")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Efficacy and safety of the CRTh2 antagonist AZD1981 as add-on therapy to inhaled corticosteroids and long-acting β2-agonists in patients with atopic asthma
Authors Bateman ED, O'Brien C, Rugman P, Luke S, Ivanov S, Uddin M
Received 26 July 2017
Accepted for publication 13 December 2017
Published 4 May 2018 Volume 2018:12 Pages 1093—1106
DOI https://doi.org/10.2147/DDDT.S147389
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Eric D Bateman,1 Christopher O’Brien,2 Paul Rugman,2 Sally Luke,2 Stefan Ivanov,2 Mohib Uddin2,3
1Department of Medicine, University of Cape Town, Cape Town, 7700, South Africa; 2Research and Development, 3Respiratory, Inflammation, and Autoimmunity, IMED Biotech Unit, AstraZeneca, Gothenburg, SE-431 83, Sweden
Objectives: To evaluate the efficacy and safety of AZD1981, a potent, specific antagonist of the CRTh2 receptor, as add-on therapy to inhaled corticosteroids (ICS) and long-acting β2-agonists (LABA), in patients with persistent asthma with an allergic component.
Patients and methods: In this placebo-controlled, parallel-group Phase IIb study, patients with persistent atopic asthma on ICS and LABA were randomized to receive 12 weeks of treatment with placebo or AZD1981 (80 mg daily, 200 mg daily, and 10 mg, 40 mg, 100 mg, or 400 mg twice daily [BID]). The primary end point was the mean change from baseline in predose, prebronchodilator forced expiratory volume in 1 second (FEV1) averaged over weeks 2, 4, 8, and 12 in the AZD1981-treatment group vs the placebo group. Secondary end points included other measures of lung function, symptoms, and asthma control, as well as standard measures of safety.
Results: In total, 1,140 patients (99.7%) received study treatment. There were improvements in the primary end point across all treatment groups over 12 weeks of treatment. However, the improvement for the highest AZD1981 dose (400 mg BID) vs placebo was not statistically significant (0.02 L, P=0.58), preventing interpretation of statistical testing for the lower doses. AZD1981 was well tolerated, and the incidence of adverse events was comparable across placebo and treatment groups.
Conclusion: In patients with allergic asthma receiving ICS and LABA therapy, the addition of AZD1981 at doses up to 400 mg BID failed to produce a clinically relevant improvement in lung function or any other measured end point, but appeared to have an acceptable safety profile. This clinical study is registered with ClinicalTrials.gov (NCT01197794).
Keywords: allergic asthma, AZD1981, CRTh2 antagonist, efficacy, safety, eosinophils
Plain-language summary
Why was the study done? CRTh2 is a receptor expressed on immune cells and has a role in driving allergic airway inflammation. Selective CRTh2 antagonism may counteract key allergic/inflammatory pathways in asthma, thereby limiting airway inflammation and potentially improving asthma control.
What did the researchers do and find? This Phase IIb study was conducted to evaluate the dose–response relationship for AZD1981, a selective CRTh2 antagonist, as an add-on to standard treatment for patients with atopic asthma. A total of 1,140 patients received either placebo or AZD1981 treatment (80 or 200 mg once daily (QD), or 10, 40, 100, or 400 mg twice daily (BID) for 12 weeks), in addition to standard treatment. Changes in lung function for the highest dose (400 mg BID) were not statistically significant at 12 weeks, so it was concluded that nothing could be inferred from the lower-dose groups. However, improvements were observed for time to treatment failure and other secondary outcomes, but with no clear dose response. AZD1981 was safe and well tolerated across the treatment groups.
What do these results mean? Treatment with AZD1981 did not result in any clinically significant effects on lung function or other outcome measures in any of the study population.
Introduction
Asthma is characterized by increased and persistent airway inflammation that often has an allergic component.1 International treatment guidelines, such as those from the Global Initiative for Asthma,2 recommend an inhaled corticosteroid (ICS) as first-line therapy for persistent asthma, with the addition of a long-acting β2-agonist (LABA) as the preferred second controller for patients whose asthma remains uncontrolled on the ICS alone. However, a substantial number of patients who have severe and/or persistent forms of asthma remain symptomatic, despite this combined treatment. Novel therapies are thus needed.3
One potential therapeutic option is the use of selective antagonists to block the chemotactic receptor CRTh2.3,4 CRTh2 is a G-protein-coupled receptor expressed on eosinophils, basophils, mast cells and T-helper (Th)-2 cells,5 as well as on the recently identified group 2 innate lymphoid cells (ILC2s),6 and CRTh2 is activated by the proinflammatory mediator prostaglandin D2 (PGD2). PGD2 is a major arachidonic acid metabolite released by airway mast cells following allergen challenge.4 Mast-cell activation and degranulation occurs in the initial phase of IgE-mediated reactions (eg, in response to allergen), and PGD2 acts as an important mediator of allergic inflammation in asthma.7,8 This prostanoid acts locally to stimulate the activation and chemotaxis of eosinophils, basophils, Th2 cells,5,8 and ILC2s9 that have the capacity to release Th2-type cytokines locally.10 Importantly, PGD2 levels and expression of the CRTh2 receptor are elevated in the airways of patients in relation to asthma disease severity,11,12 and this PGD2–CRTh2 axis may be involved in mediating allergic airway-inflammatory responses, despite ICS treatment. Uncontrolled accumulation and activation of immune cells in a PGD2–CRTh2-dependent manner may thus play an important pathobiological role in the allergic airway inflammation that typifies asthma.4 In support of this hypothesis, CRTh2 antagonism has been reported to reduce allergic airway inflammation in murine models significantly,13–15 inhibit allergic inflammation in steroid-naïve patients with asthma,8,16–18 and block eosinophilic airway inflammation in patients with persistent moderate–severe asthma on CS treatment.19
CRTh2 antagonists represent a promising new class of therapeutic agents under clinical development for the treatment of asthma.8,19–21 AZD1981 (AstraZeneca, Gothenburg, Sweden) is a potent, orally active, selective CRTh2 antagonist that has been shown consistently to block CRTh2-driven eosinophil and Th2 responses in human cell systems, and is thus a candidate for oral nonsteroidal asthma therapy.18,22 The clinical potential of AZD1981 has been evaluated in two randomized, placebo-controlled trials in asthma.23 In one, patients had their ICS treatment withdrawn prior to run-in, and in the second, three doses of AZD1981 were investigated in patients on maintenance-ICS treatment.23 The primary efficacy end point in both trials was morning peak expiratory flow after 4 weeks of treatment. In both studies, there was a nonsignificant difference in the primary end point in favor of AZD1981. In the latter study, AZD1981 was associated with a small improvement in forced expiratory volume in 1 second (FEV1) and daily asthma control by improvement in Asthma Control Questionnaire (ACQ)-5 score. In a post hoc analysis, these improvements appeared to be more pronounced in atopic patients compared with nonatopic patients, consistent with the mechanism of action for AZD1981.23
Based on these observations, we performed a randomized, placebo-controlled study to examine the efficacy and safety of AZD1981 at different doses and dose frequencies in uncontrolled patients who screened positive for specific IgE to common inhaled allergens, suggesting an allergic component to their asthma and involvement of CRTh2-driven mechanisms. In order to assess benefit and tolerability in a patient population with significant unmet need, AZD1981 was evaluated in patients symptomatic on ICS–LABA combination therapy.
