")
Back to Journals » Journal of Inflammation Research » Volume 16
Effects of Immune Cells and Cytokines on Different Cells in OA
Authors Luo P , Yuan Q , Wan X, Yang M, Xu P
Received 23 March 2023
Accepted for publication 24 May 2023
Published 30 May 2023 Volume 2023:16 Pages 2329—2343
DOI https://doi.org/10.2147/JIR.S413578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Pan Luo,* Qiling Yuan,* Xianjie Wan, Mingyi Yang, Peng Xu
Department of Joint Surgery, HongHui Hospital, Xi’an Jiaotong University, Xi’an, Shaanxi, 710054, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Peng Xu, Department of Joint Surgery, HongHui Hospital, Xi’an Jiaotong University, Xi’an, Shanxi, People’s Republic of China, Email [email protected]
Abstract: The pathological manifestations of osteoarthritis (OA) involve the destruction of articular cartilage, synovial sac thickening, subchondral osteosclerosis and osteophyte formation. The types of cells involved in OA include chondrocytes, osteoblasts, osteoclasts, synovial fibroblasts, T cells, macrophages, and mesenchymal stem cells (MSCs). There are many effects of immune cells in OA, and innate immune cells such as dendritic cells and macrophages have significant roles in the pathogenesis of OA. On the other hand, the role of adaptive immune cells such as T-cell subsets, B-cell subsets, and NK cells is important in OA pathogenesis. MSCs not only play a key role in the dynamic balance and repair of OA tissue but also inhibit the immune system. Therefore, MSCs are a new potential therapeutic agent for OA. This review mainly summarizes the effects of various immune cells and cytokines on different cells in OA to find new effective therapeutic targets for OA therapy based on the immune system and cytokine activity sites to prevent the development of OA.
Keywords: osteoarthritis, immune cells, chondrocytes, mesenchymal stem cells, osteoblasts
Introduction
Osteoarthritis (OA) is a joint degenerative disease that can cause chronic pain and motor dysfunction and affects approximately 300 million people worldwide, which adds a serious economic burden to patients and society.1 OA is caused by mechanical injury of joints and is also closely related to ageing.
Thus far, researchers have found that OA is much more complex than abrasive diseases and that metabolic and biochemical mechanisms are involved in development.2 Ageing, obesity, and joint injury are the main risk factors for the progression of OA.3 Although there have been many studies on the pathogenesis of OA, there is still no definite conclusion, and so there is no intervention to delay the progression of this disease.4 Thus, there is a need for further examination of the pathogenesis of OA to meet future clinical demands for new treatments.
The pathogenesis of OA involves the destruction of articular cartilage, synovial bursa thickening, subchondral osteosclerosis and osteophyte formation.5 The interaction between different tissues and cells is involved in these pathological manifestations. Understanding the complex interactions between different tissues and cells and their role in the initiation and progression of OA is important for clarifying the pathogenic mechanism of OA. The interaction and homeostasis between different tissues and cells in the joint system are prerequisites for the function of the joint system. Many factors affect the health of joints and bones, including the immune system, sex hormones, and the microbiota.6,7 During the occurrence and development of OA, osteochondral factors closely interact with various molecules in the immune system to maintain the health of bones and joints.8 Inflammatory mediators not only lead to abnormal tissue structure but also aggravate cartilage injury in OA.9 Therefore, examining the effect of immune cells on different tissue cells in the pathogenesis of OA may provide new treatment strategies for OA. The purpose of this paper was to explore the effect of immune cells on different tissue cells in OA and provide new ideas for the treatment of OA.
Tissue and Cell Types Involved in the Pathogenesis of OA (Excluding the Immune System)
Cartilage and Chondrocytes
Articular cartilage plays a role in lubricating joints and bearing weight associated with joint activity.10 Due to the limited supply of oxygen and nutrients, the regeneration ability of articular cartilage is low.11 Chondrocytes can form an extracellular matrix (ECM) composed of proteoglycans and type II collagen.12 In addition to chondrocytes, cartilage stem/progenitor cells (CSPCs), which account for approximately 10% of total cells, have recently been found in OA cartilage.13,14 In recent years, single-cell RNA-seq analysis has shown various groups of chondrocytes in late OA, including steady-state chondrocytes, effector chondrocytes, prehypertrophic chondrocytes, and fibrochondrocytes.15 When the metabolic state of chondrocytes changes, the synthesis and degradation of collagen become out of balance, which affects the function of joints and leads to osteoarthritis.
The development of subchondral osteosclerosis and osteophytes may gradually lead to dysfunction of the entire joint. In early-stage OA, the cartilage surface remains intact.16 The first changes occur in the molecular composition of tissues and the ECM, which are characterized by a transient chondrocyte proliferative response and an increase in ECM protein synthesis, including collagen type II (Col II) and aggrecan.16 With the development of OA, the synthesis of degrading proteases increases, leading to increased catabolic activity and decreased proteoglycan content. Subsequently, Col II is degraded, and its fragments stimulate several proteins associated with the catabolic state, such as MMPs and aggrecanases, which are members of the a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family, and this change is accompanied by increased expression levels of inflammatory cytokines (IL-1), stress and apoptosis markers (caspases-3 and −9 and Bcl-2), and transcription factors [runt-related transcription factor 2 (Runx2) and Sox9].17–19 Changes in articular cartilage composition and structure further stimulate chondrocytes to produce more catabolic factors involved in cartilage degradation. As the proteoglycan and collagen network breaks down, cartilage integrity is disrupted.20 Subsequently, apoptosis occurs in articular chondrocytes, ultimately leading to the complete loss of articular cartilage.21
Cells in Subchondral Bone
Although subchondral bone destruction occurs during cartilage degeneration, subchondral bone is not abnormal in all patients with OA.22 Moreover, in osteoarthritis in elderly individuals, the metabolic imbalance in chondrocytes occurs before abnormal subchondral bone remodelling.23 In contrast, early microdamage of subchondral bone was first detected in trauma-induced OA phenotypes.24 The microstructural changes in subchondral bone in OA include bone marrow oedema-like lesions and osteophytes caused by histopathological changes.25 Osteoblasts differentiate from mesenchymal cells. During the pathogenesis of OA, the phenotype of osteoblasts changes, as indicated by increases in transforming growth factor (TGF)-β1,26 vascular endothelial growth factor (VEGF)27 and alkaline phosphatase activity. These changes can lead to osteoclast formation. Osteoclasts are mainly responsible for bone resorption.28
The Synovium and Synovial Cells
The changes in articular cartilage and bone on X-ray are one of the diagnostic criteria of OA, and little attention is given to synovial changes. In fact, the morphology and cellular composition of the synovium is also one of the manifestations of OA. Synovial changes in OA are classified as hyperplasia (chorionic hyperplasia), fibrosis (capsular fibrosis) and inflammation (diffuse inflammation and lymphatic plasma cell infiltration and accumulation), which usually coexist.29 Synovitis has the features of fibroblast-like synoviocyte (FLS) proliferation and macrophage recruitment, leading to the proliferation of the inner layer of the synovium.30 The subsynovial layer is also rich in macrophages, mast cells (MCs), B cells, and endothelial cells.31 In addition, MCs are related to inflammation and cartilage destruction in OA.32
Adipose Cells and Adipose Cytokines
A possible source of adipocytokines in the knee joint is the subpatellar fat pad.33 As an endocrine organ, adipose tissue can produce adipose cytokines that directly cause abnormalities in the structure and function of articular cartilage in obese individuals.34 In addition to cartilage injury, the role of adipose cytokines produced in the joint can further worsen the condition of the joint. Among the many adipose cytokines, the most typical are leptin, resistin, adiponectin, vaspin, omentin, visfatin and adipsin.35
Mesenchymal Stem Cells
Pluripotent stem cells exist in bone marrow and adipose tissue,36 and bone marrow-derived mesenchymal stem cells (BMSCs) are the main pluripotent stem cells.37 In vitro, synovial mesenchymal stem cells (SMSCs) showed high cartilage differentiation abilities.38 Adipose tissue MSCs (AMSCs) can regulate inflammation. Current evidence suggests that AMSCs regulate the local microenvironment, which makes them conducive to repair, thereby delaying cartilage degeneration.39
Immune Cells
