")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Efficacy and Safety of the CFTR Potentiator Icenticaftor (QBW251) in COPD: Results from a Phase 2 Randomized Trial
Authors Rowe SM, Jones I, Dransfield MT, Haque N, Gleason S, Hayes KA, Kulmatycki K, Yates DP , Danahay H, Gosling M, Rowlands DJ, Grant SS
Received 8 April 2020
Accepted for publication 15 July 2020
Published 5 October 2020 Volume 2020:15 Pages 2399—2409
DOI https://doi.org/10.2147/COPD.S257474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Steven M Rowe,1 Ieuan Jones,2 Mark T Dransfield,1 Nazmul Haque,2 Stephen Gleason,2 Katy A Hayes,2 Kenneth Kulmatycki,2 Denise P Yates,2 Henry Danahay,3 Martin Gosling,3,4 David J Rowlands,2 Sarah S Grant2
1University of Alabama at Birmingham, Department of Medicine, Birmingham, AL, USA; 2Novartis Institutes for BioMedical Research, Cambridge, MA, USA; 3Enterprise Therapeutics, Brighton, UK; 4Sussex Drug Discovery Centre, University of Sussex, Brighton, UK
Correspondence: Sarah S Grant
Novartis Institutes for BioMedical Research, 250 Massachusetts Ave, Cambridge, MA 02139, USA
Tel +16178717812
Email [email protected]
Rationale: Excess mucus plays a key role in COPD pathogenesis. Cigarette smoke-induced cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction may contribute to disease pathogenesis by depleting airway surface liquid and reducing mucociliary transport; these defects can be corrected in vitro by potentiating CFTR.
Objective: To assess the efficacy of the CFTR potentiator icenticaftor in improving airflow obstruction in COPD patients with symptoms of chronic bronchitis.
Methods: In this double-blind, placebo-controlled study, COPD patients were randomized (2:1) to either icenticaftor 300 mg or placebo b.i.d. This non-confirmatory proof of concept study was powered for lung clearance index (LCI) and pre-bronchodilator FEV1, with an estimated sample size of 90 patients. The primary endpoint was change from baseline in LCI for icenticaftor versus placebo at Day 29; key secondary endpoints included change from baseline in pre- and post-bronchodilator FEV1 on Day 29. Key exploratory endpoints included change from baseline in sweat chloride, plasma fibrinogen levels, and sputum colonization.
Results: Ninety-two patients were randomized (icenticaftor, n=64; placebo, n=28). At Day 29, icenticaftor showed no improvement in change in LCI (treatment difference: 0.28 [19% probability of being better than placebo]), an improvement in pre-bronchodilator FEV1 (mean: 50 mL [84% probability]) and an improvement in post-bronchodilator FEV1 (mean: 63 mL [91% probability]) over placebo. Improvements in sweat chloride, fibrinogen and sputum bacterial colonization were also observed. Icenticaftor was safe and well tolerated.
Conclusion: The CFTR potentiator icenticaftor increased FEV1 versus placebo after 28 days and was associated with improvements in systemic inflammation and sputum bacterial colonization in COPD patients; no improvements in LCI with icenticaftor were observed.
Keywords: chronic obstructive pulmonary disease, chronic bronchitis, cystic fibrosis transmembrane conductance regulator potentiator, CFTR potentiator, mucociliary clearance, icenticaftor; QBW251
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease characterized by persistent airflow obstruction.1 Cigarette smoke exposure is considered the primary risk factor for COPD.2 While all COPD patients demonstrate airflow limitation, the disease is heterogeneous: at one extreme is chronic bronchitis (CB), and at the other is emphysema. The relative contributions of these two phenotypes vary from person-to-person, with many patients exhibiting features of both.1
Pathologically, small airway disease is characterized by increased numbers of goblet cells and mucus plugging, with associated smooth muscle hyperplasia, airway fibrosis, and increased inflammation.3,4 Evidence suggests that small airway disease precedes emphysematous destruction and small airway pathologic changes correlate with the degree of airway obstruction and disease severity in COPD.3–5 Excess mucus secretion is believed to play an important role in COPD pathogenesis,6,7 and is associated with a more rapid decline in pulmonary function, an increased risk of exacerbations and hospitalization, and increased risk of mortality.8–13
Smoking-induced acquired dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR), a membrane protein and anion channel located at the apical surface of epithelial cells, may contribute to COPD pathogenesis. In vitro, induced CFTR dysfunction results in a reduction of airway surface liquid (ASL) and decreased mucociliary transport (MCT).14–17 In clinical studies, cigarette smokers and COPD patients exhibit reduced CFTR function in upper and lower airways as well as systemically, which is associated with CB.14,18–21 Considering the importance of mucus hypersecretion, chronic airway inflammation and recurrent infections in both COPD and cystic fibrosis (CF), this data suggests that acquired CFTR dysfunction contributes to the pathophysiology of COPD.
CFTR potentiators, a new class of medications for CF, which correct underlying gating defects in mutant CFTR, may represent a viable therapeutic approach for COPD.20 In vitro, the CFTR potentiator ivacaftor restores CFTR-dependent chloride transport in cigarette smoke-exposed epithelial cells and improves the downstream effects of epithelial function, including MCT, ASL depth and ciliary beating.17,20 Preclinical data with roflumilast, a phosphodiesterase 4 inhibitor that reduces the risk of exacerbations in COPD patients with CB, suggest that CFTR activation may contribute to the roflumilast mechanism of action via phosphorylation of the CFTR regulatory domain and partial restoration of CFTR-dependent chloride transport in cigarette smoke-exposed epithelial cells.22 Clinically, only limited data are available for CFTR potentiators in COPD: one small pilot study of 2-weeks of ivacaftor in 12 COPD patients revealed non-statistically significant improvements in sweat chloride, nasal potential difference and symptoms score compared with placebo.23
Icenticaftor (QBW251), an orally bioavailable small molecule CFTR potentiator, can restore CFTR function in specific CFTR genotypes as well as wild-type CFTR (details in supplementary appendix). In an unpublished study in people with CF, icenticaftor improved sweat chloride and pulmonary function in individuals with particular CFTR mutations localized to the cell surface.24 This study evaluated the efficacy of 28 days of icenticaftor administration in 92 patients with moderate-to-severe COPD and CB to test the hypothesis whether CFTR potentiation with icenticaftor can improve pulmonary function and reduce air trapping in these patients.
