Back to Journals » Drug Design, Development and Therapy » Volume 15
Effects of Caffeic Acid and Its Derivatives on Bone: A Systematic Review
Authors Ekeuku SO ![]() , Pang KL
, Pang KL ![]() , Chin KY
, Chin KY ![]()
Received 17 October 2020
Accepted for publication 18 December 2020
Published 22 January 2021 Volume 2021:15 Pages 259—275
DOI https://doi.org/10.2147/DDDT.S287280
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Sophia Ogechi Ekeuku, Kok-Lun Pang, Kok-Yong Chin
Department of Pharmacology, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, Malaysia
Correspondence: Kok-Yong Chin
Department of Pharmacology, Faculty of Medicine, Universiti Kebangsaan Malaysia, Jalan Yaacob Latif, Bandar Tun Razak, 56000 Cheras, Kuala Lumpur, Malaysia
Tel +60 3-9145 9573
Email [email protected]
Purpose: Caffeic acid is a metabolite of hydroxycinnamate and phenylpropanoid, which are commonly synthesized by all plant species. It is present in various food sources that are known for their antioxidant properties. As an antioxidant, caffeic acid ameliorates reactive oxygen species, which have been reported to cause bone loss. Some studies have highlighted the effects of caffeic acid against bone resorption.
Methods: A systematic review of the literature was conducted to identify relevant studies on the effects of caffeic acid on bone. A comprehensive search was conducted from July to November 2020 using PubMed, Scopus, Cochrane Library and Web of Science databases. Cellular, animal and human studies reporting the effects of caffeic acid, as a single compound, on bone cells or bone were considered.
Results: The literature search found 226 articles on this topic, but only 24 articles met the inclusion criteria and were included in this review. The results showed that caffeic acid supplementation reduced osteoclastogenesis and bone resorption, possibly through its antioxidant potential and increased expression of osteoblast markers. However, some studies showed that caffeic acid did not affect bone resorption in ovariectomized rats and might impair bone mechanical properties in normal rats.
Conclusion: Caffeic acid potentially regulates the bone remodelling process by inhibiting osteoclastogenesis and bone resorption, as well as osteoblast apoptosis. Thus, it has medicinal values against bone diseases.
Keywords: antioxidant, bone, osteoclast, osteoblast, osteoporosis
Introduction
Bone remodelling is a tightly coupled lifelong process, whereby old bone is removed by osteoclasts (bone resorption) and new bone is formed by osteoblasts (bone formation).1,2 Osteocytes, which act as mechanosensors/endocrine cells, and bone lining cells3 are also involved in bone remodelling.4 Myriad pathophysiological factors affecting bone remodelling have been observed in skeletal diseases such as osteoporosis, arthritis and periodontal disease.5 Oxidative stress is one of the pathophysiological factors affecting bone remodelling. Oxidative stress stimulates osteoclast differentiation, thereby enhancing bone resorption.6,7 Reactive oxygen species (ROS) stimulate the apoptosis of osteoblasts and osteocytes, thus affecting bone formation. ROS also activate mitogen-activated protein kinases (MAPKs), such as extracellular signal-regulated kinases (ERK1/2), c-Jun-N terminal kinase (JNK) and p38, and enhance osteoclastogenesis and bone resorption.8–11 These phenomena skew the bone remodelling process in favour of bone loss.
Antioxidants are compounds which reduce free radicals and oxidative stress.12 Antioxidants have been reported to promote differentiation of osteoblasts, bone formation and survival of osteocytes, as well as suppressing osteoclast differentiation and activity.8,13–15 Some studies associate the age-related reduction in circulating antioxidants to osteoporosis in rats and women.16–18 A decline in antioxidant levels has been reported to promote bone loss by triggering the tumour necrosis factor-alpha (TNFα)-dependent signalling pathway,6 while administration of antioxidants, such as vitamin C, E, N-acetylcysteine and lipoic acid, have been reported to exert favourable effects in animal models of osteoporosis19–21 and individuals with osteoporosis.22–25
Caffeic acid (CA) is a metabolite of hydroxycinnamate and phenylpropanoid commonly synthesized by all plant species. It is a polyphenol present in many food sources like coffee, tea, wine, blueberries, apples, cider, honey and propolis.26 CA and its major derivatives including caffeic acid phenethyl ester (CAPE) and caffeic acid 3,4-dihydroxy-phenethyl ester (CADPE) are reported to possess potential antibacterial, antidiabetic, antioxidant, anti-inflammatory, antineoplastic and cardioprotective activities (reviewed in27–29). As a potent antioxidant, CA has been demonstrated to decrease lipoperoxyl radicals (ROO•) by donating a hydrogen atom to its corresponding hydroperoxide, which terminates the lipid peroxidation chain reaction. It also inhibits human low-density lipoprotein (LDL) oxidation induced by cupric ions.30 Furthermore, it interacts with other compounds, such as α-tocopherol, chlorogenic and caftaric acids, to exert more potent antioxidant activity in a variety of different systems.31–33 Therefore, the antioxidant activities of CA might protect against the negative effects of oxidative stress on bone cells and the skeletal system. This systematic review aims to summarise the effects of CA and its derivatives on bone cells and bone in literature.