The main role of the immune system in organisms is to remove antigens, maintain environmental stability, and play an important role in safeguarding the health of the organism.40 The innate immune system consists of cells and related mechanisms that can resist infection in a nonspecific way. Cells in the innate immune system recognize and act on pathogens nonspecifically. Unlike the acquired immune system, the innate immune system does not provide lasting protective immunity but exists in all animals and plants as a rapid response to infection. Innate immune cells include dendritic cells, macrophages and other cells. Adaptive immune cells are produced by individuals who are stimulated by antigenic foreign bodies in the external environment and include T-cell subsets, B-cell subsets and NK cells. T lymphocytes are a kind of haematopoietic cell. Studies have shown that there are changes in surface receptors of T lymphocytes, and the main factor affecting their functions is their differentiation status.41 T helper (Th) cells can interact with other immune cells (such as B lymphocytes) based on cytokines and participate in immune regulation.42 Th cells include multiple subpopulations. Th1 cells are polarized by interleukin (IL)-12 and secrete interferon-gamma (IFN γ)- and IL-2-related cytokines, providing support for the immune response.43 Tumour necrosis factor-α (TNF-α) can mediate an increase in receptor activator of nuclear factor-kappa B ligand (RANKL) expression in macrophages, thus stimulating osteoclast formation.44 B lymphocytes mature in bone marrow through C-X-C chemokine ligand 12 (CXCL12) signals, and their main function is to produce antibodies against pathogens.45 However, B lymphocytes also provide antigens for T-cell activation, such as granulocyte colony-stimulating factor (G-CSF).46 G-CSF can increase the proliferation of osteoclast progenitor cells.47 MCs are immune cells that exist in tissue and are important in allergic reactions.48 In addition, MCs can synthesize and release mediators scratched novo, including IL-6, TNF-α and VEGF. Previous research has shown that MCs are important in the regulation of bone turnover.49 Inflammatory mediators expressed and secreted by neutrophils and osteoclasts can affect MSCs in different ways.50 In addition, macrophages have two phenotypes: a proinflammatory state (called M1) and a pro-regenerative state (called M2).51 Macrophages can also affect osteocytes through paracrine signal transduction or direct intercellular contact.52
Effect of Immune Cells on Chondrocytes
Chondrocytes are the basic components of cartilage tissue and can secrete ECM to maintain normal cartilage function. Many immune cells can affect the metabolism of chondrocytes by secreting cytokines. For example, macrophages, which are innate immune cells, can secrete IL-1β. IL-1β stimulates the production of a number of inflammatory mediators implicated in OA pathology. For instance, treatment of chondrocytes with these cytokines upregulates the expression of genes encoding inducible nitric oxide synthase (iNOS), soluble phospholipase A2, cyclooxygenase 2 (COX-2) and microsomal prostaglandin E synthase 1 and stimulates the release of nitric oxide (NO) and prostaglandin E2 (PGE2). NO and PGE2 contribute to articular inflammation and destruction by enhancing the activation and production of MMPs, inhibiting the synthesis of anabolic macromolecules such as collagen and proteoglycan, inhibiting the production of IL-1ra, and promoting chondrocyte apoptosis. IL-1β also induces the production of reactive oxygen species (ROS) such as NO and the superoxide anion, that generate hydrogen peroxide, peroxy nitrate and hydroxyl radicals,53 which contribute to cartilage degradation. In addition, IL-1β downregulates the expression of antioxidant enzymes that scavenge ROS, including superoxide dismutase, catalase and glutathione peroxidase,54 thereby accelerating the damaging effects of ROS on cartilage. IL-1β affects the metabolism of chondrocytes through many signalling pathways. Most IL-1β is produced by macrophages, and some is produced by B and T cells and natural killer (NK) cells.55 Activation of extracellular signal-regulated kinase (ERK) by IL-1β can reduce the production of ECM in cartilage.56 In addition, IL-1β induces ECM degradation by inducing collagenase and aggrecanases,57 which leads to chondrocyte hypertrophy and dedifferentiation and ultimately chondrocyte apoptosis.58 Another dominant pathway in IL-1β-mediated OA progression is nuclear factor-kappa B (NF-κB), which, when activated, inhibits the expression of type II collagen and affects the metabolism of chondrocytes59 (Figure 1). NF-κB regulates chondrocyte hypertrophy mainly through SRY-box transcription factor 9 (SOX9), bone morphogenetic protein 2 (BMP2), matrix metalloproteinases (MMPs) and HIF-2α.60,61 For example, it has been reported that NF-κB is directly involved in the regulation of matrix metalloproteinase (MMP) expression, but a recent study showed that hypoxia-inducible factor-2α (HIF-2α) was mediated by NF-κB and involved in the development of OA.62 More information about the role of the NF-κB signalling pathway in OA cartilage destruction can be found in other studies.59,63,64
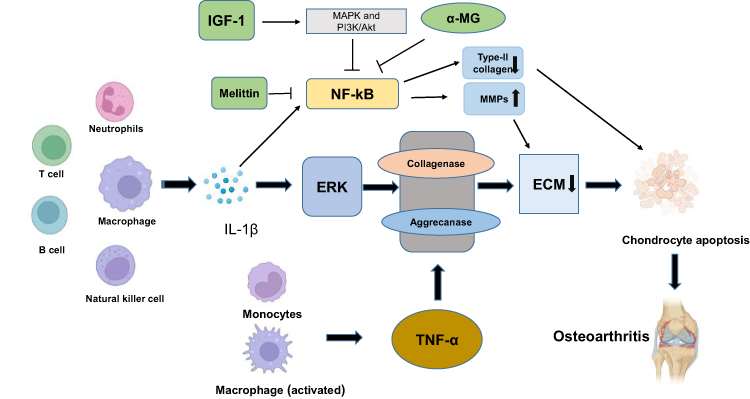 |
Figure 1 Effect of immune cells on chondrocyte apoptosis. Macrophages, T cells, B cells, NK cells and neutrophils can secrete IL-1β. IL-1β activates extracellular signal-regulated kinase (ERK) to reduce the production of cartilage ECM. TNF-α is secreted by monocytes and macrophages and results in the degradation of ECM by inducing collagenase and aggrecanase. In addition, IL-1β induces ECM degradation by inducing collagenase and aggrecanase, which leads to chondrocyte hypertrophy and dedifferentiation and eventually to chondrocyte apoptosis, resulting in osteoarthritis (OA). Another important signalling pathway by which IL-1β mediates the progression of OA is NF-κB. Activated IL-1β inhibits the expression of type II collagen and affects chondrocyte metabolism. In addition, MMPs degrade the ECM in a manner dependent on NF-κB, which leads to the degradation of chondrocytes. Insulin-like growth factor-1 (IGF-1) inhibits apoptosis in chondrocytes by regulating the MAPK and PI3K/Akt signalling pathways and inhibiting the NF-κB signalling pathway. Melittin inhibits the development of OA by inhibiting the decrease in type II collagen expression induced by NF-kB. In addition, alpha-mangostin (α-MG) inhibits the NF-kB pathway to limit the development of OA. |
TNF-α, which is mainly secreted by active macrophages and monocytes, is a pluripotent cytokine that is closely related to immune regulation. During the pathological changes of OA, these two receptors provide support for signal transduction.65 TNF-α also induces the degradation of extracellular matrix (ECM) by inducing collagenase and aggregating enzymes, which is consistent with the effects of IL-1β.66 In addition, Fujihara et al found that macrophages blocked the maturation of chondrocytes67 (Figure 1).
Activated T cells can produce IL-6, including membrane binding (mbIL-6R) and soluble (sIL-6R). The exact role of IL-6 in OA is difficult to determine because IL-6 has beneficial and harmful effects. An in vitro study of chondrocytes showed that TIMP-1 could be induced by IL-6 alone, and the effect was more obvious when chondrocytes were also treated with sIL-6R.68 Moreover, animal experiments showed that the prevalence and severity of OA in IL-6-knockout mice were higher than those in wild-type mice. Intra-articular injection of IL-6 can induce OA-like cartilage lesions.69,70 IL-6 levels in the synovial fluid of end-stage OA patients was significantly higher than that of healthy controls, and IL-6 levels in synovial fluid are known to be associated with pain in OA patients.71,72 In addition, the reduction in congenital formation of IL-6 is related to a lower risk of OA in the hand, as well as a lower risk of OA in the knee and hip joints.73
IL-17 produced by activated CD4+ lymphocytes can promote inflammation, which can activate T lymphocytes, especially in the early stage of inflammation, acting on a variety of cells and tissues.74 IL-17 can upregulate catabolic factors and anabolic factors in isolated chondrocytes and induce cartilage degradation.75 In addition, IL-1β, TNF-α and IL-6 promote the degradation of ECM and lead to the destruction of articular cartilage by stimulating the production of MMPs and aggregation proteases (ADAMTS), which are dominant in the progression of OA.76
Immune Cells Involved in Bone Remodelling in Subchondral Bone
Changes in subchondral bone caused by dynamic bone remodelling are an important link in the pathogenesis of OA. Osteoblasts and osteoclasts are important cells in bone remodelling. A recent study showed that reducing osteoclast formation and bone resorption could slow bone remodelling in subchondral bone and reverse the progression of osteoarthritis in mice.77 For example, the administration of diarylbutyl phthalate (DP) blocked the formation of overactivated osteoclasts in subchondral bone and improved articular cartilage degeneration.78
Osteoclasts are a specific type of terminally differentiated cell.79 Relevant experimental research showed that the system composed of osteoprotegerin (OPG) and RANKL plays an important role in regulating the formation of these cells and directly determines the differentiation of osteoclasts.80 The combination of RANK and RANKL can promote the maturation of these cells, while OPG can inhibit the combination of the two, thereby inhibiting the maturation process of osteoclasts.81 In OA, dysfunction of the OPG/RANK/RANKL regulatory system is closely related to histological changes in subchondral bone, and a proinflammatory osteoblast phenotype appears.82
B cells can regulate the maturation and differentiation of osteoclasts based on this system, which is also closely related to bone metabolism.83,84 IL-7 is mainly derived from BM stromal cells.85 Some experimental studies have shown that IL-7 has a significant effect on regulating bone homeostasis and is closely related to the integrity of bone structure.86 Compared with that of normal mice, the bone mass of mice with IL-7 overexpression decreased, and osteoclasts increased, which confirmed its regulatory effect.87 Toraldo’s research shows that this cytokine could not promote bone absorption in T-cell-deficient nude mice. It has been found that G-CSF secreted by B cells can increase the proliferation of osteoclast progenitor cells. Activated B cells can also activate osteoclast formation by secreting RANKL.88 Under physiological conditions, osteoprotegerin (OPG) produced by B cells can inhibit osteoclast differentiation.89 In addition, coculturing M1 macrophages and MSCs could reduce the expression of OPG, which may have an indirect effect on osteoclast formation.90 Proinflammatory cytokines secreted by macrophages may mediate direct effects on osteoclast formation, and these cytokines have been shown to increase osteoclast production91 (Figure 2).
