")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Efficacy and Safety Profile of Fostemsavir for the Treatment of People with Human Immunodeficiency Virus-1 (HIV-1): Current Evidence and Place in Therapy
Authors Muccini C, Canetti D , Castagna A, Spagnuolo V
Received 4 November 2021
Accepted for publication 14 January 2022
Published 25 January 2022 Volume 2022:16 Pages 297—304
DOI https://doi.org/10.2147/DDDT.S273660
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Camilla Muccini,1,2,* Diana Canetti,2,* Antonella Castagna,1,2 Vincenzo Spagnuolo2
1School of Medicine, Vita-Salute San Raffaele University, Milan, Italy; 2Unit of Infectious Diseases, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) San Raffaele Hospital, Milan, Italy
*These authors contributed equally to this work
Correspondence: Vincenzo Spagnuolo
Unit of Infectious Diseases, IRCCS Ospedale San Raffaele, Via Stamira d’Ancona 20, Milan, Italy, Tel +390226437907, Fax +390226437903, Email [email protected]
Abstract: Heavily-treatment-experienced people living with human immunodeficiency virus (HTE-PLWH) represent a population with limited therapeutic options and at high-risk of clinical progression, morbidity, and mortality. The development of new drugs and new drug classes for the treatment of HIV-1 infection in HTE-PLWH is critical to successfully suppress HIV-1 replication, restore the immune system, and improve quality of life. Fostemsavir is the first attachment inhibitor approved by Food and Drug Administration and European Medicines Agency for the treatment of HIV-1 infection. It is approved in combination with other antiretrovirals, for HTE-PLWH with multi-drug resistant HIV-1 after failing their current antiretroviral regimen due to resistance, intolerance, or safety considerations. In this review, we present and discuss the mechanism of action, the pharmacodynamic and pharmacokinetic properties, and the efficacy and safety of fostemsavir as an antiretroviral agent for the treatment of HIV-1 infection.
Keywords: fostemsavir, attachment inhibitor, human immunodeficiency virus, heavily treatment experienced, drug resistance
Introduction
Contemporary antiretroviral therapy (ART) has dramatically improved the prognosis of people living with HIV (PLWH) through the achievement of durable viral suppression and the consequent immune function restoration.1
However, some heavily-treatment-experienced PLWH (HTE-PLWH) are unable to achieve viral suppression because of HIV-1 multi-drug resistance and/or drug intolerance.2
For these HTE-PLWH, who are at high risk of clinical progression, morbidity, and mortality, the development of novel ART is critically important.3,4
In response to this need for novel HIV-1 treatment options, a novel drug class has emerged. Fostemsavir is a first-in-class attachment inhibitor that binds directly to the viral envelope glycoprotein 120 (gp120), close to the CD4 binding.5 This prodrug of temsavir thus prevents the binding and entry of HIV-1. Fostemsavir has been approved both by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA), in combination with other antiretrovirals, for the treatment of multi-drug resistant HIV-1 infection in HTE adults with limited antiretroviral treatment options.6,7 Here we review the mechanism of action, the pharmacodynamic and pharmacokinetic properties, and the main results of the pivotal trials that investigated the efficacy and safety of fostemsavir as an ART drug for the treatment of HIV-1.