Methods
Study Design and Participants
This was a multicenter, double-blind, randomized, placebo-controlled, Phase 2 study conducted in subjects with moderate-to-severe COPD (clinicaltrials.gov number: NCT02449018). The study included a 2-week, single-blind, placebo run-in period, to ensure the stability of the baseline COPD treatment regimen. Subjects then received either icenticaftor or placebo during a 4-week double-blind treatment period, followed by a 4-week single-blind placebo follow-up period (to assess the duration of effect), with a final efficacy assessment on Day 56 (Figure 1).
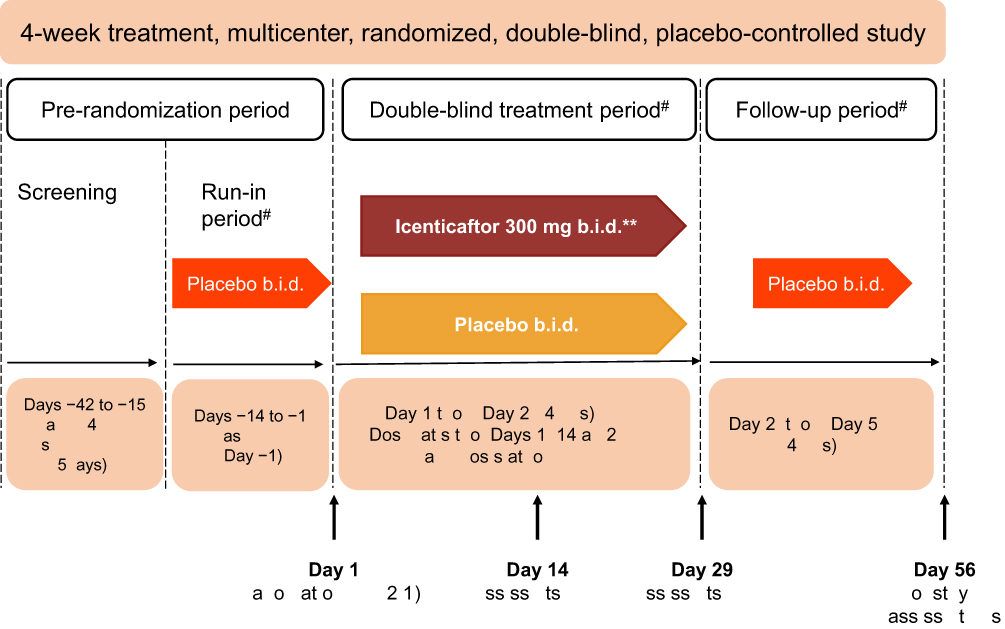 |
Figure 1 Study design. *Primary efficacy assessments on Day 29–12 hours after the last dose of icenticaftor. **A total of 92 patients were randomized. The first 4 patients in the study received icenticaftor 450 mg b.i.d. or placebo, prior to the protocol amendments reducing the dose to icenticaftor 300 mg b.i.d. #Patients continued on background COPD therapy throughout the study. During the entire study duration, subjects were maintained on stable baseline COPD therapy.Abbreviations: b.i.d., twice daily; EOS, end of study. |
Patients aged ≥35 and ≤75 years with a diagnosis of COPD and symptoms of CB and with lung clearance index (LCI) ≥8 at screening were included. Patients diagnosed with severe bronchiectasis or significant radiographic emphysema were excluded. Whole lung emphysema extent <25% (TLC % < −950 HU) and quantitative air trapping >15% (RV >15% −856 HU) were assessed by high resolution computed tomography (HRCT) for inclusion. Additional criteria are provided in the supplementary material (Table E1).
The study protocol and its amendments were reviewed by the Independent Ethics Committee and/or Institutional Review Board of each study center; please refer to the supplementary appendix for more details on participating study centres and principal investigators. The study was conducted according to the ethical principles of the Declaration of Helsinki. Informed consent was obtained from each patient in writing before any procedure.
Data Availability
Novartis will not provide access to patient-level data, if there is a reasonable likelihood that individual patients could be re-identified. In addition, clinical data, in some cases, have been collected subject to contractual or consent provisions that prohibit transfer to third parties. Such restrictions may preclude granting access under these provisions. Where co-development agreements or other legal restrictions prevent companies from sharing particular data, companies will work with qualified requestors to provide summary information where possible.
Procedures and Outcomes
The primary endpoint was the change from baseline in LCI, which represents the cumulative expired volume required to wash out nitrogen to 2.5% of the starting concentration, normalized to the functional residual capacity of the lung.25 Key secondary endpoints included the mean change from baseline in pre- and post-bronchodilator FEV1 at Day 29, as well as additional spirometry and lung volumes measurements. Spirometry was performed according to American Thoracic Society (ATS) guidelines.26
Key exploratory endpoints were changed from baseline to Day 29 in sweat chloride (to assess CFTR activity), peripheral biomarkers including plasma fibrinogen and C-reactive protein (CRP) levels, whole lung and regional air trapping quantification from expiratory phase (RV) HRCT scans, and sputum colonization. The collection of spontaneous sputum samples was attempted at all study visits, and sputum was sent to a central vendor for quantification of colony-forming units (CFUs) for key COPD pathogens.
Safety assessments included collecting all adverse events (AEs), serious AEs (SAEs), along with their severity and relationship to the study drug. The study also assessed the pharmacokinetics (PK) of icenticaftor in these patients and patient-reported outcome (PRO) questionnaires (St. George’s Respiratory Questionnaire (SGRQ) and Baseline/Transition Dyspnea Index [BDI/TDI]); full details are provided in the supplementary appendix.
Statistical Analysis
Efficacy data were analyzed in patients who received any treatment and had available pharmacodynamic data with no major relevant protocol deviations (see Supplementary appendix). The safety set included all the patients who received any study drug.
