")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Efficacy and Safety of Once-Daily Inhaled Umeclidinium in Asian Patients with COPD: Results from a Randomized, Placebo-Controlled Study
Authors Zhong N, Zheng J , Lee SH , Lipson DA , Du X, Wu S
Received 8 May 2019
Accepted for publication 10 January 2020
Published 17 April 2020 Volume 2020:15 Pages 809—819
DOI https://doi.org/10.2147/COPD.S215011
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Nanshan Zhong,1 Jinping Zheng,1 Sang Haak Lee,2 David A Lipson,3 Xin Du,4 Shunquan Wu4
1State Key Laboratory of Respiratory Disease, National Clinical Research Centre of Respiratory Disease, Guangzhou Institute of Respiratory Health, First Affiliated Hospital of Guangzhou Medical University, Guangzhou, People’s Republic of China; 2Department of Internal Medicine, Eunpyeong St. Mary’s Hospital, College of Medicine, the Catholic University of Korea, Seoul, South Korea; 3GSK, Collegeville, and Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA; 4GSK, Shanghai, People’s Republic of China
Correspondence: Jinping Zheng Email [email protected]
Purpose: Previous studies demonstrating efficacy and safety of once-daily umeclidinium (UMEC) in patients with chronic obstructive pulmonary disease (COPD) have included few Asian patients. This study evaluated efficacy and safety of UMEC 62.5 mcg versus placebo in Asian patients with COPD.
Patients and Methods: A Phase III, randomized, double-blind, parallel-group study. Patients (aged ≥ 40 years with COPD, pre-, and post-albuterol forced expiratory volume in 1 s [FEV1]/forced vital capacity ratio < 0.70 and low risk of exacerbations) were randomized 2:1 to once-daily UMEC 62.5 mcg or placebo via the ELLIPTA inhaler for 24 weeks. Primary endpoint was change from baseline (CFB) in trough FEV1 on Day 169. Secondary endpoints were weighted mean FEV1 over 0– 6 hrs post-dose on Day 1 and CFB in Transition Dyspnea Index (TDI) focal score on Day 168.
Results: A total of 306 patients were included in the modified intent-to-treat population (UMEC: 205; placebo: 101). UMEC versus placebo provided a statistically significant improvement in least squares (LS) mean trough FEV1 between baseline and Day 169 (154 mL [95% confidence interval (CI): 113, 194]; p< 0.001). A clinically meaningful difference of 125 mL in favor of UMEC (95% CI: 103, 147; p< 0.001) was also seen in LS weighted mean FEV1 0– 6 hrs post-dose on Day 1. A LS mean treatment difference in TDI focal score of 0.9 units in favor of UMEC was seen on Day 168 (95% CI: 0.3, 1.5; p=0.004). Incidence of on-treatment adverse events (AEs) was lower in the placebo (55%) versus UMEC arm (60%); non-fatal serious AEs, drug-related AEs, and AEs leading to withdrawal were similar with UMEC and placebo.
Conclusion: Once-daily UMEC 62.5 mcg resulted in statistically significant and clinically meaningful improvements in lung function and dyspnea, compared with placebo, in Asian patients with COPD, with no new safety concerns observed.
Keywords: Asia, chronic obstructive pulmonary disease, forced expiratory volume in 1 second, Transition Dyspnea Index, umeclidinium
Introduction
Chronic obstructive pulmonary disease (COPD), characterized by dyspnea, chronic cough, and sputum production is one of the leading causes of morbidity and death worldwide.1,2 In Asia, the prevalence of COPD has been reported to be between 5.4% and 13.6% and is increasing.3–8 A systematic review of patients with COPD in China showed that COPD has a high disease burden resulting in a high economic burden.9
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) strategy recommends long-acting muscarinic antagonist (LAMA) or long-acting β2-agonist (LABA) monotherapy as first-line treatment for patients with COPD in all GOLD groups, with the exception of LABA monotherapy in group D.2 The LAMA tiotropium (TIO) significantly improves lung function and health status, and reduces exacerbation rates in patients with COPD.10 However, some limitations have been reported with TIO, including delayed onset of action.11 As such, a number of LAMAs were developed to overcome some of the limitations associated with TIO in order to improve efficacy. One such LAMA is umeclidinium (UMEC), a quinuclidine derivative that is a potent, inhaled LAMA with a prolonged duration of action compared with other anticholinergic agents. Studies have reported that UMEC has similar pharmacokinetic properties in different ethnic populations, including a healthy Chinese population.11 Furthermore, UMEC has demonstrated superior efficacy on lung function compared with TIO.11,12
UMEC is approved as a maintenance bronchodilator treatment for adult patients with COPD.13,14 Previous studies have demonstrated the efficacy and safety of once-daily UMEC in COPD populations worldwide12,15,16 and one study has reported the efficacy and safety of the UMEC in combination with vilanterol in an Asian population;17 however, there are currently no studies of UMEC monotherapy in patients of Asian origin. A comparison of baseline characteristics in an international COPD trial demonstrated baseline differences between Western and Asian patients, with the Asian study population containing a higher proportion of men, had a lower body mass index, and a higher incidence of exacerbations.18 Study population differences and racial differences in socioeconomic status and clinical practice4,19,20 could influence the relative efficacy and safety of therapy in patients with COPD, and it is therefore important to assess therapies in different ethnic populations. The aim of this study was to evaluate the efficacy and safety of UMEC compared with placebo in Asian patients with COPD.
Methods
Study Design and Treatments
This 24-week, Phase III, randomized, double-blind, parallel-group study (GSK study number AC117410; NCT02184611) compared the efficacy and safety of UMEC 62.5 mcg with placebo in patients with COPD across 21 centers in China and five centers in the Republic of Korea. The study took place between May 2016 and November 2017. Following screening (Visit 1), patients entered a 7–14-day run-in period before being randomized 2:1 to receive UMEC 62.5 mcg or placebo once daily via the ELLIPTA inhaler for 24 weeks. Following randomization (Day 1, Visit 2), patients attended a further 7 outpatient clinic visits on Days 2, 28, 56, 84, 112, 168, and 169. Additionally, a follow-up telephone call took place 7 days after the end of study treatment. Rescue albuterol was provided to patients for use as needed.
Patients were assigned to treatment using a central random allocation sequence generated by the study statistician using a validated computerized system (RAMOS NG software). The block size for randomization was six, with a ratio of 4:2 for UMEC:placebo. Double-blinding was ensured by using ELLIPTA inhalers containing UMEC or placebo that were identical in appearance. Investigators enrolled the patients and assigned their intervention.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, International Council for Harmonisation Good Clinical Practice (GCP), and the applicable country-specific regulatory requirements and study materials (protocol, informed consent) were reviewed and approved by a regional, investigational center ethics committee. The study protocol is available at https://www.gsk-studyregister.com.
