Back to Journals » Journal of Inflammation Research » Volume 16
Efferocytosis: An Emerging Therapeutic Strategy for Type 2 Diabetes Mellitus and Diabetes Complications
Received 22 April 2023
Accepted for publication 24 June 2023
Published 7 July 2023 Volume 2023:16 Pages 2801—2815
DOI https://doi.org/10.2147/JIR.S418334
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xun Liu,1 Hua Liu,2 Yihui Deng1
1Hunan University of Chinese Medicine, Changsha, Hunan, 410208, People’s Republic of China; 2Southern Theater General Hospital of the Chinese People’s Liberation Army, Guangzhou, Guangdong, 510010, People’s Republic of China
Correspondence: Yihui Deng, Email [email protected]
Abstract: Increasing evidence indicates that chronic, low-grade inflammation is a significant contributor to the fundamental pathogenesis of type 2 diabetes mellitus (T2DM). Efferocytosis, an effective way to eliminate apoptotic cells (ACs), plays a critical role in inflammation resolution. Massive accumulation of ACs and the proliferation of persistent inflammation caused by defective efferocytosis have been proven to be closely associated with pancreatic islet β cell destruction, adipose tissue inflammation, skeletal muscle dysfunction, and liver metabolism abnormalities, which together are considered the most fundamental pathological mechanism underlying T2DM. Therefore, here we outline the association between the molecular mechanisms of efferocytosis in glucose homeostasis, T2DM, and its complications, and we analyzed the present constraints and potential future prospects for therapeutic targets in T2DM and its complications.
Keywords: type 2 diabetes mellitus, diabetes complications, efferocytosis, inflammation, apoptosis
Introduction
Physiologically, the recognition and elimination of apoptotic cells (ACs) comprise a host mechanism for maintaining homeostasis. Indeed, it is estimated that 100,000 cells in our bodies undergo apoptosis every second, before being engulfed by efferocytes.1 Efferocytes are cells that can specifically and efficiently eliminate ACs, including professional types (macrophages, dendritic cells) and non-professional types (monocytes, fibroblastic cells, adjacent epithelial cells).2 This clearance process is an evolutionally conserved process for removing ACs, known as efferocytosis, which inhibits secondary necrotic lysis of ACs and is crucial for restoring homeostasis after the occurrence of chronic inflammatory diseases.3 Currently, the resolution of inflammation in efferocytosis is believed to be attributed to four pathways:4 (1) inhibition of pro-inflammatory signals; (2) promotion of anti-inflammatory signals; (3) regulation of macrophage phenotype; and (4) improvement of Treg cell function. Although effective efferocytosis provokes efferocytes to exert anti-inflammatory benefits, invalid efferocytosis is responsible for secondary necrosis, causing further loss of contiguous cells.
Increasing evidence has shown that efferocytosis is essential to the development and progression of type 2 diabetes mellitus (T2DM) and its resulting complications.5 T2DM is a common chronic inflammatory illness with long-term consequences that widely affects all tissues and organs in the body.6 T2DM and its complications lead to increased morbidity, mortality, and healthcare costs.7 Although the occurrence of T2DM is closely related to genetic and environmental factors (eg, limited physical activity, bad eating habits, obesity, and aging), in general, its ultimate results from the imbalance of glucose metabolism in the body. Notably, compromised efferocytosis has been proposed to be closely related to pancreatic islet β cell destruction,8 adipose tissue inflammation,9 skeletal muscle dysfunction,10 and liver metabolism abnormalities,11 all of which largely contribute to the imbalance of glucose homeostasis. Accordingly, efferocytosis failure seems to be an initial node leading to T2DM and its complications.
Efferocytosis involves several steps, including recruitment, recognition, engulfment, and degradation, and multiple signaling molecules in each step play crucial roles in effective efferocytosis. Therefore, here we summarize the general mechanisms of efferocytosis and the role of efferocytosis failure in T2DM and its complications to provide a novel treatment approach for such diseases.
General Mechanisms of Efferocytosis
Recruitment
The effective elimination of ACs depends on the release of various “find me” signals, which are generally released at the early stage of apoptosis and lead to the migration and aggregation of efferocytes toward ACs (Figure 1A).
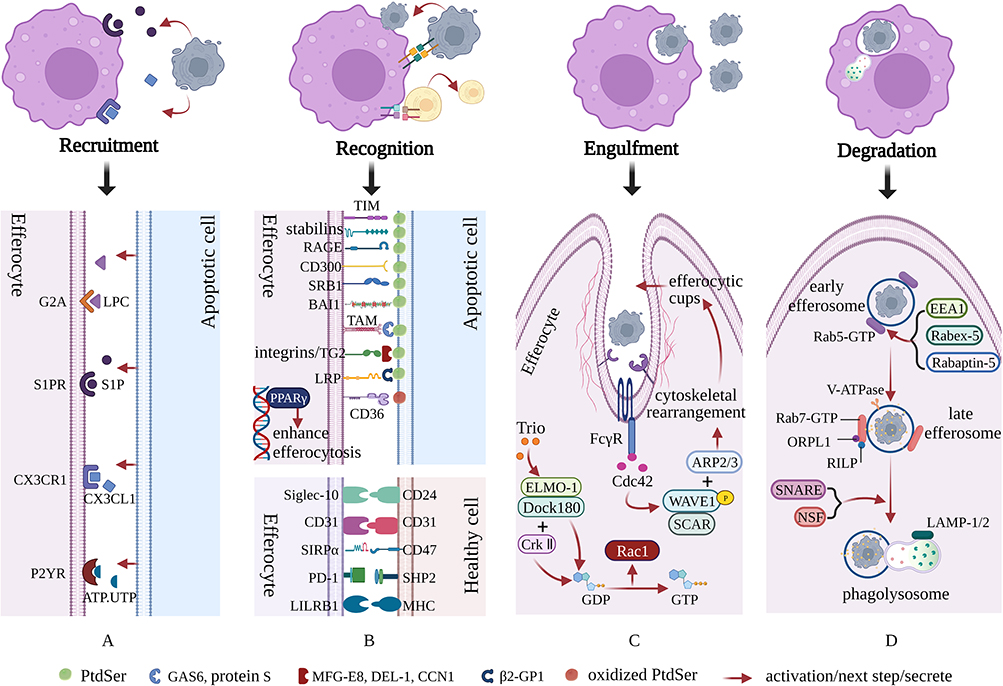 |
Figure 1 Molecular mechanisms of efferocytosis. Apoptotic cells (ACs) recruit efferocytes by releasing a series of molecules. Subsequently, efferocytes recognize ACs through unique “eat me” and “do not eat me” signals. Then, efferocytes engulf ACs through cytoskeletal recombination and the formation of efferocytic cups. Finally, ACs gradually degrade in mature phagolysosomes. (A) The effective elimination of ACs depends on the release of various “find me” signals, such as LPC, S1P, CX3CL1 and ATP/UTP, which result in the migration and aggregation of efferocytes toward ACs. (B) Once efferocytes are close enough to perform efferocytosis, “eat me” and “do not eat me” signals are used to distinguish between apoptotic cells and viable cells, while further cell-surface “eat me” and “do not eat me” signals assist with the subsequent stage of cell engulfment. (C) After recognition, efferocytes immediately initiate cytoskeletal rearrangements and the formation of efferocytic cups to rapidly complete the engulfment of ACs. This process largely relies on the Rho family of small GTPases, especially Rac-1, Rho G and Cdc42. (D) The degradation of ACs is finally completed after the newly generated efferosomes proceed through stages of early, late, and lysosomal interaction. During this process, Rab5-GTP and Rab7-GTP mediate the formation of early and late efferosomes, respectively. Additionally, these processes are accompanied by gradual acidification of the intracellular environment, which is beneficial to the degradation of ACs. Created with BioRender.com. |
These “find me” signals allow efferocytes to accurately and effectively locate ACs that need to be cleared. The following “find me” signals have been identified: lipids (lysophosphatidylcholine [LPC]12 and sphingosine 1-phosphate [S1P]13), proteins (chemokine C-X3-C motif ligand 1 [CX3CL1]),14 and extracellular nucleotides (adenosine triphosphate, [ATP] and uridine triphosphate, [UTP]).15 Specifically, LPC was the first “find me” signal to be discovered, and it is mainly formed through the hydrolysis of membrane phosphatidylcholine in ACs mediated by phospholipase A2 (PLA2), which relies on caspase-3 induction.12 Additionally, suppression of ATPase class I type 8b member 1 (Atp8b1) and ATP-binding cassette transporter A1 (ABCA1) can abolish the release of LPC, which hinders the migration of macrophages16 and monocytes17 to ACs, respectively. LPC binds and stimulates G2 accumulation (G2A),18,19 a G-protein-coupled receptor (GPCR) that is widely expressed on the surface of immune cells such as macrophages and dendritic cells. Although research on G2A is limited, its binding to LPC is a key initiator of efferocytosis and various pathophysiological reactions. S1P, derived from membrane sphingosine, is degraded by related enzymes and exerts a high affinity for S1P receptors 1–5 (S1PR1-5),20 which can orchestrate numerous physiological processes, including invasion, adhesion, and migration of macrophages. CX3CL1, a cytokine with chemotactic functions, is currently the only known member of the CX3CL subfamily.21 As a specific chemokine receptor, CX3CR1 can only bind and stimulate CX3CL1. Subsequently, CX3CL1 interacts with CX3CL1 to form the CX3CL1/CX3CR1 signal axis, which mediates the chemotaxis and migration of macrophages.14,22 ATP and UTP are also critical AC “find me” signals that can be sensed by purinergic P2 receptors of monocytes and macrophages, especially P2Y receptors (P2YRs).23 The deletion of ATP and UTP or artificial knockout of P2YRs significantly impairs the clearance of ACs.15 Notably, the release of ATP and UTP by ACs is controlled by pannexin-1 (PANX1) channels. The overexpression of PANX1 leads to extracellular ATP accumulation, eventually mediating the impairment of efferocytosis, increased inflammation of skeletal muscle, and insulin resistance (IR).24 Furthermore, some other soluble ligands, including thrombospondin 1, chemokine‐presenting apoptotic blebs, endothelial monocyte activating polypeptide II, tyrosyl tRNA synthetase, ribosomal protein S19 dime, and annexin A1 (ANXA1), can also recruit efferocytes to areas where ACs accumulate, as previously reviewed.25
Recognition
Once efferocytes are close enough to perform efferocytosis, a group of cell-surface chemicals known as “eat me” and “do not eat me” signals are used to distinguish between apoptotic and viable cells, which help with the subsequent stage of cell engulfment (Figure 1B).