Materials and Methods
Literature Review
A systematic literature search was conducted from July until November 2020 using PubMed, Scopus, Cochrane Library and Web of Science databases to identify studies on the effects of caffeic acid on bone and bone cells including osteoblasts, osteoclasts and osteocytes. The search string used was (1) caffeic acid AND (2) (bone OR osteoporosis OR osteoblasts OR osteoclasts OR osteocytes).
Selection of Articles
Studies with the following characteristics were included: (1) original research article with the primary objective of determining the effects of caffeic acid on bone and bone cells; (2) studies using cellular or animal models, or humans; (3) studies administering caffeic acid as a single compound but not in a mixture or food. Articles were excluded if they (1) do not contain original data; (2) use food rich in caffeic acid or mixtures containing caffeic acid. The bibliography of relevant review articles was traced for potential articles missed during database search. The search results were organised using EndNoteTM software (Clarivate Analytics, Philadelphia, USA). Duplicates were identified using EndNoteTM and confirmed by manual checking.
Data Extraction
Two authors (S.O.E. and K.L.P.) searched the same databases using the search string mentioned and screened the search results. All the articles that did not match the selection criteria were excluded. Next, the articles which used caffeic acid in treating models other than bone-related diseases were removed. Finally, articles which used caffeic acid in combination with other compounds were also excluded. Any disagreement on the inclusion or exclusion of articles was resolved through discussion among the two authors. The corresponding author (K.Y.C.) had the final decision on articles included if a consensus could not be reached between authors responsible for screening. This systematic review was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines and checklist.34 Steps in the selection process, from identification, screening, eligibility to the inclusion of articles, are shown in Figure 1.
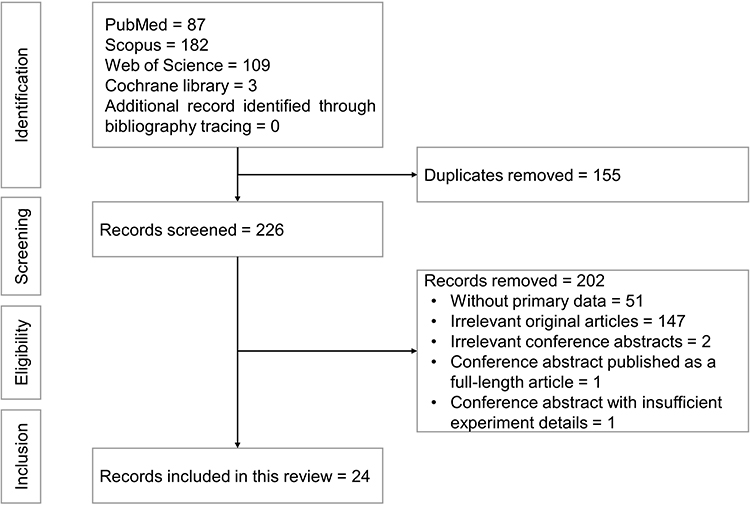 |
Figure 1 Flowchart of the article selection process. |
Results
Selection of Articles
From the literature search, 381 articles were identified, of which 87 were obtained from PubMed, 182 were from Scopus, 3 from Cochrane Library and 109 from Web of Science. A total of 155 duplicate articles were identified and removed. Of the 226 articles screened, 202 articles were excluded based on the selection criteria, whereby 51 articles did not contain primary data (3 book chapters, 2 commentary and 46 review articles), 147 articles and 2 conference abstracts presented topics irrelevant to the current review, a conference abstract had been published as a full-length research article and another conference abstract did not contain sufficient experiment details (Supplementary Material). Finally, 24 articles fulfilling all criteria mentioned were included in the review.