Statistical analysis of all continuous endpoints was performed for the comparison of icenticaftor versus placebo on Day 29 using a Bayesian model for repeated measures.27 The model included effects for age, GOLD status (GOLD 2013), smoking status, treatment-by-time interaction, and baseline-by-time interaction. Contrasts for treatment differences and time point were provided together with 90% two-sided credible intervals (CrI) under Bayesian framework and the posterior probability that the treatment effect with icenticaftor is better than the placebo was derived. Posterior probabilities ≥90% were considered as a statistically significant signal of efficacy. For prebronchodilator FEV1, a meta-analysis of similar Novartis COPD trials gave previous knowledge about week 4 placebo response (ie mean of 0.02 L worse with SD=0.05). The impact of using this weakly informative prior in a Bayesian analysis meant that 14 fewer placebo subjects were required. Non-informative priors were used otherwise. Except for Week 4 pre-bronchodilator FEV1, non-informative priors were used throughout the analysis. All efficacy analyses used SAS 9.4 PROC MCMC.
Results
Patients
From April 30th 2015 through November 30th 2016, 331 patients were screened for eligibility at 16 clinics in 2 countries (USA and Poland). Ninety-two patients were randomized, 64 to icenticaftor and 28 to placebo. Fourteen patients (placebo, n=2; icenticaftor, n=12) discontinued the study (Figure 2). Six patients (1 [4%] in placebo and 5 [8%] in icenticaftor) discontinued the study due to adverse events or meeting individual stopping rules (including COPD exacerbation). The placebo discontinuation was due to diarrhea (n=1), and the icenticaftor discontinuations were due to the following: (1) CVA (n=1) during placebo follow-up period (2) vomiting and diarrhea (n=1), and the remaining (n=3) were due to COPD exacerbations. Of note, the first 4 patients randomized in this study received icenticaftor 450 mg (n=2) or placebo (n=2) b.i.d., prior to an amendment reducing the dose to icenticaftor 300 mg b.i.d. The dose was reduced to 300 mg b.i.d. when pharmacokinetic data became available from two ongoing icenticaftor studies suggesting exposure with the 450 mg b.i.d. dose may exceed initial assessments of safe exposure coverage from short-term toxicology studies; however, subsequent assessments including long-term toxicology studies indicate that 450 mg b.i.d. is a safe dose and this dose is being studied in an ongoing Phase 2b study (see supplemental appendix for more details Figures E2, E3 and Tables E5, E6).