Mechanism of Action and Structure
The prodrug fostemsavir and its active metabolite temsavir belong to the attachment inhibitor class, and this ART class works by blocking HIV-1 entry in human cells. Temsavir specifically binds the envelope protein gp120, adjacent to the gp120-CD4 binding site, thus preventing the gp120 conformational change required for attachment to the host cell CD4 receptor and needed for viral replication process (Figure 1).8,9
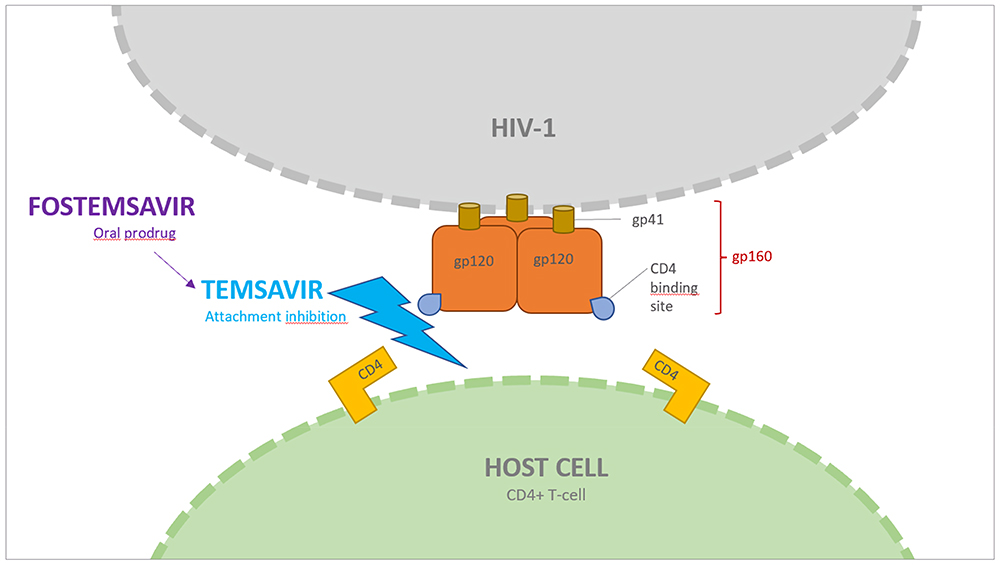 |
Figure 1 Fostemsavir mechanism of action. Temsavir, the active moiety of fostemsavir, binds the viral envelope protein gp120 on HIV-1, adjacent to the gp120-CD4+ binding site. In doing so, it prevents the conformational change required for attachment of HIV-1 to the host cell. |
BMS-488043 was the first molecule that demonstrated the efficacy and safety of this new class of antiretrovirals in an 8-day monotherapy proof-of-concept trial. It was then replaced with BMS-663068 (fostemsavir), a methyl-phosphate prodrug characterized by enhanced bioavailability.10,11 The efficacy of fostemsavir and its active form BMS-626529 (temsavir) was demonstrated in another 8-day monotherapy trial, which showed a substantial reduction of HIV-1 RNA viral load, with or without boosting agent (ritonavir).12
Synthesis of fostemsavir began with an indole glyoxamide which targeted the HIV-1 envelope and inhibited CD4 receptor binding. Then, studies of the structure-activity relationship of this compound led to the development of the derivative BMS-488043, a 4,7-dimethoxy-azaindole with a nitrogen substitution at the C6 position of the azaindole. This derivative demonstrated efficacy in reducing viral load but required a concomitant high-fat meal and a high-dose treatment twice daily.10,13,14 A more optimized compound, BMS-626529 (temsavir), was then developed, which has improved antiviral potency and pharmaceutical profile. Temsavir is characterized by a 3-methyl-1,2,4 triazole linked to the C7 position of the C6 substituted azaindole.15 The prodrug BMS-663068 (fostemsavir), which is a phosphonooxymethyl tris (hydroxymethyl)-aminomethane salt, was then developed and found to have increased solubility in the gastrointestinal tract.8,14 Fostemsavir is converted to its highly permeable derivative temsavir by alkaline phosphatase at the epithelial gut surface.16 Two-dimensional chemical structures of BMS-488043, BMS-663068 (fostemsavir) and BMS-626529 (temsavir) are reported in Figure 2.