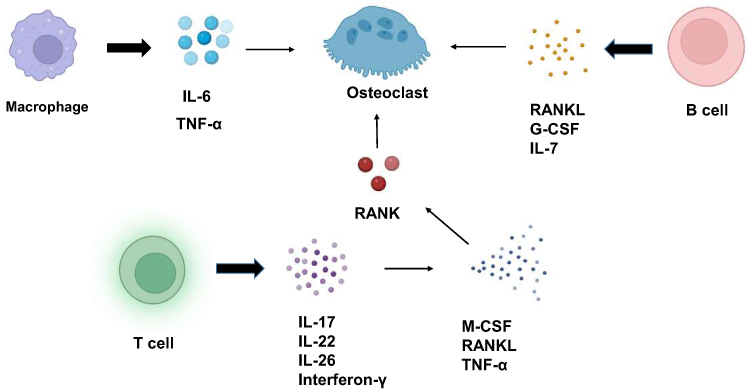 |
Figure 2 Effect of immune cells on osteoclasts. IL-7 secreted by B cells can increase osteoclasts. The G-CSF secreted by B cells can increase the proliferation of osteoclast progenitor cells and lead to an increase in osteoclasts. Activated B cells can also activate osteoclast formation by secreting RANKL. Proinflammatory cytokines secreted by macrophages can increase osteoclast production. A variety of cytokines secreted by a variety of T cells, such as IL-17 and IL-22, can further induce the expression of M-CSF, RANKL and TNF-α, thereby enhancing RANK levels in osteoclast precursors and promoting osteoclast maturation. |
The effect of T lymphocytes on bone metabolism can be determined by the regulation of osteoclasts. A variety of cytokines secreted by T helper (Th) 17 cells, Treg cells, IL-17, and IL-22 can further induce the expression of M-CSF, RANKL and TNF-α, thereby promoting osteoclast maturation92 (Figure 2). It has been reported that TH17 cells stimulate the recruitment of osteoclast progenitor cells by increasing chemokines that originate from bone marrow mesenchymal stromal cells.93 Relevant experimental studies showed that IL-17 could promote the expression of RANK in progenitor cells and increase its sensitivity to RANKL stimulation, thus playing a role in regulating bone metabolism.94 The mechanism of IL-17 in osteoclasts is still unclear, and no clear and consistent conclusion has been reached thus far. The existing experimental results are contradictory.95 Cytokines released by Th1 and Th2 subtypes can promote the development of osteoclast precursors and reduce bone resorption. Precursors of osteoclasts secrete carbon monoxide, which blocks the proliferation of T lymphocytes.96 Treg cells can also inhibit bone resorption, thereby promoting osteoclast apoptosis.97
Osteoblasts are dominant in bone synthesis and metabolism. Some scholars have found that abnormal expression of OPG and RANKL in osteoblasts may cause bone remodelling and reduced mineralization.98 Structural and functional disorders of bone in OA are typically attributed to primary osteoblast dysfunction.99 In addition, osteoblasts can produce many transcription factors, growth factors and other proteins involved in the pathogenesis of OA.100 For example, osteoblasts can synthesize indispensable ECM proteins such as type I collagen, bone sialic acid protein, and osteopontin. Osteoblasts are also responsible for mineralizing the matrix through the deposition of phosphates, leading to bone remodelling and osteophyte formation.100,101
A variety of immune cells can regulate the maturation and activation of osteoblasts, and the corresponding mechanisms are complex. Interferon-γ is secreted by CD4+ T lymphocytes; this cytokine promotes the differentiation of bone marrow mesenchymal stem cells, while the activity of transforming growth factor-β (TGF- β) secreted by CD4+ T lymphocytes is negatively correlated with osteoblast differentiation.102 IL-17F secreted by Treg cells can strongly promote osteoblastic differentiation in MSCs.103 TNF-α can indirectly stimulate osteoclast production through RANKL produced by B cells.104,105 IL-17 can induce the differentiation of MSCs into bone and promote the differentiation of osteoblasts.106 M2 cells can release osteogenic growth factors such as bone morphogenetic protein 2 (which belongs to the TGF-β family subclass) under certain conditions to promote bone formation.107 Neutrophils expanded by G-CSF in vitro can induce MSC and osteoblast apoptosis by producing reactive oxygen species (ROS).108 Moreover, C-C motif chemokine ligand 2 (CCL2) plays an important regulatory role in immune regulation. Studies have shown that this factor binds to the receptor CC-chemokine receptor-2 (CCR2), thereby regulating the status of memory T cells and basophils, which can have a negative impact on immune function.109 A large number of studies have shown that CCL2 is closely related to bone remodelling.110 For example, IL-7 secreted by osteoblasts can promote the differentiation of B cells.111
Many MC-derived mediators can regulate bone metabolism by preventing osteoblast activity and/or promoting osteoclast production (TNF, IL-6). MCs are important in maintaining the stability of the intraosseous environment.112 For example, TGF-β can stimulate osteoblast production, and IL-12 and IFN-γ can inhibit osteoclast formation113 (Figure 3). Endothelial cells and osteoblasts showed that neutrophils induced osteogenic marker expression in osteoblasts in vitro, indicating that neutrophils affected osteoblasts.114 In addition, an increase in the mineral deposition of osteoblasts was found in this model, indicating that neutrophils may undergo osteogenesis.114
 |
Figure 3 Effect of immune cells on osteoblasts. Interferon-γ can promote bone marrow mesenchymal stem cells to differentiate into osteoblasts, and the activity of TGF-β is negatively correlated with osteoblast differentiation. IL-17A and BMP-2 are secreted by Treg cells and can promote osteoblast differentiation by promoting MSC production. M2 macrophages can produce BMP-2 to promote bone formation. Neutrophils can induce apoptosis in osteoblasts by producing reactive oxygen species (ROS). |
Immune Regulation in Synovitis
Synovial fibroblasts (SFs) play an important role in synovitis. SFs secrete inflammatory cytokines and upregulate surface markers.115,116 These cells act on T cells, dendritic cells, and B cells.117,118 In addition, a study has shown that activated SFs restrict the proliferation and secretion of B cells.119 Moreover, researchers have observed a negative feedback mechanism activated by SFs in macrophages.120
Compared with healthy synovium, OA synovium has an abundant T-cell population.121,122 The proportion of T cells in OA synovium is second only to macrophages and may account for 20–25% of inflammatory cells.123 IL-17 is mainly produced by Th17 subtype of helper T cells.124 Previous studies have shown that IL-17 family cytokines induce the most effective changes in synovial transcriptional groups and chondrocytes in patients with OA.125 IL-17RA and IL-17RC are the main targets of IL-17, and IL-17RA is highly expressed in the inflamed synovial fibroblasts of patients.126 IL-4, which is secreted by Th2 cells and MCs, is an effective regulator of the immune system and is often referred to as a prototype immunoregulatory cytokine.127 The number of CD4 T cells in the subsynovial layer of OA patients was higher than that in the control group, which suggests that the production of IL-4 was mainly dependent on Th2 cells infiltrating the synovium.128 IL-4 has a strong protective effect on cartilage. This factor inhibits the secretion of MMPs, so it can inhibit the degradation of proteoglycans.129