Study Characteristics
The included studies were published between 2006 and 2020. Seven studies were in vitro experiments using mouse bone marrow macrophages (BMMs), RAW264.7, RAW D and MG63 osteoblast cell lines35–41 while 19 studies were in vivo studies using Sprague Dawley/Sprague Dawley albino rats, Wistar/Wistar albino rats, Balb/c mice, lipopolysaccharide (LPS)-resistant C3H/HEJ mice, C57BL/6J mice and ICR mice.35,38,42–58 No human studies on this topic were reported.
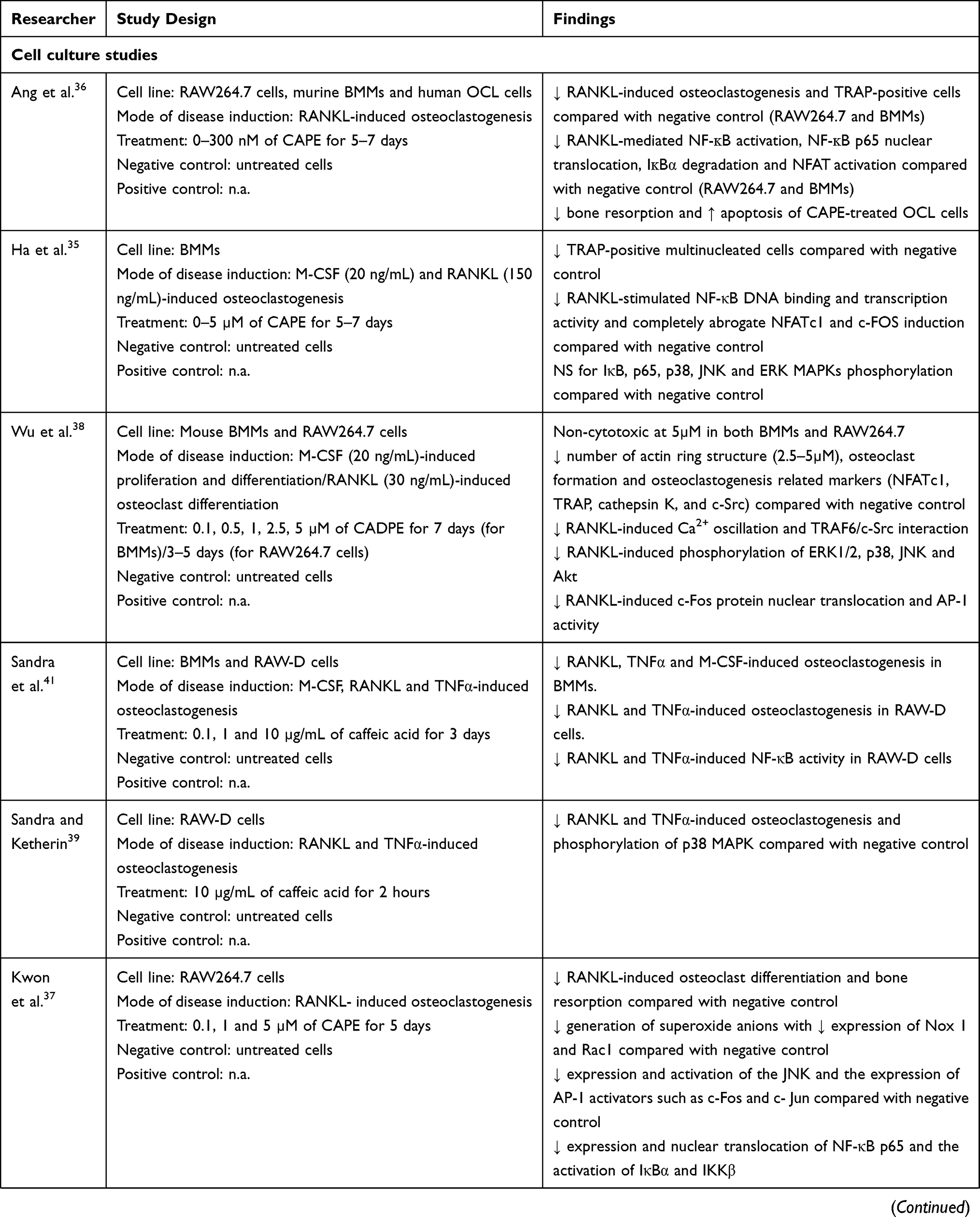
Six in vitro studies focused on the effects of CA on osteoclast differentiation from haematopoietic cells using macrophage colony-stimulating factor (M-CSF), receptor activator of NF-ĸB (RANK) ligand (RANKL) or TNF-α,35–39,41 while one in vitro study focused on the effect of CA on osteoblasts using MG63 osteoblast cell line.40 Four in vitro studies used CA doses between 0.1–5 µM.35,37,38,40 Ang et al.36 used doses between 0–0.3 µM and Sandra et al.41 and Sandra and Ketherin39 used a dose of 10 µg/mL (55.5 µM). The treatment period was 5–7 days for the differentiation of osteoclasts.