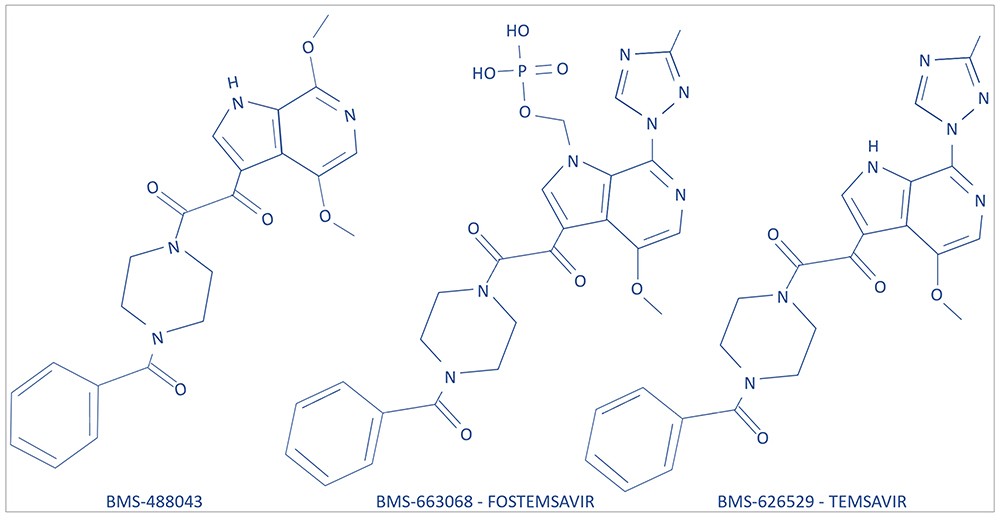 |
Figure 2 Two-dimensional chemical structures of BMS-488043, BMS-663068 (fostemsavir), and BMS-626529 (temsavir). |
Pharmacokinetics
Fostemsavir was approved as an extended-release tablet (600 mg) administered orally twice daily. It has improved aqueous solubility and stability under acidic conditions, and is rapidly metabolized to temsavir, which is the only compound detectable in plasma. It is absorbed with an absolute bioavailability of approximately 27% measured after a single-dose administration of extended-release tablets followed by intravenous infusion of [13C]-temsavir.17 Steady state was achieved in two to three days.12 Dosing with standard meals may increase fostemsavir area under the curve (AUC) by only 10%. Therefore, food intake does not significantly impact fostemsavir bioavailability.18
Temsavir is characterized by high protein binding (88.4%), mainly to serum albumin, with a distribution volume of 29.5 L at steady state.17 It is metabolized through both esterase-mediated hydrolysis and cytochrome (CY) P450-mediated oxidation. It works as a substrate of CYP3A, P-glycoprotein, and breast cancer resistance protein (BCRP), and it may inhibit OATP1B1/B3 and BCRP.12 Co-administration with strong CYP3A inducers results in decreased concentrations of temsavir. Combined use with carbamazepine, phenytoin, phenobarbital, rifampin, and St John’s wort is contraindicated due to major interactions and risk of virologic failure.17,19 No clinically relevant differences in temsavir pharmacokinetics were reported for mild to moderate hepatic impairment and mild to end-stage kidney disease, including hemodialysis.20,21
Pharmacodynamics
The antiviral activity of fostemsavir varies with 50% inhibitory concentration (IC50), trough plasma concentration, and steady-state average temsavir plasma concentrations. In particular, a low IC50 at baseline (<100 nmol) was associated with an HIV-1 RNA viral load decrease ≥1 log10, while a higher baseline IC50 (>100 nmol) was often related to a < 1 log10 viral load reduction.12
Fostemsavir demonstrated an improved inhibitory quotient and higher barrier for selecting resistance over its precursor BMS-488043.22
The occurrence of resistance to temsavir is mainly related to gp120 heterogeneity, especially among subtypes AE and O.14 The gp120 major substitutions responsible for reduced susceptibility to temsavir are S375H and M475I in subtype AE, L116Q, S375H/M/T, M426L, M434I, and M475I in subtype B, several natural polymorphisms in O, N, and P subtypes.23–26 In the CASTLE study, the activity of temsavir against gp120 subtypes from worldwide viral isolates was assessed. Excellent activity was demonstrated against most subtypes, with no geographic differences, excepted for subtype AE (common in Southeast Asia) and subtype B when characterized by a substitution of a histidine at position 375 rather than a serine and of an isoleucine at position 475.22,27,28
In a recent survey including more than one thousand isolates, Gartland et al analyzed susceptibility to temsavir in a Monogram Biosciences PhenoSenseVR Entry assay. Among twenty different subtype envelopes, they reported a broad range of variability with a prevalent high susceptibility to temsavir (IC50 <100 nmol), with the exception of CRF01_AE viruses which in all five cases exhibited IC50 >100 nmol. Nevertheless, subtype, IC50 or the presence of at-risk polymorphisms at position S375, M426, M434, and M475 seem to decrease susceptibility in a context-dependent way and do not clearly predict virological response in clinical trials.29
Viral tropism do not interfere in fostemsavir activity, as well as the combined use with other antiretrovirals, including entry inhibitors.22,30
A temsavir concentration-dependent QTc interval prolongation on electrocardiogram (ECG) was observed in some patients, which became clinically significant at four times the recommended 1200 mg daily dose. Therefore, as a precaution, patients with a history of QTc-prolongation, concomitant QTc-prolonging medications, or cardiac impairment, should be treated with caution and monitored with ECG throughout treatment.31
Clinical Data
Phase II Trials
In the last years, fostemsavir was investigated to overcome the need of new treatment regimens, in particular for heavily-treatment experienced with few options remaining.