Among all known “eat me” signals, phosphatidylserine (PtdSer) has been reported to be one of the most fundamental and pleiotropic mediators for achieving signal transduction between efferocytes and ACs.26 As a phospholipid, PtdSer is mainly located within the cell membrane under physiological conditions. However, when a cell undergoes stress or apoptosis, PtdSer turns outward to the outer surface of the cell membrane, where it serves as an “eat me” signal, which is then recognized by ligands on adjacent macrophages.27 Numerous receptors exposed on the surface of efferocytes have been associated with the PtdSer binding process on ACs. PtdSer is capable of binding efferocytes (macrophages, dendritic cells, fibroblastic cells, or endothelial cells) and strongly supporting the efferocytosis of ACs in a direct manner through receptors such as T cell immunoglobulin mucin receptors (TIMs: including TIM1,28 TIM3,29 and TIM4),30 stabilins (stabilin1 and stabilin2),31 the receptor for advanced glycation end products (RAGE),32 the CD300 family (CD300a, CD300b and CD300c),33,34 scavenger receptor class B member 1 (SRB1),35 and adhesion G protein-coupled receptor B1 (BAI1).36 However, PtdSer is also linked to efferocytes through its interactions with certain bridge molecules, such as growth arrest-specific protein 6 (GAS6), protein S, milk fat globule factor-e8 (MFG-E8), developmental endothelial locus-1 (DEL-1), and beta-2-glycoprotein 1 (β2-GP1). In brief, both GAS6 and protein S bridge PtdSer on ACs to the tumor-associated macrophage (TAM) tyrosine kinase receptors;37 MFG-E8, DEL-1 and cellular communication network factor 1 (CCN1) bridge PtdSer on ACs to the αvβ3/αvβ5 integrins;38–40 and β2-GP1 bridges PtdSer on ACs to the low-density lipoprotein receptor-related protein (LRP).41 Notably, TAM receptors are composed of Tyro3, Axl, and Mer, each with differential affinity to their ligands.42 Moreover, oxidized PtdSer rather than nonoxidized PtdSer43 is necessary for macrophage identification of ACs via the class B scavenger receptor CD36. Peroxisome-proliferator activated receptor gamma (PPARγ), a major member of the transcription factor PPAR family, is postulated as the principal driver of inflammation reduction and efferocytosis enhancement due to upregulation of molecular elements such as CD36, transglutaminase 2 (TG2), Axl, and prototypic long pentraxin 3 (PTX3).44 Notably, the recognition of ACs can be aided by additional signals. For instance, Fc receptors are capable of recognizing LPC on ACs via distinctive bridging ligand immunoglobulin M (IgM),45 while complement component C1q and mannose-binding lectin (MBL) are responsible for the interaction of calreticulin and LRP1,46,47 which are located on the membrane surfaces of ACs and macrophages, respectively.
Although recognition of the “eat me” signal is a prerequisite for efferocytosis, cells emerging from the “eat me” signal are not necessarily bound to be engulfed. The appearance of “do not eat me” signals enable viable cells to precisely avoid efferocytosis, facilitating cell survival. Moreover, CD24, CD31, CD47, Src homology region 2 domain-containing phosphatase-2 (SHP2), and major histocompatibility complex (MHC) distributed on healthy cells can interact with the inhibitory receptor sialic acid-binding Ig-like lectin-10 (Siglec-10),48 CD31,49 signal-regulatory-protein-α (SIRPα),50 programmed cell death protein 1 (PD-1),51 and leukocyte immunoglobulin-like receptor B1 (LILRB1),52 respectively, strongly suppressing the efferocytosis of viable cells.
Engulfment
Once a relatively long period of recognition between efferocytes and ACs has passed, efferocytes will immediately initiate cytoskeletal rearrangement and synthesis to rapidly complete the engulfment of ACs53 (Figure 1C).
During this process, activation of the Rho family of small GTPases (especially Rac-1, Rho G, Cdc42, and Rho A) through multiple signal pathways is a key step to synergistically facilitate cytoskeletal reorganization and the formation of efferocytic cups, completing the engulfment of ACs. The trimeric complex, composed of the phagocytic regulatory protein engulfment and cell motility protein 1 (ELMO-1), 180-kDa protein downstream of the dedicator of cytokinesis (Dock180), and Crk II appear to be favored by guanine nucleotide exchange factors (GEFs), the key regulators of Rac-1 activation via accelerating the exchange of GDP for GTP.54 Indeed, Dock180 is a linker connecting the two domains, meaning that the SH3 domain of Dock180 can bind to ELMO-1, while the SH3 domain of Crk II can interact with Dock180.55 Beyond that, Trio, the exchange agent functioning through the small GTPase Rho G, interacts with the complex of ELMO-1 and Dock180 to induce the active GTP-bound state of Rac-1.56 The activation of Rac-1 subsequently activates SCAR, an actin nucleation promoting agent that has been considered to be the upstream molecule of verprolin homology domain-containing protein (WAVE).57 Ultimately, the SCAR/WAVE1 complex nucleates the components of fresh actin filaments and forms efferocytic cups via recruiting the actin-related protein 2/3 (ARP2/3) complex.57 Notably, Rac-1 activation occurs later than that of Cdc42.58 In contrast to Rac-1, the activity of Cdc42-GEF is almost not delayed. After combining with Fc gamma receptor (FcγR), Cdc42 exhibits a strong ability to attract GEFs and promotes GDP/GTP exchange.59 Moreover, when Cdc42 is entirely active through the action of GEFs, it then concentrates on regulating actin polymerization, elongating pseudopods, and shaping efferocytic cups through stimulating the Wiskott-Aldrich syndrome protein (WASP) family, the other actin nucleation-promoting factors.59,60 Additionally, RhoA can be activated by binding to mammalian diaphanous-related formin 1 (mDia1) and Rho-associated protein kinases (ROCK), and the degree of its activation is inversely proportional to the clearance intensity of phagocytic efferocytosis.61
Although actin polymerization contributes to the formation of efferocytic cups and the accurate engulfment of ACs, the significant role of actin depolymerization in the dynamic equilibrium of efferocytosis cannot be overlooked. For instance, the presence of dynamin-2 at the plasma membrane is capable of blocking the phagosome formation.62
Degradation
Following the engulfment of ACs, efferocytes completely transform into the endocytotic vesicle (early efferosome), which is then triggered by multiple signaling events to generate a late efferosome. Subsequently, the late efferosome fuses with the lysosome to generate a mature conformation, which initiates measures to interfere with ACs, completing the entire digestion and degradation process. During this complex and precisely regulated process, it is crucial to protect normal tissues and cells from damage in an immune tolerance manner, while it is also necessary to cope with the excessive amount of proteins, nucleotides, and lipids resulting from the apoptotic process (Figure 1D).