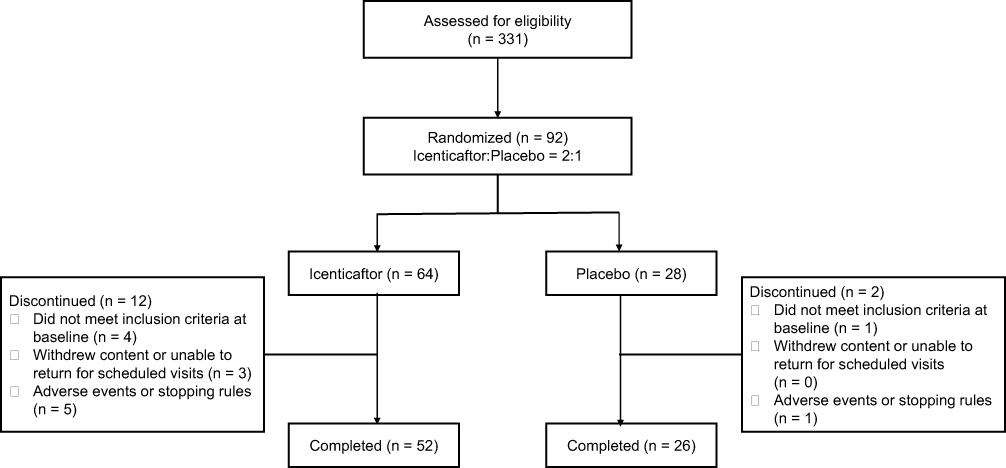 |
Figure 2 Patient disposition. |
Baseline demographics and clinical characteristics were balanced between the groups (Table 1). Patients were predominantly Caucasian (97%) with a mean age of 64 years and a baseline FEV1 of 1.5 L (51% predicted). At baseline, mean sweat chloride was 23 mmol/l, reflecting an element of acquired CFTR dysfunction in some individuals.19 Subjects continued background COPD therapy during the study. LABA/LAMA use was balanced between the groups; a minor imbalance was observed in ICS use, with 64% of placebo subjects receiving ICS versus 41% of active subjects.
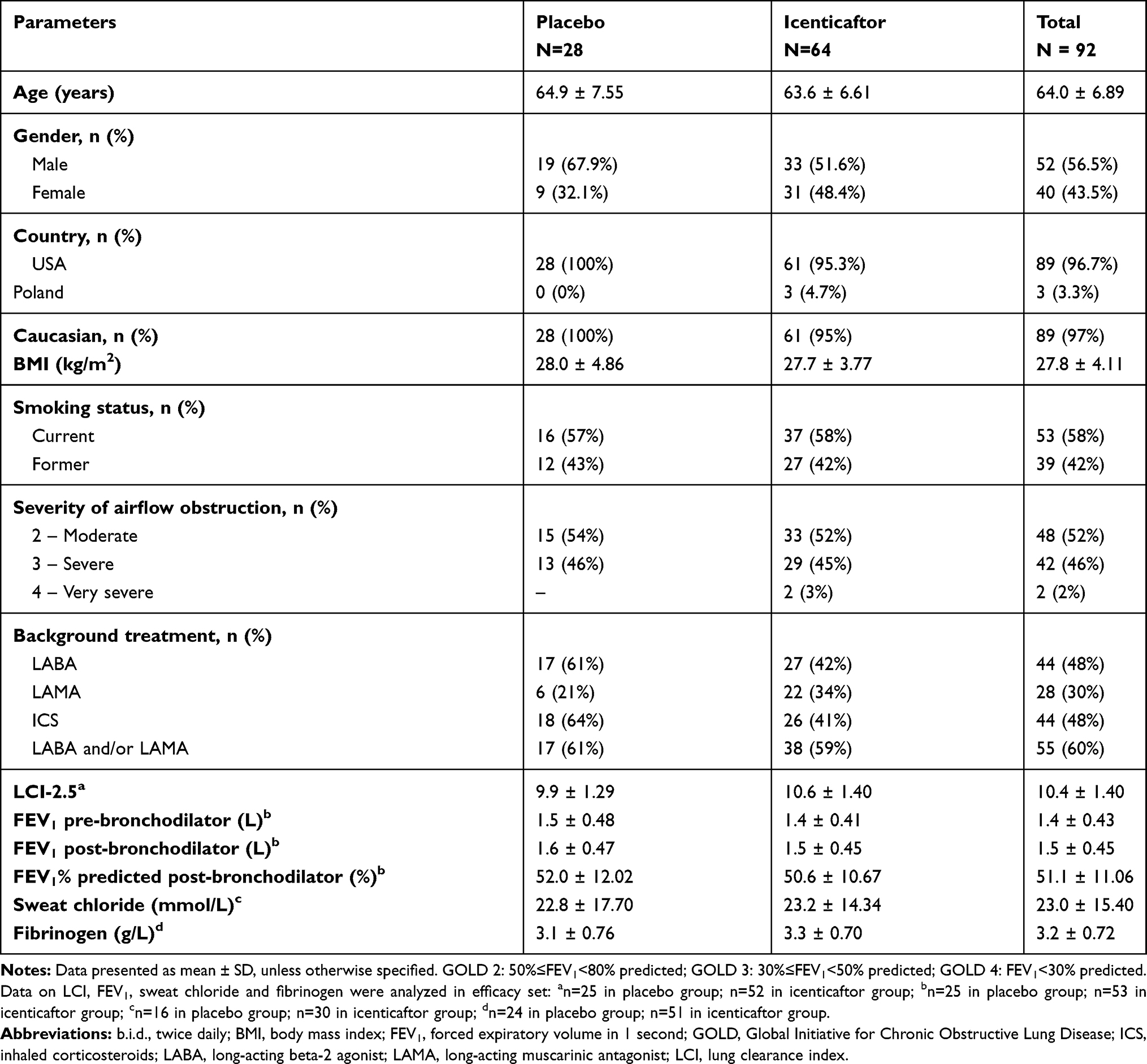 |
Table 1 Patient Demographics and Clinical Characteristics at Baseline (Safety Set) |
Efficacy
For LCI, icenticaftor showed no improvement in the change from baseline to Day 29 compared with placebo, with a treatment difference in posterior mean change from baseline of 0.28 (90% CrI: −0.24, 0.79). The probability of the icenticaftor effect being better than placebo was 19%. For pre- and post-bronchodilator FEV1, icenticaftor demonstrated an improvement in the change from baseline to Day 29 compared with placebo, with a treatment difference in posterior mean change from baseline of 0.05 L (90% CrI: −0.03, 0.13) for pre-bronchodilator FEV1 and 0.06 L (90% CrI: −0.02, 0.14) for post-bronchodilator FEV1. The observed 4-week placebo response for pre-bronchodilator FEV1 was consistent with assumptions made during the statistical design stage. The probability of the icenticaftor effect being better than placebo was 84% for pre-bronchodilator FEV1 and 91% for post-bronchodilator FEV1 with the raw mean data depicted in Figure 3 and the statistical model presented in Table 2. During the placebo follow-up period, there was a trend back towards baseline values for both the pre- and post-bronchodilator FEV1 improvements. This met predefined criteria for treatment success based on the Bayesian analysis employed
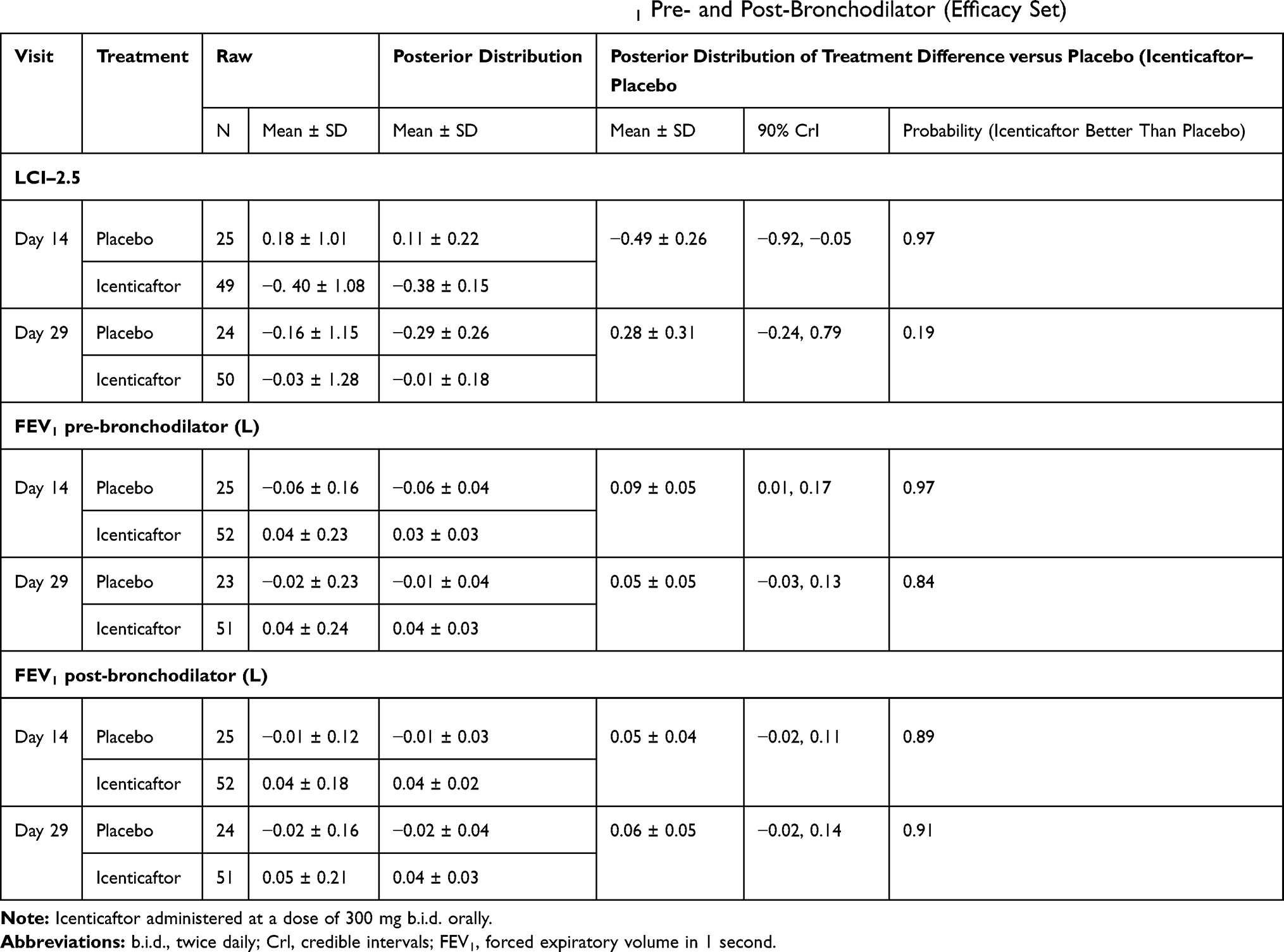 |
Table 2 Modeled Data on Change from Baseline of LCI and FEV1 Pre- and Post-Bronchodilator (Efficacy Set) |
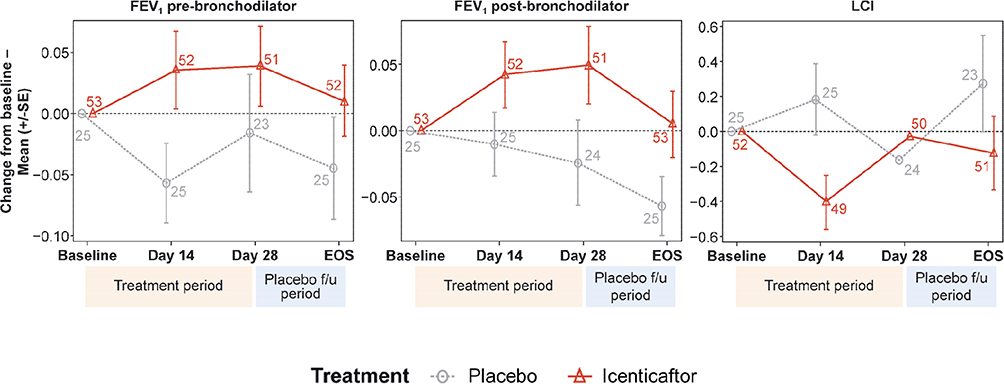 |
Figure 3 Raw mean change from baseline per treatment arm over time for pre-bronchodilator FEV1, post-bronchodilator FEV1, and lung clearance index (LCI). Numbers at each data point represent number of patients; error bars are indicative of standard error.Abbreviations: EOS, end of study; FEV1, forced expiratory volume in 1 second; LCI, lung clearance index. |
