25K could significantly inhibit the growth of SKOV3 cells (Figure 5A). Then, we evaluated the transfection efficiency of HPEI nanoparticles in SKOV3 ovarian cancer cells using the GFP plasmid as the report gene. Flow cytometric analysis suggested that the transfection efficiency of HPEI nanoparticles was 34.6%±2.5% in SKOV3 cells, which was significantly higher than PEI 2K (while PEI 25K was 42.9%±4.9%) (Figure 5B–D). The transfection efficiency of HPEI nanoparticles in SKOV3 cells is comparable to that of PEI 25K (P>0.05) (Figure 5B).
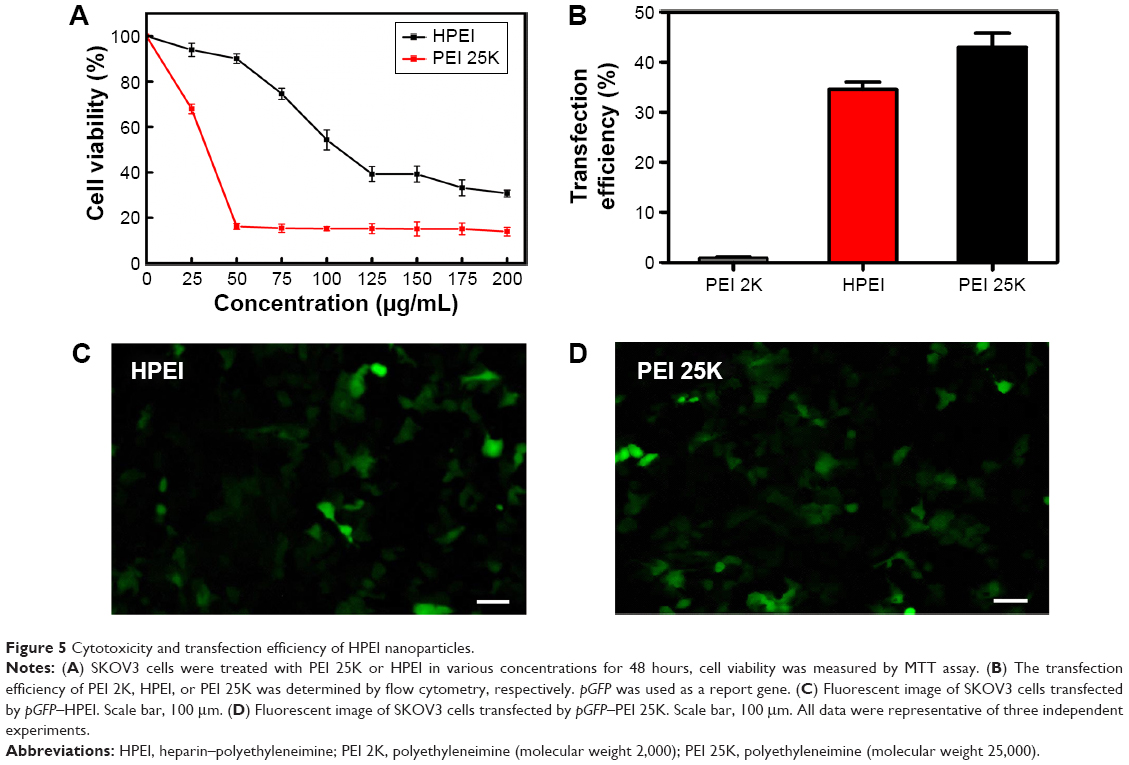 | Figure 5 Cytotoxicity and transfection efficiency of HPEI nanoparticles. |
Moreover, the stability of HPEI was also evaluated in vitro. After storage for 2 months at 4°C, HPEI nanoparticles still had a mean particle size of 67.4±2.5 nm with monodispersity (polydispersity index =0.211). The transfection efficiency of HPEI nanoparticles was also evaluated. HPEI can still effectively transfect SKOV3 cells after storage (data not shown). These results demonstrated that HPEI nanoparticles have good stability in vitro.
Anticancer activity of HPEI/hs-T34A complexes in vitro
To evaluate the anticancer effect of HPEI/hs-T34A complexes in vitro, we first constructed the pVITRO2–hs-T34A plasmid. The hs-T34A gene, which was incorporated a MluI site and a SalI site, was cloned to the expression plasmid pVITRO2. The recombinant plasmid pVITRO2–hs-T34A (Figure 6) was confirmed by restriction endonuclease analysis, PCR, and DNA sequence analysis.