A phase IIa study has assessed the antiviral activity of the HIV attachment inhibitor in an 8-day monotherapy.12
Participants (n = 50) were enrolled in five arms at different doses with or without ritonavir (1:1:1:1:1): 1) 600 mg fostemsavir plus 100 mg ritonavir BID; 2) 1200 mg fostemsavir plus 100 mg ritonavir QD; 3) 1200 mg fostemsavir plus 100 mg ritonavir BID; 4) 1200 mg fostemsavir plus 100 mg ritonavir QD; 5) 1200 mg fostemsavir BID. In both ART-naïve and ART-experienced PLWH, the maximum median decline in HIV RNA viral load from baseline varied from 1.21 log10 copies/mL in arm 5, to 1.73 log10 copies/mL in arm 3 and was observed regardless of ritonavir use. Interestingly, 4/10 (40%) subjects in arm 5 had fostemsavir IC50 values >0.1 μmol/L at baseline that could explain a poorer virologic response. During the monotherapy with fostemsavir, absolute CD4+ T cell count and CD4/CD8 ratio increased without any statistical differences between arms.12
In one phase IIb study (AI438011), the proportion of subjects with HIV RNA viral load <50 copies/mL at week 24 was the primary endpoint.32
In this trial, treatment-experienced patients were randomized in five arms (1:1:1:1:1): 1) fostemsavir 400 mg BID; 2) fostemsavir 800 mg BID; 3) fostemsavir 600 mg QD; 4) fostemsavir 1200 mg QD; 5) atazanavir 300 mg and 100 mg ritonavir 100 mg QD. The optimized background therapy for each arm consisted of raltegravir 400 mg BID and tenofovir disoproxil fumarate 300 mg QD.
Overall, 200 participants received at least one dose of fostemsavir. Among these, 80% of arm 1, 69% of arm 2, 76% of arm 3, and 72% of arm 4 achieved a viral load under the limit of detection at week 24. In comparison, 75% of arm 5, who were assigned to take ritonavir-boosted atazanavir, achieved non-detectable viral load. The median CD4+ T cells at baseline ranged from 214 cells/μL in arm 1 to 249 cells/μL in arm 5, and the median increase in CD4+ T cells at week 24 ranged from 100 cells/μL in arm 3 to 138 cells/μL in arm 1. The CD4+ T cell count trend appeared to be similar in subjects receiving fostemsavir or ritonavir-boosted atazanavir.
One of the inclusion criteria of the AI438011 study was susceptibility to fostemsavir, expressed as IC50 of less than 0.1 μmol/L; for this reason, the efficacy of the study drug in PLWH with a higher IC50 at baseline might not be comparable.
Phase III Trials
The efficacy of fostemsavir has been evaluated in one phase III trial, the BRIGHTE study, which enrolled 372 PLWH failing ART with multi-drug resistant HIV infection.33
There were 272 participants with at least one fully active agent in less than two antiretroviral classes, who were assigned to the randomized cohort, and 99 without any remaining antiretroviral option, who were assigned to the nonrandomized cohort. In the randomized cohort (3:1), PLWH received first an 8-day therapy with fostemsavir 600 mg BID or placebo in combination with their failing regimen and then open-label fostemsavir 600 mg BID in addition to an optimized background therapy. In the nonrandomized cohort, PLWH started from day 1 on fostemsavir 600 mg BID in combination with a treatment based on their individual resistance testing and ART history.
