Back to Journals » International Journal of Nanomedicine » Volume 16
Effects of Surface Charge, PEGylation and Functionalization with Dipalmitoylphosphatidyldiglycerol on Liposome–Cell Interactions and Local Drug Delivery to Solid Tumors via Thermosensitive Liposomes
Authors Petrini M ![]() , Lokerse WJM, Mach A
, Lokerse WJM, Mach A ![]() , Hossann M
, Hossann M ![]() , Merkel OM
, Merkel OM ![]() , Lindner LH
, Lindner LH ![]()
Received 8 February 2021
Accepted for publication 24 April 2021
Published 14 June 2021 Volume 2021:16 Pages 4045—4061
DOI https://doi.org/10.2147/IJN.S305106
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Matteo Petrini,1,2 Wouter JM Lokerse,1 Agnieszka Mach,1 Martin Hossann,3 Olivia M Merkel,2 Lars H Lindner1
1Department of Internal Medicine III, University Hospital, Ludwig Maximilian University, Munich, Germany; 2Department of Pharmacy, Pharmaceutical Technology and Biopharmaceutics, Ludwig Maximilian University, Munich, Germany; 3Thermosome GmbH, Planegg, Germany
Correspondence: Lars H Lindner
Ludwig Maximilian University, Department of Internal Medicine III, Klinikum Grosshadern, Marchioninistr. 15, Munich, 81377, Germany
Tel +49-89-7095-4768
Fax +49-89-7095-4776
Email [email protected]
Purpose: Previous studies demonstrated the possibility of targeting tumor-angiogenic endothelial cells with positively charged nanocarriers, such as cationic liposomes. We investigated the active targeting potential of positively charged nanoparticles in combination with the heat-induced drug release function of thermosensitive liposomes (TSL). This novel dual-targeted approach via cationic TSL (CTSL) was thoroughly explored using either a novel synthetic phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphodiglycerol (DPPG2) or a conventional polyethylene glycol (PEG) surface modification. Anionic particles containing either DPPG2 or PEG were also included in the study to highlight difference in tumor enrichment driven by surface charge. With this study, we aim to provide a deep insight into the main differences between DPPG2- and PEG-functionalized liposomes, focusing on the delivery of a well-known cytotoxic drug (doxorubicin; DOX) in combination with local hyperthermia (HT, 41– 43°C).
Materials and Methods: DPPG2- and PEG-based cationic TSLs (PG2-CTSL/PEG-CTSL) were thoroughly analyzed for size, surface charge, and heat-triggered DOX release. Cancer cell targeting and DOX delivery was evaluated by FACS, fluorescence imaging, and HPLC. In vivo particle behavior was analyzed by assessing DOX biodistribution with local HT application in tumor-bearing animals.
Results: The absence of PEG in PG2-CTSL promoted more efficient liposome–cell interactions, resulting in a higher DOX delivery and cancer cell toxicity compared with PEG-CTSL. By exploiting the dual-targeting function of CTSLs, we were able to selectively trigger DOX release in the intracellular compartment by HT. When tested in vivo, local HT promoted an increase in intratumoral DOX levels for all (C)TSLs tested, with DOX enrichment factors ranging from 3 to 14-fold depending on the type of formulation.
Conclusion: Cationic particles showed lower hemocompatibility than their anionic counterparts, which was partially mitigated when PEG was grafted on the liposome surface. DPPG2-based anionic TSL showed optimal local drug delivery compared to all other formulations tested, demonstrating the potential advantages of using DPPG2 lipid in designing liposomes for tumor-targeted applications.
Keywords: thermosensitive liposomes, mild hyperthermia, cationic liposomes, dual tumor targeting, drug delivery, liposome functionalization
Introduction
Since the 1990s, several liposomal formulations have been approved for clinical practice, and others are currently being evaluated in clinical trials for a number of different conditions.1 The liposomal encapsulation of chemotherapeutics such as doxorubicin (DOX, Doxil®), irinotecan (Onivyde®) and vincristine (Marqibo®) greatly diminishes off-target toxicity, although only modestly improving therapeutic response.2–5 The latter is mainly due to a lack of an efficient drug release mechanism and the high stability of these conventional liposomes, which strongly reduces the amount of bioavailable drug.6,7 Stimuli-responsive nanocarriers were developed to improve the biodistribution (BD) of drugs in targeted areas and thus potentially increase the therapeutic effect.8 Among these, thermosensitive liposomes (TSLs) in combination with focused mild hyperthermia (HT, 41–43°C) have been widely investigated with promising results in pre-clinical and clinical settings.9,10 The concept of TSL as a drug delivery system is to design vesicles with specific lipid components that are capable of releasing their contents exclusively upon heat application, by undergoing gel to liquid phase transition (Tm). Hence, by applying focused HT to the tumor area, the drug is released at high concentration intravascularly, inducing a gradient for extravasation into the tumor interstitium (Figure 1A).11,12
 |
Figure 1 Schematic representation of TSL (A) and CTSL (B) tumor targeting approach in combination with focused HT. TSL circulate through the vasculature after i.v. administration with negligible payload release at body temperature (37°C, NT) (A, I). External active targeting is mediated via local HT to release high concentrations of drug at tumor vascular level leading to drug extravasation (A, II). CTSL circulate in the blood stream and accumulate in tumor vasculature by means of electrostatic interaction with over-expressed negatively charged membrane proteins (B, I and II). After binding, nanoparticles are internalized into tumor endothelial cells (B, III). Regional HT applied via an external device focused on a tumor, triggers drug release from endothelium associated CTSL, delivering high concentrations of the drug intracellularly (B, IV and V). |
Interestingly, positively charged TSLs were recently tested in a dual-targeting approach involving passive accumulation in tumor vasculature and local HT application to trigger drug release.13 Targeting of inflammatory sites and angiogenic vessels have in fact been described as feasible after systemic administration with positively charged nanoparticles.14,15 The main underlying mechanism for cationic particle accumulation in tumor vessels seems to be related to an atypical phenotype characterized by tumor endothelial cells with aberrant expression of negatively charged membrane proteins.16,17 Several conventional cationic liposome formulations have already been tested in pre-clinical settings, showing promising results in inhibiting tumor vascular growth.18–22 Nevertheless, complement activation, fast clearance due to serum protein adsorption and immunological recognition strongly hampered clinical translation of these formulations.23,24 So far, to the best of our knowledge, only a single cationic liposome formulation for intravenous administration is undergoing clinical trials for pancreatic cancer (paclitaxel, EndoTAGTM).25 In order to decrease systemic toxicity and to avoid opsonization, polyethylene glycol (PEG) is often grafted onto the liposome surface with the aim of enhancing circulation time.26 PEG is responsible for increasing steric hindrance and inducing mechanical repulsions of opsonins and other serum proteins, which are responsible for particle clearance mediated by the reticuloendothelial system (RES).27–29 However, polymer-based particle shielding is in contrast with charge-based cancer cell targeting strategies. On the one hand, PEG successfully enhances circulation time of positively charge particles, while on the other hand, it might hinder electrostatic interaction with negatively charged surfaces and thereby reduce targeting efficiency. Additionally, the high steric hindrance of the PEG polymer chains was reported to interfere with drug release from the vehicle and to hinder endosomal escape from internalized particles, thereby reducing efficacy of the delivery system.30–33
Our group designed TSLs which do not contain PEG and rely on a novel synthetic phospholipid 1,2-dipalmitoyl-sn-gylcero-3-phospho-diglycerol (DPPG2) to ensure prolonged circulation time and ultra-fast drug release upon HT.34,35 Proof of concept of gemcitabine- and DOX-loaded DPPG2-TSL with increasing tumor deposition of the respective drugs and improved therapeutic efficacy has been confirmed in soft tissue sarcoma models in pre-clinical settings.36–38 Here, we designed a novel DPPG2-based cationic TSL formulation (PG2-CTSL), which was compared to a PEG-CTSL formulation to highlight possible advantages of DPPG2 versus PEG in terms of liposome–cell interactions and DOX delivery efficiency. Furthermore, we exploited the heat-triggered function in combination with the active targeting approach driven by CTSLs in vivo (Figure 1B).