Synovitis is characterized by macrophage accumulation in the intimal lining130 (Figure 4). As immune cells, synovial macrophages dominate the symptoms and structural progression of OA.131 Macrophages are also associated with the severity of OA and joint symptoms.132 In addition, macrophages can produce chemokines in inflamed joints.133 These cells can be activated by a variety of stimuli, such as proinflammatory factors (IL-1β, TNF-α and IL-6) and immunoregulatory factors.134,135 Moreover, IL-1β increases the levels of chemokines such as IL-8 and CCL2 and the expression of the cytokine IL-6, which causes synovitis and further produces more IL-1β.136 Activated macrophages are regulated by the mTOR,137 NF-κB A,138 and JNK pathways. In OA synovial tissue and synovial fluid, macrophages are polarized into the M1 or M2 subtypes.139 Relevant experimental studies showed that synovial macrophages can release large amounts of IL-1β-, TNF-α-, and IL-1-related factors after activation, and these factors can regulate the microenvironment of chondrocytes in many ways and promote the release of large amounts of MMPs, thereby degrading the ECM140 (Figure 4). The components generated after degradation can activate macrophages, leading an increase in the level of inflammation, further promoting the degradation of cartilage and aggravating the disease.141–143 The inflamed synovium promotes the release of intercellular adhesion molecule 1 (ICAM-1)- and VCAM-1-related factors. These factors have a certain adhesive effect, which leads to structural changes in the OA synovium.144
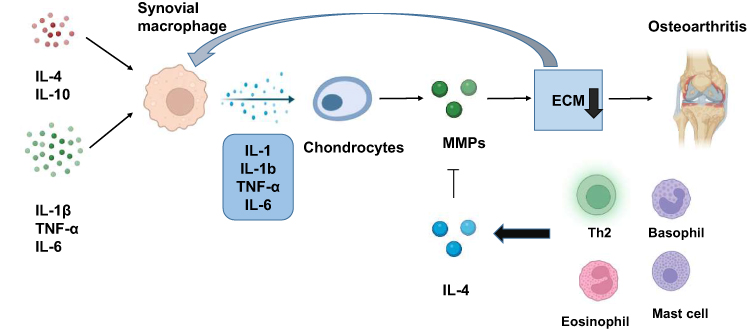 |
Figure 4 Effect of synovial macrophages on the pathogenesis of OA. Synovial macrophages can be activated by proinflammatory factors and immunoregulatory factors. Activated synovial macrophages can produce cytokines such as IL-1β, TNF-α, and IL-1, which promote chondrocytes to produce excessive matrix metalloproteinases (MMPs), leading to the degradation of ECM and the progression of OA. IL-4 is secreted by Th2 and MCs and can effectively inhibit the secretion of MMP to protect cartilage and prevent the progression of OA. |
Adipose Cells and Adipose Cytokines Involved in the Pathological Development of OA
Recent studies have revealed that adipocytokines play an important role in the metabolism of cartilage, synovium and bone.145,146 In the acute phase of inflammation, leptin has a proinflammatory effect through T-lymphocyte stimulation, resulting in the release of cytokines such as ILs and tumour necrosis factor-α (TNF-α) and increasing the activation levels of NK cells, macrophages and neutrophils.147 Leptin can increase the anabolic activity of chondrocytes by inducing the expression of IGF-1 and TGF-β at the mRNA and protein levels.148 Other studies, however, have shown a different view. Leptin upregulated the expression of ADAMTS-4 and ADAMTS-5, resulting in proteoglycan loss in the articular cartilage of rats.149 The latest data show that leptin induces the gene expression of ADAMTS in human chondrocytes through mitogen-activated protein kinase and the NF-κB signalling pathway, which leads to a subsequent increase in the inflammatory process.150 Adiponectin can protect articular cartilage by upregulating tissue inhibitor of metalloproteinase and reducing the expression of MMP-13 mediated by IL-1β.151 Visceral adiponectin is catabolized to articular cartilage. This factor is produced by OA chondrocytes and can increase the activities of MMP-3, ADAMTS-4 and ADAMTS-5 in a rat model.152 During osteophyte formation, Berry et al found that high leptin levels were related to an increase in bone formation markers.153 In addition, visceral leptin has been shown to affect osteoblast proliferation and type II collagen production.154 Adipocytokines can be partially detected in the synovium and synovial fluid of patients with OA. However, it is not clear whether adipocytokines can induce synovitis.145
Mesenchymal Stem Cells Treat OA by Regulating the Function of Immune Cells
MSCs are pluripotent cells with a high capacity for self-renewal. According to relevant research, bone marrow and adipose tissue are the most common sources of MSCs, and these stem cells can be found in skeletal muscle,155 the periosteum156 and the synovium.157 MSCs have certain inhibitory effects on the immune system.158
MSCs can inhibit the activation of inflammatory M1 macrophages through cell-to-cell contact and paracrine signalling and promote their transformation into the anti-inflammatory M2 phenotype to alleviate arthritis. Moreover, MSCs can reduce the proliferation and cytotoxicity of NK cells, prevent autoreactive antibody generation, inhibit the activation of CD4+ Th1 cells and promote the production of Tregs.159 In addition, MSCs can induce M1 macrophages to transform them into immunosuppressive cells by producing prostaglandin E2 (PGE2), thus alleviating arthritis and promoting cartilage regeneration.160 MSC-derived TSC-6 interacts with CD44 on macrophages to reduce TLR2/NF-κB signal transduction, thereby alleviating the secretion of inflammatory mediators.161,162 MSC-derived PGE2 is a cytokine that regulates effector CD4+ T cells. PGE2 inhibits the production of IFN-γ, thereby reducing inflammation driven by Th1 cells.163 Spaggiari found that MSCs could prevent the maturation of dendritic cells (DCs).164 MSC-derived interleukin-1 receptor antagonist (IL-1Ra) has anti-inflammatory effects on cartilage regeneration. When IL-1Ra binds to the corresponding IL-1R, the interaction of IL-1 and IL-1R is blocked. Therefore, chondrocyte apoptosis and the synthesis and chemokines were prevented.165 In addition to the paracrine mechanism, MSCs can regulate the function of immune cells in a contact-associated manner. For example, MSCs can inhibit the proliferation of activated T cells through cell-to-cell contact.166 At present, an increasing number of studies have shown that adipose-derived stem cells (ADSCs) can be used to treat articular cartilage injury because these cells are easy to harvest and show high cartilage production capacity. In addition, the exocrine factors derived from ADSCs have cartilage protective and anti-inflammatory properties.167,168 Exosomes encapsulate various molecular components, mediate communication between different cells and regulate a variety of biological processes, including the immune response and cell differentiation.169,170 Therefore, an extracellular vesicle (EV)-mediated delivery system can effectively and efficiently treat OA because it can reactivate ageing MSCs and can be implemented using EVs from super donors. Therefore, the exocrine system may achieve cell-free therapy and provide the same clinical benefits without the potential risks associated with live cell infusions, such as immune rejection.171 However, low production of exosomes is a great challenge in clinical practice.172 Pang et al fabricated high-yield exosome-mimicking MSC-derived nanovesicles (MSC-NVs) with enhanced regenerative and anti-inflammatory capabilities.173 The MSC-NVs were prepared using an extrusion approach and could increase chondrocyte and human bone marrow MSC differentiation, proliferation, and migration, in addition to inducing M2 macrophage polarization.173
Since the role of one cytokine in OA does not necessarily depend on the activation of another cytokine, inhibiting one cytokine may not be sufficient to prevent inflammation and the production of matrix-degrading enzymes. Therefore, the application of MSCs and other biotherapy methods that have a variety of anti-inflammatory effects and achieve significant clinical effects in many studies are worthy of future research attention to verify the effects of some anti-inflammatory factors.
Conclusion
The role of the immune system and immune cells in the pathogenesis of osteoarthritis cannot be ignored. However, little is known about the role of the immune system in osteoarthritis. Through the efforts of researchers around the world, the effects of immune cells on osteoarthritis tissue cells have gradually become clear. With the ageing of the world population, there are an increasing number of patients with osteoarthritis. Therefore, elucidating the role of immunity in the pathogenesis of osteoarthritis would be helpful to interfere with the occurrence of osteoarthritis in the context of immunology.