Eligibility Criteria
Patients were ≥40 years of age, of Asian ancestry, with a diagnosis of COPD according to the American Thoracic Society/European Respiratory Society definition and were current or former cigarette smokers with a history of ≥10 pack-years. At screening, eligible patients had pre- and post-albuterol forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) ratio of <0.70, pre- and post-albuterol FEV1 ≤70% predicted, and a Modified Medical Research Council (mMRC) Dyspnea Scale score ≥2. The following concomitant treatments were permitted during the study: study-provided salbutamol (withheld for 4 hrs prior to spirometry testing); theophylline (provided the dose remained stable throughout the study, and was withheld for 12 hrs prior to each clinic visit); ipratropium bromide (provided the dose remained stable throughout the study, andwas withheld for 4 hrs prior to each clinic visit); inhaled corticosteroids (ICS) at a dose of ≤1000 mcg/day of fluticasone propionate or equivalent; mucolytics such as acetylcysteine; medications for rhinitis (eg, intranasal corticosteroids, antihistamines, cromolyn, nedocromil, nasal decongestants); influenza or pneumonia vaccinations; short-term antibiotics; oral anticholinergics for overactive bladder; pulmonary rehabilitation in the maintenance phase; smoking cessation treatment, including a stable regimen of nicotine replacement; and positive airway pressure for sleep apnea.
Patients diagnosed with asthma were excluded, as were those who smoked but used a pipe or cigar only and those who had been hospitalized for COPD or pneumonia in the 12 weeks prior to screening or had undergone lung volume reduction surgery in the 12 months prior to screening. Patients who had clinically significant abnormalities not due to COPD shown by a chest X-ray or computed tomography scan, and those who were unable to withhold albuterol for 4 hrs prior to spirometry testing were also excluded, as were patients on long-term oxygen therapy (>12 hrs per day) or regular short-acting bronchodilator therapy via a nebulizer, and those who were in the acute phase of a pulmonary rehabilitation program in the 4 weeks prior to Visit 1.
Following the run-in period, patients eligible to be randomized to treatment had to demonstrate a normal pre-dose electrocardiogram, have had no COPD exacerbations (defined as worsening of COPD symptoms requiring additional treatment other than study treatment or rescue albuterol) or lower respiratory tract infections during the run-in period, and have maintained a stable, regular dose of ≤1000 mcg/day fluticasone propionate or equivalent during the run-in period (if using ICS). All patients provided written informed consent.
Patients experiencing a COPD exacerbation requiring treatment with systemic corticosteroids, with prescribed or non-prescribed antibiotics and/or emergency treatment or hospitalization during the study were withdrawn; however, patients with an exacerbation that required treatment with antibiotics alone could continue participating in the study.
Endpoints and Assessments
The primary efficacy endpoint was change from baseline (CFB) in trough FEV1 on Day 169. Secondary endpoints were weighted mean FEV1 over 0–6 hrs post dose (measurements obtained at 15 and 30 mins, 1, 3, and 6 hrs after administration of study medication) on Day 1 and Transition Dyspnea Index (TDI) focal score on Day 168. Other efficacy endpoints included proportion of FEV1 responders (defined as achieving a ≥100 mL increase from baseline in trough FEV1) on Day 169, CFB in trough FEV1 and trough FVC on Days 2, 28, 56, 84, 112, and 168, serial FEV1 and FVC over 0–6 hrs post-dose on Day 1. All spirometry measurements were collected in the morning, and salbutamol was withheld for at least 4 hrs prior to testing. Trough and pre-dose measurements were collected after a study medication washout period of approximately 24 hrs.
TDI focal score and proportion of TDI responders (defined as a TDI score ≥1 unit) on Days 28, 84, and 168, on-treatment COPD exacerbations, time to first COPD exacerbation and rescue albuterol use (puffs/day, recorded by patients in a diary) over Weeks 1 to 24 were also measured. Additionally, health outcomes assessments included St George’s Respiratory Questionnaire (SGRQ) and COPD Assessment Test (CAT), assessed at baseline, Day 28, Day 84, and Day 168. SGRQ responders were defined as patients achieving a decrease from baseline in SGRQ total score ≥4 units and CAT responders were defined as patients achieving a decrease from baseline in CAT score of ≥2 units.
Safety endpoints included the incidence of adverse events (AEs) and serious AEs (SAEs) over the 24-week treatment period.
Statistical Analysis
Sample size calculations estimated that 288 patients needed to be randomized (UMEC n=192, placebo n=96) to provide 156 evaluable patients in the UMEC arm and 78 evaluable patients in the placebo arm (assuming an 18% withdrawal rate) to give 94% power to detect a true treatment difference of 115 mL in trough FEV1 on Day 169.
Analyses were conducted in the modified intent-to-treat (mITT) population, which included all patients who were randomized and received at least one dose of randomized study drug.
CFB in trough FEV1, trough FVC (measurements taken on Days 2, 28, 56, 84, 112, 168, and 169), SGRQ total score, CAT score (measurements taken on Days 1, 28, 84, and 168) and TDI focal score (measurements taken on Days 28, 84, and 168) were analyzed using mixed model repeated measures, adjusting for treatment, smoking status at screening, country, day, baseline, baseline by day and treatment by day. Weighted mean FEV1 over 0–6 hrs post dose on Day 1 and puffs/day of albuterol were analyzed using an analysis of covariance (ANCOVA) model, with covariates of treatment, smoking status at screening, baseline, and country. Serial FEV1 and FVC over 0–6 hrs post-dose on Day 1 were analyzed using a repeated measures model, with covariates of treatment, baseline FEV1, country, time, time by treatment and time by baseline interactions. The proportions of FEV1, TDI, SGRQ, and CAT responders at each visit were analyzed using a logistic regression model with covariates of treatment, baseline, smoking status, and country.
Kaplan–Meier and Cox proportional hazards regression analysis with covariates of treatment, smoking status at screening and country was performed to analyze the incidence of a first COPD exacerbation.
Significance tests were performed at the two-sided 5% alpha level.
Results
Patient Disposition and Demographics
Patients were recruited between May 2016 and May 2017, and the final follow-up visit was in November 2017. The mITT population comprised 306 patients of East Asian heritage (Figure 1), with a mean age (standard deviation [SD]) of 65.7 (7.05) years; 290 (95%) patients were male; 205 patients received UMEC and 101 received placebo (Table 1). Demographic and clinical characteristics were similar between the UMEC and placebo treatment groups (Table 1). Use of on-treatment permitted concomitant medications was similar between UMEC and placebo groups (Supplementary Table 1).