For all (C)TSLs, stability at physiological temperature and the efficiency of DOX release upon HT were assessed in vitro by fluorimetry. Differences in liposome–cell interactions and drug accumulation, driven by presence of DPPG2 or PEG, were studied in depth and evaluated via fluorescence microscopy and high performance liquid chromatography (HPLC). DOX pharmacokinetic (PK) profiles were assessed in vivo after intravenous injection (i.v.) in rats. DOX biodistribution was evaluated in tumor-bearing animals in combination with local HT, to assess drug accumulation in targeted-tumors and off-target organ exposure. Using the obtained results, we aim to provide further insight into the potential of the solid tumor dual-targeting concept with the goal of defining advantages of DPPG2 in comparison with PEG in this context.
Materials and Methods
Chemicals
DPPG2 was provided by Thermosome GmbH (Planegg, Germany). 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) were purchased from Corden Pharma Switzerland LLC (Liestal, Switzerland). The 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-PE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3 benzoxadiazol-4-yl) (NBD-PE), and the cationic lipid 1,2-dipalmitoyl-3-trimethylammonium-propane (DPTAP) were obtained from Avanti Polar Lipids (Alabaster, Alabama, USA). DOX was acquired from Puren Pharma GmbH & Co. KG (München, Germany). Fetal calf serum (FCS) and Collagen G were provided by Biochrom AG (Berlin, Germany). Cell tracker green CMFDA, Lysotracker Red 99 and Hoechst 33342 were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA). ELISA kits for SC5b-9 and certified human serum for complement activation were acquired from TecoMedical GmbH (Bünde, Germany). All other chemical used were obtained either from Sigma Aldrich GmbH (München, Germany) or Carl Roth GmbH (Karlsruhe, Germany) in analytical quality. All buffers and solutions used in the study were prepared with deionized and purified water from an ultrapure water system (Milli Q Advantage, Merck Millipore, Darmstadt, Germany).
Liposome Preparation and Characterization
Lipid composition and molar ratio of each tested TSL formulation is shown in Table 1. All liposomes were prepared by lipid film hydration and extrusion method.34 Briefly, lipids were dissolved in chloroform and methanol solution (9:1, v/v) and dried under vacuum using a rotary evaporator. The lipid film was hydrated with a solution of 240 mM (NH4)2SO4 pH 5.5 for 30 minutes at 60°C. The obtained vesicles were extruded 5 times with 200 nm and 5 times with 100 nm polycarbonate filters (LipexTM Thermobarrel Extruder, Northern Lipids Inc. Burnaby, BC, Canada). Ammonium gradient for active DOX loading was applied as described in Haran et al.39 Briefly, extruded liposomes were run through a gel-exclusion chromatography (PD-10 column, GE Healthcare, Munich, Germany) and eluted with HEPES-buffered saline (HBS) pH 7.8. DOX loading was performed at 37°C for TSL and 38°C for CTSL, for a total time of 60 minutes at DOX:lipid of 0.05 (mol:mol). Unentrapped DOX was removed by centrifugation for PG2-TSL, PEG-TSL, and PEG-CTSL (75,000 x g, 1 hour, 10°C, Avanti-J26XP, Beckman Coulter, Krefeld, Germany). The liposomal pellets were thereafter resuspended in HBS pH 7.4. For PG2-CTSL and bare-CTSL, filter centrifugation via Amicon Filter 10K (Merck Millipore, Darmstadt, Germany) was preferred in order to remove unentrapped DOX and concentrate liposomes after active loading (4000 x g, 20 minutes, 10°C, repeated 3 to 5 times). Liposomes used for fluorescence imaging contained either 0.1 mol% Rho-PE or 0.3 mol% of NBD-PE and lipid film hydration was performed simply with HBS pH 7.4. Hydrodynamic diameter (Z-average), polydispersity index (PDI), and ζ-potential (ζ-POT) were measured by dynamic light scattering (DLS) using a Zeta Sizer Nano ZS (Malvern Instruments, Worcestershire, UK). Differential scanning calorimetry (DSC, Mettler Toledo DSC 821e, Giessen, Germany) was performed with freshly prepared batches after DOX loading. Hemocompatibility was investigated in vitro quantifying complement activation in certified human serum. DOX-loaded anionic and cationic liposomes were incubated for 15 minutes in human serum (1:12, v/v) and complement activation assessed by SC5b-9 ELISA test. Zymosan A (10 mg/mL) and HBS pH 7.4 were used in the assay as positive and negative control, respectively.
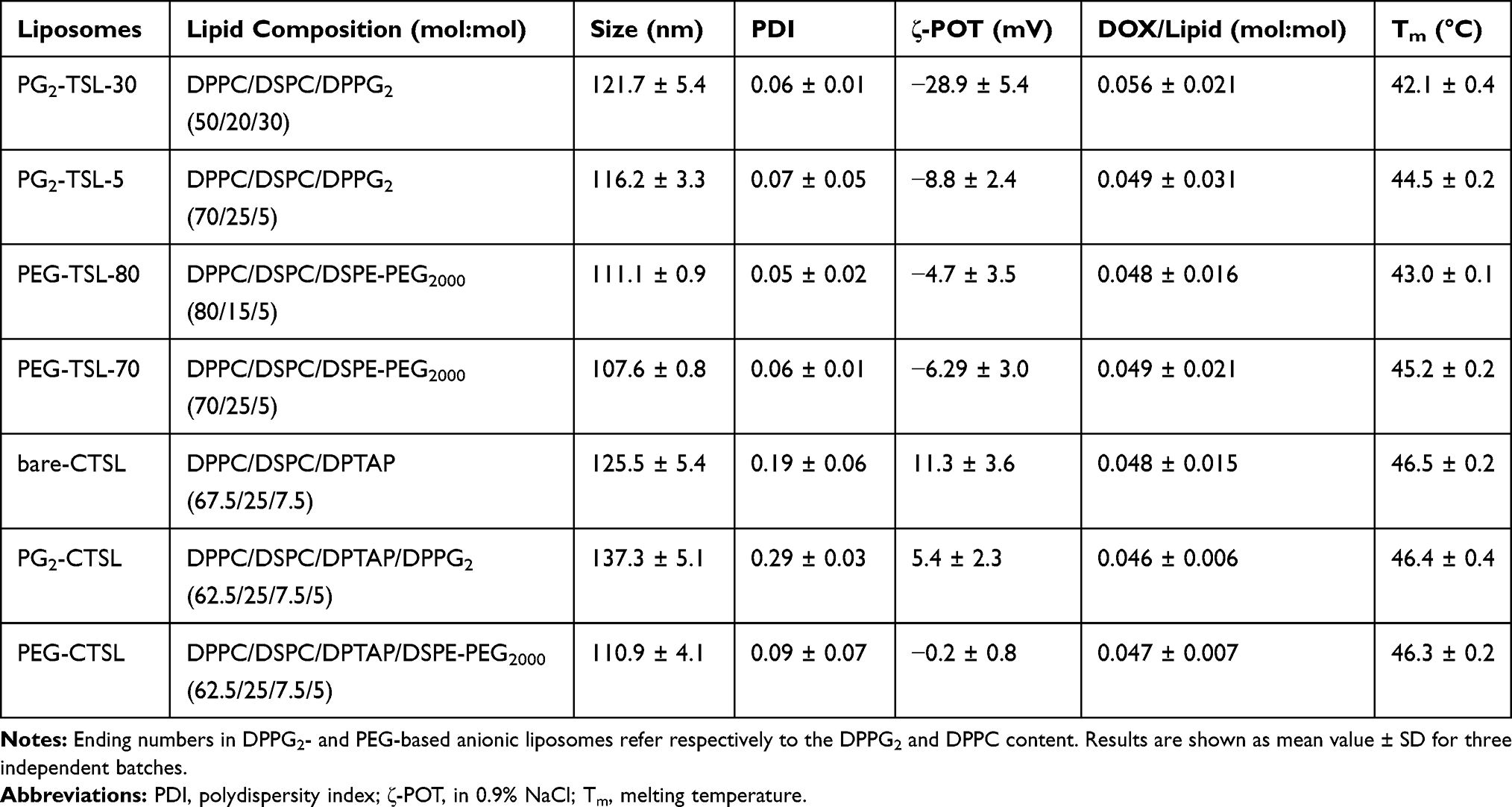 |
Table 1 Characterization of Anionic and Cationic TSLs Encapsulating DOX |
Liposomal-DOX Content and Temperature-Dependent DOX Release
DOX content analysis was conducted to calculate percentage of encapsulated DOX for each formulation. The assay was carried out either through HPLC or fluorescence spectroscopy. In the latter case, liposomes were diluted in HBS pH 7.4 at different concentrations (1:2, 1:3 and 1:4, v/v). DOX was diluted in water to create a linear reference standard ranging from 0.3 to 1.1 µM. Subsequently, 20 µL of samples and standards were mixed with 10% Triton X-100 at a 1:11 (v/v) ratio and incubated at 45°C and 750 rpm for 15 minutes in a thermoshaker. Thereafter, samples were diluted in HBS pH 7.4 (1:151, v/v) and DOX assessed via fluorescence spectroscopy at Ex/Em 470 and 555 nm. The lipid concentration was assessed by phosphorus assay as described in Eibl et al.40 The final encapsulation efficacy (EE) percentage was calculated by using the following formula (Equation 1):

where (D/L)initial and (D/L)final indicate DOX:lipid ratio (mol:mol) at the beginning of the loading process and after liposome purification, respectively. Temperature-dependent DOX release assays were performed in full FCS as previously described.35 The percentage of DOX released was calculated as follows (Equation 2):

where IT°C is the fluorescence intensity after incubation at a certain time at a specific temperature and IRT is the fluorescence baseline when the sample was incubated at room temperature (RT) for 5 minutes. Fluorescence intensity for 100% (I∞) was assessed via liposome incubation with 10% Triton X-100 (15 minutes at 45°C).