For animal studies, Duan et al.,55 Zawawi et al.,58 William et al.,51 Wu et al.,38 Zych et al.49 and Folwarczna et al.48,52 used CA or its derivatives at doses between 0.5–50 mg/kg via oral or intraperitoneal (i.p.) administration. Ucan et al.,57 Erdem et al.,53 Cicek et al.,54 Yigit et al.,45 Yildiz et al.50 and Tolba et al.56 used doses between 10–20 µmol/kg/day (2.84–5.69 mg/kg/day) via i.p. administration. Kizildağ et al.42−44 and Kazancioğlu et al.46,47 used the dose of 10 mmol/kg/day (2.843 g/kg/day) for an i.p. administration, Kazancioğlu et al.47 employed 50–100 mmol/kg/day (14.22–28.43 g/kg/day) for a localised administration, while Ha et al.35 used a collagen sponge soaked with CAPE with the final dose of 250 µg/mouse. For oral administration, first-pass effect might affect the enteric absorption of CA or its derivatives.59 For i.p. administration, the injection is commonly performed at the lower left or right quadrant of the abdomen. The peritoneum can absorb the compounds fast and reach systemic circulation with greater bioavailability with fewer handling errors.60
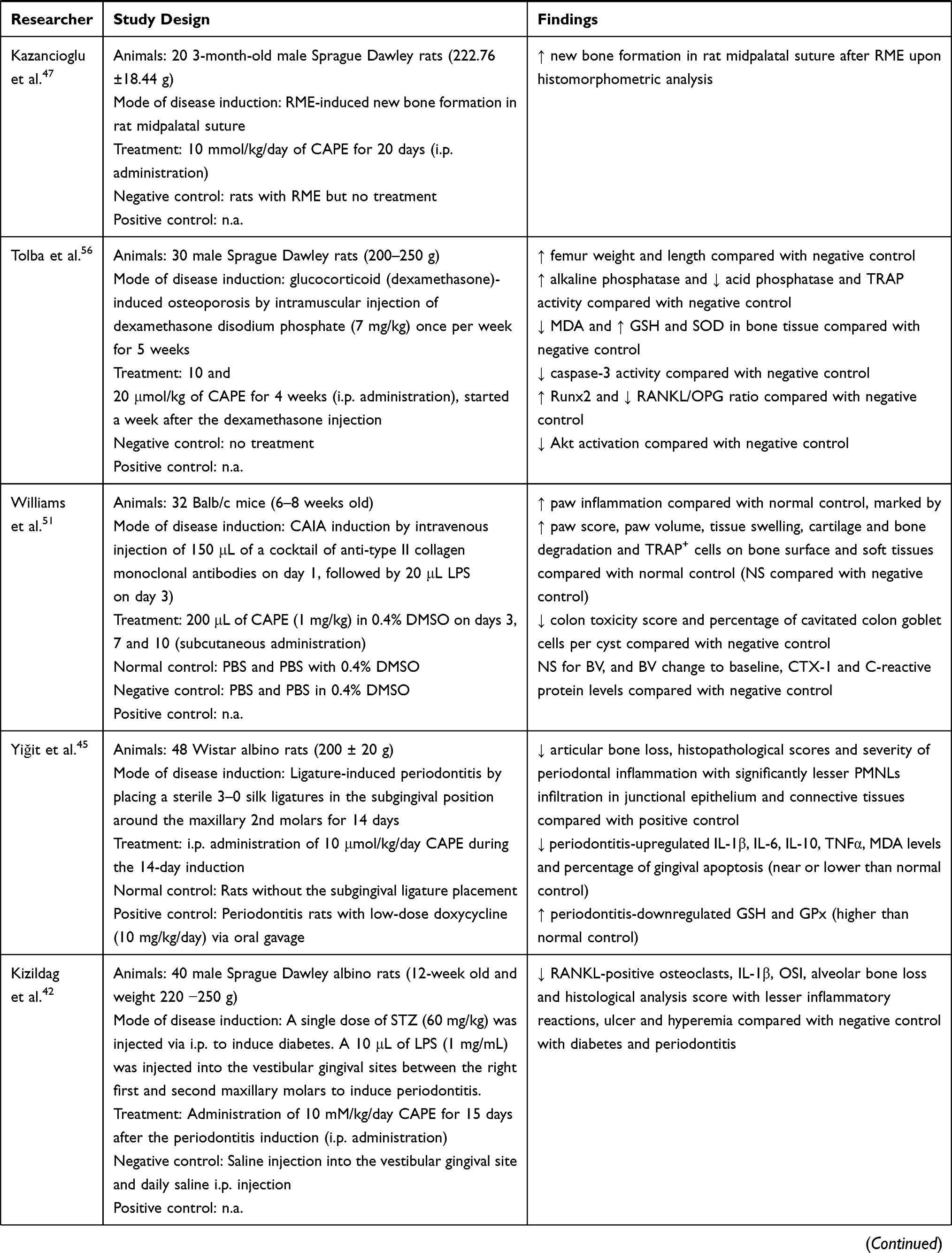
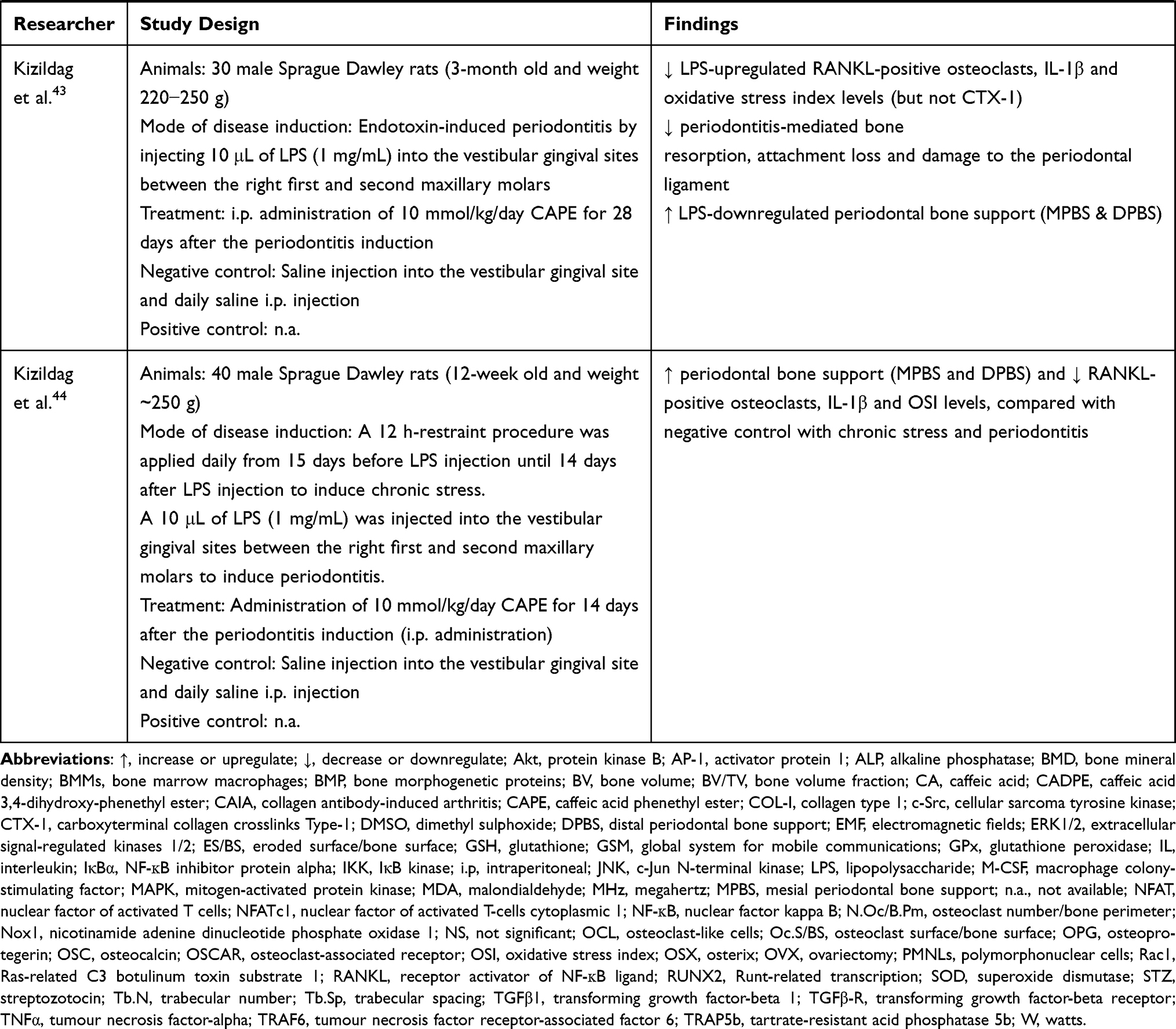
The bone-related disease models used included ovariectomy (OVX)- or glucocorticoids (dexamethasone)-induced osteoporosis, polyethylene particle-induced bone defect and osteolysis, electromagnetic force (EMF)-stimulated bone loss, osteotomy- or anti-collagen antibody-induced arthritis (CAIA) and rapid maxillary expansion (RME) and LPS-induced periodontitis. The endpoints studied included bone microstructure, histomorphometry, bone remodelling and oxidative status. The effects of CA and its derivatives on bone remodelling have been summarized in Table 1.
