")
Back to Journals » International Journal of Nanomedicine » Volume 19
Efficacy and Mechanism of a Biomimetic Nanosystem Carrying Doxorubicin and an IDO Inhibitor for Treatment of Advanced Triple-Negative Breast Cancer
Authors Hu C , Liu Y, Cao W, Li N, Gao S , Wang Z, Gu F
Received 6 October 2023
Accepted for publication 11 January 2024
Published 17 January 2024 Volume 2024:19 Pages 507—526
DOI https://doi.org/10.2147/IJN.S440332
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor R.D.K. Misra
Chuling Hu,1,* Yan Liu,2,* Wei Cao,3,* Na Li,4 Shen Gao,5 Zhuo Wang,5 Fenfen Gu2
1Department of Pharmacy, Jiaxing Maternity and Child Health Care Hospital, Affiliated Women and Children’s Hospital of Jiaxing University, Jiaxing, People’s Republic of China; 2Department of Clinical Pharmacy, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine, Shanghai, People’s Republic of China; 3Department of Neurovascular Disease, Shanghai Fourth People’s Hospital, School of Medicine, Tongji University, Shanghai, People’s Republic of China; 4Department of Pathology, Jiaxing Maternity and Child Health Care Hospital, Affiliated Women and Children’s Hospital of Jiaxing University, Jiaxing, People’s Republic of China; 5Department of Pharmacy, Changhai Hospital, Second Military Medical University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhuo Wang; Fenfen Gu, Email [email protected]; [email protected]
Introduction: Chemotherapy is still the treatment of choice for advanced triple-negative breast cancer. Chemotherapy combined with immunotherapy is being tried in patients with triple-negative breast cancer. As a kind of “cold tumor”, triple-negative breast cancer has a bottleneck in immunotherapy. Indoleamine 2, 3-dioxygenase-1 inhibitors can reverse the immunosuppressive state and enhance the immune response.
Methods: In this study, mesoporous silica nanoparticles were coated with the chemotherapeutic drug doxorubicin and indoleamine 2, 3-dioxygenase 1 inhibitor 1-Methyl-DL-tryptophan (1-MT), and then encapsulate the surfaces of a triple-negative breast cancer cell membrane to construct the tumor dual-targeted delivery system CDIMSN for chemotherapy and immunotherapy, and to investigate the immunogenic death effect of CDIMSN.
Results and discussion: The CDIMSN could target the tumor microenvironment. Doxorubicin induced tumor immunogenic death, while 1-MT reversed immunosuppression. In vivo findings showed that the tumor size in the CDIMSN group was 2.66-fold and 1.56-fold smaller than that in DOX and DIMSN groups, respectively. CDIMSN group was better than naked DIMSN in stimulating CD8+T cells, CD4+T cells and promoting Dendritic Cells(DC) maturation. In addition, blood analysis, biochemical analysis and Hematoxylin staining analysis of mice showed that the bionic nanoparticles had good biological safety.
Keywords: immunogenic cell death, doxorubicin, bionic nanoparticle, IDO1 inhibitor, triple-negative breast cancer
Graphical Abstract:
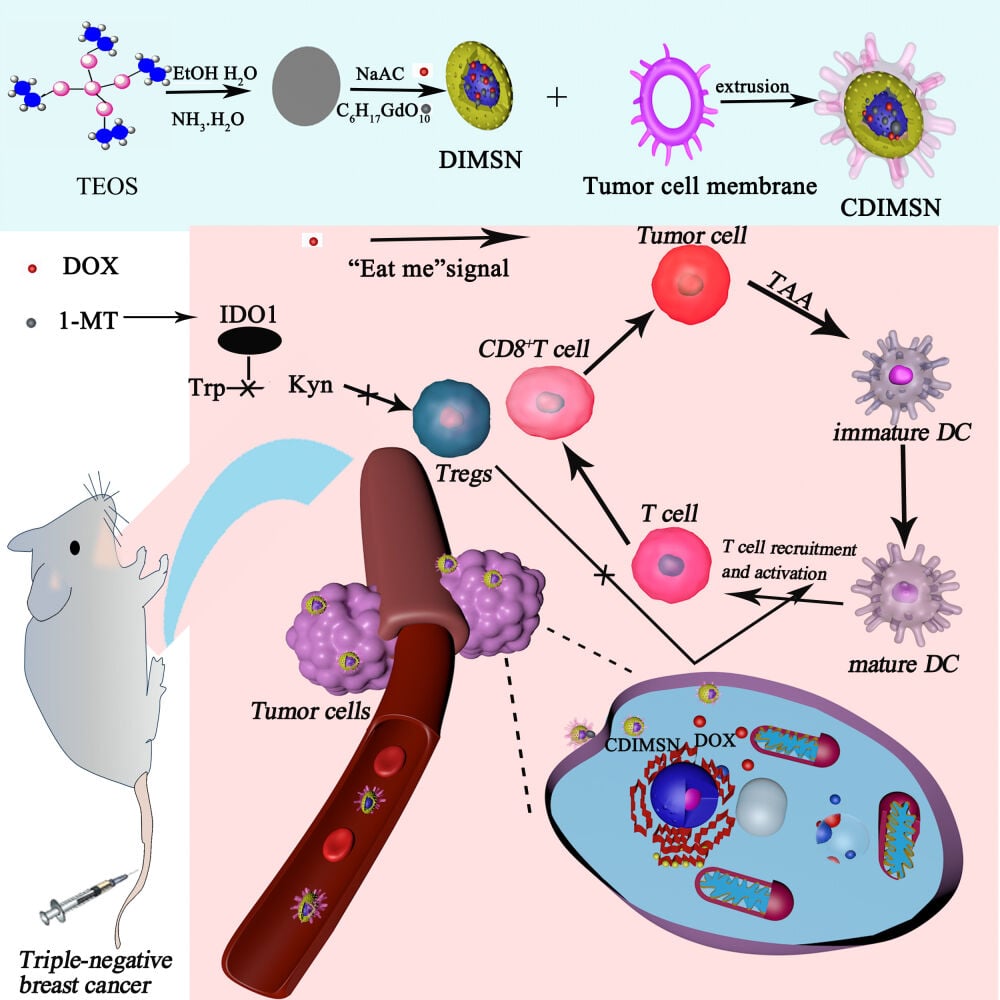
Introduction
Triple-negative breast cancer (TNBC) is a subtype of breast cancer. It is heterogeneous and does not express estrogen receptor, progesterone receptor, or human epidermal growth factor-2.1 The incidence of TNBC is approximately 10–15% of breast cancer in Western countries and 23.8% in China.2,3 TNBC is characterized by a poor prognosis and high recurrence, metastasis, and mortality rates.4–6 Cytotoxic drugs taxane and anthracycline are the main treatment for triple-negative breast cancer, but patients have became resistant to chemotherapy drugs.7–9 With the development of tumor immunotherapy, a large number of immunotherapies have been approved for clinical application. Immune checkpoint inhibitors targeting PD-1 and its ligands PD-L1 and CTLA-4 have shown promising efficacy in a wide range of solid and hematological tumors.10–13 Although tumor immunotherapy such as checkpoint inhibitors has achieved considerable therapeutic effect, it faces great challenges due to insufficient immunogenicity and immunosuppression of tumor microenvironment.14,15 In this process, tumor-associated macrophages (TAMs) play an important role in coordinating tumor immunity. TAM is a major immunosuppressive cell component in the tumor microenvironment and is involved in the production of tumor factors, recruitment of Tregs, and dysfunction of tumor killer effector cells (such as CD8+ T and NK cells). Studies have shown that targeting tumor-associated macrophages to polarize tumor-promoting M2-type TAM into anti-tumor M1-type TAM can effectively inhibit tumor growth.16–18 Therefore, knowing how to enhance tumor immunogenicity and remove the immunosuppression of tumor microenvironment to increase the recognition and presentation of tumor antigens are very important to activating the antitumor immune response and improving the effect of antitumor immunotherapy.
Indoleamine 2, 3-dioxygenase 1 (IDO1) is a rate-limiting enzyme that catalyzes catabolism of L-tryptophan through the kynurenine pathway, in which 95% of L-tryptophan is metabolized in humans.19–21 It is overexpressed in a variety of tumor cell types.22,23 IDO1-mediated tumor immune escape is the result of comprehensive regulation of various immune cell types. IDO can inhibit the activity of effector T cells, NK cells, and other tumor killing immune cells, while induce their apoptosis. Furthermore, IDO overexpression can promote the differentiation of immunotolerant Dendritic Cells(DC) meanwhile recruit immunosuppressive cells such as Treg cells and myeloid derived inhibitory cells, which synergistically induce immunosuppression and facilitate tumor cell escape from killing by the immune system.24–26 Increasing evidence indicates that expression and the reverse regulation mechanism of IDO1 are adverse to tumor immunotherapy.27,28 Inhibitors targeting IDO1 exert a therapeutic effect by inhibiting IDO1 gene expression and activity, and its effector pathway, which is an important target of tumor immunotherapy.