Cell Culture
Rat soft tissue sarcoma cells (BN175, Brown Norway rat; provided by Timo ten Hagen, Erasmus MC, Rotterdam) were cultured in RPMI 1640 medium supplemented with 10% FCS, 10 U/mL Penicillin and 100 μg/mL Streptomycin. Human umbilical vein endothelial cells (HUVECs) were kindly provided by the Department of Pharmaceutical Biology, Faculty of Chemistry and Pharmacy, Ludwig Maximilian University (LMU) Munich (pooled donors, PromoCell GmbH, Heidelberg, Germany). HUVECs were cultured in EASY Cellovation medium from Pelo Biotech (Martinsried, Germany). Cells were cultured at 37°C in humidified atmosphere at 5% CO2. Substrates for HUVEC culturing were manually coated with Collagen G in PBS (50 µg/mL).
Fluorescence Activated Cell Sorting
Cellular binding efficiency of (C)TSLs was measured by fluorescence activated cell sorting (FACS) analysis using rhodamine-labeled liposomes. Cells were seeded at 3×105 cells/well in 6-well plates and incubated for 24 hours. (C)TSLs were added at final lipid concentration of 1.1 mM in serum-free media. After 1 hour incubation at 37°C, cells were washed three times with serum-free media and manually scraped in presence of 500 μL phosphate-buffered saline (PBS) per well before FACS analysis (FACSCaliburTM, Becton Dickinson, New Jersey, USA). Forward versus side scatter gating was used to exclude debris and dead cells and red fluorescence intensity (Iso-PE channel) of the cells was evaluated with 10,000 cell counts. The data were analyzed with FlowJo software (version 10.5.0).
Live Cell Fluorescence Imaging
BN175 and HUVECs were either incubated with NBD-labeled (C)TSLs (1.1 mM final lipid concentration) or non-labeled DOX-loaded (C)TSLs (50 µM final DOX concentration) for 1 hour at 37°C. For NBD-labeled liposomes, Lysotracker red 99 was added after the first 30 minutes with a final concentration of 0.5 μM. At the end of the incubation time, cells were washed three times with serum-free media and incubated with Hoechst 33342 (5 μg/mL, 5 minutes). Cells were washed again and placed in FCS-containing media, and fluorescence microscopy was performed. Thereafter, chambers were placed in a water bath set at 41°C and fluorescence imaging performed again after 1 hour. Live cell microscopy was performed using a Leica wide field microscope provided with Optigrid technology. Images in live setting (696 x 520 pixels, 60x/1.4 Oil objective) were performed using Leica LAS X image software (Leica, Germany).
Toxicity Study on BN175 and HUVEC Cells
Toxicity of DOX-loaded (C)TSLs was assessed by Sulforhodamine B (SRB) assay.41 Cells were seeded in 96-well plate and incubated with different concentrations of liposomal DOX ranging from 0.37 to 100 μM. After 1 hour incubation at 37°C, cells were washed, and fresh serum-containing media was added. Plates were placed in a water bath set at either 37°C (NT) or 41°C (HT) for 1 hour and then incubated again at 37°C for 3 days. Cells were fixed with trichloroacetic acid 1% and placed in the fridge for at least 30 minutes. Plates were gently washed, and a solution of SRB 5% was added in each well. After 20 minutes, cells were rinsed carefully with acetic acid solution 1%. Plates were dried at 60°C for at least 3 hours and 100 μL of 10 mM Tris solution (pH 10.5) was added to each well. Plates were gently shaken for 10 minutes and absorbance analyzed by a microplate reader at 450 nm.
DOX Recovery from BN175 and HUVEC Cells
Cells were seeded in 6-well plates at 5×105 cells/well. After 24 h, cells were incubated with 100 μM of either liposomal or non-liposomal DOX. Incubation was carried out for 1 hour at 37°C in serum-free media. Next, cells were washed three times and serum-containing media was added for normothermia (NT) or HT treatments (1 hour). Medium was then removed, cells were collected and pelleted by centrifugation (2000 x g, 10 minutes, RT). Cells were resuspended in lysis buffer (1% Triton-X, 0.1% SDS, 150 mM NaCl, 20 mM Tris, pH 7.4) and placed on ice for 30 minutes. The solution was then sonicated for 1 minute on ice with a probe sonicator. Lastly, samples were centrifuged (2000 x g, 10 minutes, RT) and stored at -20°C until the HPLC DOX assessment and total protein content (Bio-Rad, DCTM protein assay).
Liposomal DOX Pharmacokinetic and Biodistribution in vivo
Animal experiments were performed according to a protocol approved by the responsible authority (Animal Care and Use Committee of the Government of Upper Bavaria, Munich, Germany) under the reference ROB-55.2–2532.Vet_02-17-2018. The European Union Directive 2010–63-EU for welfare of the laboratory animals was followed. Brown Norway rats (Charles River) were kept at appropriate conditions until experimental procedures. Animal handling and experiments were conducted in accordance to already reported methods published recently by our group.36 At specific time points, blood (200 µL) was collected in Li-heparin microvettes, vortexed and frozen. The blood concentration of DOX was fitted using the equation with a mono-exponential function (Equation 3) or a biphasic exponential function (Equation 4) via Origin software:

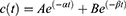
where c(t) is DOX concentration in the blood at time t (minutes) after i.v. administration, k is the constant rate of elimination and A, B, α and β are parameters of the bi-exponential model. The blood half-life (t1/2) was calculated with the following formula (Equation 5):

The area under the curve (AUC) was calculated by integrating the exponential fit from 2 to 120 minutes. DOX biodistribution was assessed in tumor-bearing rats in combination with focused HT. BN175 tumor fragments were subcutaneously inserted in both right and left hind flanks and the experiment started when one of the tumors reached a size of 0.5 cm3. The animals were placed under anesthesia (5% isoflurane for induction and 2% for maintenance) and medicated with metamizol, meloxicam and buprenorphine (100 mg/kg s.c., 0.05 mg/kg s.c. and 0.05 mg/kg s.c., respectively). One of the tumors received lamp mediated HT (41°C, 1 hour) whereas the second tumor on the opposite side was kept at physiological temperature (NT). HT-tumor temperature was measured invasively using an internal probe. Body temperature was preserved via a heat-mattress set at 37.5°C and/or a blanket and monitored throughout the study via rectal probe. (C)TSL-DOX i.v. injection via tail vein catheter was performed as soon as HT-tumor temperature of 41°C was reached. The temperature of the heated tumor was kept between 41–42°C for 1 hour after drug administration. At the end of HT application, whole body perfusion under deep state of anesthesia was performed to avoid blood contamination factors in DOX detection in tissues. Organs and tumors were excised and stored at -20°C until DOX assessment by HPLC analysis.
DOX HPLC Analysis in Aqueous Matrices, Cell Lysates and Tissues
DOX HPLC quantification in aqueous-based matrices, plasma samples, cell lysates, rat full blood, and tissues was performed using a slightly adapted method as described by Peller et al.42 Exclusively for rat full blood and tissue sample preparation, solid-phase extraction (SPE) using STRATA-X (Phenomenex Ltd., Torrance, California, USA) was performed after processing samples and standards with cold MeOH (1:11, v/v) and diluted in distilled water. Columns were washed with solutions at increasing percentage of MeOH (0–30%) and DOX elution was achieved with 2% formic acid in methanol. Depending on the matrix, calibration curves were created spiking DOX either in water, blood or liver tissue covering a range from 0.5 to 33.3 µg/mL.