Author Contributions
All authors made significant contributions to the work reported, whether that was in the conception, study design, execution, acquisition of data, analysis and interpretation or in all these areas. The authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (82072432).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bannuru RR, Osani MC, Vaysbrot EE, et al. OARSI guidelines for the non-surgical management of knee, Hip, and polyarticular osteoarthritis. Osteoarthr Cartil. 2019;27(11):1578–1589. doi:10.1016/j.joca.2019.06.011
2. Martel-Pelletier J, Barr AJ, Cicuttini FM, et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072. doi:10.1038/nrdp.2016.72
3. Felson DT, Lawrence RC, Dieppe PA, et al. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med. 2000;133(8):635–646. doi:10.7326/0003-4819-133-8-200010170-00016
4. Kloppenburg M, Berenbaum F. Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthr Cartil. 2020;28(3):242–248. doi:10.1016/j.joca.2020.01.002
5. Kulkarni P, Martson A, Vidya R, Chitnavis S, Harsulkar A. Pathophysiological landscape of osteoarthritis. Adv Clin Chem. 2021;100:37–90. doi:10.1016/bs.acc.2020.04.002
6. Tsukasaki M, Takayanagi H. Osteoimmunology: evolving concepts in bone-immune interactions in health and disease. Nat Rev Immunol. 2019;19(10):626–642. doi:10.1038/s41577-019-0178-8
7. Locantore P, Del Gatto V, Gelli S, Paragliola RM, Pontecorvi A. The interplay between immune system and microbiota in osteoporosis. Mediators Inflamm. 2020;2020:3686749. doi:10.1155/2020/3686749
8. Deng Z, Zhang Q, Zhao Z, et al. Crosstalk between immune cells and bone cells or chondrocytes. Int Immunopharmacol. 2021;101(Pt A):108179. doi:10.1016/j.intimp.2021.108179
9. Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23(5):471–478. doi:10.1097/BOR.0b013e328349c2b1
10. Wong M, Carter DR. Articular cartilage functional histomorphology and mechanobiology: a research perspective. Bone. 2003;33(1):1–13. doi:10.1016/s8756-3282(03)00083-8
11. DiDomenico CD, Lintz M, Bonassar LJ. Molecular transport in articular cartilage — what have we learned from the past 50 years? Nat Rev Rheumatol. 2018;14(7):393–403. doi:10.1038/s41584-018-0033-5
12. Bhosale AM, Richardson JB. Articular cartilage: structure, injuries and review of management. Br Med Bull. 2008;87:77–95. doi:10.1093/bmb/ldn025
13. Williams R, Khan IM, Richardson K, et al. Identification and clonal characterisation of a progenitor cell sub-population in normal human articular cartilage. PLoS One. 2010;5(10):e13246. doi:10.1371/journal.pone.0013246
14. Riegger J, Palm HG, Brenner RE. The functional role of chondrogenic stem/progenitor cells: novel evidence for immunomodulatory properties and regenerative potential after cartilage injury. Eur Cell Mater. 2018;36:110–127. doi:10.22203/eCM.v036a09
15. Ji Q, Zheng Y, Zhang G, et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann Rheum Dis. 2019;78(1):100–110. doi:10.1136/annrheumdis-2017-212863
16. Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci. 2010;1192:230–237. doi:10.1111/j.1749-6632.2009.05240.x
17. Xia B, Di C, Zhang J, Hu S, Jin H, Tong P. Osteoarthritis pathogenesis: a review of molecular mechanisms. Calcif Tissue Int. 2014;95(6):495–505. doi:10.1007/s00223-014-9917-9
18. Zhang Q, Ji Q, Wang X, et al. SOX9 is a regulator of ADAMTSs-induced cartilage degeneration at the early stage of human osteoarthritis. Osteoarthr Cartil. 2015;23(12):2259–2268. doi:10.1016/j.joca.2015.06.014
19. Tortorella MD, Malfait AM. Will the real aggrecanase(s) step up: evaluating the criteria that define aggrecanase activity in osteoarthritis. Curr Pharm Biotechnol. 2008;9(1):16–23. doi:10.2174/138920108783497622
20. Matsuo M, Nishida K, Yoshida A, Murakami T, Inoue H. Expression of caspase-3 and −9 relevant to cartilage destruction and chondrocyte apoptosis in human osteoarthritic cartilage. Acta Med Okayama. 2001;55(6):333–340. doi:10.18926/amo/32000
21. Liu S, Deng Z, Chen K, et al. Cartilage tissue engineering: from proinflammatory and anti‑inflammatory cytokines to osteoarthritis treatments (Review). Mol Med Rep. 2022;25(3):1–5.
22. Thielen NGM, van der Kraan PM, van Caam APM. TGFβ/BMP signaling pathway in cartilage homeostasis. Cells. 2019;8(9):969.
23. Peffers MJ, Chabronova A, Balaskas P, et al. SnoRNA signatures in cartilage ageing and osteoarthritis. Sci Rep. 2020;10(1):10641. doi:10.1038/s41598-020-67446-z
24. Mobasheri A, Matta C, Zákány R, Musumeci G. Chondrosenescence: definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas. 2015;80(3):237–244. doi:10.1016/j.maturitas.2014.12.003
25. Hunter DJ, Gerstenfeld L, Bishop G, et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res Ther. 2009;11(1):R11. doi:10.1186/ar2601
26. van der Kraan PM. Differential role of transforming growth factor-beta in an osteoarthritic or a healthy joint. J Bone Metabol. 2018;25(2):65–72. doi:10.11005/jbm.2018.25.2.65
27. Corrado A, Neve A, Cantatore FP. Expression of vascular endothelial growth factor in normal, osteoarthritic and osteoporotic osteoblasts. Clin Exp Med. 2013;13(1):81–84. doi:10.1007/s10238-011-0170-5
28. Katsimbri P. The biology of normal bone remodelling. Eur J Cancer Care. 2017;26(6):e12740.
29. Oehler S, Neureiter D, Meyer-Scholten C, Aigner T. Subtyping of osteoarthritic synoviopathy. Clin Exp Rheumatol. 2002;20(5):633–640.
30. Thomson A, Hilkens CMU. Synovial macrophages in osteoarthritis: the key to understanding pathogenesis? Front Immunol. 2021;12:678757. doi:10.3389/fimmu.2021.678757
31. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19(1):18. doi:10.1186/s13075-017-1229-9
32. de Lange-Brokaar BJ, Kloppenburg M, Andersen SN, et al. Characterization of synovial mast cells in knee osteoarthritis: association with clinical parameters. Osteoarthr Cartil. 2016;24(4):664–671. doi:10.1016/j.joca.2015.11.011
33. Clockaerts S, Bastiaansen-Jenniskens YM, Runhaar J, et al. The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. Osteoarthr Cartil. 2010;18(7):876–882. doi:10.1016/j.joca.2010.03.014
34. Abella V, Scotece M, Conde J, et al. Adipokines, metabolic syndrome and rheumatic diseases. J Immunol Res. 2014;2014:343746. doi:10.1155/2014/343746
35. Presle N, Pottie P, Dumond H, et al. Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. Osteoarthr Cartil. 2006;14(7):690–695. doi:10.1016/j.joca.2006.01.009
36. Pers YM, Ruiz M, Noël D, Jorgensen C. Mesenchymal stem cells for the management of inflammation in osteoarthritis: state of the art and perspectives. Osteoarthr Cartil. 2015;23(11):2027–2035. doi:10.1016/j.joca.2015.07.004
37. Wakitani S, Imoto K, Yamamoto T, Saito M, Murata N, Yoneda M. Human autologous culture expanded bone marrow mesenchymal cell transplantation for repair of cartilage defects in osteoarthritic knees. Osteoarthr Cartil. 2002;10(3):199–206. doi:10.1053/joca.2001.0504
38. Kondo S, Nakagawa Y, Mizuno M, et al. Transplantation of aggregates of autologous synovial mesenchymal stem cells for treatment of cartilage defects in the femoral condyle and the femoral groove in microminipigs. Am J Sports Med. 2019;47(10):2338–2347. doi:10.1177/0363546519859855
39. Damia E, Chicharro D, Lopez S, et al. Adipose-derived mesenchymal stem cells: are they a good therapeutic strategy for osteoarthritis? Int J Mol Sci. 2018;19(7):1926.
40. Parkin J, Cohen B. An overview of the immune system. Lancet. 2001;357(9270):1777–1789. doi:10.1016/s0140-6736(00)04904-7
41. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202–213. doi:10.1016/j.immuni.2018.01.007
42. Gerritsen B, Pandit A. The memory of a killer T cell: models of CD8(+) T cell differentiation. Immunol Cell Biol. 2016;94(3):236–241. doi:10.1038/icb.2015.118
43. Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. 2019;16(7):634–643. doi:10.1038/s41423-019-0220-6
44. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481–1488. doi:10.1172/jci11176
45. Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. 2019;177(3):524–540. doi:10.1016/j.cell.2019.03.016
46. Toni R, Di Conza G, Barbaro F, et al. Microtopography of immune cells in osteoporosis and bone lesions by endocrine disruptors. Front Immunol. 2020;11:1737. doi:10.3389/fimmu.2020.01737
47. Metcalfe DD. Mast cells and mastocytosis. Blood. 2008;112(4):946–956. doi:10.1182/blood-2007-11-078097
48. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. 2014;14(7):478–494. doi:10.1038/nri3690
49. Ragipoglu D, Dudeck A, Haffner-Luntzer M, et al. The role of mast cells in bone metabolism and bone disorders. Front Immunol. 2020;11:163. doi:10.3389/fimmu.2020.00163
50. Al-Hakami A, Alqhatani SQ, Shaik S, et al. Cytokine physiognomies of MSCs from varied sources confirm the regenerative commitment post-coculture with activated neutrophils. J Cell Physiol. 2020;235(11):8691–8701. doi:10.1002/jcp.29713
51. Clark D, Brazina S, Yang F, et al. Age-related changes to macrophages are detrimental to fracture healing in mice. Aging Cell. 2020;19(3):e13112. doi:10.1111/acel.13112
52. Chen K, Jiao Y, Liu L, et al. Communications Between Bone Marrow Macrophages and Bone Cells in Bone Remodeling. Front Cell Dev Biol. 2020;8:598263. doi:10.3389/fcell.2020.598263
53. Afonso V, Champy R, Mitrovic D, Collin P, Lomri A. Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint Bone Spine. 2007;74(4):324–329. doi:10.1016/j.jbspin.2007.02.002
54. Mathy-Hartert M, Hogge L, Sanchez C, Deby-DuPont G, Crielaard JM, Henrotin Y. Interleukin-1beta and interleukin-6 disturb the antioxidant enzyme system in bovine chondrocytes: a possible explanation for oxidative stress generation. Osteoarthr Cartil. 2008;16(7):756–763. doi:10.1016/j.joca.2007.10.009
55. Pattappa G, Schewior R, Hofmeister I, et al. Physioxia has a beneficial effect on cartilage matrix production in interleukin-1 beta-inhibited mesenchymal stem cell chondrogenesis. Cells. 2019;8(8):936.