Immunogenic cell death (ICD) is a process in which tumor cells change from non-immunogenic to immunogenic to induce anti-tumor immune response to external stimulation.29–31 ICD produce a series of signaling molecules called damage-associated molecular patterns (DAMPs), including mainly Calreticulin(CRT) which is exposed to cell surfaces, High mobility group box 1 (HMGB1) secreted by tumor cells, ATP molecules released by cells and heat shock proteins (HSP70, HSP90), etc.30,32,33 DAMPs released during ICD can bind to PRRs, a pattern recognition receptor on the surface of DC cells, and initiate a series of cytological reactions, ultimately activating innate and adaptive immune responses.34–36
The traditional view is that chemotherapy drugs will suppress the immune function while killing tumor cells, but in recent years, many studies have shown that low-dose chemotherapy drugs can induce the ICD of tumor cells, or directly stimulate the tumor-specific immune response by drugs.37–40 Some chemotherapy drugs (doxorubicin, paclitaxel and oxaliplatin, etc.) can cause ICD of tumor cells.41,42 The chemotherapeutic drug doxorubicin (DOX) induces the production of reactive oxygen species and triggers the specific apoptosis of myelogenic suppressor cells (MDSCs), thus rapidly clearing most of MDSCs.43,44 In addition, the remaining MDSCs showed overall impaired inhibitory activity, which was related to the recovery of T lymphocyte activity. It can create favorable microenvironment for tumor immunotherapy.37,45,46 In this study, we determined whether the combined use of an IDO1 inhibitor such as 1-MT with DOX could induce ICD in tumor cells as a synergistic cascade to promote immune stimulator release and enhance intratumoral infiltration of cytotoxic T lymphocytes, thereby effectively enhancing the overall antitumor immune response.
To this end, we examined whether dual delivery of DOX and 1-MT by a biomimetic mesoporous silica nanoparticle initiated an antitumor immune response in a TNBC tumor model. We constructed large specific surface area mesoporous silica nanoparticles coloaded with DOX and 1-MT, and then encapsulate the surfaces of a triple-negative breast cancer cell membrane to develop the biomimetic nanoparticle CDIMSN. We assumed that the biomimetic nanoparticle targeted the tumor microenvironment by targeting TNBC and released chemotherapeutic and immunotherapeutic drugs to inhibit TNBC cell proliferation.
Materials and Methods
Materials
The materials and reagents included 1-MT (MCE, New Jersey, USA), tetraethylorthosilicate, hexadecyl trimethyl ammonium bromide (CTAB), sodium hydroxide (Sangon Biotech, Shanghai, China), doxorubicin, 1.1’-dioctadecyltetramethyl indotricarbocyanine iodide(DIR), and 4,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Missouri, USA), Cell Counting Kit-8 (Dojindo Molecular Technologies, Tokyo, Japan), RPMI-1640 medium, penicillin-streptomycin solution (5 kU/mL), and fetal bovine serum (Gibco, Life Technologies, Grand Island, NY, USA). Anti-mouse CD4, anti-mouse CD8, anti-mouse CD3, anti-mouse CD86, anti-mouse CD11, and anti-mouse CD80 antibodies and matrigel were purchased from BD Biosciences (Maryland, USA). Mouse IL-6 and TNF-α ELISA kits were purchased from MultiSciences (LIANKE, Shangzhou, China). Alexa Fluor 647-conjugated anti-CRT was purchased from Abcam (Cambridge, England). All reagents and chemicals were of analytical grade.
Cell Lines and Animals
Mouse breast cancer cells (4T1) were purchased from the Cell Bank of the China Academy of Sciences (China) and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum(FBS) and 1% penicillin-streptomycin (10,000 U/mL) at 37 °C with 5% CO2.
Six-week-old female BALB/C mice weighing 18−19 g were purchased from Shanghai Jihui Laboratory Animal Care Co., Ltd. (Shanghai, China) and raised under specific pathogen-free conditions. All animal experiments were approved by the ethics committee of Jiaxing University.
Synthesis of MSN
Hexadecyl trimethyl ammonium Bromide (CTAB) (0.5 g) was dissolved in deionized water (240 mL). A sodium hydroxide aqueous solution (1 M, 3.5 mL) was added to the solution and stirred at 80 °C for 1 h. Tetraethylorthosilicate (2.5 mL) was added dropwise to the mixture under vigorous stirring and then stirred for another 2 h to obtain a white precipitate. The nanoparticles were centrifuged and washed with water and methanol to obtain CTAB@MSN. To remove CTAB, the CTAB@MSN nanoparticles were refluxed in a solution of methanol (200 mL) and 37% HCl (18.7 mL) for 24 h. The nanoparticles were collected by centrifugation and washed extensively with water and methanol several times. The morphology of MSN was characterized by transmission electron microscopy, and the particle size and potential of MSN were determined.
Preparation and Characterization of CDIMSN Nanoparticles
The MSN ethanol solution was centrifuged at high speed (12,000 rpm). The supernatant was discarded, and purified water was added for washing. Under stirring, DOX and 1-MT were added successively, and DIMSN was obtained by stirring while protected from light for 6 h.
The particle size and zeta potential were measured by a dynamic light-scattering instrument to investigate the stability of nanoparticles. The morphology of nanoparticles was observed by transmission microscopy. The drug loading efficiency (DLE) and drug loading content (DLC) of DOX and 1-MT were determined by an UV-vis spectroscopy in the scanning range of 200–600nm. Ultraviolet detection was performed on the supernatant after encapsulation. The detection wavelength of DOX and 1-MT were 480nm and 290nm, respectively. And the concentration of DOX and 1-MT were calculated using a standard curve. It is worth noting that since DOX also has an absorption peak at 290nm, and when the solution is adjusted to alkaline, the absorption peak at 290nm disappears, so the solution is adjusted to alkaline during 1-MT determination to eliminate the interference of DOX.
The drug loading efficiency (DLE) and drug loading content (DLC) were calculated according to Equations (1) and (2), respectively:

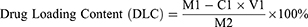
where M1: DOX input or 1-MT input, C1: DOX or 1-MT concentration measured, V1: volume of DOX or 1-MT solution, and M2: mass of carrier.
Cell membrane(CM) was mixed with drug-loaded nanoparticles DMSN at a certain ratio, and a 200 nm carbonate membrane was used for extruding and filtering to prepare CDIMSN at various membrane/core ratios. Then centrifuged at 10,000 r/min for 5min, removed excess membrane, and re-suspended the obtained CDIMSN with deionized water for use. The particle size and zeta potential were determined to ensure that the nanoparticles were covered by the cell membrane. Analysis of the cell membrane protein content was conducted to confirm that the biomimetic nanoparticles carried membrane surface antigens. The same protein concentration was added and stained with Coomassie blue. Western blotting analysis was used to detect membrane and intracellular protein markers.
Drug release of CDIMSN was determined by UV-vis spectroscopy in the scanning range of 200–600nm. To detect drug release, a 10% CDIMSN solution was treated with disodium hydrogen phosphate citrate buffer at pH 7.4, and fluorescence of DOX and 1-MT was detected at 480 nm and 290nm, respectively.
The drug-release rate was calculated as Equation (3):

where V0: total volume, Cn: DOX or 1-MT concentration outside the bag for the nth
measurement, V1: liquid volume extracted each time, Ci: DOX or 1-MT concentration
outside the bag for the ith measurement, and M: mass of DOX or 1-MT input.
Cellular Uptake
MSN was labeled with FAM, and then 4-T1 cells (1×105/well) were incubated with FAM-CDIMSN for 4 h, washed with PBS three times, and analyzed by flow cytometry to investigate uptake of the nanocarriers.
Intracellular Distribution
4-T1 cells were incubated with FAM-CDIMSN for 4 h, washed with PBS three times, fixed with paraformaldehyde for 20 min, and then analyzed by laser confocal microscopy for the intracellular distribution.
Inhibition of Kynurenine Production
4T1 cells were seeded in 96-well plates and cultured for 24 h. After discarding the medium, fresh medium containing recombinant mouse IFN-γ (50 ng/mL) was used to stimulate tumor cells and upregulate expression of IDO. After 24 hours, the medium was removed and CDIMSN was applied for 12 hours. Then, 100 μL supernatant was mixed with 75 μL of 30% trichloroacetic acid (and heated at 50 °C for 30 min to hydrolyze N-formyl kynurenine to kynurenine). The samples were then incubated at room temperature with the same volume of Ehrlich reagent (2% dimethylamino benzaldehyde glacial acetic acid solution, w/v) for 10 min. Absorbance of the reaction product at 490 nm was measured by a microplate reader. The relative inhibition rate was calculated by normalization to the untreated control group.