Statistical Analysis
The data are expressed as mean ± standard deviation (SD) of at least three independent experiments. Statistical analysis was performed via GraphPad Prism software (version 7.05) or Origin software (version 8.5). Figures were subjected to either two-tailed t-test or one-way ANOVA Bonferroni test with significance indicated when p > 0.05.
Results
Liposome Characterization
Physico-chemical properties for all formulations tested in the study are summarized in Table 1. The two cationic TSLs under analysis, PG2-CTSL and PEG-CTSL, contain a mixture of anionic (5 mol%, DPPG2 or DSPE-PEG2000) and cationic (7.5 mol%, DPTAP) lipids in the bilayer membrane. Control TSLs consist in DPPG2 and PEG-based liposomes formed by the exact same composition of cationic counterparts, besides the cationic lipid which was replaced by the zwitterionic DPPC (PG2-TSL-5 and PEG-TSL-70). Previous data have shown that DPPG2 must be present in the lipid bilayer at a minimum of 30 mol% to take full advantage of DPPG2-dependent enhancement of circulation time and heat-triggered release.34,43 Similarly, optimization studies for PEG-based TSLs confirmed that lowering the DSPC to 15 mol% showed added benefit in terms of heat-triggered release, while keeping a prolonged circulation time.44,45 Therefore, optimized DPPG2 and PEG-based TSLs consisting of a lipid composition as described in Table 1 for PG2-TSL-30 and PEG-TSL-80 were also used in the study, to investigate whether the dual-targeting approach of CTSLs could improve the overall efficiency of DOX delivery for traditional TSLs.
ζ-POT analysis in physiological saline showed a higher positive charge for PG2-CTSL (5.4 ± 1.3 mV) than for PEG-CTSL (-0.2 ± 0.8 mV) (Table 1). For DPPG2-based anionic liposomes, the surface charge significantly decreased from PG2-TSL-5 to PG2-TSL-30, in good accordance with the different percentage of the negatively charged DPPG2 (5 mol% vs 30 mol%) included in the membrane bilayer. PEG-TSL-70 and PEG-TSL-80 differed only in the amount of DPPC/DSPC, with no detectable differences in overall surface charge. All anionic TSLs were comparable in vesicle size and showed a small and monodisperse size-distribution (PDI < 0.1). Regarding cationic liposomes, a few differences in the physical properties were observed in relation to the surface functionalization. For bare-CTSL and PG2-CTSL, size and PDI after DOX loading were slightly higher in comparison with all the formulations tested (small second peak in size profiling was visible, Supplementary Figure 1A-E). On the other side, the second extra peak was not observed for PEG-CTSL and DLS analysis suggested a homogenous size-distribution similar to anionic controls. Active DOX loading was successfully completed via ammonium gradient in all liposomal formulations (>95% EE, data not shown), with no observed differences among cationic and anionic TSLs. Liposome composition analysis performed via thin layer chromatography confirmed correct lipid composition of all tested (C)TSLs and absence of lipid-degradation products (eg, lyso-lipids) after DOX remote loading (Supplementary Figure 2).
Temperature-Dependent DOX Release Profile
All tested CTSLs showed minimal content leakage after exposure to 37–38°C for 5 minutes (max 10% DOX leakage) and significant DOX release upon temperature increase, with peaks observed at around 43°C (Figure 2A). The inclusion of 5 mol% of either DPPG2 or DSPE-PEG2000 in bare-CTSL significantly improved DOX-release profile, leading up to ~80% of DOX released at 42°C (vs ~55% of bare-CTSL) (Figure 2D). The DOX temperature-curve of anionic PG2-TSL formulations was greatly affected by the amount of DPPG2 included in the liposome bilayer. At 30 mol%, liposomal Tm was 42.1 ± 0.4°C (Table 1) and the formulation showed DOX burst release at 40–41°C leading to full particle depletion within minutes (Figure 2B and E). By reducing DPPG2 to 5 mol% (PG2-TSL-5), an increase in the Tm up to 44.5 ± 0.2°C was observed (Table 1), with complete abolishment of the DOX heat-triggered release efficiency at the investigated temperature range (Figure 2B). The DOX-release profile from PEG-TSLs was also affected by the lipid composition and thereby by the according Tm. PEG-TSL-70 exhibited a sub-optimal heat-triggered profile with highest release DOX fraction of ~30% observed at 43°C (5 minutes incubation) (Figure 2C). When the DSPC content was lowered to 15 mol% in favor of DPPC, the resulting PEG-TSL-80 exhibited an improved release efficiency with DOX peaks observed at 42°C (Figure 2C), although it did not exceed ~50% of total content (Figure 2F).
 |
Figure 2 Temperature-dependent DOX release profiles. Percentage of released DOX after either 5 minutes (37–45°C) or 1 hour (37, 42°C) incubation in full FCS for CTSLs (A, D) DPPG2-TSL (B, E), and PEG-TSLs (C, F). Values are expressed as mean value + SD for three independent batches. DOX release after 1 hour incubation at 37 or 42°C was analyzed either via one-way ANOVA followed by Bonferroni test using bare-CTSL as control (D) or via two-tailed t-test (E, F). Asterisks indicate significance different between groups. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. |
In vitro Targeting of Tumor and Endothelial Cells
Positively charged DPPG2- and PEG-based TSL were compared to their anionic counterparts (PG2-TSL-5 and PEG-TSL-70). As expected, liposome targeting was drastically improved for both cell lines that were under investigation when the cationic lipid was included in the liposomal bilayer. The median fluorescence intensity (MFI) reached the highest signal after incubation with PG2-CTSL, resulting in 3.4-fold higher binding in comparison to PEG-CTSL (Figure 3). When compared to anionic vesicles, PG2-CTSL and PEG-CTSL promoted an increase in cell targeting of 13 and 10-fold, respectively. Interestingly, also the anionic PG2-TSL-5 showed significantly higher liposome–cell interactions compared with PEG-TSL-70 (2-fold), in both cell lines. Next, we investigated how adsorbed serum proteins on the liposome surface may affect particle targeting. Although the protein adsorption significantly reduced overall binding (~60%; Supplementary Figure 3), CTSLs-driven cell targeting was still observed and, remarkably, a 2-fold higher MFI was observed for PG2-CTSL when compared to PEG-CTSL.
 |
Figure 3 Fluorescence activated cell sorting (FACS) after incubation with rhodamine-labeled (C)TSLs. Iso-PE fluorescence of untreated cells was subtracted from each sample and final MFI plotted as mean value ± SD for three independent measurements. Groups were analyzed via one-way ANOVA followed by Bonferroni test, and asterisks indicate significant differences between groups. * p < 0.05, **** p < 0.0001. |
Live Targeting of Tumor and Endothelial Cells
In the next steps, we investigated the internalization rate and fate of liposomes as well as DOX-release efficiency after targeting in living cells. Anionic control formulations with the higher efficiency in DOX-release were used in the following experiments (PG2-TSL-30 and PEG-TSL-80). In good accordance with FACS results, larger fractions of liposomes (green fluorescence) were found co-localizing with target cell membranes when positively charged TSLs were tested (Figure 4A-D, second rows; Supplementary Figure 4A-B). Furthermore, co-localization with lysosomes imaged via Lysotracker Red (red fluorescence) was present to a certain extent for both PG2-CTSL and PEG-CTSL (white arrows). Green punctuate pattern in non-acidic endosomes were also moderately present in both cell lines tested. In contrast, barely any liposomes were visible after incubation with anionic PEG-TSL-80 (Figure 4C and D, first rows), whereas a measurable fraction of PG2-TSL-30 was well detected in cell cytoplasm in both BN175 and HUVEC (Figure 4A and B, first rows). To assess the kinetics of CTSL uptake and observe liposome localization after HT treatment, cells were placed in a water bath at 41°C for 1 hour and imaged again thereafter. Differences between PG2-CTSL and PEG-CTSL were detected especially on BN175 cells, as PG2-CTSL were still present on cell membranes and co-localized with lysosomes, whereas PEG-CTSL were mostly detected in a punctuate pattern in the cell cytoplasm (Supplementary Figure 5A and B).