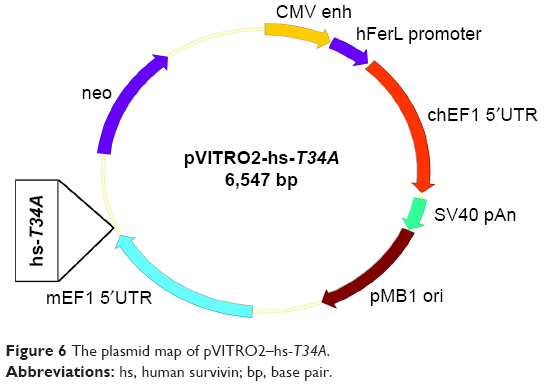 | Figure 6 The plasmid map of pVITRO2–hs-T34A. |
We then confirmed hs-T34A expression in SKOV3 cells by Western blot analysis and real time RT-PCR. As shown in Figure 7A and B, the expression of hs-T34A was markedly increased in SKOV3 cells transfected with HPEI/hs-T34A complexes compared with NS-, HPEI-, or HPEI/EP-treated cells. We then examined apoptosis using propidium iodide staining to determine the anticancer effect of HPEI/hs-T34A complexes on SKOV3 cells. As shown in Figure 7C, 37.6%±4.3% of apoptotic cells were observed in response to the treatment with HPEI/hs-T34A complexes (50 μg HPEI/5 μg DNA), while 10.9%±1.4% of apoptotic cells were observed under HPEI/EP treatment, and only 8.0%±2.8% were observed in the HPEI-treated group. These results suggested that HPEI nanoparticles could efficiently deliver the hs-T34A gene into SKOV3 cells in vitro and cause cell death by inducing apoptosis.
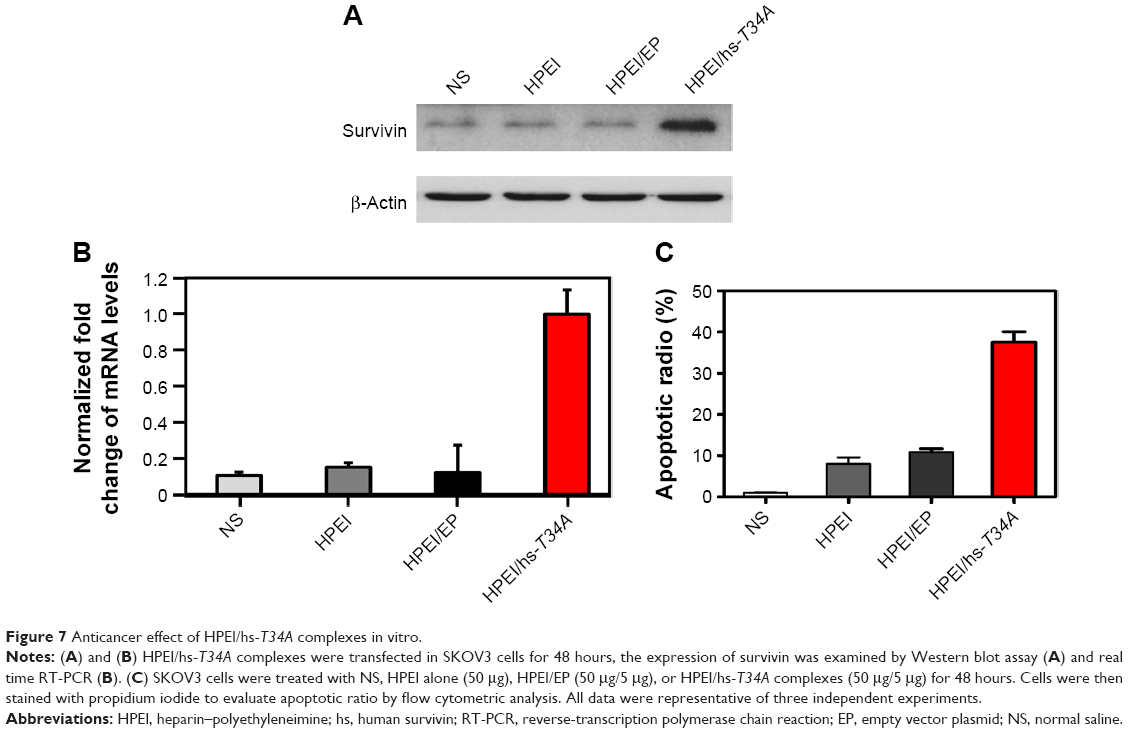 | Figure 7 Anticancer effect of HPEI/hs-T34A complexes in vitro. |
Anticancer effect of HPEI/hs-T34A complexes in vivo
To study the anticancer activity of HPEI/hs-T34A complexes on the abdominal cavity metastases of SKOV3 ovarian carcinoma, female BALB/c nude mice inoculated with SKOV3 tumor cells were treated with HPEI/hs-T34A complexes at a dose of 50 μg HPEI/5 μg DNA. The metastatic tumor tissues in each group were harvested and weighed. As shown in Figure 