The results for additional pulmonary function assessments are presented in Table E2. On Day 29, the difference of icenticaftor to placebo in estimated mean change from baseline suggests numerical improvement for multiple endpoints, including FVC, forced expiratory volume in 6 seconds (FEV6), forced expiratory flow (FEF25–75) and inspiratory capacity (IC) measured pre- and post-bronchodilator, with the FEF25–75 post-bronchodilator reaching statistical significance. There was no improvement in RV. Two patient-reported outcomes were included in this study (BDI/TDI and SGRQ) and no improvement was seen with icenticaftor as compared to placebo (supplementary appendix).
Icenticaftor decreased sweat chloride levels, a reflection of improved CFTR activity, with the estimated differences (icenticaftor [n=19] vs placebo [n=11]) in posterior mean change from baseline of −5.04 mmol/L (90% CrI: −11.12, 0.94), with a 92% probability that the icenticaftor effect is better than placebo on Day 29. This change reverted upon cessation of the study drug (Figure 4A). Fibrinogen (icenticaftor [n=48] vs placebo [n=23]) and CRP (icenticaftor [n=51] vs placebo [n=24]) were measured to assess icenticaftor impact on systemic inflammation. Icenticaftor decreased fibrinogen levels, an acute phase reactant, from baseline to Day 29 versus placebo with an estimated difference in posterior mean change from baseline of −0.39 g/L (90% CrI: −0.65, −0.13) with a 99% probability of the icenticaftor effect being better than placebo (Figure 4B). While the baseline CRP was elevated in these patients, the treatment effect of icenticaftor on CRP was not meaningfully different compared with placebo (difference: −1.20 mg/L; 90% CI: −6.19, 3.79).
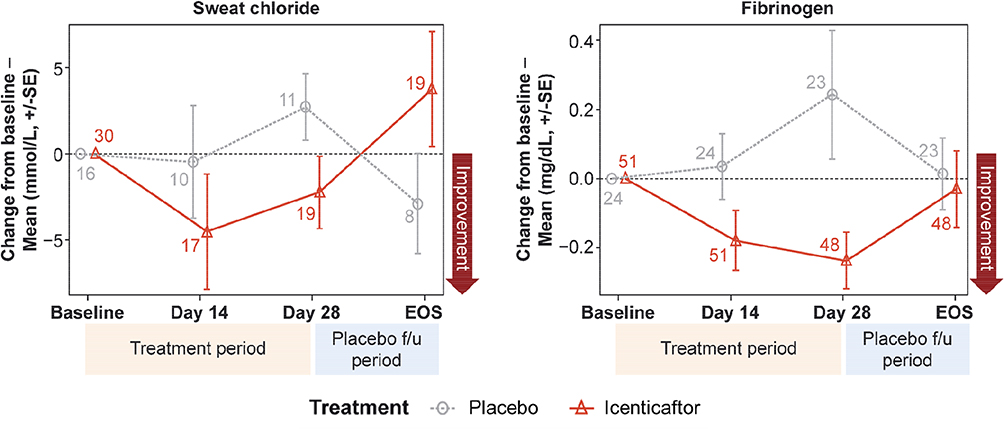 |
Figure 4 Mean change from baseline for sweat chloride (A) and fibrinogen (B) over time. Numbers at each data point represent number of patients; error bars are indicative of standard error. |
Sputum CFU analysis suggested that a higher proportion of patients with evidence of colonization at baseline were able to clear detectable bacterial colonization in the icenticaftor group on Day 29 (23% icenticaftor vs 13% placebo) and at EOS (38% icenticaftor vs 20% placebo). At EOS, the predicted odds of response (ie, no pathogen) for icenticaftor was 2.6 times that of placebo, with a 90% CrI of 0.6 (placebo better than active) to 14 (a 14-fold increase in bacterial clearance). The posterior probability that this treatment effect is better than that of placebo was 89% (See supplemental Table E3 for details on specific pathogens).