 |
Figure 4 Live cell fluorescence imaging on tumor and endothelial cells after incubation with anionic (first rows) or cationic (second rows) DPPG2- (A, B) or PEG-based TSLs (C, D). NBD-liposome were imaged using GFP filter (green color), lysosomes with DsRed filter (Lysotracker RED, red color) and nuclei with DAPI filter (Hoechst 33342, blue color). Arrows indicate co-localization (yellow) of liposomes (green) and lysosomes (red). Images were taken after 1 hour at 37°C (NT). Scale bar applied to all images is 20 µm. |
Live Cell Imaging of HT-Triggered DOX Release and Quantification of Intracellular DOX
The improved targeting with CTSLs in comparison to TSLs was further investigated by using DOX-loaded particles, to assess whether it might translate into a better DOX delivery in vitro. Live fluorescence imaging of BN175 and HUVEC after incubation with DOX-loaded (C)TSLs did not show any detectable DOX (neither in the cytoplasm nor in the nuclei) (Figure 5A-D, 1 hour NT). At this stage, DOX was entrapped and self-quenched inside liposomes, confirming good stability of the liposomal systems used in the experiment. After this initial incubation, cell chambers were placed in a water bath for 1 hour at 41°C (HT) to trigger DOX release. Upon HT application, the released DOX from PG2-CTSL was greatly noticeable in the intracellular compartments, forming a punctuate pattern (red nanobursts, Figure 5A and B, second rows). Although the majority of DOX was assessed inside the cell cytoplasm, co-localization of DOX and nuclei was observed to a certain extent (Figure 5A and B, white arrows). Cells treated with anionic DOX-loaded PG-TSL-30 also showed a detectable amount of DOX in the cellular compartments (Figure 5A and B, first rows), whereas with PEG-based TSLs, DOX was observed exclusively after incubation with positively charged liposomes (Figure 5C and D, second rows). By evaluating DOX fluorescence intensity generated in the study, we hypothesized that PG2-CTSL are a better system for DOX delivery efficiency, in comparison to PEG-CTSL.
 |
Figure 5 DOX fluorescence imaging on tumor and endothelial cells after incubation with anionic (first rows) or cationic (second rows) DPPG2- (A, B) or PEG-based TSLs (C, D). Cells were imaged using DAPI filter for Hoechst 33342 (nuclei, blue) and DsRed filter for DOX visualization (red). Images were taken after 1 hour at 37°C (NT) and after 1 hour at 41°C (HT). Arrows indicate co-localization of DOX and nuclei. Scale bar applied to all images is 20 µm. |
The hypothesis was further confirmed by quantifying the total amount of DOX delivered to cells using these delivery systems. In this case, free-DOX was also added to the tested formulations to assess maximum cellular uptake. As shown in Figure 6A, the total amount of DOX recovered from BN175 cells was 2.3-fold higher when DOX was applied via PG2-CTSL, compared with PEG-CTSL (1.02 ± 0.13 vs 0.44 ± 0.15 with HT, respectively). Remarkably, for HUVECs no difference in DOX recovery was observed between free-DOX and PG2-CTSL groups, with the latter significantly outperforming PEG-CTSL (3.3-fold higher DOX recovery) (Figure 6B). As expected, no differences in DOX uptake were observed between NT and HT groups, suggesting that in the case of extracellular HT-triggered release from membrane-bound liposomes, DOX is immediately taken up by the cells. Nevertheless, an important difference needs to be considered in relation to drug bioavailability, because after NT conditions, DOX is still encapsulated inside the liposomal core and is not intracellularly bioavailable.
 |
Figure 6 DOX recovery from tumor (A) and endothelial cell lines (B) after incubation with 100 µM DOX either formulated in (C)TSLs or as a free form. DOX and protein content in cell lysates were assessed in cell lysate via HPLC and protein assay, respectively. Results are shown as DOX/protein ratio (µg/mg) and data are presented as mean value ± SD for three independent batches. Groups were analyzed via one-way ANOVA followed by Bonferroni test and asterisks indicate significant differences between groups. *** p < 0.001, **** p < 0.0001, LOD = limit of detection. |
In vitro Cell Toxicity Investigation of DOX-Loaded (C)TSLs
Next, we investigated whether the higher DOX delivery efficiency obtained via PG2-CTSL might also translate in a potentially higher cell toxicity. After incubation with liposomal-DOX, the resulting cell viability curves were drastically affected by the type of formulation tested. In all cases, the half-maximal inhibitory concentration (IC50) of DOX was found to be lower for CTSLs compared with the anionic counterparts (Table 2). IC50 of PEG-CTSL in BN175 cell line was 3.5-fold higher compared with PG2-CTSL (5.87 ± 2.72 and 1.67 ± 4.8 µM with HT condition, respectively), suggesting a higher cancer cell toxicity for the latter. We did not observe any significant toxicity on cells driven by carriers combined with hyperthermic condition (Supplementary Figure 6A and B). Additionally, no differences between HT and NT groups were detected, independent of the liposome formulation used in the experiments (Table 2).
 |
Table 2 IC50 Values DOX of BN175 and HUVEC Treated with Different Liposomal DOX Formulations |
Pharmacokinetic, Biodistribution and Hemocompatibility Investigation
To assess if a positive charge affects particle circulation and to monitor DOX clearance from the blood stream, in vivo PK investigations were carried out. After the injection of anionic PG2-TSL-30 and PEG-TSL-80, a mono-exponential DOX elimination was observed (Figure 7), with a t1/2 of 134.3 ± 28.7 and 253.1 ± 20.8 minutes, respectively (Table 3). Additionally, maximum blood concentration (Cmax) for PG2-TSL-30 and for PEG-TSL-80 suggested a negligible DOX leakage upon TSLs injection (Table 3). In both CTSLs, Cmax was significantly lower compared with the anionic counterparts, especially with PG2-CTSL (2.7-fold). In the latter case, a significant drop in DOX plasma concentration was immediately observed upon liposome injection, with ~65% of total injected dose (ID) and a t1/2 of 11.9 ± 3.6 minutes (Table 3). After this initial rapid loss, DOX blood clearance showed a biphasic kinetic profile, with a further slower elimination rate (Figure 7). Similarly, for PEG-CTSL an initial drop in DOX blood content was also observed, but to a lesser extent than for PG2-CTSL (32.1% loss ID). Despite the significant initial loss, DOX clearance from PEG-CTSL suggested a slower elimination rate than for PG2-CTSL, with a monophasic clearance and t1/2 of 96.4 ± 13.7 minutes (Figure 7, Table 3). Liposome hemocompatibility analysis was performed via ELISA test for SC5b-9, a marker for activation of the complement system (Supplementary Figure 7). As expected, a significantly higher complement activation was observed for CTSLs compared with anionic TSLs, with the highest signal generated by PG2-CTSL.
 |
Table 3 PK Parameters of DOX-Loaded (C)TSL Formulations |
 |
Figure 7 Pharmacokinetic profile of DOX-loaded (C)TSLs in Brown Norway rats. DOX blood levels after administration via PG2-TSL-30 (black circles), PEG-TSL-80 (black squares), PEG-CTSL (white squares), PG2-CTSL (white circles), and free DOX (red triangles and dashed red line). Data were fitted via either a one-phase (Equation 3) or a two-phases (Equation 4) decay formula. Data are presented as mean value ± SD, every group consisted of three animals (n=3). DOX blood content of DPPG2- and PEG-based CTSLs at 2, 60 and 120 min was compared via two-tailed t-test and asterisks indicate significance between groups. * p < 0.05, *** p < 0.001. |
When combined with local HT, all tested (C)TSLs successfully increased DOX enrichment in heated tumors compared with untreated ones. The DOX increase in heated tumors was strongly dependent on the type of formulation tested. Remarkably, the highest DOX tumor content was observed with PG2-TSL-30 (43.1 ± 14.8 ng/mg), with a 14-fold enrichment factor compared with control tumors (NT) (Figure 8). Conversely, the biodistribution of PG2-CTSL was suboptimal and greatly affected by RES uptake, as suggested by the higher DOX values recovered in liver and spleen (Figure 8). PG2-CTSL targeting in heated tumors led to a 3.7-fold higher DOX enrichment compared with NT tumors. For PEG-based TSL, a similar amount of DOX was detected in tumors subjected to HT, without major differences observed between anionic and cationic liposomes (12.8 ± 7.3 and 8.7 ± 5.7 ng/mg, respectively). Independently from the CTSLs tested, no substantial differences in DOX delivery were observed among NT tumors. Interestingly, in the latter group, a moderately higher DOX level was generally observed when DOX was administrated via anionic TSLs, potentially due to re-distribution of the DOX in the body after HT-triggered release.
 |
Figure 8 DOX recovery in organs and tumors after i.v. administration of DOX-loaded (C)TSLs in combination with local HT. Data are presented as mean value ± SD, every group consisted of three animals (n=3). Groups were analyzed via two-tailed t-test and asterisks indicate significant differences between groups. ** p < 0.01, *** p < 0.001, **** p < 0.0001. |
Discussion
In this study, a dual tumor-targeting approach with positively charged TSLs was investigated and compared with the traditional intravascular heat-triggered release mediated by conventional TSLs. To evaluate key requirements for an efficient drug delivery strategy for cancer treatment, specific liposomal formulations with a different surface charge and lipid compositions were tested in the study. We performed a thorough analysis between PEGylated and DPPG2-based (C)TSLs, to determine potential advantages and disadvantages of DPPG2 over PEG.