8A, the tumor progress was markedly inhibited in mice treated with HPEI/hs-T34A complexes compared with those treated with saline, HPEI alone, HPEI/EP, or PEI 25K/hs-T34A. The average tumor weight in HPEI/hs-T34A complexes group was −0.335±0.076 g, while it was 1.169±0.180 g in the control group, 0.956±0.229 g in the HPEI nanoparticle group, 0.697±0.244 g in the HPEI/EP group, and 0.860±0.374 g in the HPEI/EP group, respectively. Compared with other groups, the HPEI/hs-T34A complex group showed a statistically significant reduction in tumor weight (P<0.05) (Figure 8B). In addition, the number of tumor nodules in the HPEI/hs-T34A complex group was much less, when compared with the NS, HPEI alone, HPEI/EP, or PEI 25K/hs-T34A group (P<0.05) (Figure 8C). HPEI/hs-T34A complexes also markedly decreased the ascites volume, when compared with control group (Figure 8D). These results suggested that HPEI/hs-T34A complexes can efficiently inhibit the growth of ovarian tumor cells. Furthermore, we confirmed the expression of the hs-T34A protein in tumor tissues by Western blot analysis and real time RT-PCR. As shown in Figure 9A and B, the expression of survivin was markedly increased in tumors treated with HPEI/hs-T34A complexes, when compared with NS-, HPEI-, HPEI/EP, or PEI 25K/hs-T34A-treated cells.
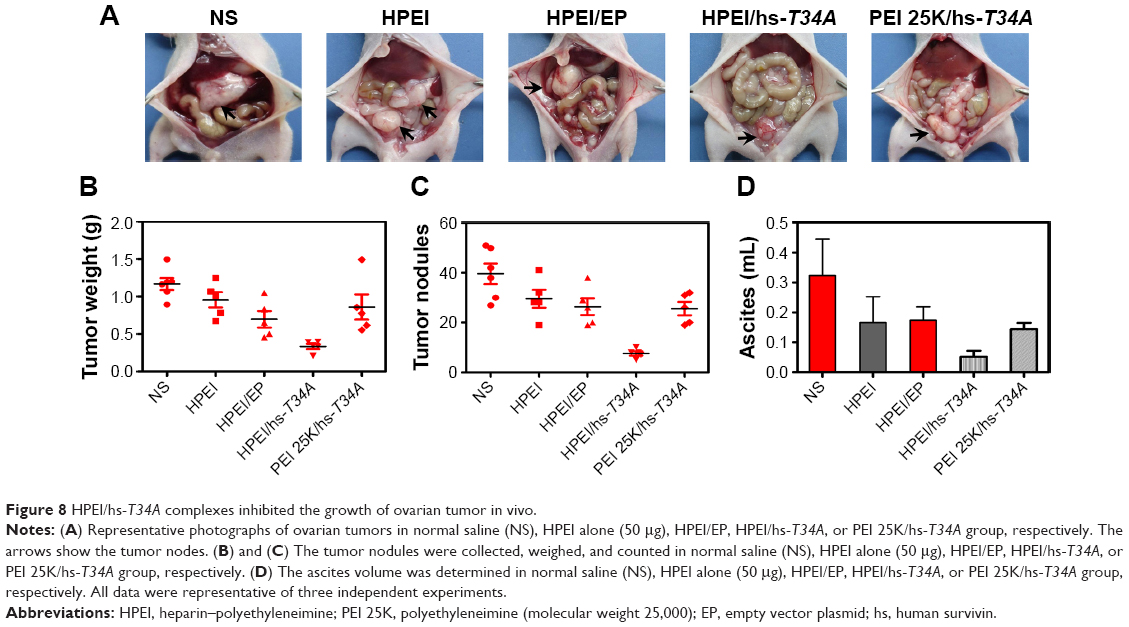 | Figure 8 HPEI/hs-T34A complexes inhibited the growth of ovarian tumor in vivo. |