Cell Proliferation Assay
Cells were seeded in 96-well plates and cultured with CDIMSN for 24 and 48 h. The medium was removed and 200 μL fresh culture medium containing 10% CCK-8 reagent was added. The cells were cultured for 1–2 h and detected at 490 nm by a microplate reader to calculate the IC50 value.
Cytokine Expression
We used co-culture method to detect cytokine expression. Briefly, BLAC mice were sacrificed for cervical vertebrae dislocation, quickly soaked in 75% ethanol for 5 minutes, then the spleen was removed with tweezers on a super-clean table and ground in a sterile petri dish containing 5mL lymphocyte separation solution. The splenic cell suspension was collected into a 15mL centrifuge tube and covered with 0.5mL RPMI 1640 medium. At room temperature, centrifuge 800g for 30min, suck out the lymphocyte layer, add 10mL RPMI 1640 medium, centrifuge 250g at room temperature for 10min to collect cells, adjust the cell density to 5.0×106/mL, and set aside. The 96-well plates were inoculated with 4-T1 cells (density 8×103/mL) and pre-prepared mouse lymphocytes (4 × 105/mL), which were shaken evenly and cultured in the incubator for 24 hours. At the end of culture, MSN, DOX, 1-MT, DMSN, IMSN, DIMSN, CDIMSN and ipopolysaccharide (LPS 10ng/mL) were given and cultured for 6h. TNF-α and IL-6 in samples were determined by ELISAs. 100 μL supernatant was added into the corresponding well and incubated at room temperature for 12 min. The OD value of the reaction product at 450 nm was measured by a microplate reader. The relative TNF-α and IL-6 concentration was calculated by normalization to the untreated control.
Efflux of CRT
CRT efflux from tumor cells induced by CDIMSN was detected by immunofluorescence. Cells seeded in 6-well plates cultured with CDIMSN for 48 h. The medium was removed, cells were washed with PBS, and 4% paraformaldehyde was applied for 20 min, followed by washing with PBS three times. Then, 3% goat serum was applied for 40 min, followed by an anti-calreticulin primary antibody (1:300 dilution) for 90 min at 37 °C. After washing with PBS twice, a fluorescently coupled secondary antibody (1:500 dilution) was applied for 1 h at 37 °C. After washing with PBS twice, DAPI (1:1000 dilution) was applied for nuclear staining at room temperature for 5 min while protected from light, followed by washing with PBS twice. The CRT distribution on the tumor cell membrane was observed by a cell imaging microplate detector.
In vivo Distribution
4-T1 cells (1×107 cells/mL) in the logarithmic growth phase were injected into the breast fat pad under the armpit of the right upper limb of female mice. After 14 days, tumors with a volume of >100 mm3 were selected for the experiment. 1.1’-dioctadecyltetramethyl indotricarbocyanine iodide (DIR), a near-infrared fluorophore, was used to investigate the vivo distribution of various nanoparticle. CDMSN (D:DIR) was injected into the caudal vein. The in vivo distribution of biomimetic nanoparticles was observed using the FX Pro in vivo imaging system (Caliper Life Sciences, Hopkinton, MA), and in vivo targeting of the drug delivery system was qualitatively evaluated.
Subsequently, the mice were sacrificed by cervical dislocation, and fluorescent substances were extracted from the heart, liver, spleen, lungs, kidneys, and tumor tissues after homogenization. The fluorescence intensity of each tissue was quantitatively investigated to assess in vivo targeting of the biomimetic nanodrug delivery system.
In vivo Efficacy and Safety Analyses
The animals were randomly divided into 8 groups and treated with CDIMSN to investigate the tumor inhibition rate, survival analysis weight loss, and therapeutic effect of biomimetic nanoparticles on TNBC in vivo. Additionally, the expression of various immune-related factors was determined, and immunohistochemistry and blood routine and liver function tests were performed to investigate safety.
Histological Analysis
After euthanasia, the heart, liver, spleen, lungs, and kidneys were collected from the mice, fixed in 4% paraformaldehyde for 24 h, and then embedded in paraffin. Tissue sections (5 μm thick) were subjected to hematoxylin and eosin staining. Tumor tissue was also analyzed by Immunofluorescence staining for CRT and other related proteins.
Statistical Analysis
Values are presented as means ± SD. Each value represents the mean of at least three repeated experiments for each group. Statistical significance was determined by analysis of variance and the unpaired Student’s t-test. Differences were considered significant at *p < 0.05 and **p< 0.01.
Results
Preparation and Characterization of CDIMSN
We investigated the size, zeta potential, drug-loading efficiency, and cell membrane coating of the nanoparticles. DOX and 1-MT were successfully encapsulated in mesoporous silica nanoparticles coated with the tumor cell membrane (CDIMSN). As shown in Figure 1A, the mesoporous silica nanoparticles had a uniform particle dispersion and particle size, and the nanoparticle size was approximately 100 nm, the surface of the regular sieve mesoporous structure can be seen. The pore of MSN was about 3nm. The obvious pores make it a good carrier for drugs. After drug loading(DIMSN), the dispersion and size of nanoparticles did not change significantly, which showed a spherical nanoparticle structure. But the pore structure of MSN could not be clearly observed in TEM images. At the same time, the feature absorption peaks disappeared compared with DOX and 1-MT, indicating that DOX and 1-MT were successfully encapsulated in MSN (Figures S1–S5, Supporting Information). When the nanoparticle was coated by the tumor cell membrane, the edge of the nanoparticle was blurred and wrinkled, indicating that the tumor cell membrane had wrapped the nanoparticle surface. The tumor cell membrane inherits the targeting function and antigen library from the source cells.47 Nanoparticles coated with the membrane prevents uptake by the endothelial reticular system in vivo and promotes long-lasting circulation of drugs.48 The proteins on the membrane targeted the tumor site. We verified the integrity of the tumor cell membrane on the nanoparticle by comparing the membrane coated nanoparticles with freshly extracted tumor cell membrane protein bands (Figures S6 and S7, Supporting Information). Both groups showed similar banding patterns, indicating that the coating process did not change the composition of tumor cell membrane. These data indicate that the proteins on the surface of tumor cell membranes are well preserved and maintain the biological functions of tumor cell membranes. Next, we verified the size and zeta potential by DLS. As shown in Figure 1B, the particle size of MSN is about 100nm, while the particle size does not change significantly after loading DOX and 1-MT (DIMSN), and still remains at about 100nm. And the particle size of the nanoparticles coated by cell membrane(CDIMSN) increases to about 120nm, which is consistent with the TEM result. As shown in Figure 1C, the zeta potential of MSN was about −6.32±1.2mV, the potential of MSN after coating DOX and 1-MT had no significant change, and remained at −6.98±1.5mV. When DIMSN was coated with negatively charged tumor cell membrane, the potential of CDIMSN changed significantly to −26.6±2.5mV, indicating the successful preparation of CDIMSN. In addition, we investigated the loading capacity of CDIMSN by dialysis method. The DOX drug loading efficiency (DLE) and drug loading content (DLC) of CDIMSN were 89.47% and 35.87% respectively. And the 1-MT drug loading efficiency (DLE) and drug loading content (DLC) of CDIMSN were 67.59% and 20.18%, respectively. These results indicated that the CDIMSN had been successfully prepared. Figure 1D shows the in vitro stability data of CDIMSN nanoparticles under pH 7.4 conditions. It can be seen that the nanoparticles can maintain no change in particle size within 2 days. Starting from the 3rd day, the particle size of the nanoparticles gradually increases to around 150nm, and starting from the 5th day, the particle size increases to around 220nm, indicating good stability of the cell membrane within 48 hours. From the 5th day, the particle size starts to increase due to clustering. DOX and 1-MT release were also measured. As shown in Figure 1E, in the first 24 hours, the DOX release of DMISN was faster than CDIMSN release. However, after 48 hours, DMISN release had stabilized, while CDIMSN release was increased, and the DOX release rates in CDIMSN group at 60 h was 78.5%. 1-MT exhibits similar release in DIMSN and CDIMSN, with the release in DIMSN occurring earlier than in CDIMSN, and finally the cumulative release in CDIMSN being greater than in DIMSN, indicating that the membrane-coated nanoparticles had relatively slow release.