The Rab GTPase family of proteins, especially Rab5 and Rab7, are key mediators in the canonical degradation cyclical process through the effectors responsible for the exchange of GTP-bound conformation and GDP-bound conformation.63 Control of early efferosome formation is governed by Rab5-GTP, which possesses fusion activity due to the activation and stabilization of several effectors, such as early endosomal antigen 1 (EEA1)64 and complex of Rabex-5 and Rabaptin-5.65 The process at this stage is relatively slow, with a half-life of approximately 15 min. Subsequently, the efferosome matures continuously from early to late stages, which is accompanied by the loss of Rab5-GTP and the acquisition of Rab7-GTP, mediated by the class C homotypic fusion and vacuole protein sorting (HOPS) complex.63 Following the establishment of late efferosomes, strong acidic efferosome lumens abundant in antimicrobial peptides and hydrolase are generated via vacuolar ATPase (V-ATPase), which results in the opening of the H+ channel and the influx of H+ into the lumen.66 Central displacement of late efferosomes and partial formation of the efferosome/lysosome (phagolysosome) are controlled by the interaction of dynactin, dynein, and Rab7-GTP, the latter of which is activated by effectors, including Rab7-interacting lysosomal protein (RILP)67 and oxysterol-binding protein–related protein 1 (ORPL1).68 Additionally, without lysosome-associated membrane protein 1 (LAMP-1) and LAMP-2, the migration of the phagolysosomes toward the microtubule-organizing centrum is hampered.69 Indeed, the formation and migration of phagolysosomes require tremendous attention to membrane fusion, which is a cumbersome and highly collaborative process. Soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptor (SNARE) complex and N-ethylmaleimide-sensitive factor (NSF) are crucial components of membrane receptors, which serve as catalysts to facilitate membrane fusion.70 Admittedly, the fusion of the membrane primarily depends on the assembly and disassembly of the SNARE complex, which incorporates (1) turning conformation of the syntaxin and manufacturing a precomplex of Q-SNARE proteins; (2) overcoming the repulsion and initiating SNARE complex formation; (3) unfolding the conformation of the clamping complex and accomplishing cis-SNARE complex establishment; (4) linking with SNAPs and NSF and recouping of the cis-SNARE complex; and (5) enzymatic hydrolysis and SNARE complex disassembly.70 Following fusion, the acidic strength further strengthens in the phagolysosome lumen via proton pump transporters, allowing for the degradation of phagocytic cargoes and the elimination of apoptotic bodies. Additionally, noncanonical degradations are mediated by LC3 and liver X receptors (LXRs), which have been reported in previous reviews.71
During degradation, efferocytes also generate cytokines such as transforming growth factor-β (TGF-β), interleukin-10 (IL-10), insulin-like growth factor-1 (IGF-1), and vascular endothelial growth factor (VEGF) to minimize inflammation, facilitate the regression of inflammation and the growth of adjacent cells, and maintain vascular integrity.72 Certainly, an explicit and accurate process of phagolysosome fusion and decomposition, as well as how the phagocytic cargoes are perfectly degraded in the phagolysosome, still merits further exploration.
Efferocytosis and Glucose Homeostasis
The normal blood glucose level is relatively stable, maintained at 3.89–6.11 mmol/L, which is the result of the dynamic balance between the production and consumption of glucose in the body. Usually, the production of glucose is highly controlled by three primary pathways: the digestion and absorption of sugary foods, hepatic glycogenolysis and gluconeogenesis of non-sugar substances. However, the consumption of glucose is governed by four main routes: aerobic oxidation; hepatic and muscle glycogen synthesis; mutation of other sugars; and transformation of adipose or amino acids. The above dynamic balance is regulated by various hormones, especially insulin and glucagon, which not only coordinate and restrict the metabolism of glucose, adipose, and amino acids, but also coordinate the metabolism of various organs and tissues, including adipose tissue, skeletal muscle and liver, to adapt to the changes in energy demand in the body through modulating the activity of critical enzymes. Once the defect appears in the insulin receptors of adipose tissue, skeletal muscle, the liver, or pancreatic islet β cells, glucose metabolism is dysregulated, resulting in the elevation of blood glucose and the occurrence of T2DM and associated complications. Accordingly, glucose homeostasis is a sophisticated and accurately mediated process, and any risk factors that affect the aforementioned metabolic pathways can contribute to the imbalance of homeostasis, among which, the failure of efferocytosis is one such risk factor that cannot be ignored.5 Below, we outline the impact of efferocytosis failure on glucose homeostasis, T2DM, and associated complications through pathological changes in different tissues (Figure 2).
 |
Figure 2 Efferocytosis failure leads to T2DM and its complications. Efferocytes have the function of clearing ACs in a non-inflammatory manner through what is called efferocytosis. Effective efferocytosis depends on the complex regulation of several processes, including recruitment, recognition, engulfment, and degradation. Defective efferocytosis triggers pancreatic islet β cell destruction, adipose tissue inflammation, skeletal muscle dysfunction, and liver metabolism abnormalities, all of which contribute to the occurrence of T2DM and complications, including diabetic macroangiopathy, diabetic microangiopathy, diabetic non-healing wounds, diabetic osteoporosis and diabetic periodontitis. Created with BioRender.com. |
Pancreatic Islet β Cell Destruction
Insulin is generated by the islet β cells and is the only hormone in the body that lowers blood glucose. Undoubtedly, apoptosis of islet β cells is a key feature of the pathogenesis of T2DM. Indeed, numerous studies on T2DM have shown that the destruction of islet β cells is accompanied by an increased proliferation of macrophages residing within the pancreas,73–75 which is conducive to the secretion of insulin after the death of islet β cells induced by intraperitoneal injection of streptozotocin (STZ).75 Furthermore, islet macrophages, which are mainly responsible for the production of IGF-1 outside the liver, also optimally exert efferocytosis to attenuate islet inflammation and reduce IR.75 Consequently, failed efferocytosis of islet macrophages can cause pancreatic islet β cell destruction, further disrupting glucose homeostasis. Moreover, the prolonged stimulation of high glucose (HG) in blood is capable of generating advanced glycation end products (AGEs), which not only cross-link with a variety of proteins, contributing to kidney, retina, and cardiovascular system damage,76 but can also be recognized by RAGE to activate multiple signal pathways to contribute to the occurrence of T2DM and its complications via enhancing oxidative stress and tissue inflammation.77 Beyond this, the concentration of AGEs is significantly increased in diabetic wounds, and exogenous addition of AGEs drives macrophage death and inflammation.78 Further evidence has provided support for the role of AGEs in suppressing the macrophage-mediated efferocytosis of islet β cells through restraining Rac1 activity and cytoskeletal rearrangement; ultimately, this facilitates the expansion of islet inflammation and further destruction of islet β cells and dramatically reduces insulin secretion,8 indicating that impaired efferocytosis is a key pathogenic element for complications of diabetes. However, Ward et al reported that during T2DM, the function of macrophages changes as a consequence of apoptotic islet β cell uptake, in that they exert beneficial anti-inflammatory effects in the early stage and play a destructive pro-inflammatory role in the late stage.79 The underlying mechanism may be related to the significant accumulation of ACs that need to be eliminated at the late stage79 or the different microenvironments in which islet macrophages reside.80 Thus, the exact mechanisms underlying efferocytosis in pancreatic islet β cell destruction have not yet been completely clarified and merit further exploration.