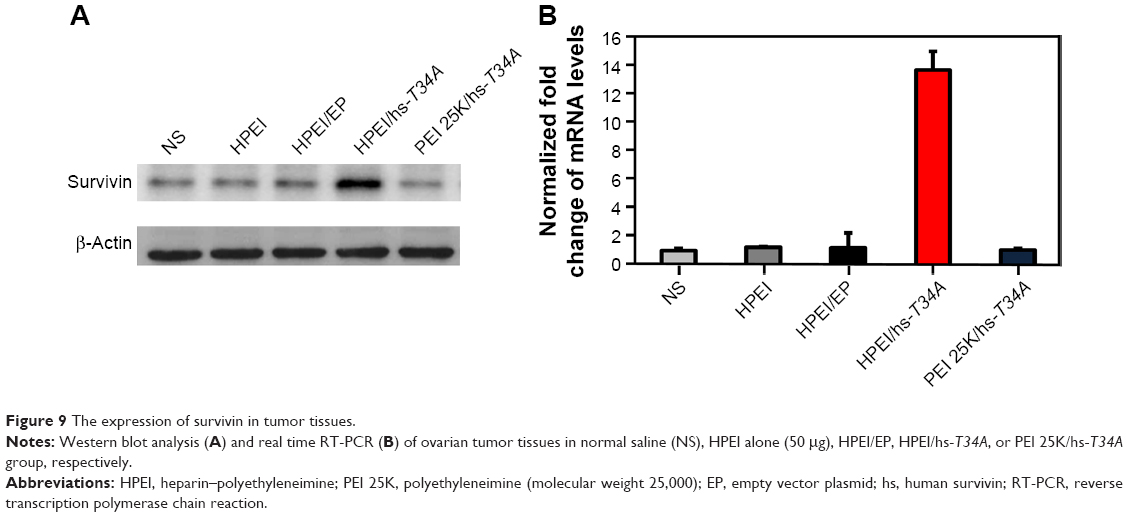 | Figure 9 The expression of survivin in tumor tissues. |
Effect of HPEI/hs-T34A complexes on ovarian tumor cell apoptosis and angiogenesis
It has been well demonstrated that the mutation of survivin’s threonine 34 to alanine (survivin-T34A) could significantly lead to apoptosis.20 Hence, to study the mechanism underlying the anticancer activity of HPEI/hs-T34A nanoparticles in vivo, TUNEL assay was performed to examine whether HPEI/hs-T34A complexes could induce apoptosis of tumor cells. As shown in Figure 10A, cells with bright green fluorescent staining (identified as apoptotic cells) could be observed in tumor tissue in the HPEI/hs-T34A-treated group, whereas the green fluorescent staining was rare in the NS, HPEI alone, or HPEI/EP group. These results implied that apoptosis was involved in the anticancer effect of HPEI/hs-T34A complexes. In addition, the inhibition of tumor growth often accompanies with compromised angiogenesis.23 To this end, we have a particular interest to examine whether HPEI/hs-T34A complexes could mitigate angiogenesis using CD31 staining. As shown in Figure 10B, the microvessel density characterized by CD31 positive staining was significantly attenuated in the HPEI/hs-T34A complex group, when compared with NS, HPEI alone, or HPEI/EP group. Together, these data suggested that HPEI/hs-T34A complexes can inhibit tumor growth by induction of apoptosis and inhibition of angiogenesis.