From the quantitative image analysis of volumetric HRCT RV scans, measures of whole lung image intensity below −856 HU at Day 29 compared to the screening value suggested a numerical improvement in global air trapping percentage [(estimated mean difference for icenticaftor [n=49] vs placebo [n=22]) of −0.50 (90% CrI: −3.74, 2.75)]. This data suggests a decrease in air trapping after 4 weeks of treatment but was not statistically significant. Lobar regional analysis did not reveal any significant local reductions in air trapping, and the range across the lobes varied from 15% to 100% suggesting regional heterogeneity despite the global threshold set for inclusion of >15%.
The overall effects of icenticaftor on different end points are presented in a radar plot (Figure E1); overall, this suggested an improvement in several domains of COPD lung disease with icenticaftor treatment as compared to placebo.
Safety
Icenticaftor was well tolerated in the study population and did not lead to any safety signals. Overall, 40.6% and 39.3% of patients in the icenticaftor and placebo groups, respectively, reported ≥1 AE. Most frequently observed AEs in both icenticaftor- and placebo-treated patients were COPD exacerbation and diarrhea (Table E4). Most AEs were of mild-to-moderate intensity with comparable incidences between the groups. Six patients discontinued the study drug due to AEs. Six SAEs were reported in four patients in the icenticaftor group, of which two SAEs (vomiting and hypokalemia) in one patient were considered possibly related to study drug. No deaths were reported during the study.
Discussion
Goblet cell hyperplasia, mucus hypersecretion, and small airway disease are central to the pathogenesis of COPD, with recent evidence suggesting that smoking-induced acquired CFTR dysfunction contributes to airway dehydration and mucus dysfunction in COPD. In this study, the CFTR potentiator icenticaftor increased FEV1 relative to placebo after 28 days of treatment by potentiating CFTR in patients with moderate-to-severe COPD and was associated with improvements in systemic inflammation and sputum bacterial colonization as assessed by the Bayesian analysis employed in this study.
The primary endpoint in this study was LCI, derived from MBNW. With heterogeneous small airway obstruction, a greater expired volume is required for nitrogen washout resulting in increased LCI. LCI has been used effectively in CF clinical trials, primarily in patients with early-stage disease and minimal airway obstruction, and is believed to be more sensitive in detecting disease in peripheral airways not detected with a measure such as FEV1.28 LCI has been shown to be elevated in COPD patients,29 but has not been used previously in COPD clinical trials to evaluate efficacy. LCI was selected as the primary endpoint assuming that variability in LCI is lower than FEV1, given the short treatment period (28 days), the icenticaftor mechanism of action (larger effect expected on small airways, which would theoretically be better reflected in LCI), and past experience with LCI with CFTR potentiator therapy in cystic fibrosis.30,31 However, icenticaftor did not demonstrate a clinically nor statistically important improvement in LCI versus placebo at the end of the 4-week treatment period.
Given the uncertainties in LCI as a primary endpoint, the study was also prospectively designed and powered to assess the effects of icenticaftor on FEV1. A Bayesian approach was employed for pre-bronchodilator FEV1, using a weakly informative prior based on existing placebo data in the literature;32 this approach allowed for a smaller placebo sample size. For pre- and post-bronchodilator FEV1, efficacy with icenticaftor over placebo was observed as early as on Day 14, and at the EOS, the mean FEV1 had returned to baseline, suggesting washout of potentiator efficacy. The magnitude of improvement in pre- and post-bronchodilator FEV1 are within the range of improvements noted for roflumilast, an oral phosphodiesterase-4 inhibitor with an anti-inflammatory mechanism of action (38 mL to 80 mL with 6 months’ to 1-year treatment duration), and was considered as evidence of a clinically relevant effect.22,33–36
Additional spirometry and lung volume secondary and exploratory efficacy endpoints also demonstrate numerical improvement for icenticaftor over placebo. Improvement was not observed in RV, but this was measured using MBNW; unlike plethysmography, MBNW cannot be effectively used to assess “trapped air” which may explain the discordance between IC and RV.37
At baseline, the sweat chloride analysis suggests CFTR dysfunction (mean 23 mmol/L), within the range of what has been reported previously in COPD patients;23 however, the sample size was lower than expected as not all patients provided samples. Despite low severity CFTR dysfunction at baseline, the sweat chloride data demonstrated significant improvement in CFTR activity on Day 29, indicating icenticaftor potentiates wild-type CFTR and confirming the mechanism of action.
Inclusion thresholds for emphysema and air trapping based on HRCT were used in this study to enrich for moderate to severe COPD subjects with small airway disease who may be more likely to demonstrate improvement with icenticaftor after 4-weeks of treatment. Despite the global thresholds applied for inclusion, significant regional heterogeneity was observed in both the emphysema and air trapping lobar quantitative analysis. For the assessment of whole lung air trapping after 4-weeks of treatment, the mean change in HRCT suggests an overall numerical reduction in air trapping for patients who received icenticaftor versus placebo, consistent with the hypothesis that icenticaftor will improve small airway obstruction.
Analysis of inflammatory biomarkers revealed a statistically significant decrease in fibrinogen with icenticaftor. Fibrinogen has been approved as a prognostic biomarker of COPD patients at high risk for exacerbations and/or all-cause mortality.38 While the clinical significance of a icenticaftor-induced decrease in fibrinogen is unclear, the change in fibrinogen supports a potential indirect anti-inflammatory effect for icenticaftor.
The exploratory analysis of bacterial colonization suggests that icenticaftor may be associated with clearance of potential pathogens. Increasing evidence suggests that bacterial colonization is associated with airway inflammation, increased symptoms, and increased risk of exacerbation in COPD patients;39 the trend towards decreased colonization supports the hypothesis that icenticaftor may be able to impact this aspect of disease pathophysiology. Improved bacterial clearance has been reported with ivacaftor in some but not all CF studies.40–42 The mechanisms for bacterial colonization clearance may be related to icenticaftor-mediated improvements in MCC; in addition, CFTR also regulates bicarbonate secretion, whose effect on airway surface liquid pH is important in the fight against pathogens,43 and macrophages exposed to cigarette smoke demonstrate altered CFTR lipid rafts, resulting in impaired bacterial phagocytosis and clearance.44 Together, these data suggest that in addition to improving MCC, icenticaftor may exert both direct and indirect host defense benefits.