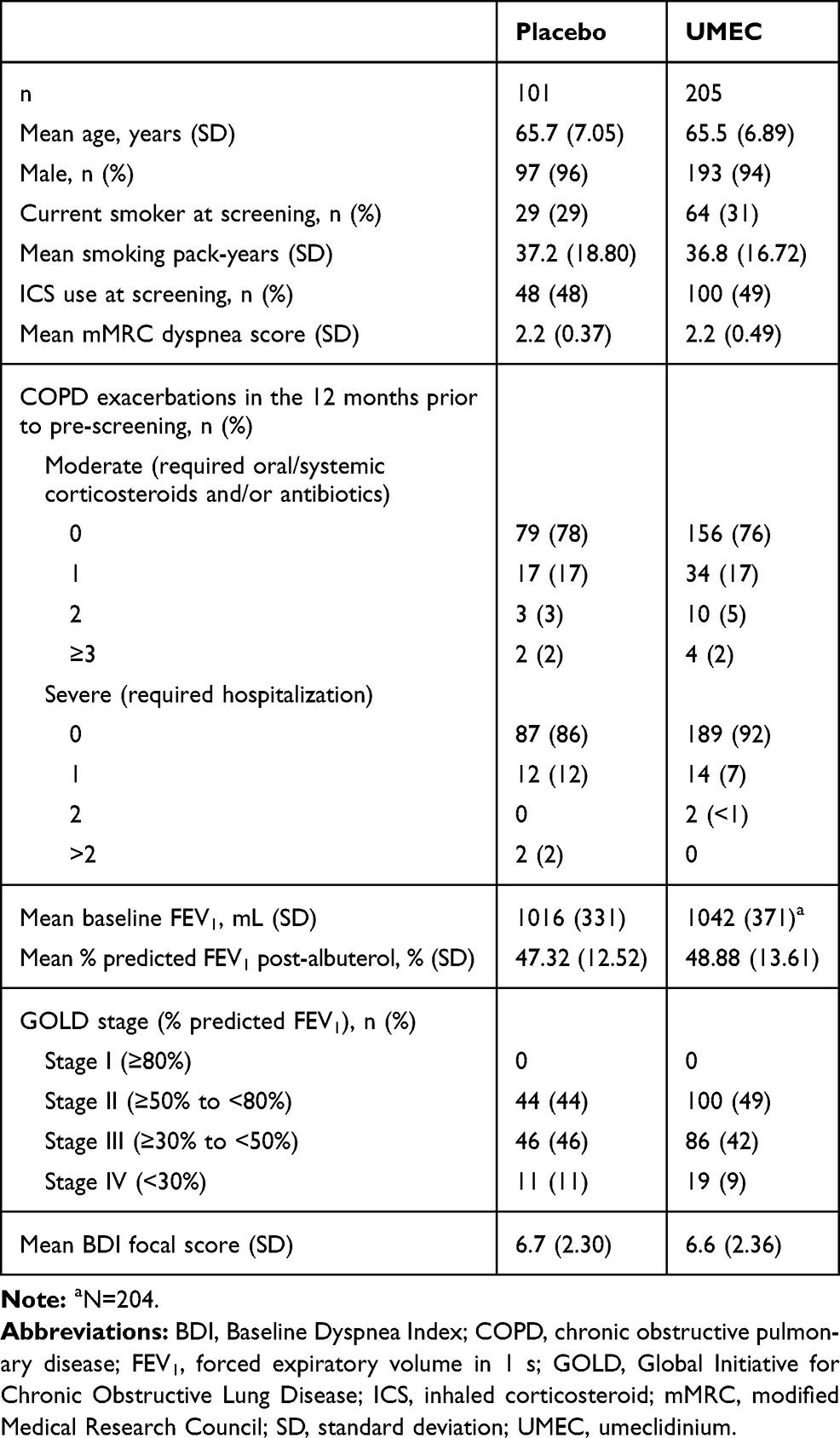 |
Table 1 Patient Demographics and Baseline Characteristics |
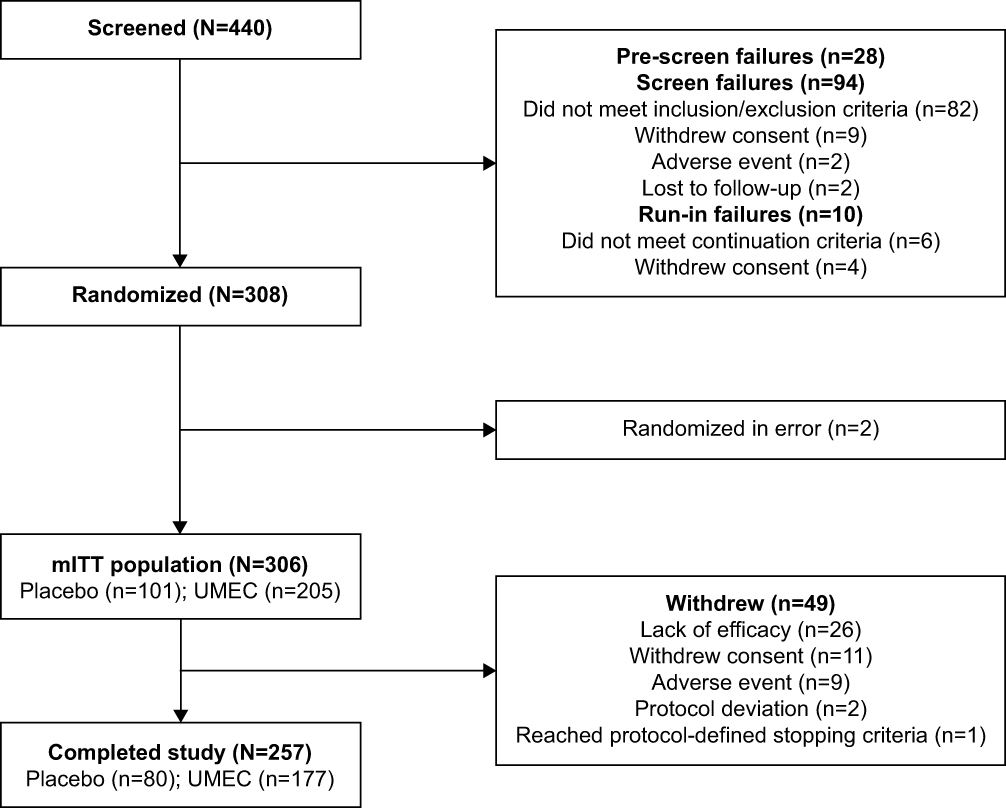 |
Figure 1 Patient disposition. Abbreviations: mITT, modified intent-to-treat; UMEC, umeclidinium. |
Overall, 257/306 (84%) patients completed the study (placebo n=80/101 [79%]; UMEC n=177/205 [86%]), and 49/306 (16%) withdrew (placebo n=21/101 [21%]; UMEC n=28/205 [14%]; Figure 1). The most common reason for withdrawal in both treatment groups was lack of efficacy (placebo n=11/101 [11%]; UMEC n=15/205 [7%]).
Efficacy Outcomes
Lung Function
The primary endpoint was met, with CFB in least squares (LS) mean trough FEV1 on Day 169 of −22 mL in the placebo arm and 131 mL in the UMEC arm, equating to a clinically meaningful (≥100 mL)21 and statistically significant improvement of 154 mL (95% confidence interval [CI]: 113, 194; p<0.001) (Table 2). At all other time points (Days 2, 28, 56, 84, 112, and 168), CFB in LS mean trough FEV1 was statistically significantly in favor of UMEC over placebo (p<0.001) and treatment differences were clinically meaningful (120–192 mL; Figure 2A). The odds of achieving a ≥100 mL increase in trough FEV1 on Day 169 were significantly higher with UMEC versus placebo (placebo n=18/101 [18%], UMEC n=101/203 [50%]; odds ratio [OR] [95% CI] 4.90 [2.71, 8.87]; p<0.001).