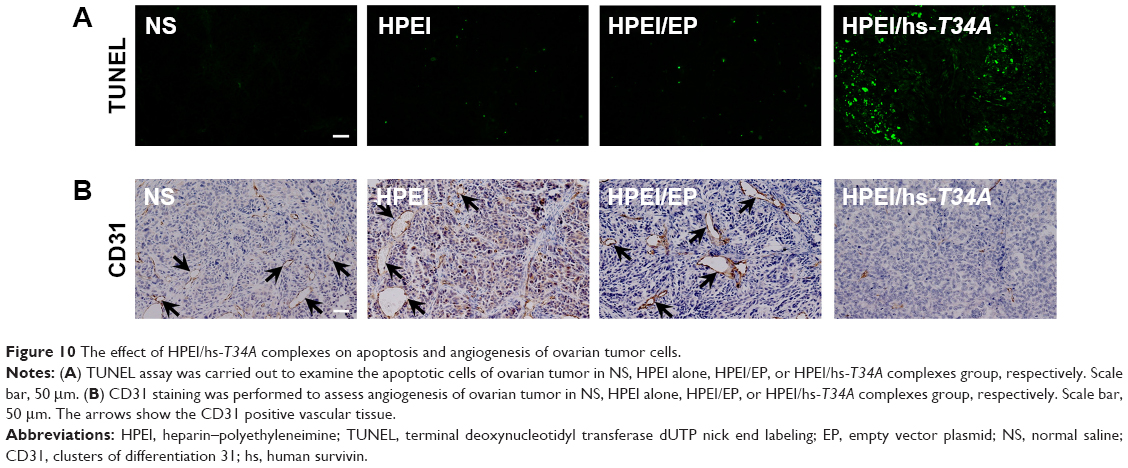 | Figure 10 The effect of HPEI/hs-T34A complexes on apoptosis and angiogenesis of ovarian tumor cells. |
Furthermore, the side effects of HPEI/hs-T34A complexes were examined in mice. Normal urine and fecal appearances were observed during HPEI/hs-T34A treatment. HE staining was performed to investigate whether HPEI/hs-T34A complexes have any toxic effect. As shown in Figure 11A, no significantly pathological changes were observed in the heart, liver, spleen, lung, or kidney, respectively. Moreover, there was no significant difference in the body weight among all groups. Overall, our data suggested that HPEI/hs-T34A complexes are capable of treating cancer by inducing apoptosis and inhibiting angiogenesis without obvious toxic side effects.
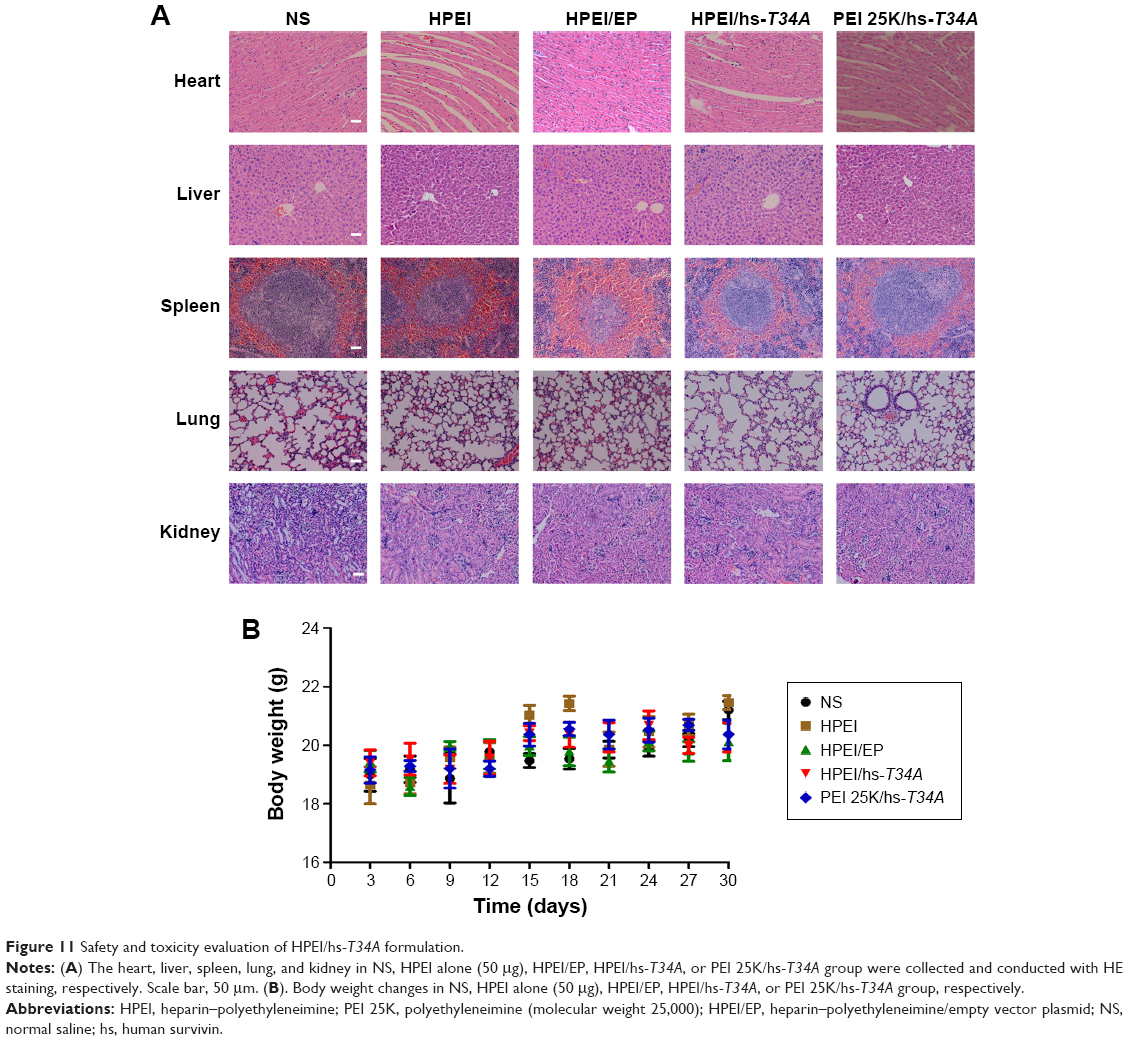 | Figure 11 Safety and toxicity evaluation of HPEI/hs-T34A formulation. |
Discussion
In this study, the biodegradable HPEI nanoparticles were used to deliver the hs-T34A gene for the treatment of ovarian cancer. The dynamic diameter of HPEI nanoparticles was ~66±4.5 nm (Figure 2A) with a zeta potential of 27.1±1.87 mV (Figure 2B). These HPEI nanoparticles had good stability with low toxicity, and were easy to prepare. Our results showed that HPEI nanoparticles were capable of transfecting the hs-T34A gene effectively in human ovarian cancer SKOV3 cells, resulting in apoptotic cell death. Moreover, HPEI/hs-T34A complexes exhibited good antitumor activity against ovarian cancer via inducing apoptosis and inhibiting angiogenesis in the SKOV3 xenograft nude mouse model. Collectively, our study suggested that HPEI-delivered hs-T34A could be a potential therapeutic strategy for ovarian cancer.
Over the past 2 decades, the clinical application of gene-based therapy has been widely investigated.8 Both viral and nonviral vectors have been used in gene delivery. Compared to viral-vectors, nonviral vectors have exhibited the potential to break through several limitations, due to their low immune response, high gene-loading capacity, handy chemical modification, large-scale production, and especially, their safety.12 PEI was one of the most effective nonviral gene carrier agents due to its strong DNA compaction capacity with an intrinsic endosomolytic activity.24,25 Thus, many PEI-based vectors have been developed. Recently, Zhang et al synthesized an N-isopropylacrylamide-modified polyethyleneimine through Michael addition reaction.26 The prepared N-isopropylacrylamide-modified polyethyleneimine could mediate the p53 gene, triggering a significant antitumor effect in vitro. Cho et al prepared a polysorbitol-based transporter to deliver the osteopontin gene for the treatment of lung cancer.27 In their study, the polysorbitol-based transporter was also prepared by Michael addition reaction between branched PEI and sorbitol diacrylate. On the other hand, in order to overcome different drawbacks of PEIs, such as nonbiodegradability and cytotoxicity,28 several strategies have been attempted