Liposome–cell interactions and fate of adsorbed liposomes (eg, fusion, endocytosis) are mediated by different factors, among which surface charge, bilayer density, lipid components, and cell morphology play an important role.31,46 In our investigation, both DPPG2- and PEG-based CTSL contained the same amount of cationic lipid DPTAP (7.5 mol%) and anionic lipid (either 5 mol% DSPE-PEG or DPPG2) incorporated in the membrane bilayer. However, the observed ζ-POT was significantly different. While PG2-CTSL showed a noticeable and well detectable positive surface charge, PEG-CTSL ζ-POT was found to be approximately neutral in saline. Hence, PEG-polymer covered the nanoparticle surface affecting the positive charge exposure to a larger extent than DPPG2. The noteworthy PEG-shielding effect of the particle surface charge has already been described in negatively charged liposomes, with a significant reduction in liposome electrophoretic mobility (5-fold loss) when liposomes were functionalized with PEG.47 Overall, the data suggest that PEGylation might hamper an effective interaction of the cationic moiety with membrane proteins.
In our tested conditions, CTSLs bearing the cationic DPTAP were observed to mediate tremendously higher binding and uptake in all cell lines tested compared with their anionic counterparts. PG2-CTSL showed a 3-fold higher targeting capability compared with PEG-CTSL, in both cancer cells and endothelial cells. Interestingly, overall, anionic DPPG2-based TSLs performed better than PEG-TSLs, as a higher liposomal uptake was observed when particles were functionalized with DPPG2. We hypothesized that two main reasons could explain the superior binding potential of DPPG2-based liposomes when compared against PEGylated particles. First, the reduced steric hindrance and the smaller size of DPPG2 head groups in comparison to DSPE-PEG2000 may contribute to a more favorable interaction with the target membrane and may result in a higher probability of cellular contact. This hypothesis is in accordance with recent studies describing the potential for PEGylation to decrease cationic liposome–cell interactions and thus hamper delivery efficiency of the encapsulated materials.48–51 Second, the specific presence of DPPG2 may promote an improved particle uptake. This assumption is supported by different studies suggesting that hydroxyl functionalities in the head group region contribute favorably to liposome-mediated in vitro transfection.52,53 Thus, it seems fair to speculate whether other factors, besides a simple reduced steric hindrance, are responsible for the higher targeting potential of DPPG2-based liposomes.
The next aspect to be investigated was the DOX delivery potential of the CTSL formulations. We have already reported that a certain mol% of DPPG2 is required in the lipid composition not only for prolonged circulation time, but also for ultra-fast drug release.35 Indeed, in our investigation on temperature-dependent release profiles, PG2-TSL-5 were not able to induce any DOX release, while, by increasing the DPPG2 amount to 30 mol%, a massive heat-triggered release was observed already at 41°C. The insertion of cationic DPTAP lipid in a DPPC/DSPC bilayer (bare-CTSL) showed a sub-optimal DOX temperature-curve with a significantly slower DOX-release rate compared with PG2-TSL-30. Remarkably, the simultaneous presence of DPTAP and either DSPE-PEG2000 or DPPG2 in PEG-TSL and PG-CTSL, respectively, induced improved heat-sensitivity. However, both CTSLs showed a similar and efficient heat-triggered DOX release, despite there not being a full particle depletion, even after prolonged incubation. Besides the HT-triggered function, we have demonstrated the possibility to selectively trigger DOX release intracellularly by exploiting the targeting function of CTSLs. After an initial incubation of liposomes with target cells at 37°C, DOX was not detectable since it was self-quenched inside liposomes, being either adsorbed on the cellular surface or endocytosed in cellular compartments. By triggering DOX release upon HT, DOX was detectable in a punctate pattern forming red nanobursts in cytoplasmic vesicles and to a certain extent in the nuclear compartment. The distinct localization of DOX in intracellular vesicles was potentially due to a temporary confinement inside the lysosomal compartments, where the acidic pH induced DOX protonation and thus slowing further transportation into the nuclei. This is in good accordance with previous findings where HT-mediated intracellular drug release was tested in combination with PEG-CTSL in a human cancer cell line.54 Qualitative information obtained via fluorescence microscopy were further confirmed by quantifying DOX via HPLC analysis in parallel experiments. Among all formulations tested, the highest DOX levels were found in cells incubated with PG2-CTSL, consistently with the higher targeting rate. It has been reported that HT significantly increases chemotherapeutic effectiveness in vitro in cancer cells or in pre-clinical settings.55,56 In our investigation, we identified HT as a potential trigger mechanism for intracellular DOX release, but without any effect on DOX uptake and cell toxicity. The latter point is likely due to the nature of the assay, with comparable findings reported when a similar experimental setting was used.36,54 Nevertheless, the higher toxicity found for PG2-CTSL is expected to be related exclusively to the higher amount of cell binding and thus to DOX delivery efficiency, since no cytotoxicity was observed when empty carriers were administrated at the same concentration.
As reported in the literature, positively charged nanoparticles bear the risk of fast clearance due to opsonization and complement activation when administrated in vivo.57 Here, we demonstrated clear effects on particle blood stability driven by PEGylation and surface charge. For instance, t1/2 of cationic DPPG2-based TSLs was drastically affected by the exposed positively charged DPTAP, with a rapid distribution and elimination of around ~65% of DOX immediately after i.v. injection. For PEG-CTSL, this effect was also observed but to a somewhat lesser extent, with an initial distribution and elimination of only ~30% ID. In both cases, a consistent reduction in AUC2h was observed, equal to 7- and 2-fold losses for PG2-CTSL and PEG-CTSL, respectively, when compared to their anionic counterparts. These findings suggest that 5 mol% of DPPG2 are not as effective as DSPE-PEG2000 in avoiding liposome opsonization. Nevertheless, due to the initial drop in DOX plasma concentration, DSPE-PEG2000 did not shield the entire surface charge, which is consistent with previous outcomes published by another group with negatively charged sterically stabilized liposomes.47 Similar results in terms of particle clearance were found with cholesterol-rich cationic liposomes (with or without PEG) vs non-cationic counterparts in studies in mice.19,20 Correlation between poor PK profiles and complement activation was further confirmed via ELISA test. In our investigation, the lack of PEG in positively charged liposomes increased complement activation, as reported in previously published studies.26,58
In accordance with the specific PK profiles, differences in DOX organ biodistribution were assessed. In all the tested formulations, whenever liposomal DOX was administrated in combination with regional HT, a significantly higher amount of DOX was recovered in heated tumors, in comparison to non-heated tumors. In the case of PG2-TSL-30, blood stability encountered in the first hour and the rapid DOX-release observed in vitro, perfectly corroborated with the higher DOX-tumor enrichment assessed in vivo (14-fold, HT vs NT tumors). For anionic PEG-TSL, modest improvements in tumor DOX-enrichment were found compared with NT tumors (7-fold). Surprisingly, although PEG-CTSL showed a significantly lower AUC2h, minimal differences in terms of tumor DOX-enrichment were detected compared with the corresponding anionic counterpart. This can be explained by the higher DOX release rate observed in vitro for PEG-CTSL vs PEG-TSL, which we hypothesized to compensate for the partial payload loss after i.v. injection. For a positively charged surface, such as PG2-CTSL, the higher clearance of particles affected BD outcome with poor DOX-tumor enrichment. These data are in good accordance with previous finding from other groups, where cationic nanoparticles were cleared in vivo faster than neutral or anionic particles with consistent differences found in tumor-drug deposition.20,59 Surprisingly, although in these studies PK and BD showed suboptimal outcomes, therapeutic effects were still preserved when tested in tumor-bearing mice. In our investigation, we have not performed a partitioning analysis between liposomal versus non-liposomal DOX in the tumor vasculature, which might be an interesting parameter to evaluate in future studies. Furthermore, BD assessment over longer periods of time could be an additional point to be evaluated, although several studies dealing with cationic liposomes for cancer therapy suggested the main interactions with tumor vasculature happen in the first hour after nanoparticle injection, with no further improvement over time.14,19
Conclusion
Our findings suggest that HT-external targeting is the main mechanism by which anionic and cationic TSLs induce DOX accumulation in tumors, with negligible or no synergistic effect due to positive charge. Functionalizing cationic liposome with DPPG2 showed interesting and promising results in vitro, whereas the in vivo readout was sub-optimal due to complement activation and significant RES uptake. While grafting PEG on cationic TSLs might improve safety and circulation properties, we observed a decrease in liposome–cell interactions and drug delivery efficiency. Anionic DPPG2-based TSL significantly outperformed PEGylated TSL in terms of solid tumor targeting, although the latter showed a more stable and prolonged circulation time in vivo. Overall, anionic DPPG2-based vesicles showed added benefit in terms of local drug delivery, representing a critical asset for the design of liposomes for tumor targeting applications with potential clinical relevance.