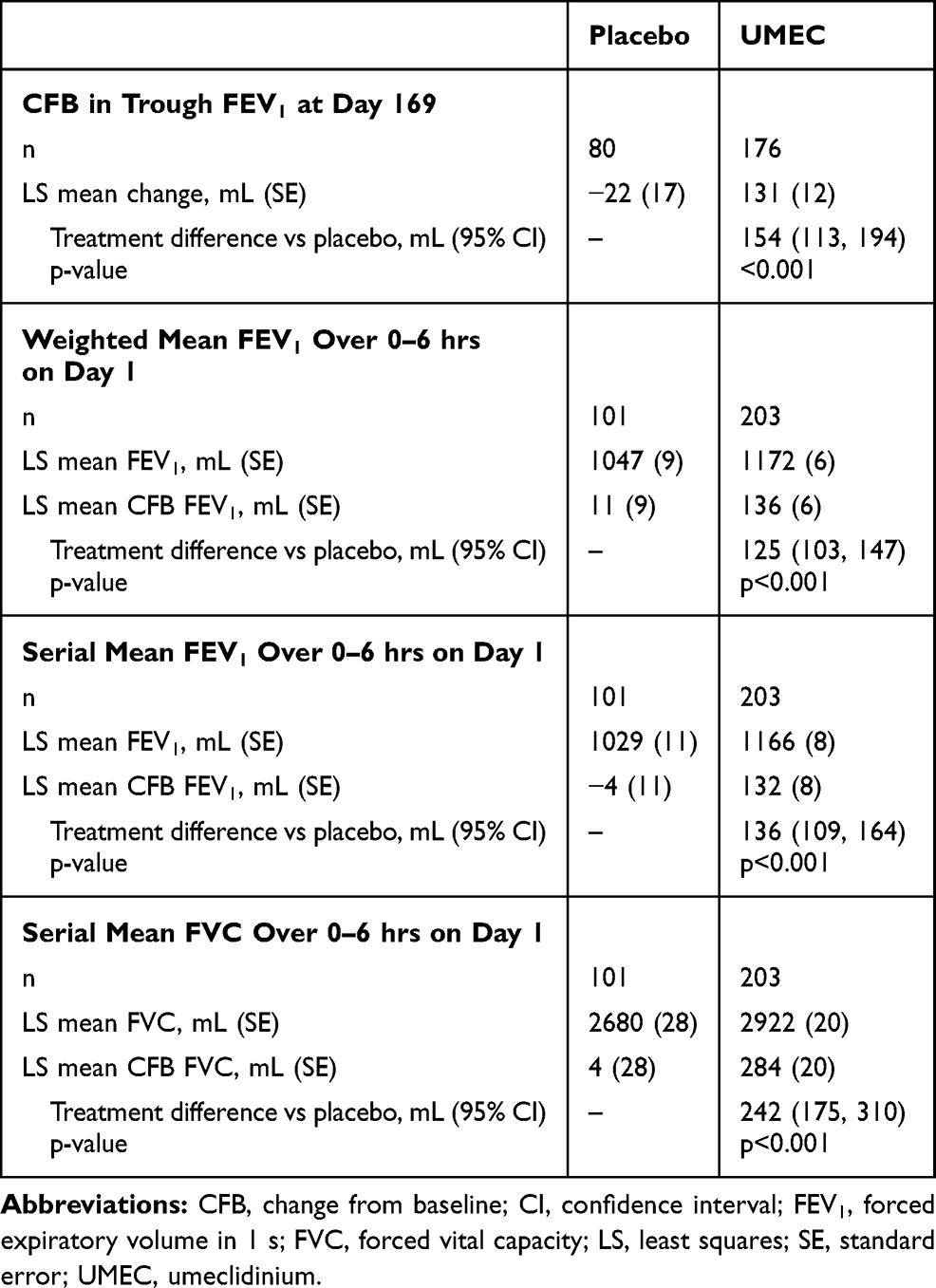 |
Table 2 Lung Function Endpoints |
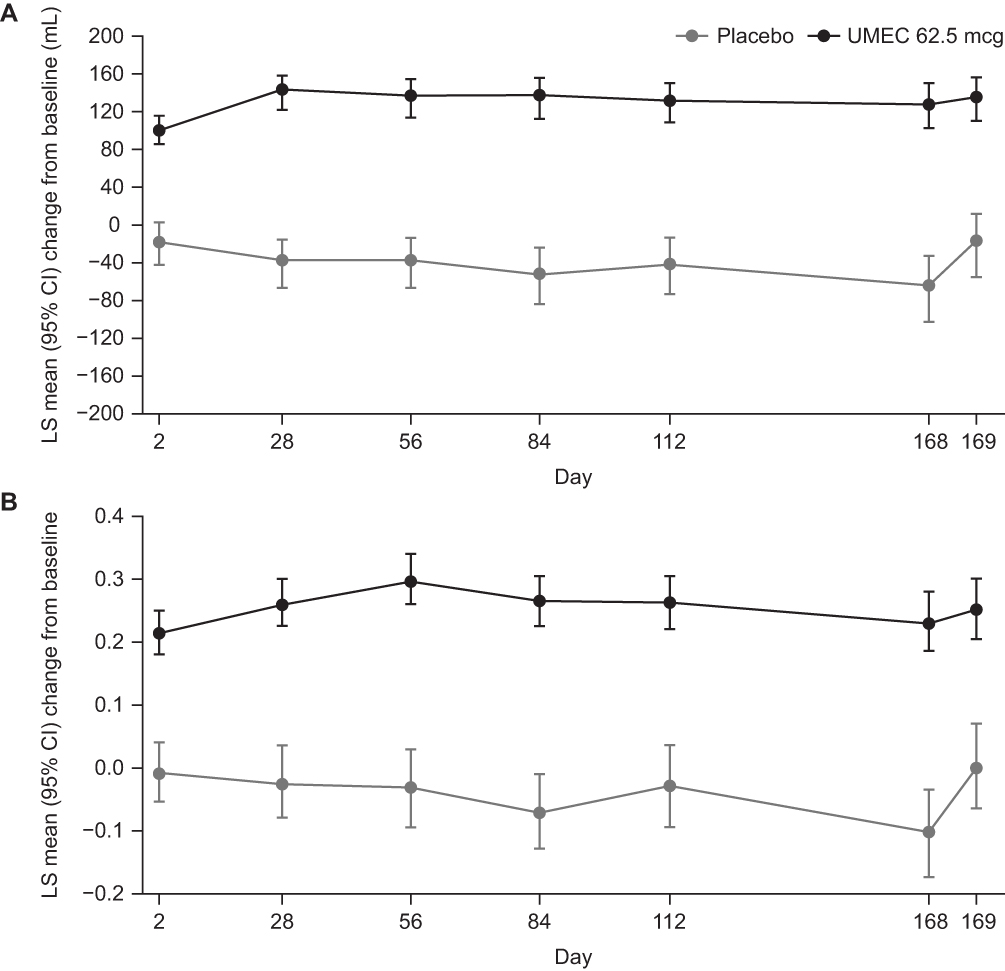 |
Figure 2 Change from baseline in (A) trough FEV1 and (B) trough FVC. Abbreviations: CI, confidence interval; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; LS, least squares; UMEC, umeclidinium. |
LS weighted mean FEV1 0–6 hrs post dose on Day 1 increased relative to baseline by 11 mL in the placebo arm and 136 mL in the UMEC arm, giving a clinically meaningful difference of 125 mL in favor of UMEC (95% CI: 103, 147; p<0.001; Table 2). Similarly, a clinically meaningful difference in favor of UMEC was seen in the CFB in serial FEV1 over 0–6 hrs post dose on Day 1, with LS mean changes of 4 mL in the placebo arm and 132 mL in the UMEC arm, equating to a between-treatment difference of 136 mL (95% CI: 109, 164; p<0.001; Table 2).