to modify PEIs, such as blocking copolymers of PEG, developing a degradable linker, or alkylating PEI. In our study, we prepared biodegradable HPEI nanoparticles with low cytotoxicity to deliver the hs-T34A gene to treat ovarian cancer. These HPEI nanoparticles were generated by covalently conjugating low molecular weight PEI 2K with heparin in room temperature. The crosslinking reaction was catalyzed with EDC/NHS in solution, forming biodegradable linkers. Compared to other studies, the preparation process of HPEI nanoparticles was much easier without any organic solvent involved. Meanwhile, according to our results, the obtained HPEI nanoparticles demonstrated strong DNA-binding ability with low cytotoxicity and were easily degraded. Besides, it could effectively deliver the HPEI/hs-T34A gene expression plasmid into SKOV3 cells in vitro and in vivo, showing great potential in gene delivery. Survivin is the smallest member of the inhibitor of apoptosis family. Previous studies proved that survivin may be a potential target for cancer therapy.29–31 Currently, several survivin-targeted drugs have successfully reached Phase I or II in human clinical trials.32,33 Accordingly, many therapeutic strategies targeting the survivin gene have been developed for cancer treatment.34 For example, Vivas-Mejia et al have used small interfering RNA to target survivin for ovarian cancer, which could suppress the expression of survivin.35 Hu et al have found survivin short hairpin RNA to treat ovarian cancer and also suppress the expression of survivin.36 Mehta et al have proved that YM155, a small molecule, could specifically inhibit survivin gene expression by suppressing its promoter activity, which has been proved to inhibit esophageal squamous-cell carcinoma growth in mice.37 In addition, dominant-negative mutant was another method of choice to suppress the antiapoptotic function of survivin. Previous studies have reported that dominant-negative mutant (threonine 34 to alanine) of survivin has been shown to attenuate the endogenous survivin expression through abolishing of cdc2-cyclin B1 complex-mediated phosphorylation.38
Herein, we incorporated the hs-T34A gene into pVITRO2 plasmid and delivered it with HPEI nanoparticles with an assumption that the expressed hs-T34A gene could effectively suppress the normal function of survivin in SKOV3 cancer cells. According to our study, the HPEI/hs-T34A formulation showed obvious anticancer activity both in vitro and in vivo. Meanwhile, our results suggested that cell apoptosis induction and angiogenesis inhibition might be important mechanisms of tumor suppression, which was consistent with previous reports.39,40 Our data suggested that HPEI/hs-T34A formulation holds the potential for the gene therapy of ovarian cancer.
Conclusion