Adipose Tissue Inflammation
Adipose tissue is the pivotal energy storage depot and is also perceived as the largest endocrine organ of the body, which can induce the overproduction of multiple cytokines, free fatty acids (FFAs), and pro-inflammatory factors. A certain amount of adipose tissue not only guarantees the supply of energy and enhances the body’s ability to adapt to food deficiency environments but also participates in the synthesis and decomposition of glucose and regulates insulin signal transduction. High accumulation of adipose tissue, such as in overweight and obese people, gives rise to adipocyte inflammation, adipocyte dysfunction, and even adipocyte apoptosis through phenotypic transformation in adipose tissue and overproduction of pro-inflammatory mediators, such as FFAs.81 Additionally, massive aggregation of macrophages is the central event in adipocyte apoptosis, which also results in a long-term concentration of low-grade inflammation in the area around the adipose tissue to eventually allow for increased adipocyte death.82 In this state, adipose tissue inflammation is responsible for the persistent decrease in insulin sensitivity, which hastens the occurrence of continuous hyperglycemia and T2DM.83 Indeed, a previous study showed that in an in vitro co-culture of apoptotic adipocytes and macrophages, apoptotic adipocytes promoted the expression of genes related to efferocytosis and were cleared by the enhanced macrophage efferocytosis.84 However, this effect was eliminated by knocking down the arachidonate 15-lipoxygenase (ALOX15) gene or suppressing the expression of ALOX15 protein by drugs.84 Furthermore, apolipoprotein E 4 (APOE4) gene replacement mice fed a high-fat diet have been shown to develop a pathological decline in insulin sensitivity, at least partly due to macrophages carrying APOE4.85 Deeper research confirmed that the dramatic defects in efferocytosis were predominantly due to the expression of APOE4 protein in macrophages destroying adipocytes and worsening adipose tissue inflammation through strengthening endoplasmic reticulum stress (ERS).86 TG2, serving as a binding partner for integrin β3, is directly linked to MFG-E8 and jointly facilitates efferocytosis of macrophages to achieve engulfment of apoptotic adipocytes.87 The loss of TG2 severely influences insulin sensitivity of mice fed high-fat diets via defective efferocytosis; however, this deficiency can be reversed by LXR agonists, thereby partially alleviating IR.88 Admittedly, efferocytosis is capable of limiting inflammation and is conducive to the restoration of impaired tissues by efficiently and precisely eliminating apoptotic adipocytes.9 Exosomes from adipose-derived stem cells stimulate macrophages to switch from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype,89 which is accompanied by the supplement of anti-inflammatory factor IL-10 and the loss of pro-inflammatory factors such as tumor necrosis factor alpha (TNF-α) and nitric oxide (NO),90 which in turn reduce inflammation in adipose tissues and improve insulin sensitivity.91 In addition, M2 macrophages also support the generation of brown adipose tissues through the non-inflammatory clearance of adipocyte remnants and the coordinated production of PPAR ligands.92
Skeletal Muscle Dysfunction
Skeletal muscles are the main organs responsible for movement in the human body, and as a consequence, they demand the utmost nutrients to supplement energy. According to statistics, 80–90% of glucose is absorbed and utilized by the skeletal muscle after glucose ingestion.93 As the dominant regulator of glucose absorption and utilization, skeletal muscle dysfunction is a pivotal point in driving disorders of glucose metabolism and systemic IR. Although exhausted islet β cells cannot be repaired, merely rescuing the function of the skeletal muscle is sufficient to improve IR and increase insulin sensitivity.94,95 Fully functional skeletal muscles are required for healthy glucose homeostasis, and several studies have demonstrated that optimum efferocytosis is necessary to maintain normal skeletal muscle function. Specifically, DEL-1, an integrin ligand, is perceived as an endogenous anti-inflammatory element that positively regulates neutrophil chemotactic recruitment and enhances macrophage efferocytosis.96 Later studies confirmed that increasing the expression of DEL-1 is a powerful solution to skeletal muscle dysfunction, as it suppresses skeletal muscle inflammation and upturns IR through inhibiting silent mating type information regulation 2 homolog-1 (SIRT1)/sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2)-mediated signaling97 and activating the AMP-activated protein kinase (AMPK)/haemoxygenase (HO)-1 signal pathway.98 ANXA1, one of the “find me” signals of efferocytosis, is considered to be a critical regulator of the elimination of inflammation in bone marrow.99 Importantly, only macrophages carrying the ANXA1 gene can clear senescent neutrophils from the bone marrow99 and promote the repair and regeneration of damaged skeletal muscles via regulating the receptor for lipoxin A4 (FPR2/ALX) and the downstream AMPK intracellular signaling pathway.100 Moreover, nuclear factor IX (NFIX), a benign macrophage profibrotic agent, is linked with the resolution of inflammation after skeletal muscle injury.101 Further probing has revealed that NFIX is required for phenotypic transformation and function of phagocytes and that inhibition of negative pathways of efferocytosis such as RhoA-ROCK1 can boost NFIX expression and restore damaged skeletal muscles to allow healing.102 As a cell-surface transmembrane receptor that binds to PtdSer, SRB1 is widely distributed on macrophages.103 The lack of SRB1 is not conducive to the recovery of impaired skeletal muscle function as it is not only a new functional molecule that converts macrophages from pro- to anti-inflammatory phenotypes but is also an upstream factor in the MAP kinase (MAPK) inflammatory signaling pathway of macrophages.104 The absence of MFG-E8, a potent PtdSer receptor-driver, is another reason why it is difficult to recover from damaged skeletal muscle as it inhibits the anti-inflammatory effect of efferocytosis105 and counteracts the proliferation of myoblasts mediated by extracellular regulated protein kinase (ERK) and protein kinase B (Akt) activation.106 Furthermore, PPARγ,107 TG2,108 or TAM109 knockdown mice all exhibit disordered macrophage phenotypic transformation, which is insufficient to eliminate damaged skeletal muscle cells, promote skeletal muscle regeneration, and restore innate skeletal muscle function. Intriguingly, neuron-derived clone 77 (Nur77), a negative inflammatory regulator, and PPARγ play opposite roles in different macrophage subpopulations, in that Nur77 is capable of antagonizing the effects of PPARγ, expanding the scope of inflammation and prolonging skeletal muscle tissue repair.110 However, the specific mechanism underlying the antagonistic effect between Nur77 and PPARγ is currently unknown.
Liver Metabolism Abnormalities
The associations between efferocytosis, glucose homeostasis, and liver metabolism are particularly sophisticated.111 As another key organ for maintaining normal glucose homeostasis, in addition to being the primary site for gluconeogenesis and glycogen synthesis, the liver also plays a crucial role in indirectly regulating blood glucose levels through governing the lipid metabolism. Furthermore, the liver is the earliest organ to receive and manage insulin,112 and it is also unique in that it has two opportunities to degrade insulin: the first through the liver’s portal vein, and the second through the systemic circulation. Approximately 75% of insulin is degraded in these ways.113 Abnormalities in liver metabolism inevitably lead to insulin metabolism disorder. Generally, glycogenolysis and adipose synthesis in the liver depend on the interaction between insulin and liver insulin receptors. However, this interaction is weakened in patients with T2DM, which leads to a high-risk combination of hyperglycemia and hyperlipidemia.114 Liver metabolism abnormalities form the other core pathological mechanism of glucose homeostasis. Importantly, defective efferocytosis has emerged as a critical risk factor for aberrant liver metabolism, even mediating chronic hepatocyte death.115 Studies have demonstrated that dead fat-laden hepatocytes are engulfed by liver macrophages carrying ANXA1, suggesting that the efferocytosis of liver macrophages contributes to regulating lipid metabolism, and preventing inflammatory expansion, thereby restoring liver metabolic function.116 Apoptotic vesicles derived from mesenchymal stem cells (MSCs) induce the efferocytosis of liver macrophages in mice with T2DM, which allows for the improvement of hepatic steatosis and the advancement of insulin sensitivity through the calreticulin “eat me” signal.111 Subsequent studies have corroborated that powerful efferocytosis confers liver macrophages with the ability to block mitochondria-derived damage-associated molecular patterns (mito-DAMPs), thereby hindering the formation of liver fibrosis and aiding in abnormal liver metabolism repair.117 Besides, as a bridge molecule between PtdSer and αvβ3/αvβ5, CCN1 is beneficial to wound healing in diabetic Lepr (db/db) mice,40 suggesting that efferocytosis is helpful for various complications caused by glucose metabolism disorder and decreased IR in patients with T2DM. CCN1 also triggers the degradation of apoptotic neutrophils and the appropriate generation of TGF-β to restore damaged liver tissue and cellular function.118
Efferocytosis in Pathologies and Therapeutic Opportunities of T2DM Development
Efferocytosis depends on the formation of efferocytic cups and phagolysosomes, which is a distinctive degradation process to clear ACs in a non-inflammatory manner. Efferocytosis is extraordinarily well coupled to the principal pathologies of T2DM, an illness with chronic low-grade inflammation that requires indispensable anti-inflammatory treatment.119 In vivo experiments have revealed an increase in the number of dead cells with IR due to defects in efferocytosis.86 Animal experiments have found that adiponectin, a susceptibility gene and therapeutic target for T2DM, is responsible for defect-free efferocytosis by linking with calreticulin on macrophages.120 Moreover, obesity is known to be a critical determinant of T2DM. Indeed, previous studies have shown that macrophage-mediated AC clearance was significantly disrupted in obese mice, which was reversed by exogenous addition of erythropoietin (EPO).121 Despite the incomplete mechanistic understanding of how glucose affects efferocytosis, the findings mentioned above may indicate ways to target the efferocytosis in obesity and even T2DM. Intriguingly, a previous animal study found that failed or defective efferocytosis in obesity and T2DM is induced by elevated levels of saturated fatty acids (FAs), whereas feeding a fish oil diet abundant in omega-3 FAs is sufficient to reverse the aforementioned dilemma.122 Additionally, physical activity, an extraordinarily useful physical therapy for T2DM, enhances the activity of macrophage efferocytosis to mitigate inflammation and enhance insulin sensitivity via the augmentation of DEL-1 expression levels.98 Despite few studies on clinical drugs targeting efferocytosis to intervene in T2DM, non-pharmacological methods, such as changing dietary habits and strengthening exercise, have verified that potent efferocytosis represents a promising therapeutic target for T2DM.