A statistically significant difference in CFB in trough FVC was seen in favor of UMEC at all time points (Figure 2B). Additionally, a clinically meaningful difference in favor of UMEC was seen in the CFB in serial FVC over 0–6 hrs post dose on Day 1, with LS mean increases of 4 mL in the placebo arm and 284 mL in the UMEC arm, equating to a between-treatment difference of 242 mL (95% CI: 175, 310; p<0.001; Table 2).
TDI Score
LS mean (SD) TDI focal score on Day 168 was 1.6 (0.26; n=80) in the placebo arm, and 2.5 (0.17; n=176) in the UMEC arm, equating to a LS mean treatment difference of 0.9 units in favor of UMEC (95% CI: 0.3, 1.5; p=0.004). The LS mean TDI focal score was also statistically significantly higher in the UMEC versus placebo groups at Day 28 but not Day 84 (Figure 3).
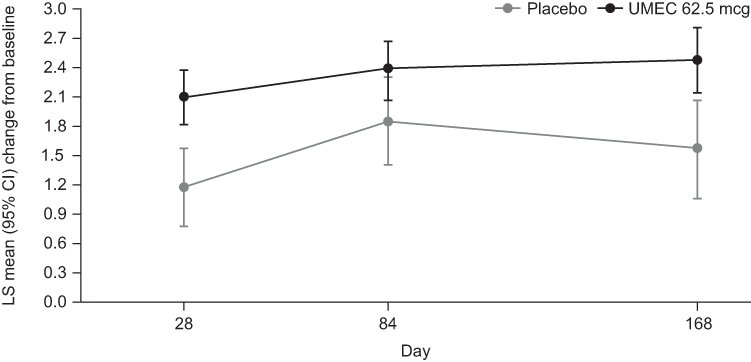 |
Figure 3 TDI focal score at Days 28, 84, and 168. Abbreviations: CI, confidence interval; LS, least squares; TDI, Transient Dyspnea Index; UMEC, umeclidinium. |
On Day 168, the proportion of TDI responders was statistically significantly higher in the UMEC group compared with the placebo group (66% vs 50%; OR 2.0 [95% CI: 1.2, 3.4]; p=0.007), whereas at Days 28 and 84 the difference between treatment groups was not statistically significant (Table 3).
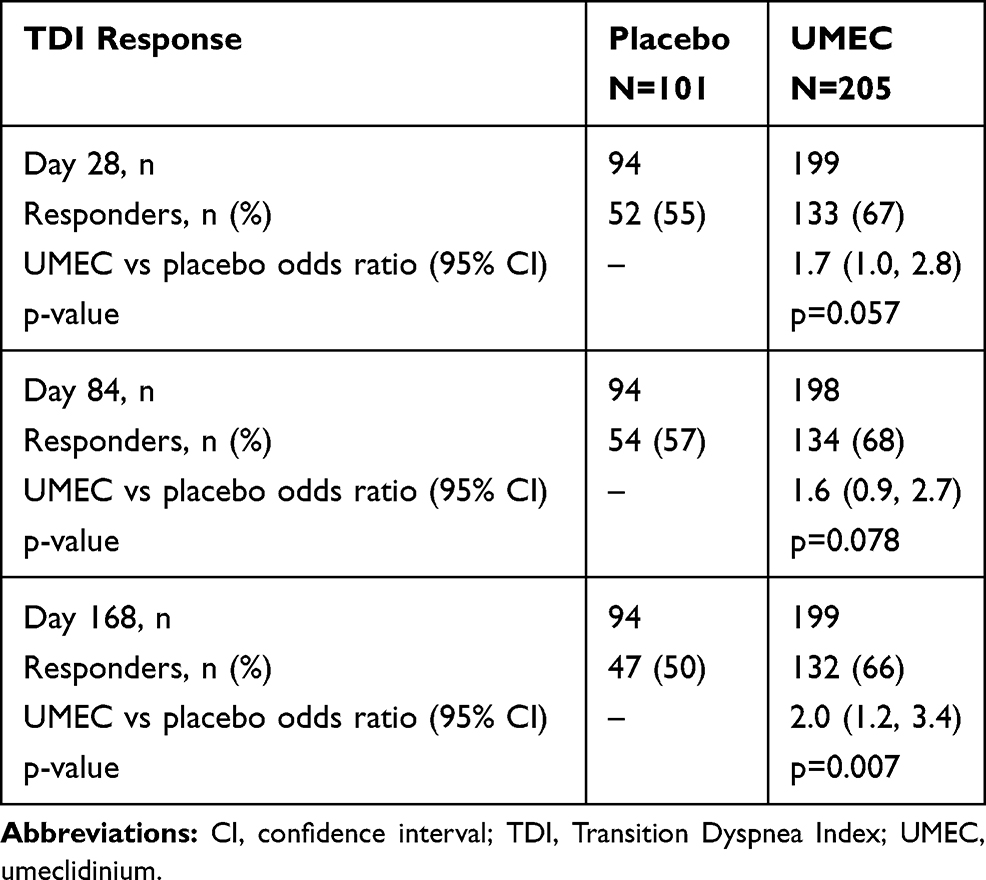 |
Table 3 Proportion of TDI Responders (Defined as a TDI Focal Score ≥1 Unit) |
COPD Exacerbations
On-treatment COPD exacerbations were experienced by 19/101 (19%) patients in the placebo arm and 24/205 (12%) patients in the UMEC arm. The probability of having a first COPD exacerbation was lower in the UMEC arm (12.5%) compared with the placebo arm (20.2%). The hazard ratio was directionally in favor of UMEC versus placebo (0.6 [95% CI: 0.3, 1.1]) but was not statistically significant (p=0.078). The majority of exacerbations were treated with antibiotics and/or steroids. Few exacerbations required hospitalization (placebo 4/21 [19%] exacerbations, UMEC 5/28 [18%] exacerbations).
Rescue Medication Use
The mean CFB in the number of puffs of rescue medication per day over Weeks 1 to 24 was statistically significantly in favor of UMEC over placebo (−0.5; 95% CI:-0.9, −0.1; p=0.008; Table 4).