Patients and methods
Study design
This was a Phase IIb, 12-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study conducted in Europe, South and North America, Japan, and South Africa (protocol D9830C00008, ClinicalTrials.gov identifier NCT01197794). The study evaluated four AZD1981 BID oral regimens (10, 40, 100, and 400 mg BID) and two AZD1981 QD oral regimens (80 and 200 mg/day) vs placebo over 12 weeks of treatment in patients with asthma also receiving the ICS budesonide and the LABA formoterol (Bud/Form 80/4.5 μg, Symbicort® pressured metered-dose inhaler, two inhalations BID throughout the study; AstraZeneca, Gothenburg, Sweden) (Figure 1). Eligible patients entered a 2-week run-in period, and those fulfilling the randomization criteria began treatment with oral tablets of AZD1981 or matching placebo, in addition to their maintenance therapy with Bud/Form 80/4.5 μg. The study was approved by local review boards/ethics committees (Table S1), and was conducted in accordance with the Declaration of Helsinki 2008 and Good Clinical Practice (GCP) guidelines.
 | Figure 1 Study design. |
Patients
Eligible patients were men and women aged ≥18 years with a documented history of persistent asthma (according to the definition of the American Thoracic Society) of ≥6 months’ duration treated with a combination of ICS–LABA for ≥3 months before the run-in period. Standard ICS doses of ≤500 μg of budesonide, fluticasone, ciclesonide, mometasone, and hydrofluoroalkane beclometasone, 1,000 μg of beclometasone dipropionate, or other ICS at equivalent doses, were permitted if on a stable regimen for ≥4 weeks before the run-in period. Other major inclusion criteria were prebronchodilator FEV1 of 40%–85% of predicted normal,24 reversible airway obstruction (≥12% increase in postbronchodilator FEV1), atopy confirmed by a positive Phadiatop test (Phadia AB, Uppsala, Sweden),25 and a total ACQ5 score ≥1.5.26 Major exclusion criteria included any respiratory infection significantly affecting asthma, a smoking history >10 pack years, and any clinically relevant medical condition/abnormal finding. Oral or parenteral CS (as well as other anti-asthma treatments) were not permitted, except for severe asthma exacerbations. Initially, only postmenopausal women were allowed to participate in the study. However, following a protocol amendment, women of childbearing potential were also permitted, provided they used a double-barrier method of contraception. All patients gave written informed consent prior to study participation.
Treatment
After a 2-week run-in period, eligible patients were randomly allocated in a 1:1:1:1:1:1:1 ratio to receive one of seven treatments: AZD1981 10 mg BID, 40 mg BID, 100 mg BID, 400 mg BID, 80 mg QD, 200 mg QD, or placebo. Eligible patients were randomized using a randomization list prepared by a validated computerized system at the study sponsor, and randomization was done in blocks of seven. Unique randomization codes were assigned strictly sequentially by AstraZeneca’s GRand system.
All patients took four tablets of AZD1981 and/or placebo in the morning and four tablets in the evening. The combination of tablets dispensed for each treatment arm is shown in Figure 2. In addition, all patients continued their maintenance treatment with two inhalations BID of Bud/Form 80/4.5 μg throughout the study and were permitted to use any inhaled short-acting β2-agonist (SABA) as reliever medication. Packaging and labeling ensured that study staff and patients were blinded to treatment with AZD1981 or placebo.
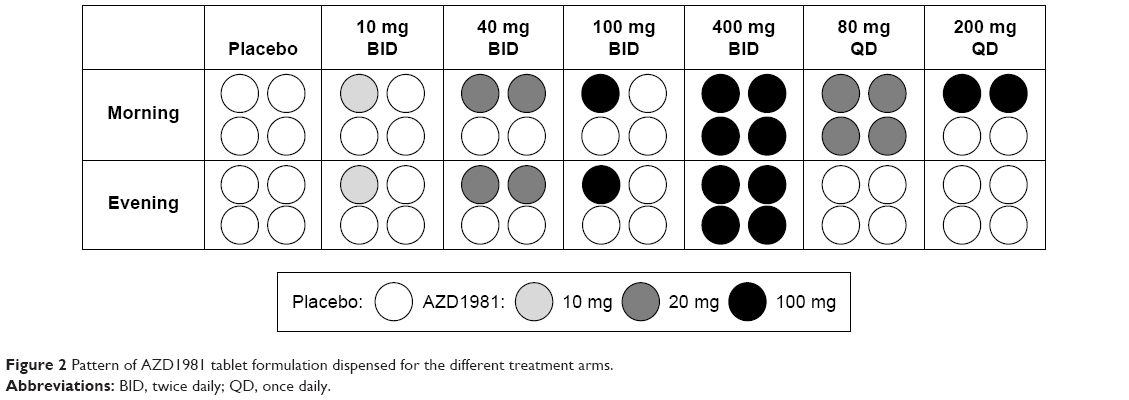 | Figure 2 Pattern of AZD1981 tablet formulation dispensed for the different treatment arms. |
End-point variables
The primary end point was the mean change from baseline in predose, prebronchodilator FEV1 averaged over weeks 2, 4, 8, and 12 in the AZD1981-treatment group vs the placebo group.27 Secondary end points included postbronchodilator FEV1, pre- and postbronchodilator forced vital capacity (FVC), time to treatment failure, and time to severe asthma exacerbation. A treatment failure was defined as the clinical need for additional ICS, as judged by the investigator and based on evaluations at clinic visits. A severe asthma exacerbation was defined as deterioration in asthma leading to oral glucocorticoid (GC) treatment for ≥3 days, as judged by the investigator and based on evaluations at the clinic and/or hospitalization or emergency-room treatment due to asthma. Patient-reported secondary efficacy end points were self-evaluated ACQ5 score and the Asthma Quality of Life Questionnaire, standardized version (AQLQ-S).28
Secondary efficacy variables, recorded in a daily electronic diary (eDiary) by the patients or derived from the eDiary recordings, included morning and evening peak expiratory flow (PEF) and FEV1, patient-assessed asthma symptoms, including total symptom score, daily and total SABA-reliever use, nights with awakenings due to asthma symptoms, number of reliever-free days, number of asthma control-free days, and number of symptom-free days.
Throughout the study, adverse events (AEs) and serious AEs (SAEs) were recorded, including their severity and their putative relationship to the study drug. Other safety evaluations included physical examinations, electrocardiography monitoring, predetermined blinded laboratory monitoring (including transaminases and total bilirubin), and assessment of vital signs.
Assessments
Clinic spirometric assessments during the active-treatment phase were performed within a 1-hour window for each study visit. Patients refrained from using their reliever medication for at least 6 hours before each evaluation. In addition, before visit 2 (reversibility screening), patients also abstained from LABA use for ≥48 hours before the clinic visit, and before visits 3–7 patients refrained from taking their ICS- and LABA-maintenance treatment within 12 hours of the clinic measurements. Postbronchodilator spirometry in the clinic was performed within 15–30 minutes of bronchodilator use.
ACQ5 and AQLQ scores were self-evaluated during clinic visits 2–7 and entered in the eDiary. An ACQ5 responder was defined as a patient with a decrease of ≥0.5 units from baseline to the end of treatment. An AQLQ responder was defined as a patient with an increase of ≥0.5 from baseline to the end of treatment. Patients completed the eDiary during the run-in and treatment periods (visits 2–7).