Abbreviations
AUC, area under the curve; BD, biodistribution; CTSL, cationic thermosensitive liposome; DLS, dynamic light scattering; DOX, doxorubicin; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DSPC, 1,2-distearoyl-sn- glycero-3-phosphocholine; DPPG2, 1,2-dipalmitoyl-sn-glycero-3-phospho-diglycerol; DPTAP; 1,2-dipalmitoyl-3-trimethylammonium-propane; DSPE-PEG2000, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]; EE, encapsulation efficacy; FACS, fluorescence activated cell sorting; FCS, fetal calf serum; HBS, HEPES-buffered saline; HPLC, high performance liquid chromatography; HT, hyperthermia; HUVEC, human umbilical vein endothelial cells; IC50, half-maximal inhibitory concentration; ID, injected dose; LOD, limit of detection; NBD-PE, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3 benzoxadiazol-4-yl); NT, normothermia; PBS, phosphate-buffered saline; PDI, polydispersity index; PEG, polyethylene glycol; PEG-TSL, polyethylene glycol-containing thermosensitive liposomes with encapsulated doxorubicin; PEG-CTSL, polyethylene glycol-containing cationic thermosensitive liposomes with encapsulated doxorubicin; PG2-TSL, phosphatidyldiglycerol-containing thermosensitive liposomes with encapsulated doxorubicin; PG2-CTSL, phosphatidyldiglycerol-containing cationic thermosensitive liposomes with encapsulated doxorubicin; PK, pharmacokinetic; RES, reticuloendothelial system; Rho-PE, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl); RT, room temperature; SDS, sodium dodecyl sulfate; SRB, Sulforhodamine B; Tm, melting temperature; Tris, trisaminomethane; TSL, thermosensitive liposome; ζ-POT, ζ-potential.
Acknowledgments
Matteo Petrini, Wouter J. M. Lokerse, and Martin Hossann were funded by the Federal Ministry of Education and Research of the Federal Republic of Germany under the grant agreements 13XP5014A and 13XP5014B (TSL-LIFU project). The authors thank Lisa Pointer (Thermosome GmbH) for technical assistance with HPLC sample preparation and analysis. The authors also thank Helga M. Schmetzer and Ujwal M. Mahajan, for assistance with FACS analysis and fluorescence microscopy.
Disclosure
Lars H Lindner and Martin Hossann hold shares in Thermosome GmbH, Planegg/Martinsried, Germany. Dr Martin Hossann reports grants from BMBF German Ministry for Education and Research for TSL-LIFU project (grant agreement 13XP5014A) during the conduct of the study and for a project independent from submitted work (grant agreeement 031B0759A). Matteo Petrini is currently employed at Thermosome GmbH; and reports grants from German Federal Ministry of Education and Research during the conduct of the study. All other authors declare no conflict of interest.
References
1. Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics. 2017;9(2):51–59. doi:10.3390/pharmaceutics9020012
2. Douer D. Efficacy and safety of vincristine sulfate liposome injection in the treatment of adult acute lymphocytic leukemia. Oncologist. 2016;21(7):840–847. doi:10.1634/theoncologist.2015-0391
3. O’Brien ME, Wigler N, Inbar M, et al. Reduced cardiotoxicity and comparable efficacy in a Phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15(3):440–449. doi:10.1093/annonc/mdh097
4. Wang-Gillam A, Li CP, Bodoky G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, Phase 3 trial. Lancet. 2016;387(10018):545–557. doi:10.1016/S0140-6736(15)00986-1
5. Judson I, Radford JA, Harris M, et al. Randomised Phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2001;37(7):870–877. doi:10.1016/S0959-8049(01)00050-8
6. Laginha KM, Verwoert S, Charrois GJ, Allen TM. Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Cli Cancer Res. 2005;11(19):6944–6949. doi:10.1158/1078-0432.CCR-05-0343
7. Zamboni WC, Gervais AC, Egorin MJ, et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother Pharmacol. 2004;53(4):329–336. doi:10.1007/s00280-003-0719-4
8. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mat. 2013;12(11):991–1003. doi:10.1038/nmat3776
9. Kneidl B, Peller M, Winter G, Lindner LH, Hossann M. Thermosensitive liposomal drug delivery systems: state of the art review. Int J Nanomedicine. 2014;16(9):4387–4398.
10. Nardecchia S, Sanchez-Moreno P, Vicente J, Marchal JA, Boulaiz H. Clinical trials of thermosensitive nanomaterials: an overview. Nanomaterials. 2019;9(2):191. doi:10.3390/nano9020191
11. Manzoor AA, Lindner LH, Landon CD, et al. Overcoming limitations in nanoparticle drug delivery: triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012;72(21):5566–5575. doi:10.1158/0008-5472.CAN-12-1683
12. Dewhirst MW, Secomb TW. Transport of drugs from blood vessels to tumour tissue. Nat Rev Cancer. 2017;17(12):738–750. doi:10.1038/nrc.2017.93
13. Dicheva BM, ten.Hagen TLM, Li L. Cationic thermosensitive liposomes: a novel dual targeted heat-triggered drug delivery approach for endothelial and tumor cells. Nano Lett. 2013;13(6):2324–2331. doi:10.1021/nl3014154
14. Krasnici S, Werner A, Eichhorn ME, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int j Cancer. 2003;105(4):561–567. doi:10.1002/ijc.11108
15. Campbell RB, Brown DF, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62(23):6831–6836.
16. Augustin HA, Braun K, Telemenakis I, Modlich U, Kuhn W. Phenotypic characterization of endothelial cells in a physiological model of blood vessel growth and regression. Am J Pathol. 1995;147(2):339–352.
17. Qu H, Nagy JA, Senger DR, Dvorak HF, Dvorak AM. Ultrastructural localization of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) to the abluminal plasma membrane and vesiculovacuolar organelles of tumor microvascular endothelium. J Histochem Cytochem. 1995;43(4):381–389. doi:10.1177/43.4.7534783
18. Campbell RB, Ying B, Kuesters GM, Hemphill R. Fighting cancer: from the bench to bedside using second generation cationic liposomal therapeutics. J Pharm Sci. 2009;98(2):411–429. doi:10.1002/jps.21458
19. Wu J, Lee A, Lu Y, Lee RJ. Vascular targeting of doxorubicin using cationic liposomes. Int J Pharm. 2007;337(1–2):329–335. doi:10.1016/j.ijpharm.2007.01.003
20. Luo D, Geng J, Li N, et al. Vessel-targeted chemophototherapy with cationic porphyrin-phospholipid liposomes. Mol Cancer Ther. 2017;16(11):2452–2461. doi:10.1158/1535-7163.MCT-17-0276
21. Wang W, Shao A, Zhang N, Fang J, Ruan JJ, Ruan BH. Cationic polymethacrylate-modified liposomes significantly enhanced doxorubicin delivery and antitumor activity. Sci Rep. 2017;7:43036. doi:10.1038/srep43036
22. Strieth S, Eichhorn ME, Sauer B, et al. Neovascular targeting chemotherapy: encapsulation of paclitaxel in cationic liposomes impairs functional tumor microvasculature. Int J Cancer. 2004;110(1):117–124. doi:10.1002/ijc.20083
23. Chonn A, Cullis PR, Devine DV. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J Immunol. 1992;146(12):4234–4241.