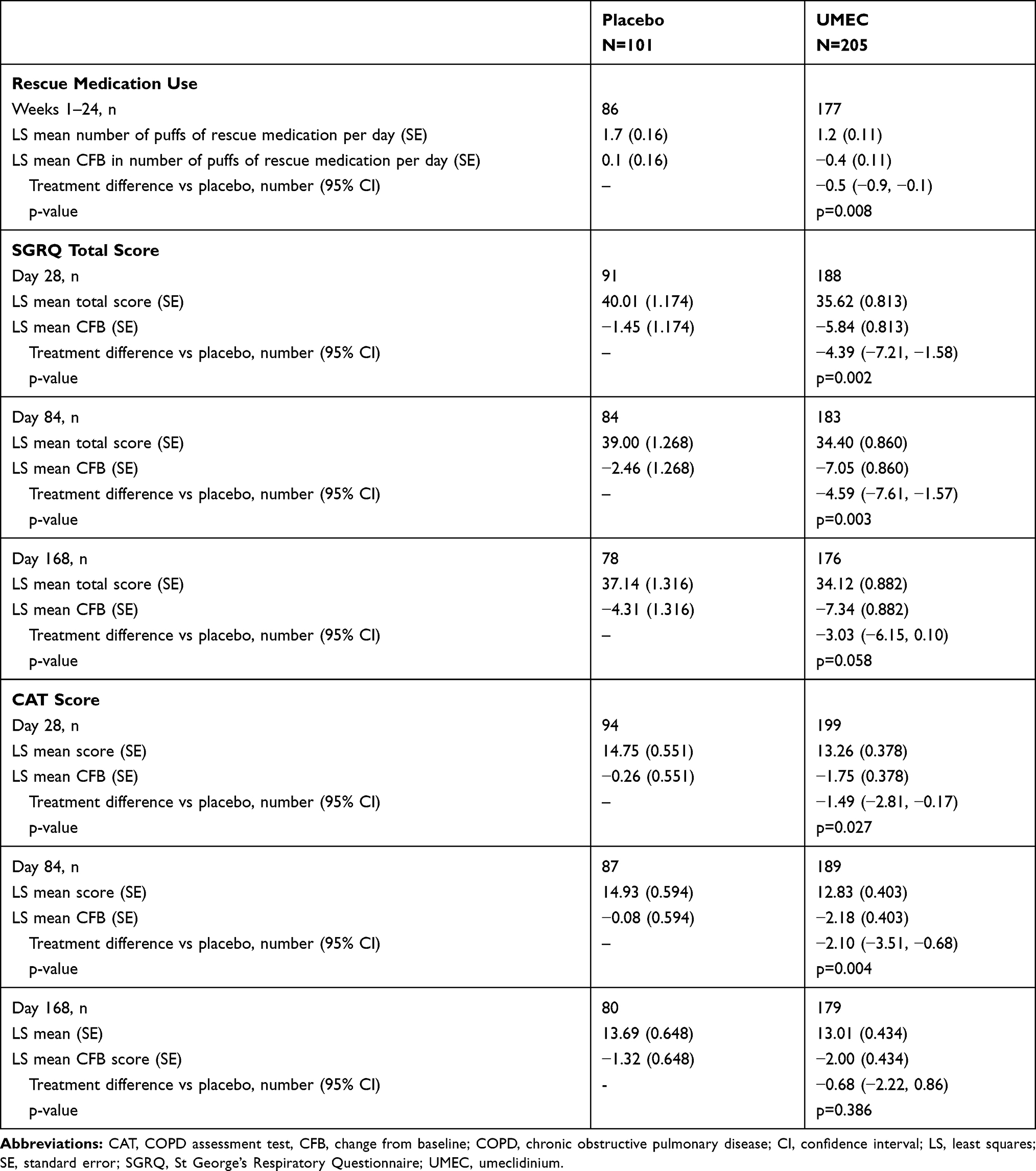 |
Table 4 Rescue Medication Use, Mean SGRQ Total Score and CAT Score |
Health Outcomes
SGRQ
CFB in SGRQ total score showed significantly greater improvements in the UMEC arm compared with the placebo arm at Days 28 and 84 (Table 4). The odds of being a responder were statistically significantly in favor of UMEC compared with placebo on Day 84 (placebo n=33/91 [36%], UMEC n=98/194 [51%]; OR 1.7; 95% CI: 1.0, 2.9; p=0.042). At Day 28 (placebo n=36/91 [40%], UMEC n=95/188 [51%]; OR 1.4; 95% CI: 0.8, 2.5; p=0.186) and Day 168 (placebo n=45/92 [49%], UMEC n=104/195 [53%]; OR 1.1; 95% CI: 0.7, 1.9; p=0.633) the odds of being a responder were not significantly different between treatment groups.
CAT
The CFB in CAT score showed statistically significant greater improvements in the UMEC arm compared with the placebo arm at Days 28 and 84 but not Day 168 (Table 4). The odds of being a responder versus a non-responder were significantly in favor of UMEC over placebo at Day 84 (placebo n=31/94 [33%], UMEC n=102/199 [51%]; OR 1.9; 95% CI: 1.1, 3.3; p=0.017) but not at Day 28 (placebo n=41/94 [44%], UMEC n=103/199 [52%]; OR 1.2; 95% CI: 0.7, 2.0; p=0.490) or Day 168 (placebo n=38/94 [40%], UMEC n=10/199 [50%]; OR 1.3; 95% CI: 0.8, 2.2; p=0.324).
Safety
The incidence of on-treatment AEs was higher in the UMEC arm (n=122/205; 60%) compared with the placebo arm (n=56/101; 55%). The incidence of non-fatal SAEs, drug-related AEs, and AEs leading to withdrawal was similar between the treatment groups (Table 5). The most common AE in both treatment groups was upper respiratory tract infections (placebo n=13/101 [13%]; UMEC n=25/205 [12%]; Table 5). There were no drug-related fatal SAEs; one (<1%) patient in the UMEC arm had a fatal SAE (sudden death), which was not considered to be drug related by the investigator.
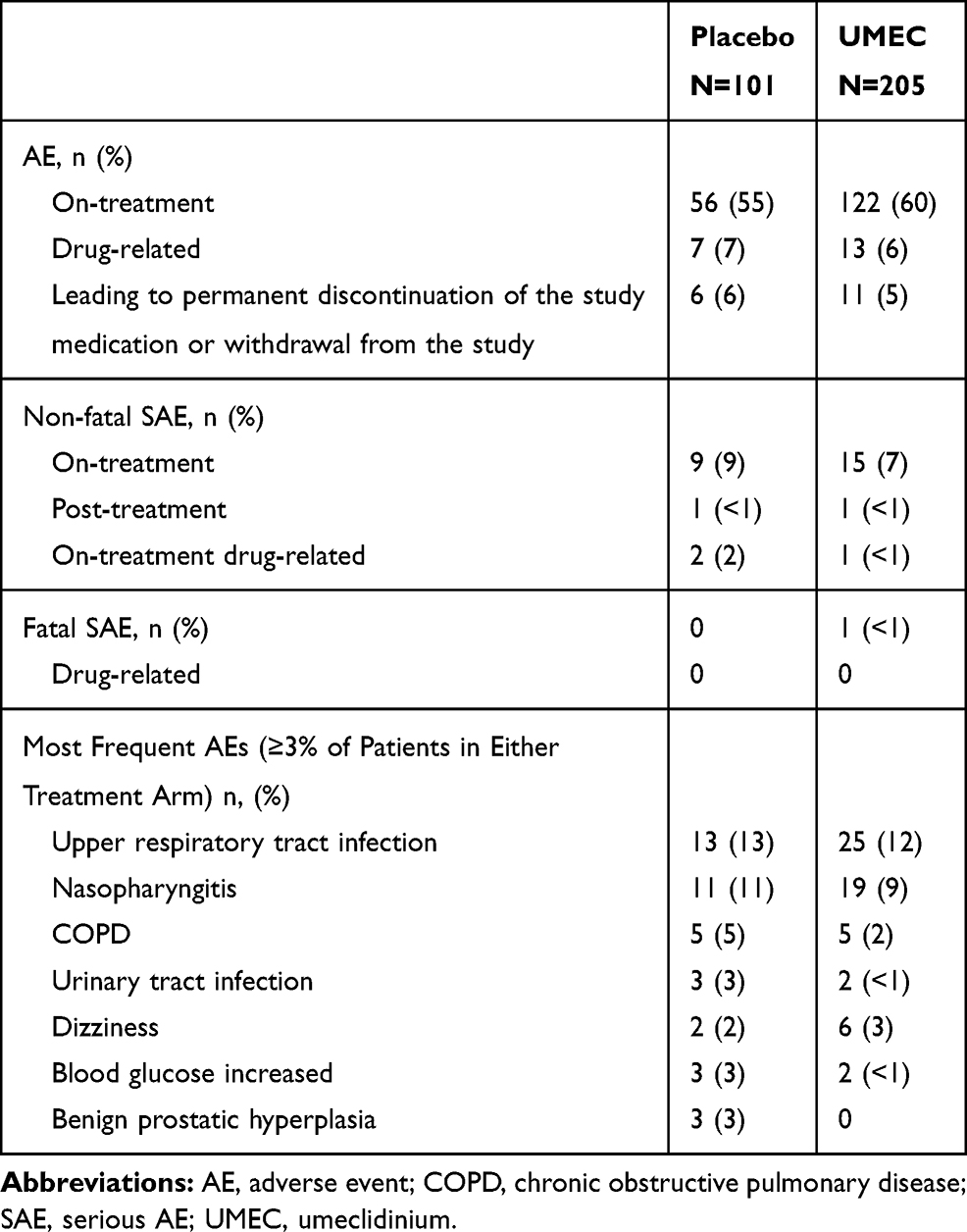 |
Table 5 Summary of on-Treatment AEs |
Discussion
This is the first study of the efficacy and safety of UMEC monotherapy in patients of Asian origin. It provides clinically important data that builds on the evidence for the use of LAMA monotherapy in other patient populations with COPD.12,15,16 In this 24 weeks, randomized study of UMEC 62.5 mcg versus placebo in Asian patients with COPD, statistically significant improvements in favor of UMEC were demonstrated for lung function parameters, dyspnea (at Days 28 and 168), rescue medication use, and SGRQ and CAT at Days 28 and 84. No new safety concerns were raised in this population. The findings of the current study confirm and extend those of previous studies that have demonstrated the efficacy and safety of once-daily UMEC 62.5 mcg in patients with COPD that have not focused on Asian patients.12,15,16
A change in trough FEV1 of 100 mL is considered to be the minimum clinically important difference perceived by patients with COPD.21 The FEV1 improvements demonstrated with UMEC in the current study exceeded this at all time points up to Day 169, and 6 hrs post dose on Day 1. In addition, statistically significant improvements were demonstrated in FVC for UMEC versus placebo, indicating a reduction in hyperinflation or air-trapping. These findings are consistent with those observed in other studies of once-daily UMEC 62.5 mcg in patients with COPD who were predominantly Caucasian.15,22–24 While our study cannot be directly compared with studies conducted in predominantly Caucasian populations due to differences in eligibility criteria and study design (eg, study length), the correlation between lung function outcomes in a number of studies suggests minimal inter-ethnic differences in response to treatment with UMEC among Asian patients and those of other ethnicities.
Importantly, statistically significant improvements in favor of UMEC were seen in other clinically relevant endpoints, including TDI focal score and the proportion of TDI responders on Day 168. Again, these findings are in line with those seen in studies with predominately Caucasian populations.24 In the present study, symptom improvement was also demonstrated by the reduction in rescue medication use, which was statistically significant in favor of UMEC over placebo. There were also fewer patients with moderate or severe COPD exacerbations during 6 months of UMEC treatment compared with placebo, which is consistent with other studies performed in predominantly Caucasian populations.15,24
Compared with placebo, improvement in health-related quality of life (HRQoL), measured by the SGRQ and CAT, was significantly greater with UMEC at Days 28 and 84 and the odds of a clinically significant response were also greater with UMEC versus placebo at Day 84. However, these clinically important differences were not maintained at Day 168 as for both HRQoL measures there was an unexpected increase in the placebo response that minimized the magnitude of the UMEC versus placebo benefit.