Sample size and statistical analysis
In the absence of previous data with AZD1981 with Bud/Form as background therapy, a standard deviation for change from baseline in FEV1 was conservatively estimated at 420 mL. Therefore, for a one-sided test at the 5% significance level, a total sample size of 1,120 patients would have 80% power to demonstrate an effect if the true group difference was 117 mL. For a two-sided test, the corresponding value was 131 mL. For the ACQ, the standard deviation was assumed to be 0.7 units, and the group difference was 0.2 units for a one-sided test at the 5% level and 0.22 units for a two-sided test. Group differences were calculated vs placebo.
Efficacy analyses were performed on the full-analysis set, consisting of all randomized patients who took at least one dose of AZD1981 and for whom postdose data were available. Most continuous end-point variables were analyzed with an analysis of covariance model with treatment and country as factors and baseline measurement as covariate. Time to first treatment failure and time to first severe asthma exacerbation were analyzed by a Cox proportional-hazard model with treatment as factor. Hierarchical statistical testing was prespecified, starting with the highest-dose comparison for the primary end point. Subsequent testing for lower AZD1981 doses was valid only if the initial comparison was found to be statistically significant, although nominal P-values are provided for descriptive purposes. Safety analyses were carried out on the safety-analysis set, which included all randomized patients who took at least one dose of study medication and for whom any data after randomization were available. This included patients affected by GCP violations.
An exploratory post hoc analysis was performed to examine the relationship between features considered to be markers of the Th2-high phenotype and clinical responses. In this analysis, patients were considered to be of a Th2 phenotype if their blood eosinophil count was ≥0.14×109 cells/L and total serum IgE >100 kU/L.29 In addition, blood eosinophil counts and serum total IgE levels were compared to the clinical end points for each of the treatment groups.
Results
Patients
The first patient was enrolled on October 19, 2010, and the last patient completed the study on February 16, 2012. A total of 2,082 patients were enrolled, 1,144 of whom were randomized to treatment (1,140 received treatment, with 1,034 patients completing the study) (Figure 3). The mean age was 46 years, 51% of patients were male, 72% were white, and the mean time since the diagnosis of asthma was 18 years (Table 1). Baseline demographic data and disease characteristics were well balanced across the treatment groups. Three patients were excluded from the efficacy analyses, due to GCP violations (Figure 3).
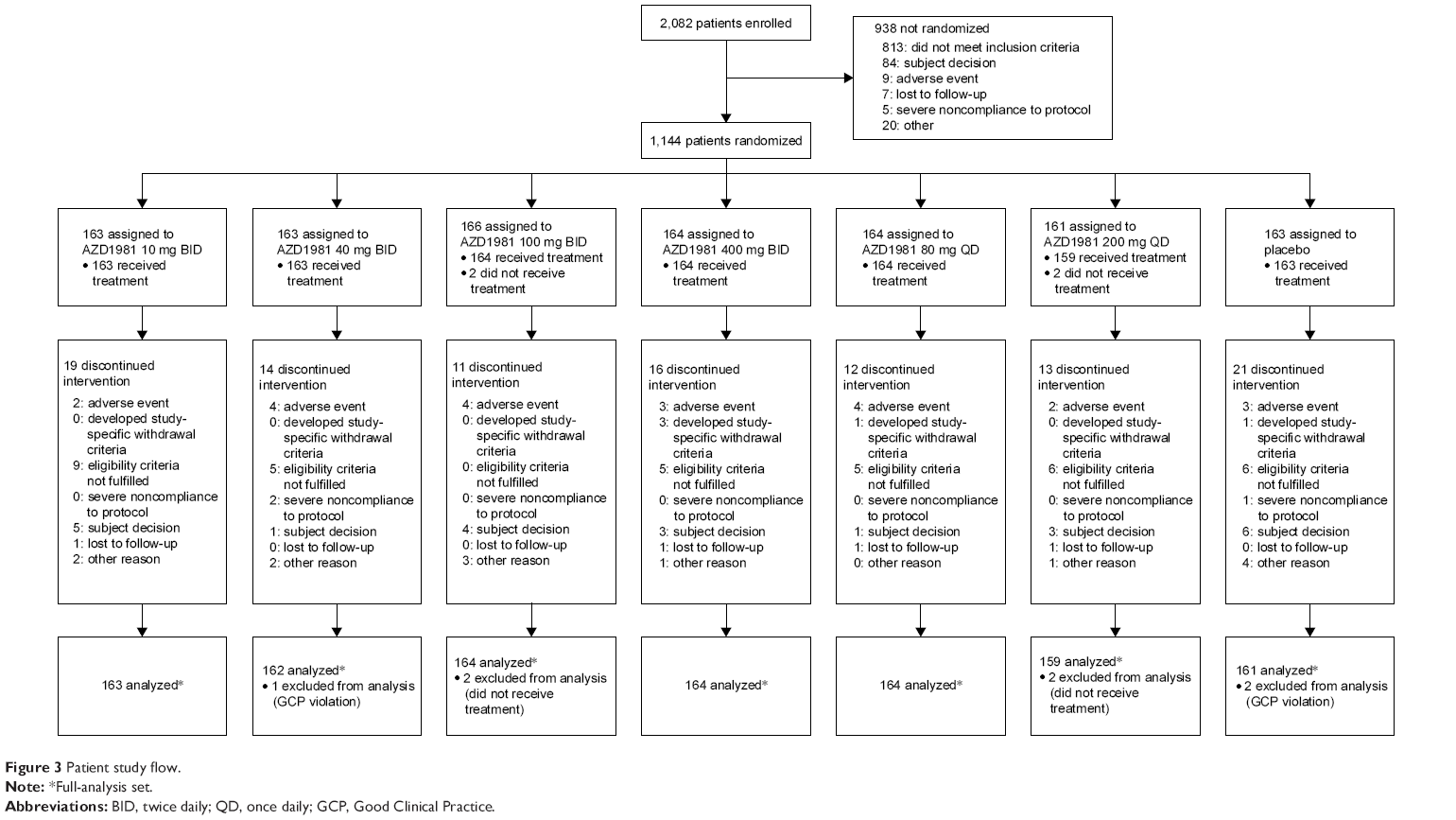 | Figure 3 Patient study flow. |
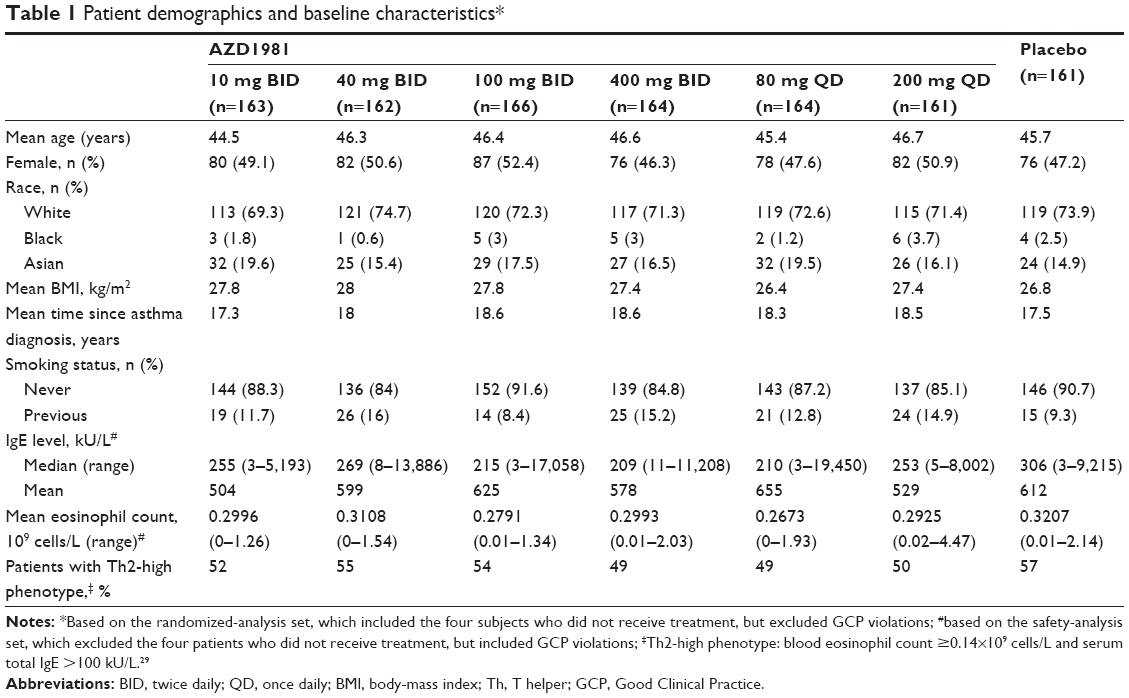 | Table 1 Patient demographics and baseline characteristics* |
Compliance
During the study, compliance with study treatment (based on tablet counts), and eDiary completion (defined as two eDiary entries for each day the patient was on treatment) was high (96.5% and 88.9% of patients, respectively, were ≥80% compliant), and this was similar across treatment groups.