This study has several limitations related to the small sample size and short treatment duration. While icenticaftor demonstrated no improvement in PROs (SGRQ and TDI) versus placebo (results presented in supplementary appendix), with only 28 days of exposure and small sample size, the usefulness of these assessments is limited. It is unknown whether longer duration icenticaftor treatment could improve PROs. Second, to maximize the ability to detect a signal with a small sample size, this study enriched the patient population specifically for symptoms of CB and absence of significant emphysema; the impact of icenticaftor in COPD subjects without these enrichment criteria is unknown. Lastly, it was not possible to collect information about exacerbations with this short study. As bacterial colonization is a risk factor for COPD exacerbations,39,45 the sputum CFU analysis is promising, but the impact of icenticaftor on exacerbations will need to be further assessed in future studies.
In summary, icenticaftor resulted in significant improvement in FEV1 over placebo in patients with moderate-to-severe COPD and CB. Analysis of the inflammatory biomarker fibrinogen demonstrated a statistically significant improvement in systemic inflammation, and exploratory analysis of sputum CFUs supported a reduction in bacterial colonization. Icenticaftor was well tolerated in these patients with a similar overall incidence of AEs between the icenticaftor and placebo groups. These results suggest that CFTR potentiation with icenticaftor represents a potential novel therapeutic approach in COPD and may provide a clinically relevant benefit on top of standard-of-care therapy, which will be further investigated in future clinical trials.
Acknowledgments
The study was supported and funded by Novartis. We thank the patients who participated in the trial. Phani Tejasvi Dantu and Rahul Lad, professional scientific writers at Novartis (Hyderabad, India), provided editorial and technical support in the preparation of the manuscript. MedQIA, including Jonathan Goldin and Grace Hyun Kim, provided assistance with imaging acquisition and analysis. We also had infrastructure support from two NIH grants, which also informed the basis of the study: R35 HL135816, P30 DK072482.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.The study was funded by Novartis Institutes for BioMedical Research, Cambridge, MA, USA.
Disclosure
Steven M Rowe reports grants and personal fees from Novartis during the conduct of the study; grants and personal fees from Bayer, grants from Translate Bio, grants from Proteostasis, grants, personal fees and non-financial support from Galapagos/Abbvie, grants, personal fees and in kind support for research from Synedgen/Synspira, grants from Eloxx, grants and personal fees from Celtaxsys, grants, personal fees, non-financial support from Vertex Pharmaceuticals Inc, grants, personal fees and stock options from Renovion, grants and personal fees from Arrowhead, grants, personal fees and in kind support for research from Ionis, grants from AstraZeneca, grants from N30/Nivalis, grants from PTC Therapeutics, personal fees from Genentech, personal fees, non-financial support from Boehringer Ingelheim, grants from Janssen, Vivus, Actelion, Johnson and Johnson, and other related entities, outside the submitted work. Mark T Dransfield reports grants from the NIH, Department of Defense and American Lung Association; consulting fees from AstraZeneca, GlaxoSmithKline, Mereo, Quark and Teva; travel support from Pulmonx; and contracted clinical trial support from AstraZeneca, Boehringer Ingelheim, Boston Scientific, Gala, Nuvaira, PneumRx/BTG, Pulmonx and Yungjin. Ieuan Jones, Katy A Hayes, David J Rowlands and Sarah S Grant have patent 8247436 issued and are employees of Novartis. Nazmul Haque, Stephen Gleason and Denise P Yates are employees of Novartis. Kenneth Kulmatycki have a patent issued and is an employee of Novartis. Martin Gosling was an employee of Novartis between 2001 and 2014. He is now affiliated with Enterprise Therapeutics. Henry Danahay was an employee of Novartis between 1997 and 2014. He is now affiliated with Enterprise Therapeutics. The authors report no other conflicts of interest in this work.
References
1. Global initiative for chronic obstructive lung disease. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease (2018 Report); 2018.
2. World health organization. Chronic Obstructive Pulmonary Disease (Copd); 2016.
3. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
4. Kim V, Kelemen SE, Abuel-Haija M, et al. Small airway mucous metaplasia and inflammation in chronic obstructive pulmonary disease. COPD. 2008;5(6):329–338. doi:10.1080/15412550802522445
5. Saetta M, Turato G, Baraldo S, et al. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with both symptoms of chronic bronchitis and chronic airflow limitation. Am J Respir Crit Care Med. 2000;161(3):1016–1021. doi:10.1164/ajrccm.161.3.9907080
6. McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575. doi:10.1056/NEJMoa1106955
7. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247. doi:10.1056/NEJMra0910061
8. Burgel PR, Nesme-Meyer P, Chanez P, et al. Initiatives bronchopneumopathie chronique obstructive scientific C. cough and sputum production are associated with frequent exacerbations and hospitalizations in COPD subjects. Chest. 2009;135(4):975–982. doi:10.1378/chest.08-2062
9. Guerra S, Sherrill DL, Venker C, Ceccato CM, Halonen M, Martinez FD. Chronic bronchitis before age 50 years predicts incident airflow limitation and mortality risk. Thorax. 2009;64(10):894–900. doi:10.1136/thx.2008.110619
10. Kim V, Han MK, Vance GB, et al. The chronic bronchitic phenotype of COPD: an analysis of the COPDgene study. Chest. 2011;140(3):626–633. doi:10.1378/chest.10-2948
11. Kim V, Zhao H, Boriek AM, et al. Persistent and newly developed chronic bronchitis are associated with worse outcomes in chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2016;13(7):1016–1025. doi:10.1513/AnnalsATS.201512-800OC
12. Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with fev1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen city heart study group. Am J Respir Crit Care Med. 1996;153(5):1530–1535. doi:10.1164/ajrccm.153.5.8630597
13. Kesimer M, Smith BM, Ceppe A, et al. Mucin concentrations and peripheral airways obstruction in COPD. Am J Respir Crit Care Med. 2018;198(11):1453–1456. doi:10.1164/rccm.201806-1016LE
14. Cantin AM, Hanrahan JW, Bilodeau G, et al. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med. 2006;173(10):1139–1144. doi:10.1164/rccm.200508-1330OC
15. Clunes LA, Davies CM, Coakley RD, et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012;26(2):533–545. doi:10.1096/fj.11-192377
16. Kreindler JL, Jackson AD, Kemp PA, Bridges RJ, Danahay H. Inhibition of chloride secretion in human bronchial epithelial cells by cigarette smoke extract. Am J Physiol Lung Cell Mol Physiol. 2005;288(5):L894–902. doi:10.1152/ajplung.00376.2004
17. Raju SV, Lin VY, Liu L, et al. The cystic fibrosis transmembrane conductance regulator potentiator ivacaftor augments mucociliary clearance abrogating cystic fibrosis transmembrane conductance regulator inhibition by cigarette smoke. Am J Respir Cell Mol Biol. 2017;56(1):99–108. doi:10.1165/rcmb.2016-0226OC
18. Dransfield MT, Wilhelm AM, Flanagan B, et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest. 2013;144(2):498–506. doi:10.1378/chest.13-0274
19. Raju SV, Jackson PL, Courville CA, et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med. 2013;188(11):1321–1330. doi:10.1164/rccm.201304-0733OC
20. Sloane PA, Shastry S, Wilhelm A, et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One. 2012;7(6):e39809. doi:10.1371/journal.pone.0039809
21. Courville CA, Tidwell S, Liu B, Accurso FJ, Dransfield MT, Rowe SM. Acquired defects in CFTR-dependent beta-adrenergic sweat secretion in chronic obstructive pulmonary disease. Respir Res. 2014;15(1):25. doi:10.1186/1465-9921-15-25
22. Lambert JA, Raju SV, Tang LP, et al. Cystic fibrosis transmembrane conductance regulator activation by roflumilast contributes to therapeutic benefit in chronic bronchitis. Am J Respir Cell Mol Biol. 2014;50(3):549–558. doi:10.1165/rcmb.2013-0228OC
23. Solomon GM, Hathorne H, Liu B, et al. Pilot evaluation of ivacaftor for chronic bronchitis. Lancet Respir Med. 2016;4(6):e32–33. doi:10.1016/S2213-2600(16)30047-9
24. Kazani S, Alcantara J, Debonnett L, et al. QBW251 is a safe and efficacious CFTR potentiator for patients with cystic fibrosis. Am J Respir Crit Care Med. 2016;193:A7789.
25. Egger B, Jost K, Anagnostopoulou P, et al. Lung clearance index and moment ratios at different cut-off values in infant multiple-breath washout measurements. Pediatr Pulmonol. 2016;51(12):1373–1381. doi:10.1002/ppul.23483
26. Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. doi:10.1183/09031936.05.00034805
27. Berry DA. Bayesian clinical trials. Nat Rev Drug Discov. 2006;5(1):27–36. doi:10.1038/nrd1927
28. Davies JC, Cunningham S, Alton EW, Innes JA. Lung clearance index in cf: a sensitive marker of lung disease severity. Thorax. 2008;63(2):96–97. doi:10.1136/thx.2007.082768
29. Fähndrich S, Lepper PM, Trudzinski F, Seibert M, Wagenpfeil S, Bals R. Lung clearance index is increased in patients with COPD - LCI measurements in the daily routine. J Pulm Respir Med. 2016;6(3):354. doi:10.4172/2161-105X.1000354
30. Davies J, Sheridan H, Bell N, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a g551d-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med. 2013;1(8):630–638. doi:10.1016/S2213-2600(13)70182-6
31. Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for f508del-CFTR: a randomised, placebo-controlled Phase 3 trial. Lancet Respir Med. 2017;5(7):557–567. doi:10.1016/S2213-2600(17)30215-1
32. Neuenschwander B, Capkun-Niggli G, Branson M, Spiegelhalter DJ. Summarizing historical information on controls in clinical trials. Clin Trial. 2010;7(1):5–18. doi:10.1177/1740774509356002
33. Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374(9691):685–694. doi:10.1016/S0140-6736(09)61255-1
34. Calverley PMA, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176(2):154–161. doi:10.1164/rccm.200610-1563OC
35. Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009;374(9691):695–703. doi:10.1016/S0140-6736(09)61252-6
36. Martinez FJ, Rabe KF, Sethi S, et al. Effect of roflumilast and inhaled corticosteroid/long-acting β2-agonist on chronic obstructive pulmonary disease exacerbations (RE2SPOND). A randomized clinical trial. Am J Respir Crit Care Med. 2016;194(5):559–567. doi:10.1164/rccm.201607-1349OC
37. Sue DY. Measurement of lung volumes in patients with obstructive lung disease. A matter of time (constants). Ann Am Thorac Soc. 2013;10(5):
38. Duvoix A, Dickens J, Haq I, et al. Blood fibrinogen as a biomarker of chronic obstructive pulmonary disease. Thorax. 2013;68(7):670–676. doi:10.1136/thoraxjnl-2012-201871
39. Finney LJ, Ritchie A, Pollard E, Johnston SL, Mallia P. Lower airway colonization and inflammatory response in COPD: a focus on haemophilus influenzae. Int J Chron Obstruct Pulmon Dis. 2014;9:1119–1132. doi:10.2147/COPD.S54477
40. Heltshe SL, Mayer-Hamblett N, Burns JL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with g551d-CFTR treated with ivacaftor. Clin Infect Dis. 2015;60(5):703–712. doi:10.1093/cid/ciu944
41. Rowe SM, Heltshe SL, Gonska T, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in g551d-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014;190(2):175–184. doi:10.1164/rccm.201404-0703OC
42. Hisert KB, Heltshe SL, Pope C, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. 2017;195(12):1617–1628. doi:10.1164/rccm.201609-1954OC
43. Pezzulo AA, Tang XX, Hoegger MJ, et al. Reduced airway surface ph impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–113. doi:10.1038/nature11130
44. Ni I, Ji C, Vij N, Chu HW. Second-hand cigarette smoke impairs bacterial phagocytosis in macrophages by modulating CFTR dependent lipid-rafts. PLoS One. 2015;10(3):e0121200. doi:10.1371/journal.pone.0121200
45. Patel IS, Seemungal TA, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax. 2002;57(9):759–764. doi:10.1136/thorax.57.9.759
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.