Diabetic Macroangiopathy
Theoretically, the impairment of efferocytosis may contribute to specific compromises of diabetic macroangiopathy owing to the fact that the accumulation of ACs and persistent inflammation are unfavorable for endothelial function, clearance of apoptotic foam cells, and smooth muscle cell survival, all of which eventually lead to the development of atherosclerosis.123 Defective efferocytosis is postulated as a predominantly hazardous player in atherosclerosis,124 and reinforcement of efferocytosis aids in the reduction of atherosclerosis.125,126 Alternatively, the favorable remediation effect of cardiac function after myocardial infarction (MI) is attributed to cardiac macrophage efferocytosis, which generates VEGF in a CD36-dependent manner to reduce inflammation and promote lymphangiogenesis.127 The deletion of legumain, a gene encoding endolysosomal cysteine protease that is highly expressed in macrophages, has a prejudicial impact on cardiac function recovery after MI as it causes efferocytosis failure, widespread inflammation, and accumulation of apoptotic cardiomyocytes.128 Although how efferocytosis affects specific diabetic macroangiopathy remains an unanswered research question, evidence shows that efferocytosis plays a positive role in both T2DM and macroangiopathy individually, implying that it also contributes to the pathogenesis of diabetic macroangiopathy, highlighting the direction for future research.
Diabetic Microangiopathy
Partial mechanisms underlying diabetic cardiomyopathy (DCM) are closely associated with the loss of efferocytosis. Experimental studies have shown that salvage of defective efferocytosis in diabetes can promote the degradation of apoptotic cardiomyocytes and repair the damage of adjacent cardiomyocytes through overexpression of microRNAs-126.129 Furthermore, mice with DCM appear to show a reduction in LC3-mediated canonical autophagy and a high accumulation of apoptotic cardiomyocytes.130 LC3 is the critical driver of phagolysosome formation as LC3 depletion prevents efferosome-lysosome fusion,131,132 suggesting that the accumulation of apoptotic cardiomyocytes may be partially attributed to the defect in efferocytosis induced by the suppression of LC3-associated phagocytosis.
Diabetic Non-Healing Wounds
Non-healing wounds are the most common and distressing chronic complications of T2DM, and efficient efferocytosis is one of the decisive methods that positively regulates the wound-healing process.133,134 Both in vivo and in vitro experiments have confirmed that enhancing the efferocytosis of dendritic cells is beneficial for wound healing through inhibition of solute carrier family 7 member 11 (SLC7A11) function to increase aerobic glycolysis.135 Additionally, increasing the expression of MFG-E8136 achieves the same therapeutic effect. Single-cell RNA sequencing has revealed that the upregulation of Axl contributes to wound closure independent of efferocytosis,137 suggesting the involvement of other mechanisms. Interestingly, a new hybrid biomaterial composed of formyl-met-leu-phe (fMLP), FasL-conjugated silica nanoparticles (SiO2-FasL),138 and ANXA1 N-terminal peptide Ac2-26139 were found to successfully reduce inflammation and promote wound healing by accelerating efferocytosis to generate anti-inflammatory factors and eliminate excess neutrophils. Additionally, low-dose aspirin,140 insulin,141 modified collagen gel142 for external use, and Naoxintong (NXT)143 for oral administration, a Chinese patent medicine, are known to facilitate wound repair owing to effective efferocytosis via promoting macrophages to switch from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype.
Diabetic Osteoporosis
Osteoporosis is a common disease in elderly people, with osteoporosis caused by T2DM considered the most detrimental.144 Experiments in mice models have revealed that the PPARβ/δ agonist145 and exendin-4 synergized with vitamin D146 aid in the improvement of diabetic osteoporosis due to the correction of macrophage dysfunction via facilitating M1/M2 macrophage polarization. Co-culture of osteoclasts (OCs) and neutrophils in the HG environment revealed an increase in apoptotic neutrophils and a loss of efferocytosis of OCs, which could be reversed by an efferocytosis activator such as lipoxin A4 and insulin.147 Alternatively, lipoxin A4 and insulin significantly increased the number and efferocytosis of OCs and increased the bone absorption markers and bone mineral content in rats with STZ-induced diabetic osteoporosis.147
Diabetic Periodontitis
Diabetic periodontitis is a serious oral disease, which results from accumulation of apoptotic neutrophils and is characterized by chronic inflammation and, in severe cases, may result in periodontal abscess, tooth loss and even other more pernicious diabetes complications.148 Some studies have identified persistent cellular inflammation and severe periodontal damage due to the decreased efferocytosis of macrophages lacking SIRT6 after HG stimulation via the indirect inhibition of the DEL-1/CD36 “eat me” signaling axis,149 suggesting that reinforcement of efferocytosis may aid in the recovery of diabetic periodontitis.
Conclusion
The prompt clearance of ACs is indispensable for the preservation of homeostasis and recovery from inflammatory injury.3 Here, we provide an overview of the available research showing that massive accumulation of ACs and the proliferation of persistent inflammation due to defective efferocytosis are closely associated with pancreatic islet β cell destruction, adipose tissue inflammation, skeletal muscle dysfunction, and liver metabolism abnormalities, all of which are fundamental pathological mechanisms of T2DM. However, the research on efferocytosis in T2DM and its associated complications is still emerging. Indeed, the exploration of efferocytosis in diabetic wound healing is fairly widespread, but research on diabetic angiopathy, such as diabetic cardiocerebrovascular disease, diabetic nephropathy, and diabetic retinopathy, remains rare. Additionally, some animal experiments have confirmed that reasonable diet, physical exercise, and insulin-targeted enhancement of efferocytosis are beneficial to T2DM and its complications; however, the treatment measures applied to clinical research and research on other glucose-lowering medications such as metformin remain unclear. The research trajectory aimed at investigating efferocytosis in individuals with T2DM and its complications remains extensive. However, it is undeniable that efferocytosis has become a promising research direction for T2DM and its complications.
Acknowledgments
We thank LetPub for its linguistic assistance during the preparation of this manuscript. Figures were created with BioRender.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the Science and Technology Innovation Team Project of Hunan Province (2020RC4050), Hunan Province Traditional Chinese Medicine Research Program Project (E2022010) and Hunan Province Graduate Research Innovation Project (QL20210175).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147(4):742–758. doi:10.1016/j.cell.2011.10.033
2. Ge Y, Huang M, Yao YM. Efferocytosis and Its Role in Inflammatory Disorders Front Cell Dev Biol. 2022;10:839248.
3. Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20(4):254–267. doi:10.1038/s41577-019-0240-6
4. Zhang J, Ding W, Zhao M, et al. Mechanisms of efferocytosis in determining inflammation resolution: therapeutic potential and the association with cardiovascular disease. Br J Pharmacol. 2022;179(23):5151–5171. doi:10.1111/bph.15939
5. Mahmoudi A, Firouzjaei AA, Darijani F, et al. Effect of diabetes on efferocytosis process. Mol Biol Rep. 2022;49(11):10849–10863. doi:10.1007/s11033-022-07725-2
6. Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP. Diabetes mellitus and inflammation. Curr Diab Rep. 2013;13(3):435–444. doi:10.1007/s11892-013-0375-y
7. Tomic D, Shaw JE, Magliano DJ. The burden and risks of emerging complications of diabetes mellitus. Nat Rev Endocrinol. 2022;18(9):525–539. doi:10.1038/s41574-022-00690-7
8. Mao QY, He SY, Hu QY, et al. Advanced Glycation End Products (AGEs) inhibit macrophage efferocytosis of apoptotic β cells through binding to the receptor for AGEs. J Immunol. 2022;208(5):1204–1213. doi:10.4049/jimmunol.2100695
9. Tajbakhsh A, Gheibihayat SM, Karami N, et al. The regulation of efferocytosis signaling pathways and adipose tissue homeostasis in physiological conditions and obesity: current understanding and treatment options. Obes Rev. 2022;23(10):e13487. doi:10.1111/obr.13487
10. Juban G, Chazaud B. Efferocytosis during skeletal muscle regeneration. Cells. 2021;10(12):3267. doi:10.3390/cells10123267
11. Horst AK, Tiegs G, Diehl L. Contribution of macrophage efferocytosis to liver homeostasis and disease. Front Immunol. 2019;10:2670. doi:10.3389/fimmu.2019.02670
12. Lauber K, Bohn E, Kröber SM, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113(6):717–730. doi:10.1016/s0092-8674(03)00422-7
13. Gude DR, Alvarez SE, Paugh SW, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB j. 2008;22(8):2629–2638. doi:10.1096/fj.08-107169
14. Truman LA, Ford CA, Pasikowska M, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112(13):5026–5036. doi:10.1182/blood-2008-06-162404
15. Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–286. doi:10.1038/nature08296
16. Yang WJ, Cao RC, Xiao W, et al. Acinar ATP8b1/LPC pathway promotes macrophage efferocytosis and clearance of inflammation during chronic pancreatitis development. Cell Death Dis. 2022;13(10):893. doi:10.1038/s41419-022-05322-6
17. Peter C, Waibel M, Keppeler H, et al. Release of lysophospholipid ‘find-me’ signals during apoptosis requires the ATP-binding cassette transporter A1. Autoimmunity. 2012;45(8):568–573. doi:10.3109/08916934.2012.719947
18. Zhu K, Baudhuin LM, Hong G, et al. Sphingosylphosphorylcholine and lysophosphatidylcholine are ligands for the G protein-coupled receptor GPR4. J Biol Chem. 2001;276(44):41325–41335. doi:10.1074/jbc.M008057200
19. Peter C, Waibel M, Radu CG, et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem. 2008;283(9):5296–5305. doi:10.1074/jbc.M706586200
20. Cartier A, Hla T. Sphingosine 1-phosphate: lipid signaling in pathology and therapy. Science. 2019;366(6463). doi:10.1126/science.aar5551
21. Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385(6617):640–644. doi:10.1038/385640a0
22. Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91(4):521–530. doi:10.1016/s0092-8674(00)80438-9
23. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509(7500):310–317. doi:10.1038/nature13085
24. Jorquera G, Meneses-Valdés R, Rosales-Soto G, et al. High extracellular ATP levels released through pannexin-1 channels mediate inflammation and insulin resistance in skeletal muscle fibres of diet-induced obese mice. Diabetologia. 2021;64(6):1389–1401. doi:10.1007/s00125-021-05418-2
25. Tajbakhsh A, Yousefi F, Abedi SM, et al. The cross-talk between soluble “Find me” and “Keep out” signals as an initial step in regulating efferocytosis. J Cell Physiol. 2022;237(8):3113–3126. doi:10.1002/jcp.30770
26. Segawa K, Nagata S. An apoptotic ‘Eat Me’ signal: phosphatidylserine exposure. Trends Cell Biol. 2015;25(11):639–650. doi:10.1016/j.tcb.2015.08.003
27. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148(7):2207–2216.