In this study, the biodegradable HPEI nanoparticles were used to deliver hs-T34A gene to treat ovarian cancer. The prepared HPEI nanoparticles showed high stability, low toxicity, and high transfection ability. The HPEI/hs-T34A complexes exhibited obvious antitumor activity against SKOV3 ovarian cancer by inducing apoptosis and inhibiting angiogenesis. Collectively, our study suggested that HPEI-delivered hs-T34A could be a potential strategy for ovarian cancer gene therapy.
Acknowledgments
This work was supported by the National Science and Technology Major Project (2013ZX09301-304-008) and the National Natural Science Foundation (81422025, 81572990, 81201785), and 863 Program (2014AA020509).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res. 2013;19(5):961–968. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Leboulch P. Gene therapy: primed for take-off. Nature. 2013;500(7462):280–282. | ||
Walther W, Schlag PM. Current status of gene therapy for cancer. Curr Opin Oncol. 2013;25(6):659–664. | ||
O’Connor DM, Boulis NM. Gene therapy for neurodegenerative diseases. Trends Mol Med. 2015;21(8):504–512. | ||
Bongianino R, Priori SG. Gene therapy to treat cardiac arrhythmias. Nat Rev Cardiol. 2015;12(9):531–546. | ||
Ginn SL, Alexander IE, Edelstein ML, et al. Gene therapy clinical trials worldwide to 2012 – an update. J Gene Med. 2013;15(2):65–77. | ||
Yin H, Kanasty RL, Eltoukhy AA, et al. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555. | ||
Giacca M, Zacchigna S. Virus-mediated gene delivery for human gene therapy. J Control Release. 2012;161(2):377–388. | ||
Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358. | ||
Ramamoorth M, Narvekar A. Non viral vectors in gene therapy – an overview. J Clin Diagn Res. 2015;9(1):GE01–GE06. | ||
Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. 2009;109(2):259–302. | ||
Kunath K, von Harpe A, Fischer D, et al. Low-molecular-weight polyethylenimine as a non-viral vector for DNA delivery: comparison of physicochemical properties, transfection efficiency and in vivo distribution with high-molecular-weight polyethylenimine. J Control Release. 2003;89(1):113–125. | ||
Gou M, Men K, Zhang J, et al. Efficient inhibition of C-26 colon carcinoma by VSVMP gene delivered by biodegradable cationic nanogel derived from polyethyleneimine. ACS Nano. 2010;4(10):5573–5584. | ||
Liu P, Gou M, Yi T, et al. Efficient inhibition of an intraperitoneal xenograft model of human ovarian cancer by HSulf-1 gene delivered by biodegradable cationic heparin-polyethyleneimine nanogels. Oncol Rep. 2012;27(2):363–370. | ||
Yang F, Gou M, Deng H, et al. Efficient inhibition of ovarian cancer by recombinant CXC chemokine ligand 10 delivered by novel biodegradable cationic heparin-polyethyleneimine nanogels. Oncol Rep. 2012;28(2):668–676. | ||
Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3(8):917–921. | ||
Stauber RH, Mann W, Knauer SK. Nuclear and cytoplasmic survivin: molecular mechanism, prognostic, and therapeutic potential. Cancer Res. 2007;67(13):5999–6002. | ||
Aspe JR, Wall NR. Survivin-T34A: molecular mechanism and therapeutic potential. Onco Targets Ther. 2010;3:247–254. | ||