At baseline, median HIV RNA viral load was 4.7 log10 copies/mL (interquartile range, IQR = 1.6, 6.6) in the randomized cohort and 4.3 log10 copies/mL (IQR = 1.6, 6.6) in the nonrandomized cohort. The median baseline CD4+ T cell count was 99 cell/mcL (IQR = 0, 1160) in the randomized cohort and 41 cell/mcL (IQR = 0, 641) in the nonrandomized cohort.
Fostemsavir was effective both in decreasing HIV RNA and increasing CD4+ T cells. The proportion of participants who achieved virologic suppression was 53% in the randomized cohort and 37% in the nonrandomized cohort at week 24, then 54% and 38% at week 48, and 60% and 37% at week 96.33,34 Over the 96-week period, the CD4+ T cell count increased by 205 cell/mcL from baseline (standard deviation, SD = 191) in the randomized cohort and by 119 cell/mcL (SD = 202) in the nonrandomized cohort.34
In the BRIGHTE trial, virologic failure was defined as: 1) HIV RNA viral load ≥400 copies/mL after prior confirmed viral suppression to <400 copies/mL, 2) ≥400 copies/mL at last available prior to discontinuation, or 3) >1 log10 copies/mL increase in HIV RNA above nadir level (≥40 copies/mL). Using these criteria, virologic failure was found at week 48 in 49/272 (18%) participants in the randomized arm and 46/99 (46%) participants in the nonrandomized arm, and at week 96 in 63/272 (23%) and 49/99 (49%), respectively.33,34
Dosage, Administration, and Drug Interactions
Each extended-release tablet contains 600 mg fostemsavir. It can be taken with or without food, but it should not be crushed, chewed, or split. The recommended dosage is 600 mg PO BID, combined with an individually optimized background regimen. Fostemsavir should not be co-administered with strong inducers of CYP3A4, such as rifampin, because plasma levels of temsavir could be decreased, leading to a reduction in therapeutic effect.35
Safety and Tolerability
Fostemsavir has been well‐tolerated in phase II and III trials conducted in PLWH. Nausea, diarrhea, and headache were the most common drug-related adverse events observed. No significant laboratory abnormalities were seen associated with the study drug.
Severe adverse events were more frequent in people with a compromised immune system; among these, an atypical mycobacterial infection secondary to immune reconstitution inflammatory syndrome was considered to be due to fostemsavir in the phase III trial.33
Moreover, the occurrence of acute infections and AIDS-related disease were the major causes of death of participants in the BRIGHTE study, especially in PLWH with CD4+ T cell count <20 cell/mcL.33,36
No sufficient safety data are available on the effects of fostemsavir in pregnant women to assess teratogenicity in humans. Therefore, it is preferable to avoid its use during pregnancy and breastfeeding or to monitor strictly patients if it is not possible to modify their current antiretroviral regimen.36
Self-administered patient questionnaires have demonstrated good tolerability. They reveal that treatment with fostemsavir has led to improvements in health-related quality of life (HRQOL) in a population of HTE-PLWH.37 A better HRQOL may lead to an increase in adherence to treatment in people with limited therapeutic options and play a role in maintaining a long-lasting virologic response.
Emergence of Resistance
A scarce compliance is not the only factor associated with virological failure: gp120 substitutions, that are likely related to a decreased susceptibility to temsavir, were reported in 70% of nonrandomized group participants in the BRIGHTE trial, even though these genotypic substitutions were not present in a portion greater than 50% of randomized group participants with a detectable viremia. In the randomized cohort, 51% of participants experiencing virological failure had emergent substitutions in gp120 in four key sites (S375, M426, M434, and M475).33
To better evaluate the role of gp120 substitutions, sequences both from ART-naïve and ART-treated (but naïve to fostemsavir) people with HIV were investigated; although the high variability of gp120 described, the frequency of mutations was limited. The most frequent substitution emerged was S375T that has a marginal impact on the efficacy of fostemsavir.24,25
However, our attention may be focused in identifying new possible polymorphisms that could interfere with the susceptibility to temsavir through further in-depth in vitro and clinical studies.