28. Kobayashi N, Karisola P, Peña-Cruz V, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27(6):927–940. doi:10.1016/j.immuni.2007.11.011
29. DeKruyff RH, Bu X, Ballesteros A, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184(4):1918–1930. doi:10.4049/jimmunol.0903059
30. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. doi:10.1038/nature06307
31. Lee SJ, Park SY, Jung MY, Bae SM, Kim IS. Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood. 2011;117(19):5215–5223. doi:10.1182/blood-2010-10-313239
32. He M, Kubo H, Morimoto K, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011;12(4):358–364. doi:10.1038/embor.2011.28
33. Lankry D, Rovis TL, Jonjic S, Mandelboim O. The interaction between CD300a and phosphatidylserine inhibits tumor cell killing by NK cells. Eur J Immunol. 2013;43(8):2151–2161. doi:10.1002/eji.201343433
34. Murakami Y, Tian L, Voss OH, Margulies DH, Krzewski K, Coligan JE. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ. 2014;21(11):1746–1757. doi:10.1038/cdd.2014.86
35. Tao H, Yancey PG, Babaev VR, et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res. 2015;56(8):1449–1460. doi:10.1194/jlr.M056689
36. Lala T, Doan JK, Takatsu H, Hartzell HC, Shin HW, Hall RA. Phosphatidylserine exposure modulates adhesion GPCR BAI1 (ADGRB1) signaling activity. J Biol Chem. 2022;298(12):102685. doi:10.1016/j.jbc.2022.102685
37. Vago JP, Amaral FA, van de Loo FAJ. Resolving inflammation by TAM receptor activation. Pharmacol Ther. 2021;227:107893. doi:10.1016/j.pharmthera.2021.107893
38. Kourtzelis I, Li X, Mitroulis I, et al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol. 2019;20(1):40–49. doi:10.1038/s41590-018-0249-1
39. Akakura S, Singh S, Spataro M, et al. The opsonin MFG-E8 is a ligand for the alphavbeta5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res. 2004;292(2):403–416. doi:10.1016/j.yexcr.2003.09.011
40. Jun JI, Kim KH, Lau LF. The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat Commun. 2015;6:7386. doi:10.1038/ncomms8386
41. Maiti SN, Balasubramanian K, Ramoth JA, Schroit AJ. Beta-2-glycoprotein 1-dependent macrophage uptake of apoptotic cells. Binding to lipoprotein receptor-related protein receptor family members. J Biol Chem. 2008;283(7):3761–3766. doi:10.1074/jbc.M704990200
42. McShane L, Tabas I, Lemke G, Kurowska-Stolarska M, Maffia P. TAM receptors in cardiovascular disease. Cardiovasc Res. 2019;115(8):1286–1295. doi:10.1093/cvr/cvz100
43. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203(12):2613–2625. doi:10.1084/jem.20060370
44. Majai G, Sarang Z, Csomós K, Zahuczky G, Fésüs L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. 2007;37(5):1343–1354. doi:10.1002/eji.200636398
45. Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med. 2002;196(5):655–665. doi:10.1084/jem.20020542
46. Gardai SJ, McPhillips KA, Frasch SC, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–334. doi:10.1016/j.cell.2005.08.032
47. Ogden CA, deCathelineau A, Hoffmann PR, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194(6):781–795. doi:10.1084/jem.194.6.781
48. Bradley CA. CD24 - a novel ‘don’t eat me’ signal. Nat Rev Cancer. 2019;19(10):541. doi:10.1038/s41568-019-0193-x
49. Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418(6894):200–203. doi:10.1038/nature00811
50. Kojima Y, Volkmer JP, McKenna K, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536(7614):86–90. doi:10.1038/nature18935
51. Marasco M, Berteotti A, Weyershaeuser J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv. 2020;6(5):eaay4458. doi:10.1126/sciadv.aay4458
52. Barkal AA, Weiskopf K, Kao KS, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19(1):76–84. doi:10.1038/s41590-017-0004-z
53. Richards DM, Endres RG. The mechanism of phagocytosis: two stages of engulfment. Biophys J. 2014;107(7):1542–1553. doi:10.1016/j.bpj.2014.07.070
54. Gumienny TL, Brugnera E, Tosello-Trampont AC, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107(1):27–41. doi:10.1016/s0092-8674(01)00520-7
55. Hasegawa H, Kiyokawa E, Tanaka S, et al. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16(4):1770–1776. doi:10.1128/mcb.16.4.1770
56. deBakker CD, Haney LB, Kinchen JM, et al. Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr Biol. 2004;14(24):2208–2216. doi:10.1016/j.cub.2004.12.029
57. Evans IR, Ghai PA, Urbančič V, Tan KL, Wood W. SCAR/WAVE-mediated processing of engulfed apoptotic corpses is essential for effective macrophage migration in Drosophila. Cell Death Differ. 2013;20(5):709–720. doi:10.1038/cdd.2012.166
58. Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell. 2004;15(8):3509–3519. doi:10.1091/mbc.e03-11-0847
59. Park H, Cox D. Cdc42 regulates Fc gamma receptor-mediated phagocytosis through the activation and phosphorylation of Wiskott-Aldrich syndrome protein (WASP) and neural-WASP. Mol Biol Cell. 2009;20(21):4500–4508. doi:10.1091/mbc.e09-03-0230
60. Dart AE, Donnelly SK, Holden DW, Way M, Caron E. Nck and Cdc42 co-operate to recruit N-WASP to promote FcγR-mediated phagocytosis. J Cell Sci. 2012;125(Pt 12):2825–2830. doi:10.1242/jcs.106583
61. Bros M, Haas K, Moll L, Grabbe S. RhoA as a key regulator of innate and adaptive immunity. Cells. 2019;8(7):733. doi:10.3390/cells8070733
62. Marie-Anaïs F, Mazzolini J, Herit F, Niedergang F. Dynamin-actin cross talk contributes to phagosome formation and closure. Traffic. 2016;17(5):487–499. doi:10.1111/tra.12386
63. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–525. doi:10.1038/nrm2728
64. Rubino M, Miaczynska M, Lippé R, Zerial M. Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J Biol Chem. 2000;275(6):3745–3748. doi:10.1074/jbc.275.6.3745
65. Horiuchi H, Lippé R, McBride HM, et al. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell. 1997;90(6):1149–1159. doi:10.1016/s0092-8674(00)80380-3
66. Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry. 2010;49(23):4715–4723. doi:10.1021/bi100397s
67. Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003;23(18):6494–6506. doi:10.1128/mcb.23.18.6494-6506.2003
68. Johansson M, Lehto M, Tanhuanpää K, Cover TL, Olkkonen VM. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol Biol Cell. 2005;16(12):5480–5492. doi:10.1091/mbc.e05-03-0189
69. Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26(2):313–324. doi:10.1038/sj.emboj.7601511
70. Yoon TY, Munson M. SNARE complex assembly and disassembly. Curr Biol. 2018;28(8):R397–r401. doi:10.1016/j.cub.2018.01.005
71. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21(7):398–414. doi:10.1038/s41580-020-0232-1
72. Han CZ, Juncadella IJ, Kinchen JM, et al. Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature. 2016;539(7630):570–574. doi:10.1038/nature20141
73. Ying W, Fu W, Lee YS, Olefsky JM. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat Rev Endocrinol. 2020;16(2):81–90. doi:10.1038/s41574-019-0286-3
74. Lundberg M, Seiron P, Ingvast S, Korsgren O, Skog O. Insulitis in human diabetes: a histological evaluation of donor pancreases. Diabetologia. 2017;60(2):346–353. doi:10.1007/s00125-016-4140-z
75. Nackiewicz D, Dan M, Speck M, et al. Islet macrophages shift to a reparative state following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience. 2020;23(1):100775. doi:10.1016/j.isci.2019.100775
76. Yamagishi S, Fukami K, Matsui T. Crosstalk between advanced glycation end products (AGEs)-receptor RAGE axis and dipeptidyl peptidase-4-incretin system in diabetic vascular complications. Cardiovasc Diabetol. 2015;14:2. doi:10.1186/s12933-015-0176-5