 |
Figure 1 Characterization of CDIMSN. (A) Representative TEM images of MSN, DMSN and CDIMSN. (B) Particle size of the CDIMSN as determined by dynamic light scattering (DLS). (C) The zeta potential of the CDIMSN as determined by DLS. (D) Stability Study of CDIMSN Placed at pH 7.4 room temperature for 7 Days. (E) The profiles of doxorubicin (DOX) and 1-MT release from CDIMSN in PBS at pH 7.4 at 37°C. |
Synergistic Delivery of DOX and 1-MT
The cell absorption efficiency of drugs is an important factor affecting their activity and clinical application.49 The cellular uptake capacity of MSN, DOX, DMSN, DIMSN, and CDIMSN for 4 h was determined by flow cytometry. DOX-positive cells accounted for 96.5%, 97.43%, 97.20%, and 90.40% for DOX, DMSN, DIMSN, and CDIMSN, respectively (Figure 2A). DOX uptake of the membrane-coated CDIMSN group was lower than that of the other DOX groups. Cellular uptake of free DOX was close to that of DOX wrapped by nanoparticles, which may be related to the strong cell penetration ability of doxorubicin, and the membrane envelope prevented drug escape.50
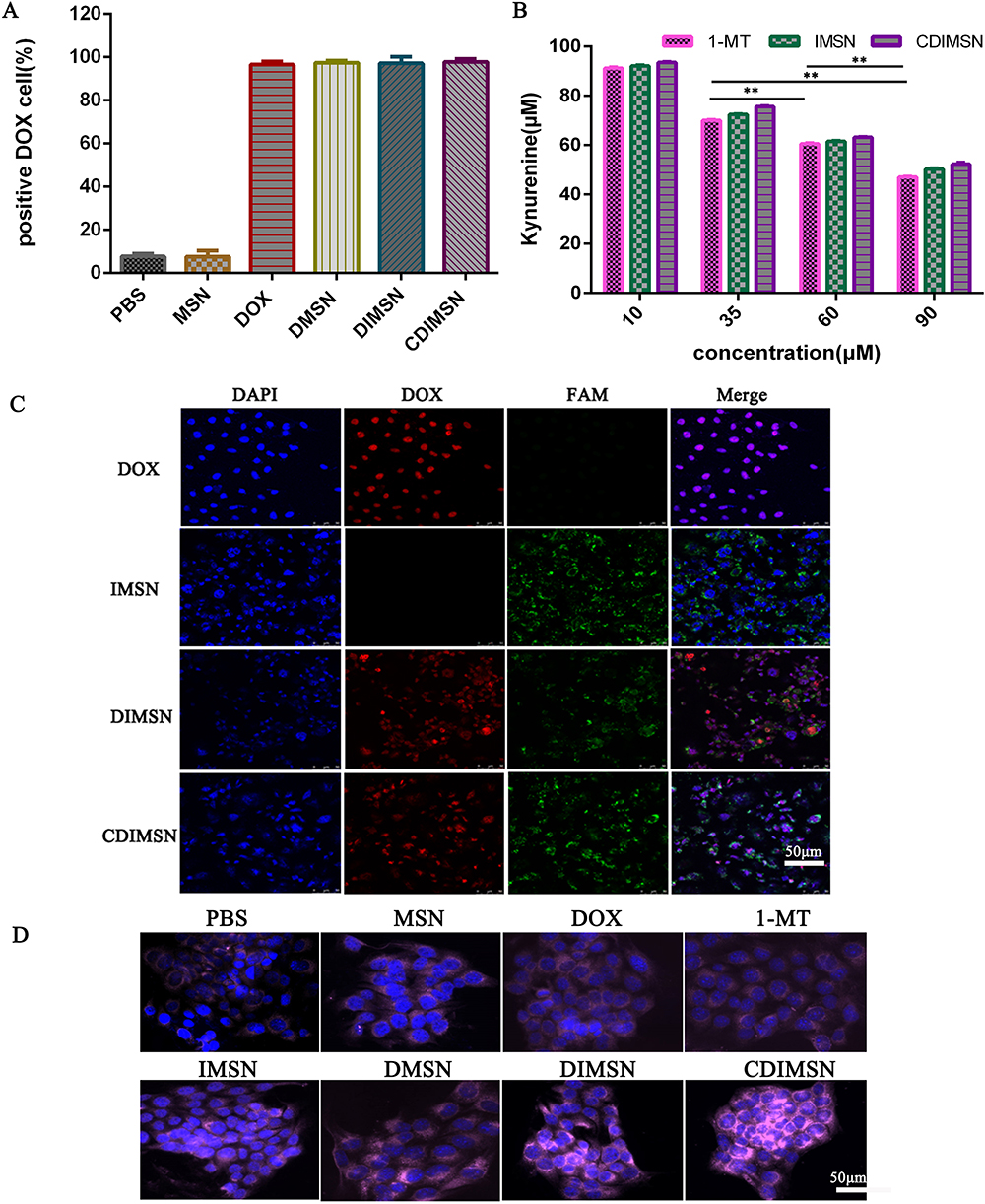 |
Figure 2 The cellular uptake of doxorubicin (DOX) and IDO inhibitory effect in vitro. (A) Quantitative analysis of DOX uptake in 4-T1 cells after treatment with Bionic nanoparticle (CDIMSN) for 4h. (B) IDO inhibitory effect on the level of Kyn of different treated 4-T1 cells (n=3).(C) Confocal microscopic images of 4-T1 cells incubated with DOX, IMSN, DIMSN CDIMSN for 4 h. Green fluorescence presents FAM-labeled MSN, red fluorescence represents DOX, and blue fluorescence represents the cell nucleus. Scale bar is 50 μm. (D) fluorescence microscopy images of CRT exposure of different treated 4-T1 cells. Scale bar is 50 μm. **p <0.01. |
To determine the intracellular location of DOX, IMSN, DIMSN, and CDIMSN, 4-T1 cells were observed by CLSM. Nuclei were marked with DAPI, and MSN was marked with FAM. Figure 2C shows the intracellular distribution of nanoparticles in 4-T1 cells after incubation for 4 h. In 4-T1 cells incubated with free DOX, red fluorescence was distributed in the nucleus and cytoplasm. In the IMSN group, green fluorescence was distributed in the cytoplasm, indicating that the nanoparticles successfully entry into cells. In 4-T1 cells incubated with DIMSN, red and green fluorescence were uniformly distributed in the nucleus and cytoplasm, and the fluorescence intensity was brighter than that of the free DOX group, indicating that DOX and 1-MT were transported into the cells by the nanoparticles, and DOX had escaped from endosomes and entered the nucleus to exert its cytotoxic effect via nanostructures. These results suggested that the co-delivery system promoted endosomal escape and carried the chemotherapeutic drugs into the nucleus. In the CDIMSN group, red fluorescence was also distributed in the nucleus and cytoplasm, and the fluorescence intensity was relatively lower than that of the other DOX groups, which may be related to the sustained drug release after membrane encapsulation.
IDO Inhibitory Effect in vitro
The inhibitory effect of IDO was examined by measuring the content of IDO metabolite kynurenine (kyn). As shown in Figure 2B, the inhibitory effect of IDO was enhanced with the increase of the 1-MT concentration. IMSN and CDIMSN showed similar but slightly weaker inhibitory effects compared with free 1-MT, and CDIMSN showed the weakest inhibitory effect. This was because although nanoparticles had a strong cell internalization effect and high intracellular content, they needed to release 1-MT to exert an effect. In this experiment, the cells were co-incubated with the nanoparticles for 2 days. According to the in vitro release experiment, during 48 h, 1-MT release from nanoparticles was <100%. Therefore, the IDO inhibitory effect of IMSN and CDIMSN was slightly weaker than the free 1-MT inhibitory effect in vitro, but no significant difference was observed among the three groups. Coated 1-MT retained strong IDO inhibition, which is expected to be used in the immunological microenvironment for tumor regulation, further confirming the feasibility of the combined treatment strategy.
Efflux of Calreticulin
Calreticulin (CRT) is usually present in the endoplasmic reticulum, but everts onto the membrane very quickly in cells undergoing immunogenic cell death and further activates the immune response.51 As shown in Figure 2D, CRT was externalized in the free DOX group, which was more obvious in the CDIMSN group, and CRT outburst in the other groups was between the DOX group and the CDIMSN group. Anthracyclines translocate calreticulin to the cell membrane and cluster. Nanoparticles coated with the cell membrane activated the immune response through ICD, thereby enhancing the effect of immunotherapy.