56. Wang X, Li F, Fan C, Wang C, Ruan H. Effects and relationship of ERK1 and ERK2 in interleukin-1β-induced alterations in MMP3, MMP13, type II collagen and aggrecan expression in human chondrocytes. Int J Mol Med. 2011;27(4):583–589. doi:10.3892/ijmm.2011.611
57. Jenei-Lanzl Z, Meurer A, Zaucke F. Interleukin-1β signaling in osteoarthritis - chondrocytes in focus. Cell Signal. 2019;53:212–223. doi:10.1016/j.cellsig.2018.10.005
58. Hwang HS, Kim HA. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int J Mol Sci. 2015;16(11):26035–26054. doi:10.3390/ijms161125943
59. Choi MC, Jo J, Park J, Kang HK, Park Y. NF-κB signaling pathways in osteoarthritic cartilage destruction. Cells. 2019;8(7):734.
60. Choi MC, MaruYama T, Chun CH, Park Y. Alleviation of murine osteoarthritis by cartilage-specific deletion of IκBζ. Arthrit Rheumatol. 2018;70(9):1440–1449. doi:10.1002/art.40514
61. Singh P, Marcu KB, Goldring MB, Otero M. Phenotypic instability of chondrocytes in osteoarthritis: on a path to hypertrophy. Ann N Y Acad Sci. 2019;1442(1):17–34. doi:10.1111/nyas.13930
62. Saito T, Tanaka S. Molecular mechanisms underlying osteoarthritis development: notch and NF-κB. Arthritis Res Ther. 2017;19(1):94. doi:10.1186/s13075-017-1296-y
63. Chang SH, Mori D, Kobayashi H, et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-κB pathway. Nat Commun. 2019;10(1):1442. doi:10.1038/s41467-019-09491-5
64. Jimi E, Fei H, Nakatomi C. NF-κB signaling regulates physiological and pathological chondrogenesis. Int J Mol Sci. 2019;20(24):6275.
65. Westacott CI, Barakat AF, Wood L, et al. Tumor necrosis factor alpha can contribute to focal loss of cartilage in osteoarthritis. Osteoarthr Cartil. 2000;8(3):213–221. doi:10.1053/joca.1999.0292
66. Xue J, Wang J, Liu Q, Luo A. Tumor necrosis factor-α induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Mol Med Rep. 2013;8(6):1755–1760. doi:10.3892/mmr.2013.1729
67. Fujihara Y, Takato T, Hoshi K. Macrophage-inducing FasL on chondrocytes forms immune privilege in cartilage tissue engineering, enhancing in vivo regeneration. Stem Cells. 2014;32(5):1208–1219. doi:10.1002/stem.1636
68. Latourte A, Cherifi C, Maillet J, et al. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis. 2017;76(4):748–755. doi:10.1136/annrheumdis-2016-209757
69. Ryu JH, Yang S, Shin Y, Rhee J, Chun CH, Chun JS. Interleukin-6 plays an essential role in hypoxia-inducible factor 2α-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011;63(9):2732–2743. doi:10.1002/art.30451
70. de Hooge AS, van de Loo FA, Bennink MB, Arntz OJ, de Hooge P, van den Berg WB. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthr Cartil. 2005;13(1):66–73. doi:10.1016/j.joca.2004.09.011
71. Beekhuizen M, Gierman LM, van Spil WE, et al. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr Cartil. 2013;21(7):918–922. doi:10.1016/j.joca.2013.04.002
72. Li L, Li Z, Li Y, Hu X, Zhang Y, Fan P. Profiling of inflammatory mediators in the synovial fluid related to pain in knee osteoarthritis. BMC Musculoskelet Disord. 2020;21(1):99. doi:10.1186/s12891-020-3120-0
73. Goekoop RJ, Kloppenburg M, Kroon HM, et al. Low innate production of interleukin-1beta and interleukin-6 is associated with the absence of osteoarthritis in old age. Osteoarthr Cartil. 2010;18(7):942–947. doi:10.1016/j.joca.2010.03.016
74. Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. 2013;2(9):e60. doi:10.1038/emi.2013.58
75. Sinkeviciute D, Aspberg A, He Y, Bay-Jensen AC, Önnerfjord P. Characterization of the interleukin-17 effect on articular cartilage in a translational model: an explorative study. BMC Rheumatol. 2020;4:30. doi:10.1186/s41927-020-00122-x
76. Glyn-Jones S, Palmer AJ, Agricola R, et al. Osteoarthritis. Lancet. 2015;386(9991):376–387. doi:10.1016/s0140-6736(14)60802-3
77. Gong Y, Qiu J, Ye J, et al. AZ-628 delays osteoarthritis progression via inhibiting the TNF-α-induced chondrocyte necroptosis and regulating osteoclast formation. Int Immunopharmacol. 2022;111:109085. doi:10.1016/j.intimp.2022.109085
78. Fang C, Guo JW, Wang YJ, et al. Diterbutyl phthalate attenuates osteoarthritis in ACLT mice via suppressing ERK/c-fos/NFATc1 pathway, and subsequently inhibiting subchondral osteoclast fusion. Acta Pharmacol Sin. 2022;43(5):1299–1310. doi:10.1038/s41401-021-00747-9
79. Ponzetti M, Rucci N. Updates on osteoimmunology: what’s new on the cross-talk between bone and immune system. Front Endocrinol. 2019;10:236. doi:10.3389/fendo.2019.00236
80. Udagawa N, Koide M, Nakamura M, et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J Bone Miner Metab. 2021;39(1):19–26. doi:10.1007/s00774-020-01162-6
81. Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther. 2007;9(Suppl 1):S1. doi:10.1186/ar2165
82. Kovács B, Vajda E, Nagy EE. Regulatory effects and interactions of the Wnt and OPG-RANKL-RANK signaling at the bone-cartilage interface in osteoarthritis. Int J Mol Sci. 2019;20(18):4653.
83. Horowitz MC, Fretz JA, Lorenzo JA. How B cells influence bone biology in health and disease. Bone. 2010;47(3):472–479. doi:10.1016/j.bone.2010.06.011
84. Chen X, Wang Z, Duan N, Zhu G, Schwarz EM, Xie C. Osteoblast-osteoclast interactions. Connect Tissue Res. 2018;59(2):99–107. doi:10.1080/03008207.2017.1290085
85. Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110(11):1643–1650. doi:10.1172/jci15687
86. Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R. Interleukin-7 stimulates osteoclast formation by up-regulating the T-cell production of soluble osteoclastogenic cytokines. Blood. 2000;96(5):1873–1878.
87. Salopek D, Grcević D, Katavić V, Kovacić N, Lukić IK, Marusić A. Increased bone resorption and osteopenia are a part of the lymphoproliferative phenotype of mice with systemic over-expression of interleukin-7 gene driven by MHC class II promoter. Immunol Lett. 2008;121(2):134–139. doi:10.1016/j.imlet.2008.10.002
88. Weitzmann MN. The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica. 2013;2013:125705. doi:10.1155/2013/125705
89. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol. 2014;5:511. doi:10.3389/fimmu.2014.00511
90. Zhang Y, Böse T, Unger RE, Jansen JA, Kirkpatrick CJ, van den Beucken J. Macrophage type modulates osteogenic differentiation of adipose tissue MSCs. Cell Tissue Res. 2017;369(2):273–286. doi:10.1007/s00441-017-2598-8
91. Gong L, Zhao Y, Zhang Y, Ruan Z. The macrophage polarization regulates MSC osteoblast differentiation in vitro. Ann Clin Lab Sci. 2016;46(1):65–71.