24. Knudsen KB, Northeved H, Kumar PE, et al. In vivo toxicity of cationic micelles and liposomes. Nanomedicine. 2015;11(2):467–477. doi:10.1016/j.nano.2014.08.004
25. Lohr JM, Haas SL, Bechstein WO, et al. Cationic liposomal paclitaxel plus gemcitabine or gemcitabine alone in patients with advanced pancreatic cancer: a randomized controlled phase II trial. Ann Oncol. 2012;23(5):1214–1222. doi:10.1093/annonc/mdr379
26. Semple SC, Chonn A, Cullis PR. Interactions of liposomes and lipid-based carrier systems with blood proteins: relation to clearance behaviour in vivo. Adv Drug Deliv Rev. 1988;32(1–2):3–17. doi:10.1016/S0169-409X(97)00128-2
27. Salmaso S, Caliceti P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J Drug Deliv. 2013;2013:1–19. doi:10.1155/2013/374252
28. Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):237–251. doi:10.1016/0014-5793(90)81016-H
29. Maruyama K, Vuda T, Okamoto A, Kojima S, Suginaka A, lwatsuru M. Prolonged circulation time in vivo of large unilamellar liposomes composed of distearoyl phosphatidylcholine and cholesterol containing amphipathic poly(ethylene glycol). Biochim Biophys Acta. 1992;1128(1):44–49. doi:10.1016/0005-2760(92)90255-T
30. Pozzi D, Colapicchioni V, Caracciolo G, et al. Effect of polyethyleneglycol (PEG) chain length on the bio-nano-interactions between PEGylated lipid nanoparticles and biological fluids: from nanostructure to uptake in cancer cells. Nanoscale. 2014;6(5):2782–2792. doi:10.1039/c3nr05559k
31. Dan N. Effect of liposome charge and PEG polymer layer thickness on cell–liposome electrostatic interactions. Biochim Biophys Acta. 2002;1564(2):343–348. doi:10.1016/S0005-2736(02)00468-6
32. Sanchez L, Yi Y, Yu Y. Effect of partial PEGylation on particle uptake by macrophages. Nanoscale. 2017;9(1):288–297. doi:10.1039/C6NR07353K
33. Degors IMS, Wang C, Rehman ZU, Zuhorn IS. Carriers break barriers in drug delivery: endocytosis and endosomal escape of gene delivery vectors. Acc Chem Res. 2019;52(7):1750–1760. doi:10.1021/acs.accounts.9b00177
34. Lindner LH, Eichhorn ME, Eibl H, et al. Novel temperature-sensitive liposomes with prolonged circulation time. Clin Cancer Res. 2004;10(6):2168–2178. doi:10.1158/1078-0432.CCR-03-0035
35. Hossann M, Wiggenhorn M, Schwerdt A, et al. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochim Biophys Acta. 2007;1768(10):2491–2499. doi:10.1016/j.bbamem.2007.05.021
36. Limmer S, Hahn J, Schmidt R, et al. Gemcitabine treatment of rat soft tissue sarcoma with phosphatidyldiglycerol-based thermosensitive liposomes. Pharm Res. 2014;31(9):2276–2286. doi:10.1007/s11095-014-1322-6
37. Zimmermann K, Hossann M, Hirschberger J. et al. A pilot trial of doxorubicin containing phosphatidyldiglycerol based thermosensitive liposomes in spontaneous feline soft tissue sarcoma. Int J Hyperthermia;2016. 1–13. doi:10.3109/02656736.2015.1131338
38. Hossann M, Hirschberger J, Schmidt R, et al. A heat-activated drug delivery platform based on phosphatidyl-(oligo)-glycerol nanocarrier for effective cancer treatment. Adv NanoBiomed Res. 2021:200089. doi:10.1002/anbr.202000089.
39. Haran H, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. doi:10.1016/0005-2736(93)90105-9
40. Eibl H, Lands WEM. A new, sensitive determination of phosphate. Anal Biochem. 1969;30(1):51–57. doi:10.1016/0003-2697(69)90372-8
41. Vichai V, Sulforhodamine KK. B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. doi:10.1038/nprot.2006.179
42. Peller M, Willerding L, Limmer S, et al. Surrogate MRI markers for hyperthermia-induced release of doxorubicin from thermosensitive liposomes in tumors. J Control Release. 2016;237:138–146. doi:10.1016/j.jconrel.2016.06.035
43. Lokerse WJM, Lazarian A, Kleinhempel A, et al. Mechanistic investigation of thermosensitive liposome immunogenicity and understanding the drivers for circulation half-life: a polyethylene glycol versus 1,2-dipalmitoyl-sn-glycero-3-phosphodiglycerol study. J Control Release. 2021;333:1–15. doi:10.1016/j.jconrel.2021.03.014
44. Li L, Ten Hagen TL, Hossann M, et al. Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J Control Release. 2013;168(2):142–150. doi:10.1016/j.jconrel.2013.03.011
45. Lokerse WJM, Kneepkens ECM, Ten Hagen TLM, Eggermont AMM, Grüll H, Koning GA. In depth study on thermosensitive liposomes: optimizing formulations for tumor specific therapy and in vitro to in vivo relations. Biomaterials. 2016;82:138–150. doi:10.1016/j.biomaterials.2015.12.023
46. Miller CR, Bondurant B, McLean SD, McGovern KA, O’Brien DF. Liposome-cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry. 1998;37(5):12875–12883. doi:10.1021/bi980096y
47. Webb MS, Saxon D, Wong FM, et al. Comparison of different hydrophobic anchors conjugated to poly(ethylene glycol): effects on the pharmacokinetics of liposomal vincristine. Biochim Biophys Acta. 1998;1372(2):272–282. doi:10.1016/S0005-2736(98)00077-7
48. Majzoub RN, Chan CL, Ewert KK, et al. Uptake and transfection efficiency of PEGylated cationic liposome-DNA complexes with and without RGD-tagging. Biomaterials. 2014;35(18):4996–5005. doi:10.1016/j.biomaterials.2014.03.007
49. Kono Y, Iwasaki A, Fujita T. Effect of surface charge, particle size, and modification by polyethylene glycol of liposomes on their association with Caco-2 cells across an unstirred water layer. Pharmazie. 2018;73(1):3–8. doi:10.1691/ph.2018.7110
50. Kono Y, Jinzai H, Kotera Y, Fujita T. Influence of physicochemical properties and PEG modification of magnetic liposomes on their interaction with intestinal epithelial Caco-2 cells. Biol Pharm Bull. 2017;40(12):2166–2174. doi:10.1248/bpb.b17-00563
51. Song LY, Ahkong QF, Rong Q, et al. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim Biophys Acta. 2002;1558(1):1–13. doi:10.1016/S0005-2736(01)00399-6
52. Lindner LH, Brock R, Arndt-Jovin D, Eibl H. Structural variation of cationic lipids: minimum requirement for improved oligonucleotide delivery into cells. J Control Release. 2006;110(2):444–456. doi:10.1016/j.jconrel.2005.10.009
53. Banerjee R, Mahidhar YV, Chaudhuri A, Gopal V, Rao NM. Design, synthesis, and transfection biology of novel cationic glycolipids for use in liposomal gene delivery. J Med Chem. 2001;44(24):4176–4185. doi:10.1021/jm000466s
54. Dicheva BM, Ten Hagen TL, Schipper D, et al. Targeted and heat-triggered doxorubicin delivery to tumors by dual targeted cationic thermosensitive liposomes. J Control Release. 2014;195:37–48. doi:10.1016/j.jconrel.2014.07.058
55. Lindner LH, Hossann M, Vogeser M, et al. Dual role of hexadecylphosphocholine (miltefosine) in thermosensitive liposomes: active ingredient and mediator of drug release. J Control Release. 2008;125(2):112–120. doi:10.1016/j.jconrel.2007.10.009
56. Overgaard J. Combined adriamycin and hyperthermia treatment of a murine mammary carcinoma in vivo. Cancer Res. 1976;36(9):3077–3081.
57. Chonn A, Cullis PR, Devine DV. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J Immunol. 1991;146(12):4234–4241.
58. Shan X, Yuan Y, Liu C, Tao X, Sheng Y, Xu F. Influence of PEG chain on the complement activation suppression and longevity in vivo prolongation of the PCL biomedical nanoparticles. Biomed Microdevices. 2009;11(6):1187–1194. doi:10.1007/s10544-009-9336-2
59. Zhao W, Zhuang S, Qi XR. Comparative study of the in vitro and in vivo characteristics of cationic and neutral liposomes. Int J Nanomedicine. 2011;6:3087–3098. doi:10.2147/IJN.S25399
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.