Toxicity of CMSN
The toxicity and biocompatibility of membrane-coated mesoporous silica were evaluated by monitoring changes in blood as well as liver and kidney functions in mice after repeated injection of CMSN through the caudal vein. There were no significant changes in white blood cells, granulocytes, lymphocytes, hemoglobin, or platelets after CMSN injection (Figure 3A). Biochemical indexes showed no obvious abnormalities in liver functions, kidney functions, or the myocardial enzyme profile after CMSN injection (Figure 3B–D). These results indicated that membrane-coated MSN had good biocompatibility in vivo and was almost non-toxic under our experimental conditions.
 |
Figure 3 The biosafety and in vitro anti-tumor effect of biomimetic nanoparticles. The biosafety of CMSN: The Routine blood test (A), Liver function (B), Renal function index (C) and Myocardial zymogram index (D). (E) The viability of 4-T1 cells after treatment with doxorubicin (DOX), DMSN, DIMSN and CDIMSN at different DOX concentrations for 24h. (F) The viability of 4-T1 cells after treatment with doxorubicin (DOX), DMSN, DIMSN and CDIMSN at different DOX concentrations for 48h.(G) Cell apoptosis assay. Cell apoptosis analysis of 4-T1 cells treated as above for 48 h was measured by flow cytometry using Annexin V–APC and Phycoerythrin (PE) staining (DOX: 0.5 μg/mL; 1-MT:100nM). Data are expressed as the mean ± SD (n = 3). |
Cytotoxicity
Next, we investigated the killing effect of the various drug delivery systems in 4-T1 cells. As shown in Figure 3E and F, the cell survival rate decreased with the increase in DOX concentration. The proliferation of 4-T1 cells treated with free DOX, DMSN, DIMSN, and CDIMSN was inhibited in a DOX concentration-dependent manner. CCK-8 assays demonstrated that the IC50 values of free DOX, DMSN, DIMSN, and CDIMSN in 4-T1 cells at 48 h were 1.76, 1.05, 0.59, and 0.34 μg/mL, respectively. The IC50 value of CDIMSN was separately approximately 5.2, 3.1 and 1.7-fold lower than that of free DOX, DMSN and DIMSN (p < 0.05) to evaluate the effects of CDIMSN on apoptosis in 4-T1 cells, 4-T1 cells were co-cultured with mouse lymphocytes and cell apoptosis was measured by flow cytometry after staining with APC-Annexin V/PE. As shown in Figure 3G, there was almost no apoptosis in the blank control group, the single vector group and free 1-MT group. IMSN showed higher apoptotic cell percentage while 1-MT and blank MSN had no significant effects, may be due to the hydrophobicity of 1-MT, it is difficult to enter the cell, but the nanoparticle encapsulated drug can successfully enter the cell and release the drug. However, apoptosis was significantly enhanced in the CDIMSN group. In the CDIMSN group, 20.83% of cells were apoptotic compared with 17.09% in the DIMSN group, 14.27% in the DMSN group, 13.36% in the DOX group and 7.74% in the IMSN group, which suggested that CDIMSN group had a better synergistic antitumor activity due to the targeting of tumor cell membranes. In addition, the strongest apoptosis induced by CDIMSN may be the result of the synergistic effect of DOX and 1-MT.
Upregulated Cytokine Expression
TNF-α and IL-6 secretion in the co-culture system of 4-T1 cells and lymphocytes were measured after various treatments. As shown in Figure 4A and B, compared with the control group, there was no significant expression of TNF-α or IL-6 in the MSN group, indicating that MSN nanoparticles had no immunomodulatory ability. The 1-MT group showed only slight increases in TNF-α and IL-6, suggesting that the IDO inhibitor alone could not effectively activate the immune response. TNF-α and IL-6 were highly expressed in CDIMSN and DIMSN groups. These results indicated that DOX and 1-MT acted together to enhance the antitumor immune response. Importantly, the CDMISN group showed the highest activity, indicating that the membrane coating increased the affinity of the nanoparticles for tumor cells. These results indicated that our membrane-coated nano-preparation can effectively regulate the immune response of the body.
 |
Figure 4 In vitro immune activity of the Bionic nanoparticle (CDIMSN) and Biodistribution of CDIMSN in vivo. (A) IL-6 production in Lymphocytes which were co-cultured with 4-T1 cells determined by ELISA after varies treatment for 24 h. (B) TNF-α production in Lymphocytes which were co-cultured with 4-T1 cells determined by ELISA after varies treatment for 8 h. (C) In vivo imaging of CDMSN in 4-T1 cells tumor-bearing nude mice after tail vein injection. (D) Ex vivo imaging of tumors and organs collected from mice administered DIR, DMSN or CDMSN for 6, 12, 24h (DIR 5mg/kg). (E) Quantitatively analyzed of the fluorescence intensity of EX tumor. *p<0.05, **p <0.01. |
In vivo Distribution
Investigation of the in vivo distribution is essential to evaluate the safety and effectiveness of nano-micellar delivery. DIR is used to reduce interference of animal-related autofluorescence. To assess the biodistribution of DIR, DMSN (D:DIR), and CDMSN (D:DIR), tumors and major organs were excised from treated mice at 2, 6, 12, and 24 h post-administration and imaged (Figure 4C–E). In the free DIR group, a fluorescence signal was observed in the liver, lungs, kidneys, and tumor. In the DMSN and CDMSN groups, a significant fluorescence signal was observed in the tumor site because of the EPR effect. Quantitative analysis indicated that the DIR signal intensity in the tumor tissue of the CDMSN group was 2.19-fold higher than that in the free DIR treatment group (P<0.01) and 1.67-fold higher than that in the DMSN group (P<0.01). This could be explained by the EPR effect and avoidance of the reticuloendothelial system (RES) owing to the nanosize of the particles52 and homologous-targeting tumor accumulation from cancer membranes in vivo.53 Collectively, these results indicated that CDIMSN nanoparticles efficiently delivered drugs into tumor tissues, likely via the EPR effect and the functionalization of the homologous binding adhesion molecules from cancer cell membranes.
Antitumor Effects in vivo
The antitumor effects of CDIMSN biomimetic nanoparticles were observed in the 4-T1 mouse tumor transplantation model. We investigated the in vivo therapeutic efficacy and tumor volume of PBS, MSN, DOX, 1-MT, DMSN, IMSN, DIMSN, and CDIMSN groups. Systemic toxicity was assessed by measuring changes in mouse body weight. Figure 5A showed a direct view of free tumors in each group. As shown in Figure 5C, except for the DOX group, there was no significant weight loss in other treatment groups, indicating that 1-MT, DMSN, IMSN, DIMSN, and CDIMSN did not cause significant systemic toxicity. Compared with the PBS group, the 1-MT group showed no obvious inhibition of tumor growth (Figure 5B). DOX, DMSN, DIMSN and CDIMSN groups showed various degrees of anti-tumor growth activity. After sacrificing the mice on day 21, the tumor size of the CDIMSN group was approximately 2.66, 3.13, 2.25, 3.03 and 1.56-fold smaller than those of the DOX, 1-MT, DMSN, IMSN, and DIMSN groups (Figure 5B) (p<0.01). There was also a similar trend in the tumor weight. In addition to changes in tumor size, survival rates were also recorded for each group of mice. The survival curves of tumor-bearing mice are shown in Figure 5D. The survival rate on day 40 in PBS, MSN, DOX, 1-MT, DMSN, IMSN, DIMSN and CDIMSN groups was 0%, 0%, 0%, 0%, 16.67%, 0%, 33.33%, and 50%, respectively. Histological analysis and TUNEL staining assay were conducted to investigate antitumor effects in vivo. TUNEL staining showed that tumor cell apoptosis in the CDIMSN group was the most obvious, which was consistent with the results of HE staining (Figure 5E). As shown in Figure 5F, tumor necrotic areas in the CDIMSN group were more obvious than those in the control, DOX, 1-MT, DMSN, IMSN, and DIMSN groups. These results indicated that the IDO inhibitor alone did not produce a strong antitumor immune response. In the CDIMSN group, DOX induced tumor cell apoptosis, activated immunogenic cell death, and released tumor-related antigens or “danger” signals, which further improved recognition by the immune system. Additionally, the antitumor activity of the CDIMSN group was stronger than that of the DIMSN group, which may be due to targeting of the cell membrane, thereby enhancing the antitumor effect. Therefore, the CDIMSN group showed a synergistic antitumor effect.