92. Walsh MC, Choi Y. Regulation of T cell-associated tissues and T cell activation by RANKL-RANK-OPG. J Bone Miner Metab. 2021;39(1):54–63. doi:10.1007/s00774-020-01178-y
93. Ciucci T, Ibáñez L, Boucoiran A, et al. Bone marrow Th17 TNFα cells induce osteoclast differentiation, and link bone destruction to IBD. Gut. 2015;64(7):1072–1081. doi:10.1136/gutjnl-2014-306947
94. Adamopoulos IE, Chao CC, Geissler R, et al. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res Ther. 2010;12(1):R29. doi:10.1186/ar2936
95. Lee Y. The role of interleukin-17 in bone metabolism and inflammatory skeletal diseases. BMB Rep. 2013;46(10):479–483. doi:10.5483/bmbrep.2013.46.10.141
96. Charles JF, Aliprantis AO. Osteoclasts: more than ‘bone eaters’. Trends Mol Med. 2014;20(8):449–459. doi:10.1016/j.molmed.2014.06.001
97. Hu Y, Ek-Rylander B, Wendel M, Andersson G. Reciprocal effects of Interferon-γ and IL-4 on differentiation to osteoclast-like cells by RANKL or LPS. Oral Dis. 2014;20(7):682–692. doi:10.1111/odi.12189
98. Maruotti N, Corrado A, Cantatore FP. Osteoblast role in osteoarthritis pathogenesis. J Cell Physiol. 2017;232(11):2957–2963. doi:10.1002/jcp.25969
99. Lajeunesse D, Reboul P. Subchondral bone in osteoarthritis: a biologic link with articular cartilage leading to abnormal remodeling. Curr Opin Rheumatol. 2003;15(5):628–633. doi:10.1097/00002281-200309000-00018
100. Findlay DM, Atkins GJ. Osteoblast-chondrocyte interactions in osteoarthritis. Curr Osteoporos Rep. 2014;12(1):127–134. doi:10.1007/s11914-014-0192-5
101. Titorencu I, Pruna V, Jinga VV, Simionescu M. Osteoblast ontogeny and implications for bone pathology: an overview. Cell Tissue Res. 2014;355(1):23–33. doi:10.1007/s00441-013-1750-3
102. Won HY, Lee J-A, Park ZS, et al. Prominent bone loss mediated by RANKL and IL-17 produced by CD4+ T cells in TallyHo/JngJ mice. PLoS One. 2011;6(3):e18168. doi:10.1371/journal.pone.0018168
103. Croes M, Öner FC, van Neerven D, et al. Proinflammatory T cells and IL-17 stimulate osteoblast differentiation. Bone. 2016;84:262–270. doi:10.1016/j.bone.2016.01.010
104. Zhang W, Dang K, Huai Y, Qian A. Osteoimmunology: the regulatory roles of T lymphocytes in osteoporosis. Front Endocrinol. 2020;11:465. doi:10.3389/fendo.2020.00465
105. Du D, Zhou Z, Zhu L, et al. TNF-α suppresses osteogenic differentiation of MSCs by accelerating P2Y2 receptor in estrogen-deficiency induced osteoporosis. Bone. 2018;117:161–170. doi:10.1016/j.bone.2018.09.012
106. Nam D, Mau E, Wang Y, et al. T-lymphocytes enable osteoblast maturation via IL-17F during the early phase of fracture repair. PLoS One. 2012;7(6):e40044. doi:10.1371/journal.pone.0040044
107. Li C-J, Xiao Y, Yang M, et al. Long noncoding RNA Bmncr regulates mesenchymal stem cell fate during skeletal aging. J Clin Invest. 2018;128(12):5251–5266. doi:10.1172/jci99044
108. Singh P, Hu P, Hoggatt J, Moh A, Pelus LM. Expansion of bone marrow neutrophils following G-CSF administration in mice results in osteolineage cell apoptosis and mobilization of hematopoietic stem and progenitor cells. Leukemia. 2012;26(11):2375–2383. doi:10.1038/leu.2012.117
109. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–621. doi:10.1056/NEJMra052723
110. Binder NB, Niederreiter B, Hoffmann O, et al. Estrogen-dependent and C-C chemokine receptor-2–dependent pathways determine osteoclast behavior in osteoporosis. Nat Med. 2009;15(4):417–424. doi:10.1038/nm.1945
111. Walsh MC, Takegahara N, Kim H, Choi Y. Updating osteoimmunology: regulation of bone cells by innate and adaptive immunity. Nat Rev Rheumatol. 2018;14(3):146–156. doi:10.1038/nrrheum.2017.213
112. Kroner J, Kovtun A, Kemmler J, et al. Mast cells are critical regulators of bone fracture-induced inflammation and osteoclast formation and activity. J Bone Miner Res. 2017;32(12):2431–2444. doi:10.1002/jbmr.3234
113. Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Föger-Samwald U, Ellinger I. Immunology of Osteoporosis: a Mini-Review. Gerontology. 2016;62(2):128–137. doi:10.1159/000431091
114. Herath TDK, Larbi A, Teoh SH, Kirkpatrick CJ, Goh BT. Neutrophil-mediated enhancement of angiogenesis and osteogenesis in a novel triple cell co-culture model with endothelial cells and osteoblasts. J Tissue Eng Regen Med. 2018;12(2):e1221–e1236. doi:10.1002/term.2521
115. Georganas C, Liu H, Perlman H, Hoffmann A, Thimmapaya B, Pope RM. Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-kappa B but not C/EBP beta or c-Jun. J Immunol. 2000;165(12):7199–7206. doi:10.4049/jimmunol.165.12.7199
116. Yang CM, Luo SF, Hsieh HL, et al. Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: involvement of ERK, JNK, AP-1, and NF-kappaB. J Cell Physiol. 2010;224(2):516–526. doi:10.1002/jcp.22153
117. Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. 2000;1(6):510–514. doi:10.1038/82763
118. Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity. 2004;20(5):589–599. doi:10.1016/s1074-7613(04)00105-0
119. Störch H, Zimmermann B, Resch B, et al. Activated human B cells induce inflammatory fibroblasts with cartilage-destructive properties and become functionally suppressed in return. Ann Rheum Dis. 2016;75(5):924–932. doi:10.1136/annrheumdis-2014-206965
120. Donlin LT, Jayatilleke A, Giannopoulou EG, Kalliolias GD, Ivashkiv LB. Modulation of TNF-induced macrophage polarization by synovial fibroblasts. J Immunol. 2014;193(5):2373–2383. doi:10.4049/jimmunol.1400486
121. de Lange-Brokaar BJ, Ioan-Facsinay A, van Osch GJ, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthr Cartil. 2012;20(12):1484–1499. doi:10.1016/j.joca.2012.08.027
122. Li YS, Luo W, Zhu SA, Lei GH. T cells in osteoarthritis: alterations and beyond. Front Immunol. 2017;8:356. doi:10.3389/fimmu.2017.00356
123. Pessler F, Chen LX, Dai L, et al. A histomorphometric analysis of synovial biopsies from individuals with Gulf War Veterans’ Illness and joint pain compared to normal and osteoarthritis synovium. Clin Rheumatol. 2008;27(9):1127–1134. doi:10.1007/s10067-008-0878-0
124. Wu X, Tian J, Wang S. Insight into non-pathogenic Th17 cells in autoimmune diseases. Front Immunol. 2018;9:1112. doi:10.3389/fimmu.2018.01112
125. Mimpen JY, Baldwin MJ, Cribbs AP, et al. Interleukin-17A causes osteoarthritis-like transcriptional changes in human osteoarthritis-derived chondrocytes and synovial fibroblasts in vitro. Front Immunol. 2021;12:676173. doi:10.3389/fimmu.2021.676173
126. Mimpen JY, Carr AJ, Dakin SG, Snelling SJ. Inhibition of interleukin-17-induced effects in osteoarthritis-an in vitro study. Osteoarthr Cartil. 2018;26:S118. doi:10.1016/j.joca.2018.02.258
127. Brown MA, Hural J. Functions of IL-4 and control of its expression. Crit Rev Immunol. 2017;37(2–6):181–212. doi:10.1615/CritRevImmunol.v37.i2-6.30
128. Ishii H, Tanaka H, Katoh K, Nakamura H, Nagashima M, Yoshino S. Characterization of infiltrating T cells and Th1/Th2-type cytokines in the synovium of patients with osteoarthritis. Osteoarthr Cartil. 2002;10(4):277–281. doi:10.1053/joca.2001.0509
129. Doi H, Nishida K, Yorimitsu M, et al. Interleukin-4 downregulates the cyclic tensile stress-induced matrix metalloproteinases-13 and cathepsin B expression by rat normal chondrocytes. Acta Med Okayama. 2008;62(2):119–126. doi:10.18926/amo/30956
130. Sun AR, Friis T, Sekar S, Crawford R, Xiao Y, Prasadam I. Is synovial macrophage activation the inflammatory link between obesity and osteoarthritis? Curr Rheumatol Rep. 2016;18(9):57. doi:10.1007/s11926-016-0605-9
131. Mushenkova NV, Nikiforov NG, Shakhpazyan NK, Orekhova VA, Sadykhov NK, Orekhov AN. Phenotype diversity of macrophages in osteoarthritis: implications for development of macrophage modulating therapies. Int J Mol Sci. 2022;23(15):8381.
132. Kraus VB, McDaniel G, Huebner JL, et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthr Cartil. 2016;24(9):1613–1621. doi:10.1016/j.joca.2016.04.010
133. Wu CL, Harasymowicz NS, Klimak MA, Collins KH, Guilak F. The role of macrophages in osteoarthritis and cartilage repair. Osteoarthr Cartil. 2020;28(5):544–554. doi:10.1016/j.joca.2019.12.007
134. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614. doi:10.3389/fimmu.2014.00614
135. Dutta B, Arya RK, Goswami R, Alharbi MO, Sharma S, Rahaman SO. Role of macrophage TRPV4 in inflammation. Lab Invest. 2020;100(2):178–185. doi:10.1038/s41374-019-0334-6
136. Molnar V, Matišić V, Kodvanj I, et al. Cytokines and chemokines involved in osteoarthritis pathogenesis. Int J Mol Sci. 2021;22(17):9208.