Primary efficacy analysis
An improvement in mean change from baseline predose, prebronchodilator FEV1 values over the treatment period was seen across all treatment groups, with values ranging from 0.14 L for placebo (95% CI 0.08–0.19) to 0.2 L for AZD1981 40 mg BID (95% CI 0.15–0.26) (Figure 4A). There was an improvement in the placebo-adjusted change from baseline in treatment-average prebronchodilator FEV1, ranging from 0.02 L with AZD1981 400 mg BID to 0.07 L with AZD1981 40 mg BID. However, the improvement in mean change from baseline predose, prebronchodilator FEV1 value observed with AZD1981 400 mg BID was not statistically significantly different vs placebo (least-square mean difference 0.02 L, 95% CI −0.06 to 0.1; P=0.58), preventing interpretation of statistical testing for the lower doses. No dose–response relationship was observed across the active-treatment groups for predose, prebronchodilator FEV1. The largest treatment difference was observed in the AZD1981 40 mg BID group compared with placebo (least-square mean difference 0.07, 95% CI −0.01 to 0.14). Similarly, there was no statistically significant difference in the effect of QD vs BID dosing for the primary end point (least-square mean difference for 200 mg QD vs 100 mg BID 0, 95% CI −0.07 to 0.08, P=0.91; least-square mean difference for 80 mg QD vs 40 mg BID −0.05, 95% CI −0.13 to 0.03, P=0.215).
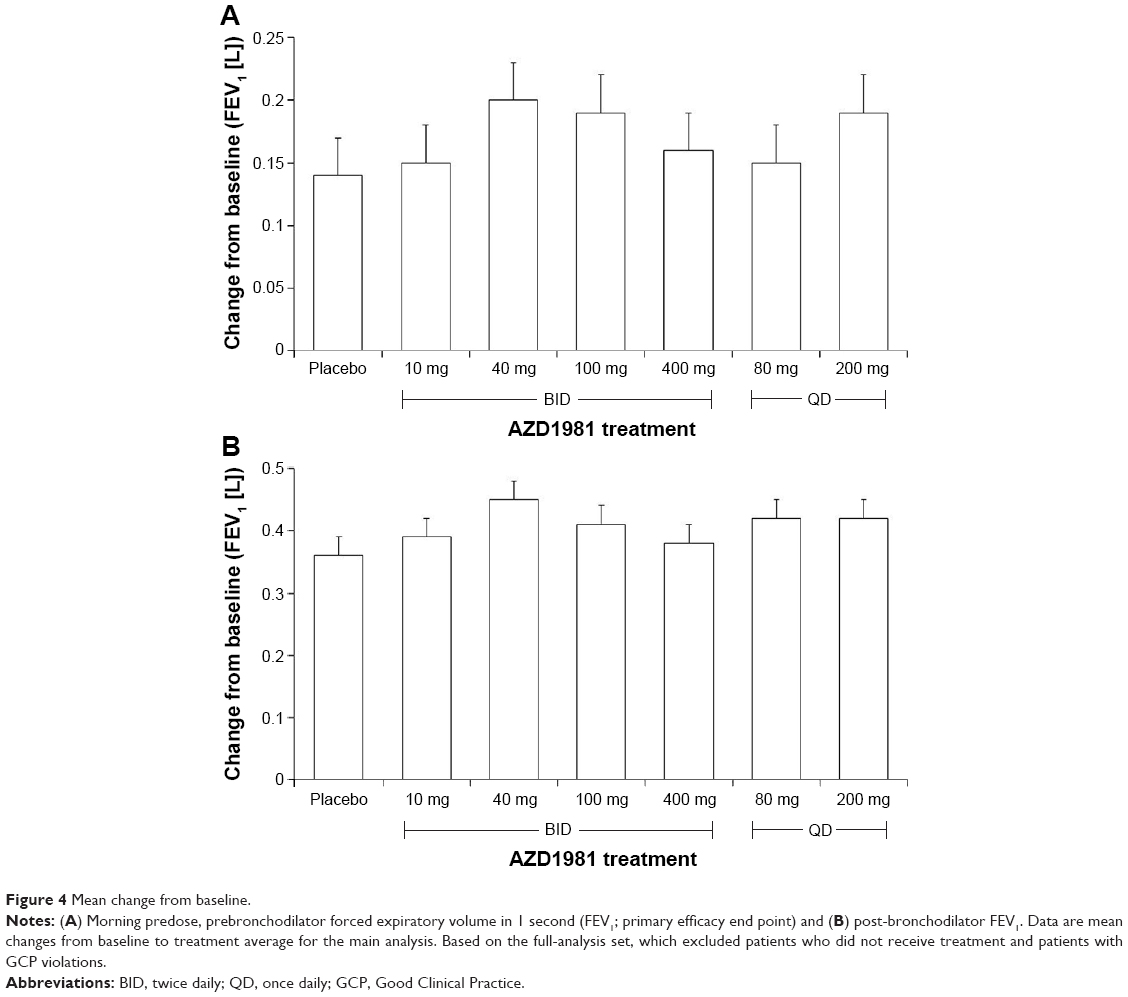 | Figure 4 Mean change from baseline. |
Secondary end points
Lung function
An improvement in mean change from baseline postbronchodilator FEV1 measurements over the treatment period average was seen in all treatment groups, with values ranging from 0.36 L for placebo (95% CI 0.3–0.41) to 0.45 L for AZD1981 40 mg BID (95% CI 0.4–0.5) (Figure 4B). Improvements in mean average change from baseline pre- and postbronchodilator FVC measurements over the treatment period were seen in all treatment groups. For prebronchodilator FVC, values ranged from 0.13 L for placebo (95% CI 0.06–0.21) to 0.2 L for AZD1981 100 mg BID (95% CI 0.12–0.27). For postbronchodilator FVC, values ranged from 0.34 L for placebo (95% CI 0.27–0.4) to 0.42 L for AZD1981 40 mg BID (95% CI 0.35–0.49). No dose–response relationship was observed across the treatment groups for pre- or postbronchodilator FVC.