The mechanism of action of fostemsavir results in blocking gp120-mediated viral entry, which is similar to ibalizumab, which binds CD4 to interfere with CD4-gp120 interactions, and maraviroc, which binds CCR5 to interfere with CCR5-gp120 interactions. However, recent studies have demonstrated that there is no cross-resistance between fostemsavir and these agents, which further emphasizes the importance of this drug in the HTE-PLWH patients.38,39
Place in Therapy
Fostemsavir has been approved both by the FDA and the EMA for the treatment of HTE-PLWH harboring an HIV-1 MDR infection and failing their current antiretroviral regimen.6,7 In this population, clinical trials have demonstrated the antiviral activity and safety of fostemsavir.33,34
In addition, the recent approval of fostemsavir and of the post-attachment inhibitor ibalizumab, together with the probable, future availability of islatravir, the first-in-class nucleoside reverse transcriptase translocation inhibitor, and lenacapavir, the first-in-class capsid inhibitor, may help to fill the need for effective ART for HTE-PLWH.40–42
Moreover, FTR has been tested as a possible ART component in HIV eradication strategies both in humanized mice in vivo and in peripheral blood mononuclear cells of HIV-1-infected patients ex vivo.43
Finally, fostemsavir has shown a reassuring safety profile without a significant impact on metabolic parameters.33,34 These characteristics may be useful in switch strategies for virologically suppressed HTE-PLWH or even for PLWH in earlier therapeutic lines. However, further clinical data are needed to evaluate the possible use of fostemsavir in these clinical settings.
Conclusion
Fostemsavir is the first attachment inhibitor approved for the treatment of HIV in HTE-PLWH with virologic failure. In this population, fostemsavir showed good efficacy and a reassuring safety profile. However, clinicians should be aware of possible drug–drug interactions due to the role of CYP3A4 in its metabolism (eg, coadministration of fostemsavir with strong CYP3A inducers is contraindicated). The approval of fostemsavir and ibalizumab and the probable, future availability of islatravir and lenacapavir are poised to revolutionize the treatment of HTE-PLWH in the coming years.
Acknowledgments
We would like to thank JournalEdit for reviewing this manuscript for English language.
Disclosure
V.S. received speaking fees by Gilead Sciences, ViiV Healthcare, Merck Sharp and Dohme, Janssen-Cilag. A.C. received speaking fees by Gilead Sciences, ViiV Healthcare, Merck Sharp and Dohme, Janssen-Cilag, and Theratechnologies. The authors report no other conflicts of interest in this work.
References
1. Samji H, Cescon A, Hogg RS, et al. North American AIDS Cohort Collaboration on Research and Design (NA-ACCORD) of IeDEA. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One. 2013;8(12):e81355. doi:10.1371/journal.pone.0081355
2. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents with HIV. Department of Health and Human Services. Available from: https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/AdultandAdolescentGL.pdf.
3. Galli L, Parisi MR, Poli A, et al; PRESTIGIO Study Group. Burden of disease in PWH harboring a multidrug-resistant virus: data from the PRESTIGIO Registry. Open Forum Infect Dis. 2020;7(11):ofaa456. doi:10.1093/ofid/ofaa456
4. Hsu R, Fusco J, Henegar C, et al. Clinical outcomes of heavily treatment experienced individuals in the OPERA cohort.
5. Pancera M, Lai Y-T, Bylund T, et al. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat Chem Biol. 2017;13:1115–1122. doi:10.1038/nchembio.2460
6. US Food and Drug Administration. FDA approves new HIV treatment for patients with limited treatment options; 2020. Available from: https://www.fda.gov/news-events/press-announcements/fda-approvesnew-hiv-treatment-patients-limitedtreatment-options.
7. European medicines agency. EMA/CHMP/613669/2020. Rukobia (fostemsavir); 2020.
8. Meanwell NA, Krystal MR, Nowicka-Sans B, et al. Inhibitors of HIV-1 attachment: the discovery and development of temsavir and its prodrug fostemsavir. J Med Chem. 2018;61:62–80. doi:10.1021/acs.jmedchem.7b01337
9. Cahn P, Fink V, Patterson P. Fostemsavir: a new CD4 attachment inhibitor. Curr Opin HIV AIDS. 2018;13:341–345. doi:10.1097/COH.0000000000000469
10. Hanna GJ, Lalezari J, Hellinger JA, et al. Antiviral activity, pharmacokinetics, and safety of BMS-488043, a novel oral small-molecule HIV-1 attachment inhibitor, in HIV-1-infected subjects. Antimicrob Agents Chemother. 2011;55:722–728. doi:10.1128/AAC.00759-10
11. Wang T, Ueda Y, Connolly TP, et al. Use of a phosphonoxymethyl prodrug approach to successfully improve the oral delivery of HIV-1 attachment inhibitors: design, preclinical profile, and human exposure [abstract MEDI-346].