137. Byles V, Covarrubias AJ, Ben-Sahra I, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4:2834. doi:10.1038/ncomms3834
138. Amos N, Lauder S, Evans A, Feldmann M, Bondeson J. Adenoviral gene transfer into osteoarthritis synovial cells using the endogenous inhibitor IkappaBalpha reveals that most, but not all, inflammatory and destructive mediators are NFkappaB dependent. Rheumatology. 2006;45(10):1201–1209. doi:10.1093/rheumatology/kel078
139. Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage “Switch” phenotype. Biomed Res Int. 2015;2015:341308. doi:10.1155/2015/341308
140. Bondeson J, Wainwright SD, Lauder S, Amos N, Hughes CE. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res Ther. 2006;8(6):R187. doi:10.1186/ar2099
141. Chen X, Liu Y, Wen Y, et al. A photothermal-triggered nitric oxide nanogenerator combined with siRNA for precise therapy of osteoarthritis by suppressing macrophage inflammation. Nanoscale. 2019;11(14):6693–6709. doi:10.1039/c8nr10013f
142. Ashraf S, Mapp PI, Shahtaheri SM, Walsh DA. Effects of carrageenan induced synovitis on joint damage and pain in a rat model of knee osteoarthritis. Osteoarthr Cartil. 2018;26(10):1369–1378. doi:10.1016/j.joca.2018.07.001
143. Siebelt M, van der Windt AE, Groen HC, et al. FK506 protects against articular cartilage collagenous extra-cellular matrix degradation. Osteoarthr Cartil. 2014;22(4):591–600. doi:10.1016/j.joca.2014.02.003
144. Bellucci F, Cucchi P, Catalani C, Giuliani S, Meini S, Maggi CA. Novel effects mediated by bradykinin and pharmacological characterization of bradykinin B2 receptor antagonism in human synovial fibroblasts. Br J Pharmacol. 2009;158(8):1996–2004. doi:10.1111/j.1476-5381.2009.00511.x
145. de Boer TN, van Spil WE, Huisman AM, et al. Serum adipokines in osteoarthritis; comparison with controls and relationship with local parameters of synovial inflammation and cartilage damage. Osteoarthr Cartil. 2012;20(8):846–853. doi:10.1016/j.joca.2012.05.002
146. Migliaccio S, Greco EA, Wannenes F, Donini LM, Lenzi A. Adipose, bone and muscle tissues as new endocrine organs: role of reciprocal regulation for osteoporosis and obesity development. Horm Mol Biol Clin Investig. 2014;17(1):39–51. doi:10.1515/hmbci-2013-0070
147. Fernández-Riejos P, Najib S, Santos-Alvarez J, et al. Role of leptin in the activation of immune cells. Mediators Inflamm. 2010;2010:568343. doi:10.1155/2010/568343
148. Dumond H, Presle N, Terlain B, et al. Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. 2003;48(11):3118–3129. doi:10.1002/art.11303
149. Bao JP, Chen WP, Feng J, Hu PF, Shi ZL, Wu LD. Leptin plays a catabolic role on articular cartilage. Mol Biol Rep. 2010;37(7):3265–3272. doi:10.1007/s11033-009-9911-x
150. Yaykasli KO, Hatipoglu OF, Yaykasli E, et al. Leptin induces ADAMTS-4, ADAMTS-5, and ADAMTS-9 genes expression by mitogen-activated protein kinases and NF-ĸB signaling pathways in human chondrocytes. Cell Biol Int. 2015;39(1):104–112. doi:10.1002/cbin.10336
151. Chen TH, Chen L, Hsieh MS, Chang CP, Chou DT, Tsai SH. Evidence for a protective role for adiponectin in osteoarthritis. Biochim Biophys Acta. 2006;1762(8):711–718. doi:10.1016/j.bbadis.2006.06.008
152. Gosset M, Berenbaum F, Salvat C, et al. Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum. 2008;58(5):1399–1409. doi:10.1002/art.23431
153. Berry PA, Jones SW, Cicuttini FM, Wluka AE, Maciewicz RA. Temporal relationship between serum adipokines, biomarkers of bone and cartilage turnover, and cartilage volume loss in a population with clinical knee osteoarthritis. Arthritis Rheum. 2011;63(3):700–707. doi:10.1002/art.30182
154. Xie H, Tang SY, Luo XH, et al. Insulin-like effects of visfatin on human osteoblasts. Calcif Tissue Int. 2007;80(3):201–210. doi:10.1007/s00223-006-0155-7
155. Okumachi E, Lee SY, Niikura T, et al. Comparative analysis of rat mesenchymal stem cells derived from slow and fast skeletal muscle in vitro. Int Orthop. 2015;39(3):569–576. doi:10.1007/s00264-014-2569-6
156. De bari C, Dell’Accio F, Luyten FP. Human periosteum-derived cells maintain phenotypic stability and chondrogenic potential throughout expansion regardless of donor age. Arthritis Rheum. 2001;44(1):85–95. doi:10.1002/1529-0131(200101)44:1<85::aid-anr12>3.0.co;2-6
157. De Bari C, Dell’Accio F, Vandenabeele F, Vermeesch JR, Raymackers JM, Luyten FP. Skeletal muscle repair by adult human mesenchymal stem cells from synovial membrane. J Cell Biol. 2003;160(6):909–918. doi:10.1083/jcb.200212064
158. Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol. 2018;14(8):493–507. doi:10.1038/s41581-018-0023-5
159. Harrell CR, Markovic BS, Fellabaum C, Arsenijevic A, Volarevic V. Mesenchymal stem cell-based therapy of osteoarthritis: current knowledge and future perspectives. Biomed Pharmacother. 2019;109:2318–2326. doi:10.1016/j.biopha.2018.11.099
160. Németh K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15(1):42–49. doi:10.1038/nm.1905
161. Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-κB signaling in resident macrophages. Blood. 2011;118(2):330–338. doi:10.1182/blood-2010-12-327353
162. Melief SM, Geutskens SB, Fibbe WE, Roelofs H. Multipotent stromal cells skew monocytes towards an anti-inflammatory interleukin-10-producing phenotype by production of interleukin-6. Haematologica. 2013;98(6):888–895. doi:10.3324/haematol.2012.078055
163. Duffy MM, Pindjakova J, Hanley SA, et al. Mesenchymal stem cell inhibition of T-helper 17 cell- differentiation is triggered by cell-cell contact and mediated by prostaglandin E2 via the EP4 receptor. Eur J Immunol. 2011;41(10):2840–2851. doi:10.1002/eji.201141499
164. Spaggiari GM, Abdelrazik H, Becchetti F, Moretta L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: central role of MSC-derived prostaglandin E2. Blood. 2009;113(26):6576–6583. doi:10.1182/blood-2009-02-203943
165. Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Induction of cartilage damage by overexpression of T cell interleukin-17A in experimental arthritis in mice deficient in interleukin-1. Arthritis Rheum. 2005;52(3):975–983. doi:10.1002/art.20885
166. Volarevic V, Al-Qahtani A, Arsenijevic N, Pajovic S, Lukic ML. Interleukin-1 receptor antagonist (IL-1Ra) and IL-1Ra producing mesenchymal stem cells as modulators of diabetogenesis. Autoimmunity. 2010;43(4):255–263. doi:10.3109/08916930903305641
167. Maumus M, Manferdini C, Toupet K, et al. Adipose mesenchymal stem cells protect chondrocytes from degeneration associated with osteoarthritis. Stem Cell Res. 2013;11(2):834–844. doi:10.1016/j.scr.2013.05.008
168. Manferdini C, Maumus M, Gabusi E, et al. Adipose-derived mesenchymal stem cells exert anti inflammatory effects on chondrocytes and synoviocytes from osteoarthritis patients through prostaglandin E2. Arthritis Rheum. 2013;65(5):1271–1281. doi:10.1002/art.37908
169. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. doi:10.1146/annurev-cellbio-101512-122326
170. Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16(21):2813–2828. doi:10.1101/gad.1017802
171. Zhou X, Hong Y, Zhang H, Li X. Mesenchymal stem cell senescence and rejuvenation: current status and challenges. Front Cell Dev Biol. 2020;8:364. doi:10.3389/fcell.2020.00364
172. Zhou D, Zhou F, Sheng S, Wei Y, Chen X, Su J. Intra-articular nanodrug delivery strategies for treating osteoarthritis. Drug Discov Today. 2023;28(3):103482. doi:10.1016/j.drudis.2022.103482
173. Pang L, Jin H, Lu Z, et al. Treatment with mesenchymal stem cell-derived nanovesicle-containing gelatin methacryloyl hydrogels alleviates osteoarthritis by modulating chondrogenesis and macrophage polarization. Adv Healthcare Mater. 2023:e2300315. doi:10.1002/adhm.202300315
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.