Patient-reported outcomes: ACQ5 and AQLQ-S scores
Compared with placebo, AZD1981 had no significant effect on ACQ5 score (Figure 5). The proportion of patients with an ACQ5 response (defined as a decrease of ≥0.5 units from baseline to the end of treatment) ranged from 61% in the placebo group to ~70% in three of the AZD1981 treatment groups (200 mg QD, 80 mg QD, and 100 mg BID). Similarly, improvements were observed in all treatment groups in AQLQ-S scores, with response rates (defined as an increase of ≥0.5 from baseline to the end of treatment) ranging from 52% for placebo to 63% for AZD1981 100 mg BID.
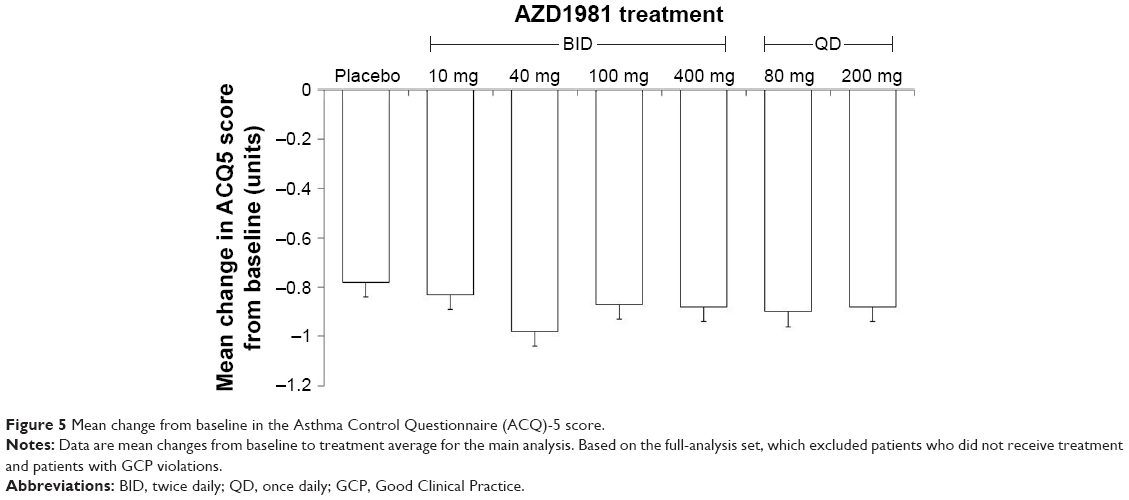 | Figure 5 Mean change from baseline in the Asthma Control Questionnaire (ACQ)-5 score. |
eDiary and eDiary-derived end points
Compared with placebo, AZD1981 had no significant effect on lung-function parameters assessed at home vs placebo, including morning and evening PEF and FEV1 (Table 2), patient-assessed total symptom score, total daily SABA-reliever use, and the percentage of reliever-free days, asthma-control days, and incidence of symptom-free days. For all these secondary end points, there was no evidence of a dose–response relationship.
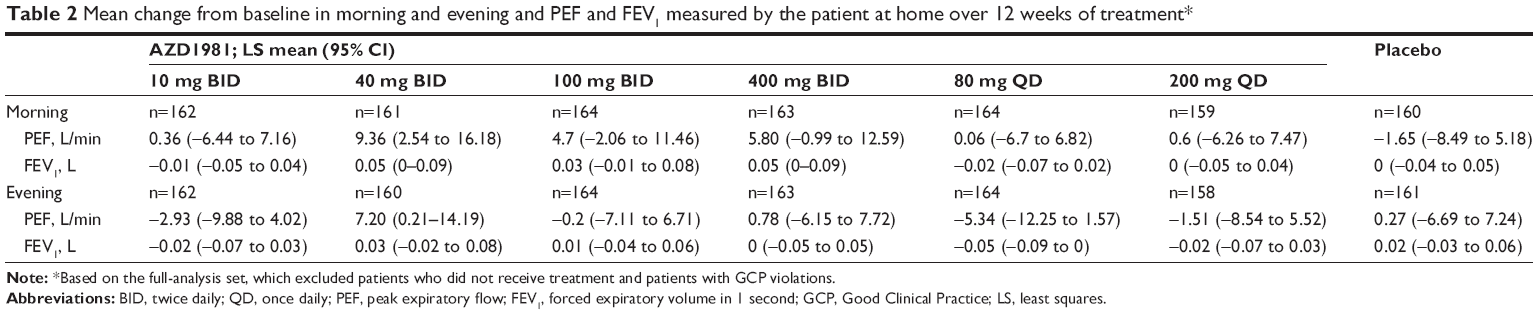 | Table 2 Mean change from baseline in morning and evening and PEF and FEV1 measured by the patient at home over 12 weeks of treatment* |
Severe asthma exacerbations
The number of patients with one or more severe asthma exacerbations was low across the treatment groups: six (3.7%), five (3.1%), seven (4.3%), four (2.4%), seven (4.3%), three (1.9%), and five (3.1%) in the AZD1981 10 mg BID, 40 mg BID, 100 mg BID, 400 mg BID, 80 mg QD, 200 mg QD, and placebo groups, respectively. Patients typically experienced a single event during the treatment period, with few patients experiencing two events; these occurred in the AZD1981 200 mg QD (one patient), AZD1981 80 mg QD (two patients), and placebo (two patients) groups. Cox regression analysis revealed no statistically significant difference in the time to first severe asthma exacerbation between any of the AZD1981-treatment groups relative to placebo.
Time to treatment failure
The number of patients with a protocol-defined treatment failure was low across the treatment groups: 12 (7.4%), ten (6.2%), three (1.8%), four (2.4%), eight (4.9%), nine (5.7%), and 12 (7.5%) in the AZD1981 10 mg BID, 40 mg BID, 100 mg BID, 400 mg BID, 80 mg QD, 200 mg QD, and placebo groups, respectively. Analysis of time to first treatment failure generally showed no difference between AZD1981 and placebo; however, compared with placebo, the risk of an event was significantly lower for the AZD1981 400 mg BID and the AZD1981 100 mg BID groups (Table 3).