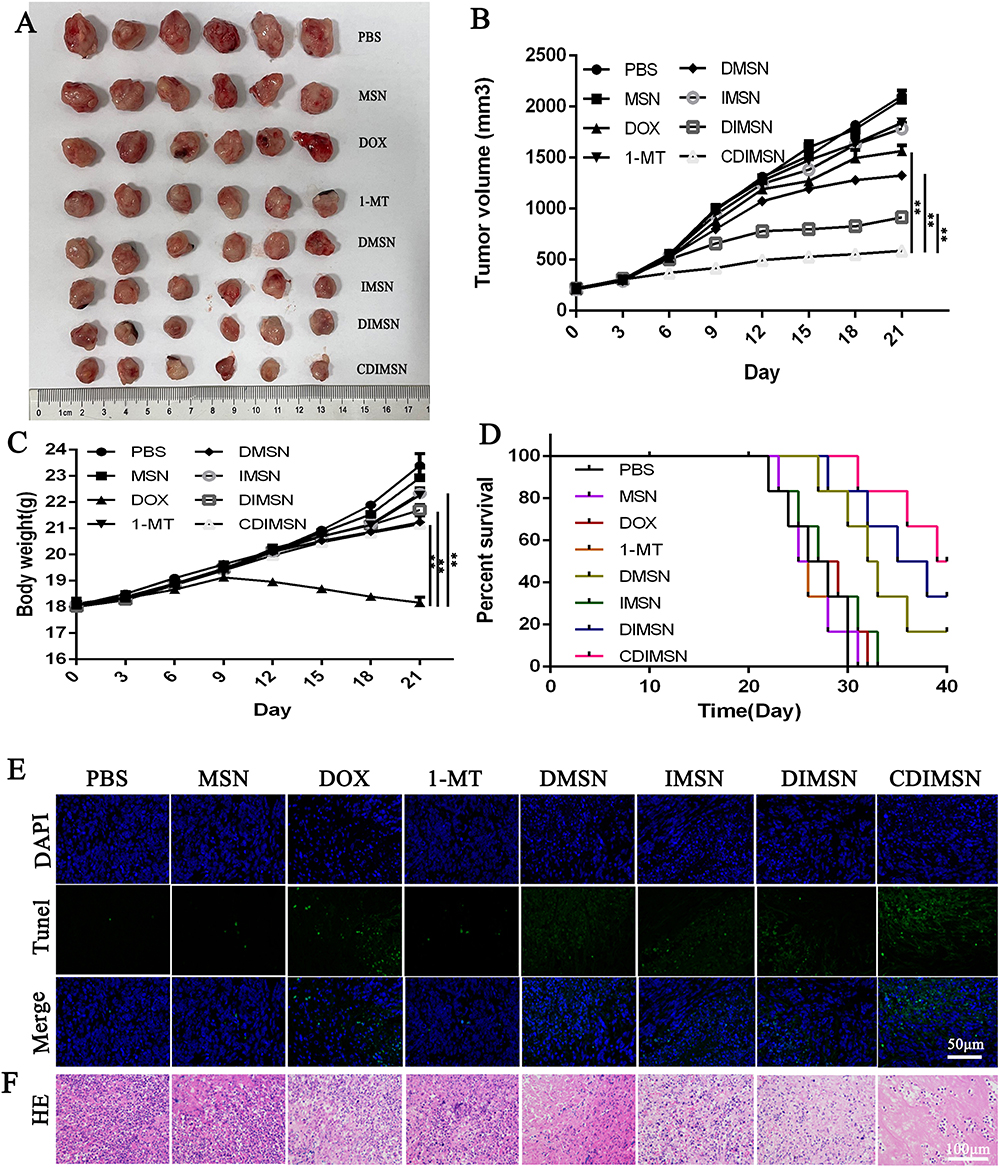 |
Figure 5 Antitumor efficacy of CDIMSN in vivo. (A) Image of EX tumor at the time of sacrifice after 21 days post-treatment. (B) Tumor growth curves of mice receiving different therapeutic regimens. Data are expressed as means ±SD (n=6). (C) The body weight of tumor-bearing mice treated with PBS, MSN, DOX, 1-MT, DMSN, IMSN, DIMSN and CDIMSN. Data are given as the mean±SD (n=6). (D) Survival rate of 4-T1 tumor-bearing BALB/c mice. (E) TUNEL staining of tumor tissue. Scale bar is 50 μm. (F) The histological characteristics of 4-T1 tumor tissue after treatment with phosphate-buffered saline (PBS), MSN, DOX, 1-MT, DMSN, IMSN, DIMSN and CDIMSN. **p<0.01. |
Safety in vivo
We performed myocardial enzyme profile analysis to verify the biocompatibility of CDIMSN in vivo. As shown in Figure 6A, when comparing with the free DOX group, we found no obvious differences in the treatment groups. Furthermore, hematoxylin and eosin staining verified no major organ damage (Figure 6B). These results indicated that CDIMSN had high biocompatibility and a great potential for clinical application.
 |
Figure 6 In vivo safety of biomimetic nanoparticles and CRT valgus promotion. (A) Myocardial zymogram index of CDIMSN. **p<0.01. (B) Histological characteristics of organ histology after treatment with phosphate-buffered saline (PBS), MSN, DOX, 1-MT, DMSN, IMSN, DIMSN and CDIMSN. Scale bar is 100 μm. (C) Promoting CRT efflux in tumor-bearing BALB/c mice. Scale bar is 50 μm. |
CRT Efflux in vivo
An in vivo CRT efflux assay showed more obvious CRT was in the CDIMSN group then that in the DOX, 1-MT, DMSN, IMSN and DIMSN group which results were consistent with in vitro results (Figure 6C). It can be seen that bionic nanoparticles co-carrying DOX and 1-MT release TAAs and DAMPs while killing tumor cells, and turn on the “switch” of immune response sequence through ICD, thus enhancing the effect of immunotherapy.
Detection of CD4+ and CD8+ T Cell in Mice Spleen
CD4+ and CD8+ T cells are the main T cell subtypes. T cell responses to PBS, DOX, IDO, DMSN, IMSN, DIMSN, and CDIMSN were assessed by flow cytometry. As shown in Figure 7A, CD4+ T cells were activated in all experimental groups (DOX, 1-MT, DMSN, IMSN, DIMSN and CDIMSN). As shown in Figure 7A and B, there were significantly more CD4+ T cells in the CDIMSN group (36.0%) than in the DOX (19.2%, p<0.01), 1-MT (8.81%, p<0.01), DMSN (20.3%, p<0.01), IMSN (10.6%, p<0.01), and DIMSN groups (28.0%, p<0.01), and significantly more CD8+T cells in the CDIMSN group (45.5%, p<0.01) than in the DOX (28.7%, p<0.01), 1-MT (6.96%, p<0.01), DMSN (30.5%, p<0.01), IMSN (7.34%, p<0.01), and DIMSN groups (32.8%, p<0.01)(Figure 7C and D). These results indicated that combined administration of DOX and 1-MT effectively activated the immune response.
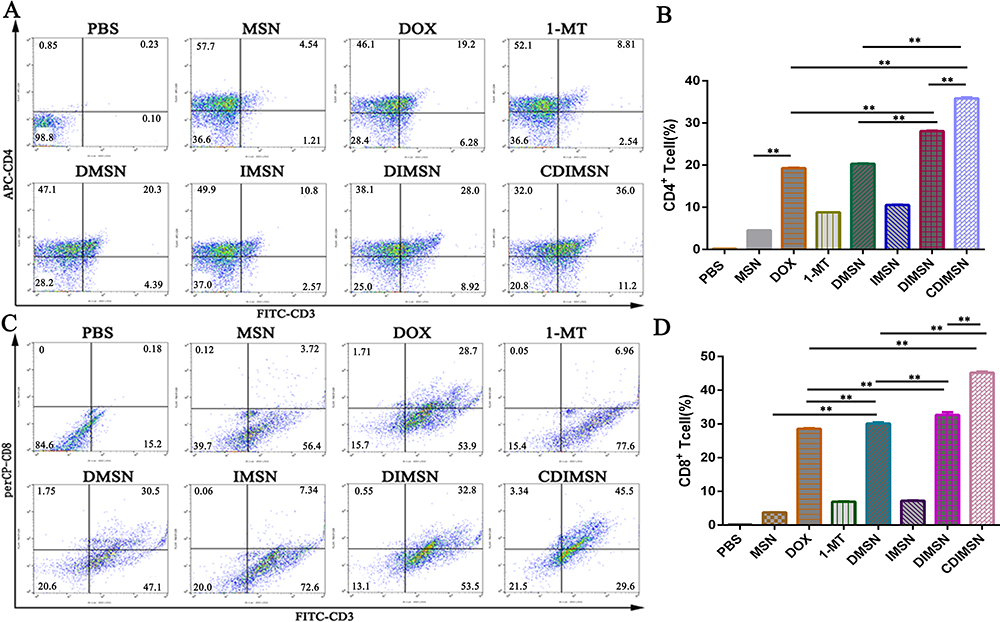 |
Figure 7 Expression of CD4+ and CD8+ T-cells after treatment with PBS, MSN, DOX, 1-MT, DMSN, IMSN, DIMSN and CDIMSN. The expression of splenocyte surface molecules was determined by flow cytometry. (A) Representative result CD4+ T cell of independent experiments and (B) Quantitative analysis of the percentage of positive CD4+ T cells. (C) Representative result CD8+ T cell of independent experiments and (D) Quantitative analysis of the percentage of positive CD8+ T cells. The data shown as means±SD (n =5). The statistical significance of the results was analyzed and indicated as **p <0.01. |
DCs Maturation in Tumor Infiltration
The spleen regulates T lymphocyte immunity, which is an important part of tumor immunity. DCs in spleen was collected and labeled and then analyzed by flow cytometry. CD80-PEcy7 and CD86-APC positive number can be used to reflect the maturity of DCs. As shown in Figure 8A and B, the mature degree of CD80 was significantly higher (23.6%) than the DOX (10.7%, p<0.01), 1-MT (7.93%, p<0.01), DMSN (13.3%, p<0.01), IMSN (9.5%, p<0.01), DIMSN (15.9%, p<0.01) group. And significantly more CD86 in the CDIMSN group (49.4%, p<0.01) than in the DOX (37.2%, p<0.01), 1-MT (33.0%, p<0.01), DMSN (42.1%, p<0.01), IMSN (34.8%, p<0.01), and DIMSN groups (44.4%, p<0.01) (Figure 8C and D). In conclusion, ICD induced by biomimetic nanoparticles stimulated the immune response of tumor cells, and the release of TAAs and DAMPs effectively improved the maturation level of DCs, contributing to the process of anti-tumor immune response.