McKay TR, Bell S, Tenev T, et al. Procaspase 3 expression in ovarian carcinoma cells increases survivin transcription which can be countered with a dominant-negative mutant, survivin T34A; a combination gene therapy strategy. Oncogene. 2003;22(23):3539–3547. | ||
Sun IC, Eun DK, Na JH, et al. Heparin-coated gold nanoparticles for liver-specific CT imaging. Chemistry. 2009;15(48):13341–13347. | ||
Bono F, De Smet F, Herbert C, et al. Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Cancer Cell. 2013;23(4):477–488. | ||
Boussif O, Lezoualc’h F, Zanta MA, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92(16):7297–7301. | ||
Lungwitz U, Breunig M, Blunk T, et al. Polyethylenimine-based non-viral gene delivery systems. Eur J Pharm Biopharm. 2005;60(2):247–266. | ||
Zhang J, Wu D, Xing Z, et al. N-isopropylacrylamide-modified polyethylenimine-mediated p53 gene delivery to prevent the proliferation of cancer cells. Colloids Surf B Biointerfaces. 2015;129:54–62. | ||
Cho WY, Hong SH, Singh B, et al. Suppression of tumor growth in lung cancer xenograft model mice by poly(sorbitol-co-PEI)-mediated delivery of osteopontin siRNA. Eur J Pharm Biopharm. 2015;94:450–462. | ||
Nguyen HK, Lemieux P, Vinogradov SV, et al. Evaluation of polyether-polyethyleneimine graft copolymers as gene transfer agents. Gene Ther. 2000;7(2):126–138. | ||
Mita AC, Mita MM, Nawrocki ST, et al. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14(16):5000–5005. | ||
Samarasinghe RM, Gibbons J, Kanwar RK, et al. Nanotechnology based platforms for survivin targeted drug discovery. Expert Opin Drug Discov. 2012;7(11):1083–1092. | ||
Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8(1):61–70. | ||
Miyazaki A, Kobayashi J, Torigoe T, et al. Phase I clinical trial of survivin-derived peptide vaccine therapy for patients with advanced or recurrent oral cancer. Cancer Sci. 2011;102(2):324–329. | ||
Giaccone G, Zatloukal P, Roubec J, et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27(27):4481–4486. | ||
Mobahat M, Narendran A, Riabowol K. Survivin as a preferential target for cancer therapy. Int J Mol Sci. 2014;15(2):2494–2516. | ||
Vivas-Mejia PE, Rodriguez-Aguayo C, Han HD, et al. Silencing survivin splice variant 2B leads to antitumor activity in taxane – resistant ovarian cancer. Clin Cancer Res. 2011;17(11):3716–3726. | ||
Hu Q, Li W, Hu X, et al. Synergistic treatment of ovarian cancer by co-delivery of survivin shRNA and paclitaxel via supramolecular micellar assembly. Biomaterials. 2012;33(27):6580–6591. | ||
Mehta A, Zhang L, Boufraqech M, et al. Inhibition of survivin with YM155 induces durable tumor response in anaplastic thyroid cancer. Clin Cancer Res. 2015;21(18):4123–4132. | ||
Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3(1):46–54. | ||
Pan L, Peng XC, Leng F, et al. Therapeutic effects of survivin dominant negative mutant in a mouse model of prostate cancer. J Cancer Res Clin Oncol. 2011;137(1):19–28. | ||
O’Connor DS, Schechner JS, Adida C, et al. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol. 2000;156(2):393–398. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.