12. Nettles RE, Schürmann D, Zhu L, et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J Infect Dis. 2012;206(7):1002–1011. doi:10.1093/infdis/jis432
13. Lin PF, Blair W, Wang T, et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A. 2003;100(19):11013–11018. doi:10.1073/pnas.1832214100
14. Kadow JF, Ueda Y, Meanwell NA, et al. Inhibitors of human immunodeficiency virus type 1 (HIV-1) attachment 6. Preclinical and human pharmacokinetic profiling of BMS-663749, a phosphonooxymethyl prodrug of the HIV-1 attachment inhibitor 2-(4-benzoyl-1-piperazinyl)-1-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)-2-oxoethanone (BMS-488043). J Med Chem. 2012;55(5):2048–2056. doi:10.1021/jm201218m
15. Swidorski JJ, Liu Z, Yin Z, et al. Inhibitors of HIV-1 attachment: the discovery and structure-activity relationships of tetrahydroisoquinolines as replacements for the piperazine benzamide in the 3-glyoxylyl 6-azaindole pharmacophore. Bioorg Med Chem Lett. 2016;26(1):160–167. doi:10.1016/j.bmcl.2015.11.009
16. Brown J, Chien C, Timmins P, et al. Compartmental absorption modeling and site of absorption studies to determine feasibility of an extended-release formulation of an HIV-1 attachment inhibitor phosphate ester prodrug. J Pharm Sci. 2013;102(6):1742–1751. doi:10.1002/jps.23476
17. Food and Drug Administration: RUKOBIA (fostemsavir) extended-release tablets, for oral use. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212950s000lbl.pdf. Accessed on January 20, 2022.
18. Sevinsky H, Magee M, Ackerman P, et al. The effect of food on the pharmacokinetics of the HIV-1 attachment inhibitor temsavir, the active moiety of the prodrug fostemsavir.
19. Moore KP, Mageau AS, Magee M, Gorycki PD, Ackerman P, Llamoso C. 2500. Fostemsavir drug–drug interaction profile, an attachment inhibitor and oral prodrug of temsavir, for heavily treatment experienced HIV-1-infected patients. Open Forum Infect Dis. 2019;6(Suppl2):S867. doi:10.1093/ofid/ofz360.2178
20. Sevinsky H, Magee M, Ackerman P, et al. Pharmacokinetics of temsavir, the active moiety of the prodrug fostemsavir, in subjects with hepatic impairment. Open Forum Infect Dis. 2017;4(Suppl 1):S430. doi:10.1093/ofid/ofx163.1086
21. Hiryak K, Koren DE. Fostemsavir: a novel attachment inhibitor for patients with multidrug-resistant HIV-1 infection. Ann Pharmacother. 2021;55(6):792–797. doi:10.1177/1060028020962424
22. Nowicka-Sans B, Gong YF, McAuliffe B, et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob Agents Chemother. 2012;56(7):3498–3507. doi:10.1128/AAC.00426-12
23. Zhou N, Nowicka-Sans B, McAuliffe B, et al. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J Antimicrob Chemother. 2014;69:573–581. doi:10.1093/jac/dkt412
24. Bouba Y, Berno G, Fabeni L, et al. Identification of gp120 polymorphisms in HIV-1 B subtype potentially associated with resistance to fostemsavir. J Antimicrob Chemother. 2020;75:1778–1786. doi:10.1093/jac/dkaa073
25. Lepore L, Fabrizio C, Bavaro DF, et al. Gp120 substitutions at positions associated with resistance to fostemsavir in treatment-naive HIV-1-positive individuals. J Antimicrob Chemother. 2020;75:1580–1587. doi:10.1093/jac/dkaa034
26. Alessandri-Gradt E, Charpentier C, Leoz M, Mourez T, Descamps D, Plantier JC. Impact of natural polymorphisms of HIV-1 nongroup M on genotypic susceptibility to the attachment inhibitor fostemsavir. J Antimicrob Chemother. 2018;73:2716–2720. doi:10.1093/jac/dky271