77. Tóbon-Velasco JC, Cuevas E, Torres-Ramos MA. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol Disord Drug Targets. 2014;13(9):1615–1626. doi:10.2174/1871527313666140806144831
78. Wang Q, Zhu G, Cao X, Dong J, Song F, Niu Y. Blocking AGE-RAGE signaling improved functional disorders of macrophages in diabetic wound. J Diabetes Res. 2017;2017:1428537. doi:10.1155/2017/1428537
79. Ward MG, Li G, Hao M. Apoptotic β-cells induce macrophage reprogramming under diabetic conditions. J Biol Chem. 2018;293(42):16160–16173. doi:10.1074/jbc.RA118.004565
80. Parv K, Westerlund N, Merchant K, Komijani M, Lindsay RS, Christoffersson G. Phagocytosis and efferocytosis by resident macrophages in the mouse pancreas. Front Endocrinol. 2021;12:606175. doi:10.3389/fendo.2021.606175
81. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185. doi:10.1038/nature21363
82. Lindhorst A, Raulien N, Wieghofer P, et al. Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages. Cell Death Dis. 2021;12(6):579. doi:10.1038/s41419-021-03872-9
83. Zatterale F, Longo M, Naderi J, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol. 2019;10:1607. doi:10.3389/fphys.2019.01607
84. Kwon HJ, Kim SN, Kim YA, Lee YH. The contribution of arachidonate 15-lipoxygenase in tissue macrophages to adipose tissue remodeling. Cell Death Dis. 2016;7(6):e2285. doi:10.1038/cddis.2016.190
85. Arbones-Mainar JM, Johnson LA, Altenburg MK, Kim HS, Maeda N. Impaired adipogenic response to thiazolidinediones in mice expressing human apolipoproteinE4. FASEB J. 2010;24(10):3809–3818. doi:10.1096/fj.10-159517
86. Cash JG, Kuhel DG, Basford JE, et al. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J Biol Chem. 2012;287(33):27876–27884. doi:10.1074/jbc.M112.377549
87. Tóth B, Garabuczi E, Sarang Z, et al. Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J Immunol. 2009;182(4):2084–2092. doi:10.4049/jimmunol.0803444
88. Sághy T, Köröskényi K, Hegedűs K, et al. Loss of transglutaminase 2 sensitizes for diet-induced obesity-related inflammation and insulin resistance due to enhanced macrophage c-Src signaling. Cell Death Dis. 2019;10(6):439. doi:10.1038/s41419-019-1677-z
89. Zhu D, Johnson TK, Wang Y, et al. Macrophage M2 polarization induced by exosomes from adipose-derived stem cells contributes to the exosomal proangiogenic effect on mouse ischemic hindlimb. Stem Cell Res Ther. 2020;11(1):162. doi:10.1186/s13287-020-01669-9
90. Ghahremani Piraghaj M, Soudi S, Ghanbarian H, Bolandi Z, Namaki S, Hashemi SM. Effect of efferocytosis of apoptotic mesenchymal stem cells (MSCs) on C57BL/6 peritoneal macrophages function. Life Sci. 2018;212:203–212. doi:10.1016/j.lfs.2018.09.052
91. Zhao H, Shang Q, Pan Z, et al. Exosomes from adipose-derived stem cells attenuate adipose inflammation and obesity through polarizing M2 macrophages and beiging in white adipose tissue. Diabetes. 2018;67(2):235–247. doi:10.2337/db17-0356
92. Lee YH, Kim SN, Kwon HJ, Maddipati KR, Granneman JG. Adipogenic role of alternatively activated macrophages in β-adrenergic remodeling of white adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2016;310(1):R55–65. doi:10.1152/ajpregu.00355.2015
93. Ferrannini E, Simonson DC, Katz LD, et al. The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metabolism. 1988;37(1):79–85. doi:10.1016/0026-0495(88)90033-9
94. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(2):S157–163. doi:10.2337/dc09-S302
95. Merz KE, Thurmond DC. Role of skeletal muscle in insulin resistance and glucose uptake. Compr Physiol. 2020;10(3):785–809. doi:10.1002/cphy.c190029
96. Choi EY, Chavakis E, Czabanka MA, et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science. 2008;322(5904):1101–1104. doi:10.1126/science.1165218
97. Sun JL, Park J, Lee T, Jeong JH, Jung TW. DEL-1 ameliorates high-fat diet-induced insulin resistance in mouse skeletal muscle through SIRT1/SERCA2-mediated ER stress suppression. Biochem Pharmacol. 2020;171:113730. doi:10.1016/j.bcp.2019.113730
98. Kwon CH, Sun JL, Kim MJ, Abd El-Aty AM, Jeong JH, Jung TW. Clinically confirmed DEL-1 as a myokine attenuates lipid-induced inflammation and insulin resistance in 3T3-L1 adipocytes via AMPK/HO-1- pathway. Adipocyte. 2020;9(1):576–586. doi:10.1080/21623945.2020.1823140
99. Dalli J, Jones CP, Cavalcanti DM, Farsky SH, Perretti M, Rankin SM. Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J. 2012;26(1):387–396. doi:10.1096/fj.11-182089
100. McArthur S, Juban G, Gobbetti T, et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J Clin Invest. 2020;130(3):1156–1167. doi:10.1172/jci124635
101. Saclier M, Angelini G, Bonfanti C, Mura G, Temponi G, Messina G. Selective ablation of Nfix in macrophages attenuates muscular dystrophy by inhibiting fibro-adipogenic progenitor-dependent fibrosis. J Pathol. 2022;257(3):352–366. doi:10.1002/path.5895
102. Saclier M, Lapi M, Bonfanti C, Rossi G, Antonini S, Messina G. The transcription factor nfix requires RhoA-ROCK1 dependent phagocytosis to mediate macrophage skewing during skeletal muscle regeneration. Cells. 2020;9(3). doi:10.3390/cells9030708
103. Chinetti G, Gbaguidi FG, Griglio S, et al. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation. 2000;101(20):2411–2417. doi:10.1161/01.cir.101.20.2411
104. Zhang J, Qu C, Li T, Cui W, Wang X, Du J. Phagocytosis mediated by scavenger receptor class BI promotes macrophage transition during skeletal muscle regeneration. J Biol Chem. 2019;294(43):15672–15685. doi:10.1074/jbc.RA119.008795
105. Ikemoto-Uezumi M, Zhou H, Kurosawa T, et al. Increased MFG-E8 at neuromuscular junctions is an exacerbating factor for sarcopenia-associated denervation. Aging Cell. 2022;21(1):e13536. doi:10.1111/acel.13536
106. Li H, Guan K, Liu D, Liu M. Identification of mitochondria-related hub genes in sarcopenia and functional regulation of MFG-E8 on ROS-mediated mitochondrial dysfunction and cell cycle arrest. Food Funct. 2022;13(2):624–638. doi:10.1039/d1fo02610k
107. Varga T, Mounier R, Patsalos A, et al. Macrophage PPARγ, a lipid activated transcription factor controls the growth factor GDF3 and skeletal muscle regeneration. Immunity. 2016;45(5):1038–1051. doi:10.1016/j.immuni.2016.10.016
108. Budai Z, Al-Zaeed N, Szentesi P, et al. Impaired skeletal muscle development and regeneration in transglutaminase 2 knockout mice. Cells. 2021;10(11). doi:10.3390/cells10113089
109. Al-Zaeed N, Budai Z, Szondy Z, Sarang Z. TAM kinase signaling is indispensable for proper skeletal muscle regeneration in mice. Cell Death Dis. 2021;12(6):611. doi:10.1038/s41419-021-03892-5
110. Garabuczi É, Tarban N, Fige É, et al. Nur77 and PPARγ regulate transcription and polarization in distinct subsets of M2-like reparative macrophages during regenerative inflammation. Front Immunol. 2023;14:1139204. doi:10.3389/fimmu.2023.1139204
111. Zheng C, Sui B, Zhang X, et al. Apoptotic vesicles restore liver macrophage homeostasis to counteract type 2 diabetes. J Extracell Vesicles. 2021;10(7):e12109. doi:10.1002/jev2.12109
112. Song SH, McIntyre SS, Shah H, Veldhuis JD, Hayes PC, Butler PC. Direct measurement of pulsatile insulin secretion from the portal vein in human subjects. J Clin Endocrinol Metab. 2000;85(12):4491–4499. doi:10.1210/jcem.85.12.7043
113. Meier JJ, Veldhuis JD, Butler PC. Pulsatile insulin secretion dictates systemic insulin delivery by regulating hepatic insulin extraction in humans. Diabetes. 2005;54(6):1649–1656. doi:10.2337/diabetes.54.6.1649
114. Mu W, Cheng XF, Liu Y, et al. Potential nexus of non-alcoholic fatty liver disease and type 2 diabetes mellitus: insulin resistance between hepatic and peripheral tissues. Front Pharmacol. 2019;9:1566. doi:10.3389/fphar.2018.01566
115. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15(12):738–752. doi:10.1038/s41575-018-0065-y
116. Jindal A, Bruzzì S, Sutti S, et al. Fat-laden macrophages modulate lobular inflammation in nonalcoholic steatohepatitis (NASH). Exp Mol Pathol. 2015;99(1):155–162. doi:10.1016/j.yexmp.2015.06.015
117. An P, Wei LL, Zhao S, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun. 2020;11(1):2362. doi:10.1038/s41467-020-16092-0
118. Kim KH, Cheng N, Lau LF. Cellular communication network factor 1-stimulated liver macrophage efferocytosis drives hepatic stellate cell activation and liver fibrosis. Hepatol Commun. 2022;6(10):2798–2811. doi:10.1002/hep4.2057
119. Donath MY. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia. 2016;59(4):679–682. doi:10.1007/s00125-016-3873-z
120. Takemura Y, Ouchi N, Shibata R, et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. 2007;117(2):375–386. doi:10.1172/jci29709
121. Luo B, Wang Z, Zhang Z, Shen Z, Zhang Z. The deficiency of macrophage erythropoietin signaling contributes to delayed acute inflammation resolution in diet-induced obese mice. Biochim Biophys Acta Mol Basis Dis. 2019;1865(2):339–349. doi:10.1016/j.bbadis.2018.10.005
122. Li S, Sun Y, Liang CP, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res. 2009;105(11):1072–1082. doi:10.1161/circresaha.109.199570
123. Van Vré EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(4):887–893. doi:10.1161/atvbaha.111.224873
124. Yurdagul A Jr, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2017;4:86. doi:10.3389/fcvm.2017.00086
125. Doddapattar P, Dev R, Ghatge M, et al. Myeloid Cell PKM2 deletion enhances efferocytosis and reduces atherosclerosis. Circ Res. 2022;130(9):1289–1305. doi:10.1161/circresaha.121.320704
126. Kumar D, Pandit R, Yurdagul A Jr. Mechanisms of continual efferocytosis by macrophages and its role in mitigating atherosclerosis. Immunometabolism. 2023;5(1):e00017. doi:10.1097/in9.0000000000000017
127. Glinton KE, Ma W, Lantz C, et al. Macrophage-produced VEGFC is induced by efferocytosis to ameliorate cardiac injury and inflammation. J Clin Invest. 2022;132(9). doi:10.1172/jci140685
128. Jia D, Chen S, Bai P, et al. Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation. 2022;145(20):1542–1556. doi:10.1161/circulationaha.121.057549
129. Suresh Babu S, Thandavarayan RA, Joladarashi D, et al. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci Rep. 2016;6:36207. doi:10.1038/srep36207
130. Zhang M, Lin J, Wang S, et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J Pineal Res. 2017;63(2):e12418. doi:10.1111/jpi.12418
131. Nakamura S, Yoshimori T. New insights into autophagosome-lysosome fusion. J Cell Sci. 2017;130(7):1209–1216. doi:10.1242/jcs.196352
132. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology. 2020;158(7):1913–1928. doi:10.1053/j.gastro.2019.11.311
133. Aitcheson SM, Frentiu FD, Hurn SE, Edwards K, Murray RZ. Skin wound healing: normal macrophage function and macrophage dysfunction in diabetic wounds. Molecules. 2021;26(16):4917. doi:10.3390/molecules26164917
134. Khanna S, Biswas S, Shang Y, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5(3):e9539. doi:10.1371/journal.pone.0009539
135. Maschalidi S, Mehrotra P, Keçeli BN, et al. Targeting SLC7A11 improves efferocytosis by dendritic cells and wound healing in diabetes. Nature. 2022;606(7915):776–784. doi:10.1038/s41586-022-04754-6
136. Das A, Ghatak S, Sinha M, et al. Correction of MFG-E8 resolves inflammation and promotes cutaneous wound healing in diabetes. J Immunol. 2016;196(12):5089–5100. doi:10.4049/jimmunol.1502270
137. Justynski O, Bridges K, Krause W, et al. Apoptosis recognition receptors regulate skin tissue repair in mice. bioRxiv. 2023. doi:10.1101/2023.01.17.523241
138. Liu X, Dou G, Li Z, et al. Hybrid biomaterial initiates refractory wound healing via inducing transiently heightened inflammatory responses. Adv Sci. 2022;9(21):e2105650. doi:10.1002/advs.202105650
139. Huang JJ, Xia CJ, Wei Y, et al. Annexin A1-derived peptide Ac2-26 facilitates wound healing in diabetic mice. Wound Repair Regen. 2020;28(6):772–779. doi:10.1111/wrr.12860
140. Dardenne C, Salon M, Authier H, et al. Topical aspirin administration improves cutaneous wound healing in diabetic mice through a phenotypic switch of wound macrophages toward an anti-inflammatory and proresolutive profile characterized by LXA4 release. Diabetes. 2022;71(10):2181–2196. doi:10.2337/db20-1245
141. Yang P, Wang X, Wang D, et al. Topical insulin application accelerates diabetic wound healing by promoting anti-inflammatory macrophage polarization. J Cell Sci. 2020;133(19). doi:10.1242/jcs.235838
142. Das A, Abas M, Biswas N, et al. A modified collagen dressing induces transition of inflammatory to reparative phenotype of wound macrophages. Sci Rep. 2019;9(1):14293. doi:10.1038/s41598-019-49435-z
143. Fang L, Chen L, Song M, et al. Naoxintong accelerates diabetic wound healing by attenuating inflammatory response. Pharm Biol. 2021;59(1):252–261. doi:10.1080/13880209.2021.1877735
144. Jiao H, Xiao E, Graves DT. Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep. 2015;13(5):327–335. doi:10.1007/s11914-015-0286-8
145. Chen M, Lin W, Ye R, Yi J, Zhao Z. PPARβ/δ Agonist alleviates diabetic osteoporosis via regulating M1/M2 macrophage polarization. Front Cell Dev Biol. 2021;9:753194. doi:10.3389/fcell.2021.753194
146. Lu Y, Liu S, Yang P, et al. Exendin-4 and eldecalcitol synergistically promote osteogenic differentiation of bone marrow mesenchymal stem cells through M2 macrophages polarization via PI3K/AKT pathway. Stem Cell Res Ther. 2022;13(1):113. doi:10.1186/s13287-022-02800-8
147. An Y, Zhang H, Wang C, et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019;33(11):12515–12527. doi:10.1096/fj.201802805RR
148. Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7(12):738–748. doi:10.1038/nrendo.2011.106
149. Li B, Xin Z, Gao S, et al. SIRT6-regulated macrophage efferocytosis epigenetically controls inflammation resolution of diabetic periodontitis. Theranostics. 2023;13(1):231–249. doi:10.7150/thno.78878
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Metabolic Characteristics of Gestational Diabetes Mellitus and the Effects on Pregnancy Outcomes
Luo P, Fan Y, Xiong Y, Zhang C, Yang Z, Sun F, Mei B
Diabetes, Metabolic Syndrome and Obesity 2023, 16:15-29
Published Date: 11 January 2023
The Association Between Sarcopenia and Diabetes: From Pathophysiology Mechanism to Therapeutic Strategy
Chen H, Huang X, Dong M, Wen S, Zhou L, Yuan X
Diabetes, Metabolic Syndrome and Obesity 2023, 16:1541-1554
Published Date: 30 May 2023
Influence of Fibrinogen/Albumin Ratio and Fibrinogen/Pre-Albumin Ratio on Cardiac Autonomic Neuropathy in Type 2 Diabetes
Zhao S, Yang Z, Yu M, Xiang L, Lv Y, Tian C, Li R
Diabetes, Metabolic Syndrome and Obesity 2023, 16:3249-3259
Published Date: 18 October 2023
A Novel Inflammatory Marker: Relationship Between Red Cell Distribution Width/Albumin Ratio and Vascular Complications in Patients with Type 2 Diabetes Mellitus
Yu M, Pei L, Liu H, Wang J, Wen Y, Yang X, Ma C, Zhang X, Wu L, Wang L
Journal of Inflammation Research 2024, 17:6265-6276
Published Date: 10 September 2024
Interplay Between Insulin Resistance and Immune Dysregulation in Type 2 Diabetes Mellitus: Implications for Therapeutic Interventions
Berbudi A, Khairani S, Tjahjadi AI
ImmunoTargets and Therapy 2025, 14:359-382
Published Date: 3 April 2025