 |
Figure 8 Expression of DC cells maturation after treatment with PBS, MSN, DOX, 1-MT, DMSN, IMSN, DIMSN and CDIMSN. The expression of splenocyte surface molecules was determined by flow cytometry. (A) Representative result CD80+ cell of independent experiments and (B) Quantitative analysis of the percentage of positive CD80+ cells. (C) Representative result CD86+ cell of independent experiments and (D) Quantitative analysis of the percentage of positive CD86+ cells. The data shown as means±SD (n =5). The statistical significance of the results was analyzed and indicated as **p <0.01. |
Discussion
In this study, biomimetic nanoparticles using mesoporous silica as carrier were constructed to co-carry chemotherapy drug DOX and IDO1 inhibitor 1-MT, and their properties, targeting ability and antitumor activity in vivo and in vitro were investigated through a series of experiments. The biomimetic nanoparticles can target the delivery of DOX and immune adjuvants to the tumor site, achieving pH-sensitive drug release in the tumor microenvironment. Importantly, the bionic nanoparticles greatly reduced the leakage of DOX into the blood circulation. The results showed that the nanoparticles had a good anti-TNBC effect in vivo and in vitro, and could stimulate the tumor immune regeneration ability of mice. More importantly, nanoparticles showed a good killing effect on tumor cells in the blood circulation. Our study shows that for immune “cold tumor” TNBC, chemotherapeutic drugs can be used in combination with immune-activating drugs to obtain better antitumor effects. The results show that the synthesized bionic nanoparticles are intelligent and multifunctional antitumor drugs.
In this study, the cellular uptake among different treatment groups were very similar (above 90%), nevertheless, “in vitro cytotoxicity” and “CRT expression” are different. We think since we only took the drug at a certain time point in the cell uptake experiment, we may just see no significant difference in the four groups, but free DOX may be caused by P-glycoprotein efflux and other factors,54,55 while the endocytosis and retention time of nanoparticles in the cell are longer,56 and their action time is longer. At the same time, the membrane coated nanoparticles compete with the surface antigens of homologous cancer cells to obtain immune escape and homologous targeting ability,57 so as to obtain the best anti-proliferation effect and CRT expression.
Compared with traditional nanoparticles, from cell-derived peptide modifications to cell-based drug delivery systems,58 developments in biology are changing the design of drug delivery systems. Targeting drug delivery systems (TDDS) have developed as a promising delivery strategy due to low immunogenicity, innate mutation rate,59 long cycle time, non-neurotoxicity or tumorigenicity, and innate targeting ability. Tumor cell membranes inherit homologous targets and antigens from the source cell, has been applied in tumor targeted therapy and immunotherapy. In this study, we reported a bionic nanoparticles coated with TNBC tumor cell membrane. The results showed that the bionic nanoparticles had the characteristics of sustained release compared with the bare nanoparticles. Importantly, bionic nanoparticles have better targeting and longer cycles than bare nanoparticles. In vivo, the antitumor activity of CDIMSN was stronger than DIMSN, and the stimulation of DCs maturation was also better than DIMSN, indicating that the cell membrane coated nanoparticles have a potential application prospect.
Conclusion
Reversal of the immunosuppressive state and tumor immunogenic death promoted by chemotherapeutic drugs were combined, and the tumor microenvironment was targeted with biomimetic nanoparticles to activate the antitumor immune response and inhibit advanced TNBC. Mesoporous silica nanoparticles (MSN) loaded with DOX and an IDO1 inhibitor were constructed. The carrier had good biocompatibility and low toxicity, its porous structure provided a large surface area and high loading efficiency for drugs, and encapsulation of MSN reduced toxic side-effects of drugs. To prevent recognition and clearance of MSN by the immune system, the TNBC cell membrane was coated on the surface of MSN. Because of the low immunogenicity of the cell membrane, uptake of the endothelial reticular system in vivo was prevented to promote the long circulation of nanomaterials in vivo. Moreover, accumulation of MSN loaded with chemotherapeutic drugs in tumor tissues was realized by targeting of the membrane proteins to the tumor site, thereby effectively inhibiting tumor growth. The biomimetic nano-delivery system constructed for combined application of chemotherapy and immunotherapy targeted DOX and immune activator 1-MT to the tumor site. DOX induced tumor immunogenic death and directly killed tumor cells. 1-MT reversed tumor immunosuppression and suppressed tumor immune escape, thereby restarting the normal immune response to fight the tumor.
Ethics Approval and Informed Consent
Animal experiments were approved by the Jiaxing University ethics Committee. All animals were operated in accordance with the Guidelines for The Care and Use of Experimental Animals of Jiaxing University.
Acknowledgments
We wish to thank the timely help given by Chao Li in the supplementary experiment.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Funding
The work was funded by the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (NO.2023RC100), Medical and Industrial Cross Research Foundation of Shanghai Jiao Tong University(NO. YG2023 QNA15), the Science and Technology Project of Jiaxing, Zhejiang, China(Grants 2019AD32255).
Disclosure
The authors declare that they have no competing interests.
References
1. Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013;73(7):202. doi:10.1158/0008-5472.CAN-12-1699
2. Howard FM, Olopade OI. Triple-negative breast cancer therapy: current and future perspectives (Review). Int J Oncol. 2020;57(6):1245–1261. doi:10.3892/ijo.2020.5135
3. Won KA, Spruck C. Triple-negative breast cancer therapy: current and future perspectives (Review). Int J Oncol. 2020;57(6):1245–1261.
4. Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi:10.1172/JCI45014
5. Nedeljković M, Damjanović A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells. 2019;8(9):957. doi:10.3390/cells8090957
6. Yin L, Duan JJ, Bian XW, et al. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020;22(1):61. doi:10.1186/s13058-020-01296-5
7. Hwang S-Y, Park S, Kwon Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol Ther. 2019;199:30–57. doi:10.1016/j.pharmthera.2019.02.006
8. Li Y, Zhang H, Merkher Y, et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J Hematol Oncol. 2022;15(1):121. doi:10.1186/s13045-022-01341-0
9. MacDonald I, Nixon NA, Khan OF. Triple-negative breast cancer: a review of current curative intent therapies. Curr Oncol. 2022;29(7):4768–4778. doi:10.3390/curroncol29070378
10. Criscitiello C, Esposito A, Trapani D, et al. Prognostic and predictive value of tumor infiltrating lymphocytes in early breast cancer. Cancer Treat Rev. 2016;50:205–207. doi:10.1016/j.ctrv.2016.09.019
11. Loi S, Drubay D, Adams S, et al. Tumor-infiltrating lymphocytes and prognosis: a pooled individual patient analysis of early-stage triple-negative breast cancers. J Clin Oncol. 2019;37(7):559–569. doi:10.1200/JCO.18.01010
12. Benci JL, Johnson LR, Choa R, et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell. 2019;178(4):933–948. doi:10.1016/j.cell.2019.07.019
13. Liu Y, Zheng P. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci. 2020;41(1):4–12. doi:10.1016/j.tips.2019.11.003
14. Retecki K, Seweryn M, Graczyk-Jarzynka A, et al. The immune landscape of breast cancer: strategies for overcoming immunotherapy resistance. Cancers. 2021;13(23):6012. doi:10.3390/cancers13236012
15. Wang A, Yang X, Li R, et al. Immunomodulator-mediated suppressive tumor immune microenvironment remodeling nanoplatform for enhanced immuno/chemo/photothermal combination therapy of triple negative breast cancer. ACS Appl Mater Interfaces. 2023;15(46):53318–53332. doi:10.1021/acsami.3c14137
16. Jing ZH, Li YF, Song JX, et al. Efficient TNBC immunotherapy by dual reprogramming tumor-infiltrating dendritic cells and tumor-associated macrophages with stimulus-responsive miR155 nanocomplexes. Int J Biol Macromol. 2023;253(Pt 3):126912. doi:10.1016/j.ijbiomac.2023.126912
17. Mo XP, Zheng ZN, He Y, et al. Antiglioma via regulating oxidative stress and remodeling tumor-associated macrophage using lactoferrin-mediated biomimetic codelivery of simvastatin/fenretinide. J Control Release. 2018;287:12–23. doi:10.1016/j.jconrel.2018.08.012
18. Yang YC, Tian Q, Wu SQ, et al. Blue light-triggered Fe2+-release from monodispersed ferrihydrite nanoparticles for cancer iron therapy. Biomaterials. 2021;271:120739.