27. Molina JM, Andrade-Villanueva J, Echevarria J, et al; CASTLE Study Team. Once-daily atazanavir/ritonavir compared with twice-daily lopinavir/ritonavir, each in combination with tenofovir and emtricitabine, for management of antiretroviral-naive HIV-1-infected patients: 96-week efficacy and safety results of the CASTLE study. J Acquir Immune Defic Syndr. 2010;53(3):323–332. doi:10.1097/QAI.0b013e3181c990bf
28. Zhou N, Nowicka-Sans B, Zhang S, et al. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob Agents Chemother. 2011;55(2):729–737. doi:10.1128/AAC.01173-10
29. Gartland M, Zhou N, Stewart E, et al. Susceptibility of global HIV-1 clinical isolates to fostemsavir using the PhenoSense® Entry assay. J Antimicrob Chemother. 2021;76:648–652. doi:10.1093/jac/dkaa474
30. Li Z, Zhou N, Sun Y, et al. Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors. Antimicrob Agents Chemother. 2013;57:4172–4180. doi:10.1128/AAC.00513-13
31. Lagishetty C, Moore K, Ackerman P, Llamoso C, Magee M. Effects of temsavir, active moiety of antiretroviral agent fostemsavir, on QT interval: results from a Phase I study and an exposure-response analysis. Clin Transl Sci. 2020;13:769–776. doi:10.1111/cts.12763
32. Lalezari JP, Latiff GH, Brinson C, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a Phase 2b, randomised controlled trial. Lancet HIV. 2015;2:e427–e437. doi:10.1016/S2352-3018(15)00177-0
33. Kozal M, Aberg J, Pialoux G, et al. Fostemsavir in adults with multidrug-resistant HIV-1 infection. N Engl J Med. 2020;382:1232–1243. doi:10.1056/NEJMoa1902493
34. Lataillade M, Lalezari JP, Kozal M, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug fostemsavir in heavily treatment-experienced individuals: week 96 results of the Phase 3 BRIGHTE study. Lancet HIV. 2020;7:e740–e751. doi:10.1016/S2352-3018(20)30240-X
35. Rukobia. Package insert. ViiV healthcare; 2020.
36. Ackerman P, Thompson M, Molina JM, et al. Long-term efficacy and safety of fostemsavir among subgroups of heavily treatment-experienced adults with HIV-1. AIDS. 2021;35:1061–1072. doi:10.1097/QAD.0000000000002851
37. Anderson SJ, Murray M, Cella D, et al. Patient-reported outcomes in the Phase III BRIGHTE trial of the HIV-1 attachment inhibitor prodrug fostemsavir in heavily treatment-experienced individuals. Patient. 2021;15:131–143. doi:10.1007/s40271-021-00534-y
38. Ronald R, Gartland M, Li Z, et al. Clinical evidence for a lack of cross-resistance between temsavir and ibalizumab or maraviroc. AIDS. 2021. doi:10.1097/QAD.0000000000003097
39. Saladini F, Giannini A, Giammarino F, et al. In vitro susceptibility to fostemsavir is not affected by long-term exposure to antiviral therapy in MDR HIV-1-infected patients. J Antimicrob Chemother. 2020;75(9):2547–2553. doi:10.1093/jac/dkaa178
40. Food and Drug Administration. Ibalizumab-UIYK. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=761065.
41. Molina JM, Segal-Maurer S, Stellbrink HJ, et al. Efficacy and safety of long-acting subcutaneous lenacapavir in phase 2/3 in heavily treatment-experienced people with HIV: week 26 results (Capella study). J Int AIDS Soc. 2021;24(S4):75.
42. Molina JM, Yazdanpanah Y, Afani Saud A, et al. Islatravir in combination with doravirine maintains HIV-1 viral suppression through 96 weeks. Ethnicity. 2020;24:77–74.
43. Li M, Liu W, Bauch T, et al. Clearance of HIV infection by selective elimination of host cells capable of producing HIV. Nat Commun. 2020;11(1):4051. doi:10.1038/s41467-020-17753-w
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.