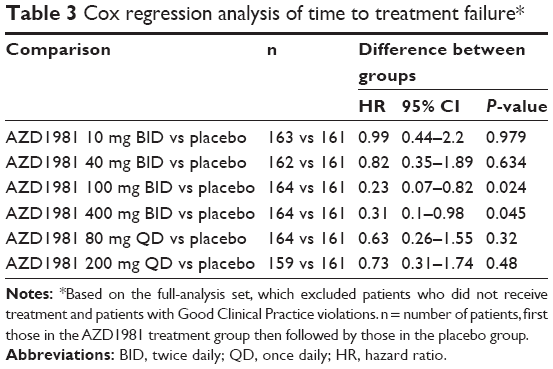 | Table 3 Cox regression analysis of time to treatment failure* |
Analysis of Th2 phenotype and clinical responses
Among the active-treatment groups the 40 mg-treatment group had both the highest mean baseline eosinophil count (0.31×109 cells/L [SD 0.2565]) and median total IgE level (269 kU/L) and exhibited the greatest responses to AZD1981 treatment for several asthma-control measures, which prompted a post hoc analysis of results based on the level of Th2-driven immunity. Across treatment groups, 52% (562) patients were categorized as Th2 high and 48% (513) as Th2 low (Table 1). However, in different treatment groups, patients characterized as Th2 high did not consistently demonstrate greater improvements in predose, prebronchodilator FEV1 responses compared to Th2-low patients, and neither subgroup demonstrated a consistent dose response (data not shown).
Safety and tolerability
The incidence of AEs was similar across all treatment groups, with the AE profile for AZD1981 similar to that of placebo (Table 4). Most AEs were mild or moderate in severity. Only a small proportion of patients treated with AZD1981 or placebo experienced SAEs during the study. AEs and SAEs leading to study-drug discontinuation were low in number and consistent across treatment groups. The incidence of AEs that were judged by the investigator to be drug-related was low, with the highest incidence in the AZD1981 400 mg BID group. There were no deaths during the study.
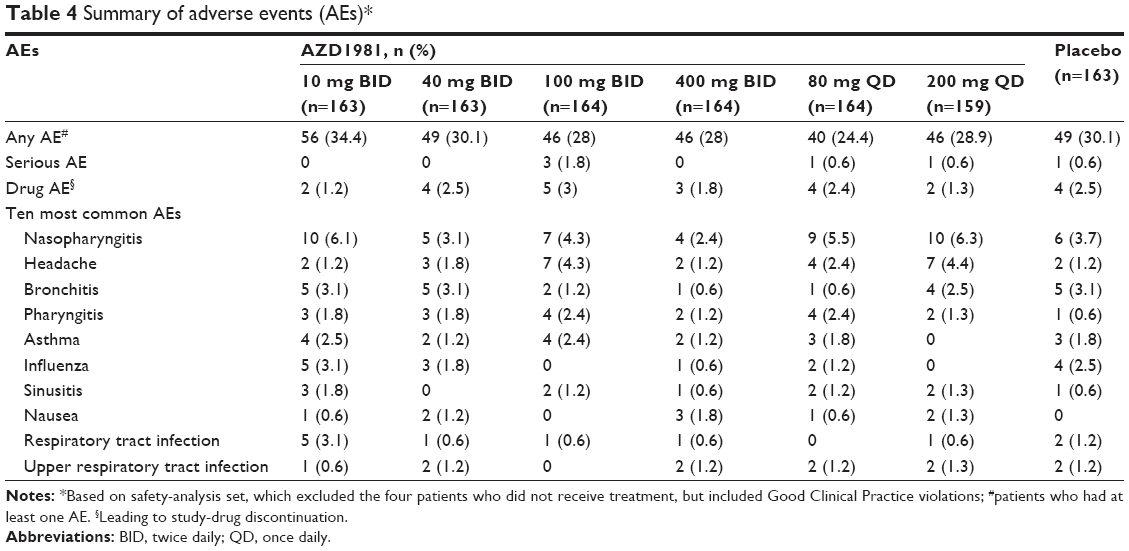 | Table 4 Summary of adverse events (AEs)* |
Compared with placebo, there were no clinically relevant changes with regard to clinical laboratory assessments, physical examination, electrocardiography, or vital signs. A small proportion of patients had levels of alanine aminotransferase or aspartate aminotransferase ≥3× the upper limit of normal (ULN), the highest proportion of which occurred with AZD1981 400 mg BID. There were four cases of transaminases elevated to ≥3× ULN or total bilirubin ≥2× ULN that may have been related to the study drug (one patient treated with AZD1981 80 mg QD, one patient treated with AZD1981 100 mg BID, and two patients treated with AZD1981 400 mg BID). Transaminase levels returned to baseline values after AZD1981 was stopped.
Discussion
This randomized, placebo-controlled, parallel-group Phase IIb study evaluated the efficacy and safety of add-on treatment with AZD1981, a selective orally active CRTh2 antagonist, in patients who were symptomatic despite maintenance therapy with ICS–LABA. Only asthma patients with evidence of atopy defined by a positive Phadiatop test were included in the study, as it was expected that they would have allergic airway inflammation as a component of their asthma and would show greater improvements with AZD1981 than excluded nonatopic patients. At the highest of the AZD1981 doses tested (400 mg BID), no statistically significant effect was observed vs placebo in the primary efficacy end point (mean change from baseline in predose, prebronchodilator FEV1). Similarly, results for the secondary variables, including postbronchodilator FEV1, pre- and postbronchodilator FVC, patient-assessed parameters (ACQ5, AQLQ-S, and total asthma-symptom scores), and PEF and FEV1 measured at home were not significantly different from those for placebo. For most measures, the largest treatment difference from placebo was observed in the AZD1981 40 mg BID group, but no consistent dose–response effects were observed. In general, the smallest differences between placebo and AZD1981 across different variables were in the 10 mg BID group, but since the prespecified statistical testing procedure failed at the highest dose comparison for predose, prebronchodilator FEV1, definitive conclusions could not be drawn from the lower-dose arms. AZD1981 was generally well tolerated. With the exception of elevated transaminase values in a few patients (that returned to baseline after AZD1981 had been stopped), the AE profile was similar to that of placebo. These safety results are consistent with the findings of previous clinical studies with AZD1981.23
Reasons for the failure of AZD1981 to demonstrate clinical efficacy in our study could relate to the drug (potency, dose, and posology) or the inclusion of a study population (eg, patients on ICS–LABA) in which CRTh2 mechanisms may be less clinically relevant for measures of lung function or signs and symptoms (eg, ACQ5). The study of CRTh2-mediated mechanisms in airway diseases has been limited by the absence of a measurable biomarker that reflects their presence and activity. This deficiency has affected both the evaluation and selection of candidate CRTh2-antagonist drugs and patients in whom to test them. Currently, the most widely used surrogate for CRTh2 activity is inhibition of PGD2-induced shape change of peripheral blood eosinophils ex vivo.30 This biomarker has recently been used in pharmacokinetic/pharmacodynamic models to identify doses and dosing regimens leading to 90% of maximum blockade of CRTh2 at trough by two selective and potent CRTh2 antagonists currently in clinical development: ACT453859 (Actelion Pharmaceuticals) and setipiprant.7 A sustained target pharmacodynamic-effect level of 90% CRTh2 blockade was defined as clinically relevant.