Current guidelines recommend LAMA or LABA monotherapy alone as initial treatment for symptomatic patients at a low risk of exacerbations.2 Similar to the current study, benefits in Asian patients with COPD have also been reported in studies of other LAMAs, such as TIO and glycopyrronium. In the 4-year UPLIFT study, patients receiving once-daily TIO 18 mcg demonstrated improvements in FEV1 compared with patients receiving placebo10 and in a subanalysis of Asian patients, the response to TIO was generally comparable between Asian patients and the total cohort, with improvements in lung function, HRQoL, and reductions in exacerbation frequency among those treated with TIO compared with control.25 Improvements with LAMA monotherapy in an Asian population were also shown in a 26-week, multicenter, double-blind, placebo-controlled, parallel-group study in predominantly Chinese patients with moderate-to-severe COPD, in which once-daily glycopyrronium 50 mcg significantly improved lung function, dyspnea, and health status compared with placebo.26
The strength of this study lies in its robust design and the fact that it was conducted in a large, well-characterized Asian population with COPD at low risk of exacerbations. It therefore provides much needed evidence for how UMEC 62.5 mcg performs in this specific population. However, there are some limitations to consider. For example, there was a higher rate of withdrawals in the placebo arm compared with the UMEC arm, which may have led to a loss of differentiation between treatments. In addition, TDI response in the placebo group was unexpectedly high, although similar placebo effects have been demonstrated for TDI in previous studies.17,24 The underlying cause of these effects remains unclear, but may be related to the Hawthorne effect, a process by which study participants experience improved outcomes regardless of their treatment allocation.27 In addition, 95% of the patients in this study were male, which is consistent with a previous study in Asian patients with COPD18 but may limit generalizability of the findings to the broader patient population. Despite this, the study provides valuable data on the efficacy and safety of UMEC 62.5 mcg compared with placebo in a population of Asian patients with COPD, providing useful and relevant information for prescribing physicians in Asia.
Conclusions
In Asian patients with COPD, compared with placebo, UMEC 62.5 mcg administered once-daily via the ELLIPTA inhaler over a 24-week treatment period resulted in statistically significant improvements in lung function and dyspnea throughout the study and HRQoL at Days 24 and 84, with no new safety concerns observed.
Ethics Approval and Informed Consent
The study protocol, any amendments, the informed consent form, and any other materials requiring pre-approval were reviewed and approved by the ethics committee at each investigational center. The investigational centers in China were 3rd Xiangya Hospital, Central South University, Changsha; Affiliated Hospital of Guangdong Medical College, Guangdong, Zhanjiang; The First Hospital of Shanxi Medical University, Shanxi, Taiyuan; Hangzhou First People`s Hospital, Zhejiang, Hangzhou; The Second Hospital of Jilin University, Jilin, Changchun; Wuxi People’s Hospital, Jiangsu, Wuxi; Daping Hospital, Third Military Medical University, Chongqing; Southwest Hospital, Third Military Medical University, Chongqing; The First Affiliated Hospital of Nanchang University, Jiangxi, Nanchang; Shanghai Pulmonary Hospital, Shanghai; Hai Nan Provincial People’s Hospital, Hainan, Haikou; The First Affiliated Hospital of Guangzhou Medical College, Guangzhou; General Hospital of Shenyang Military Command, Liaoning, Shenyang; Shengjing Hospital of China Medical University, Liaoning, Shenyang; General Hospital of Ningxia Medical University, Ningxia, Yinchuan; Jinan Central Hospital Affiliated to Shandong University, Shandong, Jinan; Qingdao Municipal Hospital, Shandong, Qingdao; Peking University First Hospital, Beijing; 1st Affiliated Hospital of Jinan University, Guangdong, Guangzhou; 2nd Affiliated Hospital to Nanchang University, Nanchang; Inner Mongolia People’s Hospital, Inner Mongolia, Huhhot. The investigational centers in the Republic of Korea were: Konkuk University Chungju Hospital, Seoul; The Catholic University of Korea St. Vincent’s Hospital, Suwon; St. Paul’s Hospital, Catholic University of Korea, Seoul; Kyung Hee University Medical Center, Seoul; National Health Insurance Corporation Ilsan Hospital, Gyeonggi-do. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, International Council for Harmonisation Good Clinical Practice (GCP), and the applicable country-specific regulatory requirements. All patients provided written informed consent to participate in the study. Patient consent is not required for publication of the results as all data are anonymized.