19. Tang K, Wang B, Yu B, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors and PROTAC-based degraders for cancer therapy. Eur J Med Chem. 2022;227:113967. doi:10.1016/j.ejmech.2021.113967
20. Zhao Y, Wang B, Liu J, et al. An overview on the methods of determining the activity of Indoleamine 2, 3-Dioxygenase 1. J Drug Target. 2019;27:724–731.
21. Zhai L, Ladomersky E, Lenzen A, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol. 2018;15(5):447–457. doi:10.1038/cmi.2017.143
22. Newman AC, Falcone M, Huerta Uribe A, et al. Immune-regulated IDO1-dependent tryptophan metabolism is source of one-carbon units for pancreatic cancer and stellate cells. Mol Cell. 2021;81:2290–2302.e7.
23. Li F, Zhang R, Li S, et al. IDO1: an important immunotherapy target in cancer treatment. Int Immunopharmacol. 2017;47:70–77. doi:10.1016/j.intimp.2017.03.024
24. Kraehenbuehl L, Weng CH, Eghbali S, et al. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol. 2022;19:37–50.
25. Li F, Zhao Y, Wei L, et al. Tumor-infiltrating Treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol Ther. 2018;19(8):695–705. doi:10.1080/15384047.2018.1450116
26. Fujiwara Y, Kato S, Nesline MK, et al. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat Rev. 2022;110:102461. doi:10.1016/j.ctrv.2022.102461
27. Song X, Si Q, Qi R, et al. Indoleamine 2, 3-Dioxygenase 1: a promising therapeutic target in malignant tumor. Front Immunol. 2021;12:800630. doi:10.3389/fimmu.2021.800630
28. Kenski JCN, Huang X, Vredevoogd DW, et al. An adverse tumor-protective effect of IDO1 inhibition. Cell Rep Med. 2023;4(2):100941. doi:10.1016/j.xcrm.2023.100941
29. Kroemer G, Galassi C, Zitvogel L, et al. Immunogenic cell stress and death. Nat Immunol. 2022;23(4):487–500. doi:10.1038/s41590-022-01132-2
30. Ahmed A, Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol. 2020;14(12):2994–3006. doi:10.1002/1878-0261.12851
31. Zhou J, Wang G, Chen Y, et al. Immunogenic cell death in cancer therapy: present and emerging inducers. J Cell Mol Med. 2019;23(8):4854–4865. doi:10.1111/jcmm.14356
32. Zhu M, Yang M, Zhang J, et al. Immunogenic cell death induction by ionizing radiation. Front Immunol. 2021;12:705361. doi:10.3389/fimmu.2021.705361
33. Alzeibak R, Mishchenko TA, Shilyagina NY, et al. Targeting immunogenic cancer cell death by photodynamic therapy: past, present and future. J Immunother Cancer. 2021;9(1):e001926. doi:10.1136/jitc-2020-001926
34. Krysko DV, Garg AD, Kaczmarek A, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875. doi:10.1038/nrc3380
35. Xu M, Lu JH, Zhong YZ, et al. Immunogenic cell death-relevant damage-associated molecular patterns and sensing receptors in triple-negative breast cancer molecular subtypes and implications for immunotherapy. Front Oncol. 2022;12:870914. doi:10.3389/fonc.2022.870914
36. Hayashi K, Nikolos F, Lee YC, et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat Commun. 2020;11(1):6299. doi:10.1038/s41467-020-19970-9
37. Jiang M, Zeng J, Zhao L, et al. Chemotherapeutic drug-induced immunogenic cell death for nanomedicine-based cancer chemo-immunotherapy. Nanoscale. 2021;13(41):17218–17235. doi:10.1039/D1NR05512G
38. Qi J, Jin F, You Y, et al. Synergistic effect of tumor chemo-immunotherapy induced by leukocyte-hitchhiking thermal-sensitive micelles. Nat Commun. 2021;12(1):4755. doi:10.1038/s41467-021-24902-2
39. Lau TS, Chan LKY, Man GCW, et al. Paclitaxel induces immunogenic cell death in ovarian cancer via TLR4/IKK2/SNARE-dependent exocytosis. Cancer Immunol Res. 2020;8(8):1099–1111. doi:10.1158/2326-6066.CIR-19-0616
40. Abdel-Bar HM, Walters AA, Lim Y, et al. An “eat me” combinatory nano-formulation for systemic immunotherapy of solid tumors. Theranostics. 2021;11(18):8738–8754. doi:10.7150/thno.56936
41. Song JX, Cheng MY, Xie Y, et al. Efficient tumor synergistic chemoimmunotherapy by self-augmented ROS-responsive immunomodulatory polymeric nanodrug. J Nanobiotechnology. 2023;21(1):93.
42. Banstola A, Poudel K, Kim JO, et al. Recent progress in stimuli-responsive nanosystems for inducing immunogenic cell death. J Control Release. 2021;337:505–520. doi:10.1016/j.jconrel.2021.07.038
43. Zuo H, Tao J, Wang M, et al. A novel immunochemotherapy based on immunogenicity-activated and immunosuppression-reversed biomimetic nanoparticles. RSC Adv. 2022;12(43):28104–28112. doi:10.1039/D2RA04326B
44. Li Y, Qiao K, Zhang X, et al. Targeting myeloid-derived suppressor cells to attenuate vasculogenic mimicry and synergistically enhance the anti-tumor effect of PD-1 inhibitor. iScience. 2021;24(12):103392. doi:10.1016/j.isci.2021.103392
45. Xia C, Li M, Ran G, et al. Redox-responsive nanoassembly restrained myeloid-derived suppressor cells recruitment through autophagy-involved lactate dehydrogenase A silencing for enhanced cancer immunochemotherapy. J Control Release. 2021;335:557–574. doi:10.1016/j.jconrel.2021.05.034
46. Birmpilis AI, Paschalis A, Mourkakis A, et al. Immunogenic cell death, DAMPs and prothymosin α as a putative anticancer immune response biomarker. Cells. 2022;11(9):1415. doi:10.3390/cells11091415
47. Chen Z, Zhao P, Luo Z, et al. Cancer cell membrane-biomimetic nanoparticles for homologous-targeting dual-modal imaging and photothermal therapy. ACS Nano. 2016;10(11):10049–10057. doi:10.1021/acsnano.6b04695
48. Jiang Y, Krishnan N, Zhou J, et al. Engineered cell-membrane-coated nanoparticles directly present tumor antigens to promote anticancer immunity. Adv Mater. 2020;32(30):e2001808. doi:10.1002/adma.202001808
49. Serpooshan V, Sheibani S, Pushparaj P, et al. Effect of cell sex on uptake of nanoparticles: the overlooked factor at the nanobio interface. ACS Nano. 2018;12(3):2253–2266. doi:10.1021/acsnano.7b06212
50. Kang T, Cho Y, Park C, et al. Effect of biomimetic shear stress on intracellular uptake and cell-killing efficiency of doxorubicin in a free and liposomal formulation. Int J Pharm. 2016;510(1):42–47. doi:10.1016/j.ijpharm.2016.06.017
51. Fucikova J, Spisek R, Kroemer G, et al. Calreticulin and cancer. Cell Res. 2021;31(1):5–16. doi:10.1038/s41422-020-0383-9
52. Shi Y, van der Meel R, Chen X, et al. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10(17):7921–7924. doi:10.7150/thno.49577
53. Zhen X, Cheng P, Pu K. Recent advances in cell membrane-camouflaged nanoparticles for cancer phototherapy. Small. 2019;15(1):e1804105. doi:10.1002/smll.201804105
54. An D, Yu X, Jiang L, et al. Reversal of multidrug resistance by apolipoprotein A1-modified doxorubicin liposome for breast cancer treatment. Molecules. 2021;26(5):1280. doi:10.3390/molecules26051280
55. Wang J, Yeung BZ, Cui M, et al. Exosome is a mechanism of intercellular drug transfer: application of quantitative pharmacology. J Control Release. 2017;268:147–158. doi:10.1016/j.jconrel.2017.10.020
56. Liu X, Chen Y, Li H, et al. Enhanced retention and cellular uptake of nanoparticles in tumors by controlling their aggregation behavior. ACS Nano. 2013;7(7):6244–6257. doi:10.1021/nn402201w
57. Wathiong B, Deville S, Jacobs A, et al. Role of nanoparticle size and sialic acids in the distinct time-evolution profiles of nanoparticle uptake in hematopoietic progenitor cells and monocytes. J Nanobiotechnology. 2019;17(1):62. doi:10.1186/s12951-019-0495-x
58. Rodriguez PL, Harada T, Christian DA, et al. Minimal “self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339(6122):971–975. doi:10.1126/science.1229568
59. Krueger TEG, Thorek DLJ, Denmeade SR, et al. Concise review: mesenchymal stem cell-based drug delivery: the good, the bad, the ugly, and the promise. Stem Cells Transl Med. 2018;7(9):651–663. doi:10.1002/sctm.18-0024
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.