There is a growing body of information on CRTh2 antagonists and molecules with dual antagonism of CRTh2 and human D-prostanoid receptors in allergic asthma.7,17,19,20,23,31–33 These entities have been shown to inhibit allergic inflammation in asthma patients subjected to bronchial antigens,16 and the CRTh2 antagonist OC000459 (Oxagen) has been reported to improve lung function and symptoms in a placebo-controlled trial conducted in steroid-naïve patients with moderate persistent asthma.17 OC000459 treatment was also associated with a reduction in sputum eosinophils (geometric mean). In addition, there was a trend (albeit nonsignificant) of decrement in serum total IgE. In studies evaluating the CRTh2 antagonist BI671800 (Boehringer Ingelheim), small improvements in spirometry were observed in steroid-naïve patients, but these were inferior to those achieved with low-dose fluticasone propionate.8,33 When added to inhaled fluticasone (88 μg BID) in adult patients with symptomatic asthma, BI671800 (400 mg) resulted in nonsignificant improvements in trough FEV1% predicted, but no statistically or clinically significant changes in Asthma Control Test score, after 4 weeks of treatment.34 In a further study in patients receiving low-dose ICS, small improvements in lung function and asthma control similar to or numerically greater than that achieved with montelukast were seen.32 In contrast to these results, a recent study of AMG853 (Amgen) a small-molecule dual antagonist of the human D-prostanoid and CRTh2 receptors, with ACQ as the primary end point, reported negative findings.31
An initial concern in studies with weak or negative results is lack of potency or suboptimal dosing. In an attempt to avoid the latter, this study used a wide range of doses and both QD and BID dosing regimens, which on best evidence should have provided sufficient drug exposure. However, the AZD1981 80 mg QD-regimen results for the primary and secondary lung-function parameters were numerically smaller than those for 40 mg BID, and consistently with this, trough plasma concentrations of AZD1981 were lower for the QD regimen compared with the BID regimen at the same total daily dose (data not shown). These findings suggest that QD dosing of AZD1981 may not have been optimal. Increased potency and prolonged residence time at CRTh2 receptors are key pharmacological properties actively being sought by other pharmaceutical companies, as it is believed this will improve the clinical efficacy of CRTh2 antagonists in asthma.8,35,36
Selection of the optimal target-patient group in whom Th2-driven allergic airway inflammation is predominant has been approached differently in various studies. For example, studies examining the efficacy of ICS or targeted Th2-cytokine inhibition differentiated their target populations according to specific Th2-associated biomarkers.29,37–40 Predictive biomarkers employed include the degree of atopy in terms of number of positive skin tests, total IgE, airway or peripheral blood eosinophilia, fractional excretion of nitric oxide, and certain anti-IL13-inducible gene products. Notably, the proportion of asthmatic patients with the Th2-high phenotype (blood eosinophil count ≥0.14×109 cells/L and total serum IgE >100 kU/L) constituted approximately 50% of the total patient population who completed this study. This is similar to the proportion of patients with mild–moderate asthma characterized by degree of activation of Th2-inflammatory pathways.40
Emerging studies support the concept of a responding-patient phenotype defined by targeting of immunopathways that extend beyond simple allergic sensitization.29,37–40 Importantly, while atopic sensitization may serve to identify a potentially susceptible population, atopic status in and of itself has not consistently predicted efficacy in a number of studies of Th2-directed therapies.
Busse et al reported that a subgroup of asthmatics with blood eosinophilia >6% were more responsive to AMG853.31 Sputum eosinophilia of >2% has been used as a primary inclusion criterion in a current trial of a CRTh2 antagonist, QAW039 (now fevipiprant; Novartis), in patients with persistent asthma.19,32 In this study, all patients were positive for allergen-specific IgE by Phadiatop, with mean eosinophil numbers 0.267–0.321×109 cells/L (Table 1), and median total serum IgE 209–306 kU/L (Table 1). The post hoc analysis of group responses according to Th2 status failed to account for the negative results, and did not suggest a consistent differential benefit between Th2-high and Th2-low subjects vs placebo across doses or a dose response within the Th2-high subgroup.
Nevertheless, the importance of patient stratification has been illustrated in recent clinical studies assessing lung function in several predefined subgroups with elevated sputum eosinophil count (≥2%),19 blood eosinophilia (≥250 cells/μL),8 positive skin-prick tests,21 and more severe disease (baseline FEV1 <70%).20 Along these lines, we speculate that patients with more severe asthma (defined both by baseline measures of impairment and prior asthma treatment) with an eosinophil-dominant phenotype are most likely to respond to CRTh2 antagonists and the potential positioning of this Th2-directed nonbiological therapy. A more recent study showed that QAW039 improved lung function and asthma control in a subset of patients with greater severity of airflow limitation (FEV1 <70%), but had no significant effect in the general study population.20 These were patients with mild–moderate uncontrolled allergic asthma (n=170) who were either without or weaned off ICS and LABA. Further, in a dose-ranging study of fevipiprant, significant improvements in predose FEV1 were observed, albeit similar to those achieved with montelukast in patients with allergic asthma uncontrolled by low-dose ICS.41
Our study enrolled patients with inadequately controlled moderate–severe asthma with an FEV1 40%–85% of predicted normal during maintenance ICS–LABA therapy. Patients enrolled in the OC000459 study had an FEV1 that was 60%–80% of predicted normal. Therefore, in both studies, there was room for improvement in FEV1. The significant difference vs placebo for risk of an event in time to treatment failure analysis also indicates some possible stabilization of asthma at the higher doses (100 and 400 mg BID).
Randomized clinical trials evaluating the efficacy of selective CRTh2 antagonists in patients with asthma and persistent eosinophilia are beginning to be conducted. Recently, fevipiprant was shown to be effective at inhibiting eosinophilic airway inflammation in patients with persistent eosinophilic asthma.19 Therefore, the integration of biomarkers into patient subphenotyping may enable the identification of defined endotypes in asthma and the future potential for a precision-medicine approach, as seen with anti-IL5-targeted biologic therapies in patients with severe eosinophilic asthma.
However, a more relevant difference between the OC000459 and BI671800 studies and both the AMG953 and current study is use of concurrent therapy. Patients in the current study and the AMG953 study continued maintenance therapy with ICS–LABA and ICS, respectively; whereas patients in the OC000459 and BI671800 studies were steroid-naïve at study entry and received no regular controller treatment. Prior and background controller treatment may influence the outcomes in several ways. First, as seen in our study, the “placebo”-treated patients improved significantly. This is a common finding in clinical trials, and is most likely due to improved adherence to ICS and other controllers during the study.31 Such a response in the placebo group would reduce the possibility of detecting a treatment effect. Second, there was a high level of symptoms in spite of one or more controller therapies, which suggests that the patients in the study may have been relatively refractory to treatment. These patients might thus also have been less responsive or changes in lung function might be less prominent in all treatments, including CRTh2 antagonists.
Conclusion
In conclusion, this study demonstrated that at doses up to 400 mg BID, AZD1981 was well tolerated, but did not provide any clinically relevant beneficial effects on lung function or other efficacy variables in the full-study patient population with allergic asthma receiving combination ICS–LABA maintenance therapy.
Acknowledgments
The authors would like to thank the following people for their contributions to the manuscript: Christer Hultquist and Carin Jorup from AstraZeneca, who contributed to the study design; Jennifer McElhatten from AstraZeneca, who was the statistician for the study and completed the analyses; and Dr Elizabeth Hutchinson from Fishawack Communications Ltd and David Candlish from inScience Communications, who provided medical writing services, funded by AstraZeneca. This study was sponsored by AstraZeneca.