Data Sharing Statement
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
Acknowledgments
ELLIPTA is owned by or licensed to the GSK group of companies. Editorial support (in the form of writing assistance during development of the initial draft, assembling tables and figures, collating authors comments, grammatical editing, and referencing) was provided by Joanne Ashworth and Katie White, PhD, of Fishawack Indicia Ltd, UK, and was funded by GSK. This study was previously presented at the 23rd Congress of the Asian Pacific Society of Respirology, 29 November–2 December 2018, Taipei, Taiwan.
Author Contributions
All authors contributed to study conception and design, acquisition of data or data analysis and interpretation; drafting and revising the article; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Funding
GSK (study number AC117410, clinicaltrials.gov ID NCT02184611).
Disclosure
JZ has participated in advisory boards and speaker’s bureaus for AstraZeneca and Boehringer Ingelheim and has conducted a study supported by GSK. DAL and XD are employees of GSK and hold stocks and shares in the company. SW is an employee of GSK. The authors report no other conflicts of interest in this work.
References
1. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi:10.1016/S0140-6736(12)61728-0
2. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease; 2020.
3. Kim C, Yoo KH, Rhee CK, et al. Health care use and economic burden of patients with diagnosed chronic obstructive pulmonary disease in Korea. Int J Tuberc Lung Dis. 2014;18(6):737–743. doi:10.5588/ijtld.13.0634
4. Tan WC, Ng TP. COPD in Asia: where east meets west. CHEST. 2008;133(2):517–527. doi:10.1378/chest.07-1131
5. Zhong N, Wang C, Yao W, et al. Prevalence of chronic obstructive pulmonary disease in China: a large, population-based survey. Am J Respir Crit Care Med. 2007;176(8):753–760. doi:10.1164/rccm.200612-1749OC
6. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi:10.1371/journal.pmed.0030442
7. Yoo KH, Kim YS, Sheen SS, et al. Prevalence of chronic obstructive pulmonary disease in Korea: the fourth Korean National Health and Nutrition Examination Survey, 2008. Respirology. 2011;16(4):659–665. doi:10.1111/res.2011.16.issue-4
8. Fang L, Gao P, Bao H, et al. Chronic obstructive pulmonary disease in China: a nationwide prevalence study. Lancet Respir Med. 2018;6(6):421–430. doi:10.1016/S2213-2600(18)30103-6
9. Zhu B, Wang Y, Ming J, Chen W, Zhang L. Disease burden of COPD in China: a systematic review. Int J Chron Obstruct Pulmon Dis. 2018;13:1353–1364. doi:10.2147/COPD
10. Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543–1554. doi:10.1056/NEJMoa0805800
11. Babu KS, Morjaria JB. Umeclidinium in chronic obstructive pulmonary disease: latest evidence and place in therapy. Ther Adv Chronic Dis. 2017;8(4–5):81–91. doi:10.1177/2040622317700822
12. Feldman G, Maltais F, Khindri S, et al. A randomized, blinded study to evaluate the efficacy and safety of umeclidinium 62.5 mug compared with tiotropium 18 mug in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:719–730. doi:10.2147/COPD.S102494
13. Incruse Ellipta® (umeclidinium bromide) [summary of product characteristics]. Dublin, Ireland: GlaxoSmithKline; 2019.
14. Incruse Ellipta® (umeclidinium bromide) [prescribing information]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
15. Donohue JF, Anzueto A, Brooks J, Mehta R, Kalberg C, Crater G. A randomized, double-blind dose-ranging study of the novel LAMA GSK573719 in patients with COPD. Respir Med. 2012;106(7):970–979. doi:10.1016/j.rmed.2012.03.012
16. Donohue JF, Niewoehner D, Brooks J, O’Dell D, Church A. Safety and tolerability of once-daily umeclidinium/vilanterol 125/25 mcg and umeclidinium 125 mcg in patients with chronic obstructive pulmonary disease: results from a 52-week, randomized, double-blind, placebo-controlled study. Respir Res. 2014;15:78. doi:10.1186/1465-9921-15-78
17. Zheng J, Zhong N, Newlands A, Church A, Goh AH. Efficacy and safety of once-daily inhaled umeclidinium/vilanterol in Asian patients with COPD: results from a randomized, placebo-controlled study. Int J Chron Obstruct Pulmon Dis. 2015;10:1753–1767.
18. Kim KY, Miravitlles M, Sliwinski P, et al. Comparison of clinical baseline characteristics between Asian and Western COPD patients in a prospective, international, multicenter study. Int J Chron Obstruct Pulmon Dis. 2019;14:1595–1601.
19. Martin A, Badrick E, Mathur R, Hull S. Effect of ethnicity on the prevalence, severity, and management of COPD in general practice. Br J Gen Pract. 2012;62(595):e76–e81. doi:10.3399/bjgp12X625120
20. Wedzicha JA, Zhong N, Ichinose M, et al. Indacaterol/glycopyrronium versus salmeterol/fluticasone in Asian patients with COPD at a high risk of exacerbations: results from the FLAME study. Int J Chron Obstruct Pulmon Dis. 2017;12:339–349. doi:10.2147/COPD.S125058
21. Donohue JF. Minimal clinically important differences in COPD lung function. COPD. 2005;2(1):111–124. doi:10.1081/COPD-200053377
22. Decramer M, Maltais F, Feldman G, et al. Bronchodilation of umeclidinium, a new long-acting muscarinic antagonist, in COPD patients. Respir Physiol Neurobiol. 2013;185(2):393–399. doi:10.1016/j.resp.2012.08.022
23. Trivedi R, Richard N, Mehta R, Church A. Umeclidinium in patients with COPD: a randomised, placebo-controlled study. Eur Respir J. 2014;43(1):72–81. doi:10.1183/09031936.00033213
24. Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62. 5/25mcg in COPD. Respir Med. 2013;107(10):1538–1546. doi:10.1016/j.rmed.2013.06.001
25. Fukuchi Y, Fernandez L, Kuo HP, et al. Efficacy of tiotropium in COPD patients from Asia: a subgroup analysis from the UPLIFT trial. Respirology. 2011;16(5):825–835. doi:10.1111/j.1440-1843.2011.01982.x
26. Wang C, Sun T, Huang Y, et al. Efficacy and safety of once-daily glycopyrronium in predominantly Chinese patients with moderate-to-severe chronic obstructive pulmonary disease: the GLOW7 study. Int J Chron Obstruct Pulmon Dis. 2015;10:57–68. doi:10.2147/COPD.S72650
27. Sedgwick P, Greenwood N. Understanding the Hawthorne effect. BMJ. 2015;351:h4672. doi:10.1136/bmj.h4672
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.