Author contributions
EDB served as international principal investigator, and all authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
EDB has been remunerated for participation in advisory boards, as a consultant to AstraZeneca, and for delivering lectures. His institution had received payment for involvement in clinical trials, and postgraduate students have received research grants from AstraZeneca. He has served as a consultant for and/or on advisory boards for ALK-Abello, Almirall, Actelion, Amgen, Boehringer Ingelheim, Elevation Pharma, Forest, GlaxoSmithKline, Pfizer, Napp Pharma, Novartis, Merck, and Takeda, and has received lecture fees from Boehringer Ingelheim, Chiesi, Forest, GlaxoSmithKline, Takeda, Teva, and Pfizer. He holds no stocks in AstraZeneca, nor has he received conference or travel support. CO, SI, PR, SL, and MU are employees of AstraZeneca. PR, MU, and CO also hold shares in AstraZeneca. The authors report no other conflicts of interest in this work.
References
Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18:673–683. | ||
Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. Bethesda (MD): GINA; 2016. | ||
O’Byrne PM, Naji N, Gauvreau GM. Severe asthma: future treatments. Clin Exp Allergy. 2012;42:706–711. | ||
Schuligoi R, Sturm E, Luschnig P, et al. CRTH2 and D-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology. 2010;85:372–382. | ||
Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–261. | ||
Mjösberg JM, Trifari S, Crellin NK, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–1062. | ||
Krause A, Zisowsky J, Strasser DS, et al. Pharmacokinetic/pharmacodynamic modelling of receptor internalization with CRTH2 antagonists to optimize dose selection. Clin Pharmacokinet. 2016;55:813–821. | ||
Hall IP, Fowler AF, Gupta A, et al. Efficacy of BI 671800, an oral CRTH2 antagonist, in poorly controlled asthma as sole controller and in the presence of inhaled corticosteroid treatment. Pulm Pharmacol Ther. 2015;32:37–44. | ||
Xue L, Salimi M, Panse I, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133:1184–1194. | ||
Chang YJ, DeKruyff RH, Umetsu DT. The role of type 2 innate lymphoid cells in asthma. J Leukoc Biol. 2013;94:933–940. | ||
Balzar S, Fajt ML, Comhair SA, et al. Mast cell phenotype, location, and activation in severe asthma: data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183(3):299–309. | ||
Fajt ML, Gelhaus SL, Freeman B, et al. Prostaglandin D2 pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol. 2013;131:1504–1512. | ||
Uller L, Mathiesen JM, Alenmyr L, et al. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir Res. 2007;8:16. | ||
Lukacs NW, Berlin AA, Franz-Bacon K, et al. CRTH2 antagonism significantly ameliorates airway hyperreactivity and downregulates inflammation-induced genes in a mouse model of airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2008;295:L767–L779. | ||
Stebbins KJ, Broadhead AR, Correa LD, et al. Therapeutic efficacy of AM156, a novel prostanoid DP2 receptor antagonist, in murine models of allergic rhinitis and house dust mite-induced pulmonary inflammation. Eur J Pharmacol. 2010;638:142–149. | ||
Singh D, Cadden P, Hunter M, et al. Inhibition of the asthmatic allergen challenge response by the CRTH2 antagonist OC000459. Eur Respir J. 2013;41:46–52. | ||
Barnes N, Pavord I, Chuchalin A, et al. A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. 2012;42:38–48. | ||
Luker T, Bonnert R, Schmidt J, et al. Substituted indole-1-acetic acids as potent and selective CRTH2 antagonists: discovery of AZD1981. Bioorg Med Chem Lett. 2011;21:6288–6292. | ||
Gonem S, Berair R, Singapuri A, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. 2016;4:699–707. | ||
Erpenbeck VJ, Popov TA, Miller D, et al. The oral CRTH2 antagonist QAW039 (fevipiprant): a phase II study in uncontrolled allergic asthma. Pulm Pharmacol Ther. 2016;39:54–63. | ||
Pettipher R, Hunter MG, Perkins CM, et al. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy. 2014;69:1223–1232. | ||
Schmidt JBF, Akam E. Biochemical and pharmacological characterization of AZD1981, an orally available selective DP2 antagonist in clinical development for asthma. Br J Pharmacol. 2013;168:162616–38. | ||
Kuna P, Bjermer L, Tornling G. Two phase II randomized trials on the CR Th2 antagonist AZD1981 in adults with asthma. Drug Des Devel Ther. 2016;10:2759–2770. | ||
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilatory flows. Eur Respir J. 1993;6 Suppl 16:5–40. | ||
Merrett J, Merrett TG. Phadiatop: a novel IgE antibody screening test. Clin Allergy. 1987;17:409–416. | ||
Juniper EF, O’Byrne PM, Roberts JN. Measuring asthma control in group studies: do we need airway calibre and rescue β2-agonist use? Respir Med. 2001;95:319–323. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–338. | ||
Juniper EF, Buist AS, Cox FM, Ferrie PJ, King DR. Validation of a standardized version of the Asthma Quality of Life Questionnaire. Chest. 1999;115:1265–1270. | ||
Corren J, Lemanske RF, Hanania NA, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–1098. | ||
Pettipher R, Hansel TT, Armer R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat Rev Drug Discov. 2007;6:313–325. | ||
Busse WW, Wenzel SE, Meltzer EO, et al. Safety and efficacy of the prostaglandin D2 receptor antagonist AMG 853 in asthmatic patients. J Allergy Clin Immunol. 2013;131:339–345. | ||
Hall I, Sarno M, Disse B, et al. Efficacy and safety of BI 671800, an oral CRTH2 antagonist, as add on therapy in poorly controlled asthma patients prescribed an inhaled corticosteroid. Eur Respir J. 2012;40 Suppl 56:3085. | ||
Sutherland R, Tetzlaff K, Nivens C, et al. Efficacy and safety of BI 671800, an oral CRTH2 antagonist in controller naïve patients with poorly-controlled asthma. Eur Respir J. 2012;40 Suppl 56:3084. | ||
Miller D, Wood C, Bateman E, et al. A randomized study of BI 671800, a CRTH2 antagonist, as add-on therapy in poorly controlled asthma. Allergy Asthma Proc. 2017;38:157–164. | ||
Calbet M, Andrés M, Armengol C, et al. Pharmacological characterization of CRTH2 antagonist LAS191859: long receptor residence time translates into long-lasting in vivo efficacy. Pharmacol Res. 2016;111:208–216. | ||
Sykes DA, Bradley ME, Riddy DM, et al. Fevipiprant (QAW039), a slowly dissociating CRTH2 antagonist with the potential for improved clinical efficacy. Mol Pharmacol. 2016;89:593–605. | ||
Otulana B, Bowden A, Yen Y, Puthukkeril S. Development of pitrakinra: an interleukin receptor antagonist to treat eosinophilic asthma. Respir Drug Deliv. 2012;1:21–28. | ||
Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–659. | ||
Szefler SJ, Phillips BR, Martinez FD, et al. Characterization of within-subject responses to fluticasone and montelukast in childhood asthma. J Allergy Clin Immunol. 2005;115:233–242. | ||
Woodruff PG, Modrek B, Choy DF, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. | ||
Bateman E, Guerreros A, Brockhaus F, et al. Fevipiprant, an oral prostaglandin DP2 receptor (CRTH2) antagonist, in allergic asthma uncontrolled on low-dose inhaled corticosteroids. Eur Respir Med. 2017;50:1700670. |
Supplementary material
 | Table S1 Institutional review boards and ethics committees by country |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.