 |
 |
 |
 |
 |
 |
Table 1 Effects of CA and Its Derivatives in Bone Remodelling |
Evidence from Cell Culture Studies
Melguizo-Rodríguez et al. reported that 24-hour CA (1 µM) incubation increased the number of MG63 osteoblast cells compared with control.40 Gene expression studies revealed that CA increased the expression of osteoblast-related genes such as bone morphogenetic protein-2 and -7 (BMP-2 and BMP-7), transforming growth factor-beta 1 (TGF-β1), transforming growth factor-beta receptor 1, 2 and 3 (TGF-βR1, TGF-βR2 and TGF-βR3) and osteoblastogenesis genes including Runt-related transcription (RUNX-2), alkaline phosphatase (ALP), collagen type 1 (COL-I), osterix (OSX) and osteocalcin (OSC).40 Additionally, pretreatment of CA (10 µg/mL or 55.5 µM) on RAW D cells for 2 h also significantly inhibited the RANKL and TNFα-induced osteoclastogenesis with the suppression of p38 MAPK phosphorylation and tartrate-resistant acid phosphatase (TRAP)-positive osteoclast-like cells (OCLs) formation.39 Similarly, pretreatment of CA (0.1, 1 and 10 µg/mL or 0.555, 5.55 and 55.5 µM) on RAW D cells and BMMs for 3 days significantly inhibited the RANKL and TNFα-induced osteoclastogenesis and NF-κB activity in RAW-D cells and RANKL, TNFα and M-CSF-induced osteoclastogenesis in BMMs.41
On the other hand, CAPE treatment (0–0.3 µM; 5–7 days) suppressed the formation of TRAP-positive OCLs on RANKL-treated RAW264.7 cells and BMMs.36 Apoptosis occurred in CAPE-treated RAW264.7 cells with the disruption of the microtubule network in OCLs.36 Similarly, Kwon et al. reported that CAPE treatment (0.1–5 µM) for 5 days suppressed OCLs formation from RANKL-stimulated RAW264.7 cells.37 Another study by Ha et al. treating M-CSF and RANKL-stimulated BMMs with CAPE (0–5 µM for 5–7 days) also showed decreased OCLs formation in a concentration-dependent manner.35 The amount of TRAP-positive OCLs was decreased upon 0.1 and 0.5 μM CAPE treatment by 30% and 95% respectively.35 No OCL formation was observed upon 1 μM CAPE treatment.35 The anti-osteoclastogenic activities of CAPE are mainly contributed by its anti-inflammatory and antioxidant properties. Mechanistically, CAPE reduces superoxide anion generation by downregulating the nicotinamide adenine dinucleotide phosphate oxidase 1 (Nox1) expression through the interruption of nuclear factor-kappa B (NF-κB) and c-Jun N-terminal kinase (JNK) signalling pathways.37 CAPE suppresses RANKL-mediated activation of the NF-κB pathway by downregulating NF-κB p65 subunit expression and its nuclear translocation,37 suppressing nuclear factor of activated T cells (NFAT) activities36 and degradation of NF-κB inhibitor (IκBα),36,37 as well as inducing the degradation of IĸB kinase (IKK).37 CAPE also suppresses the expression and activation of JNK and its downstream transcription factors, such as c-Fos and c-Jun, which subsequently interrupt the protein activator-1 (AP-1) complex formation.37 Additionally, CAPE suppressed RANKL-induced activation of the Nox1 by inhibiting the Nox p47PHOX subunit translocation to the cell membrane and downregulation of Ras-related C3 botulinum toxin substrate 1 (Rac1) expression.37
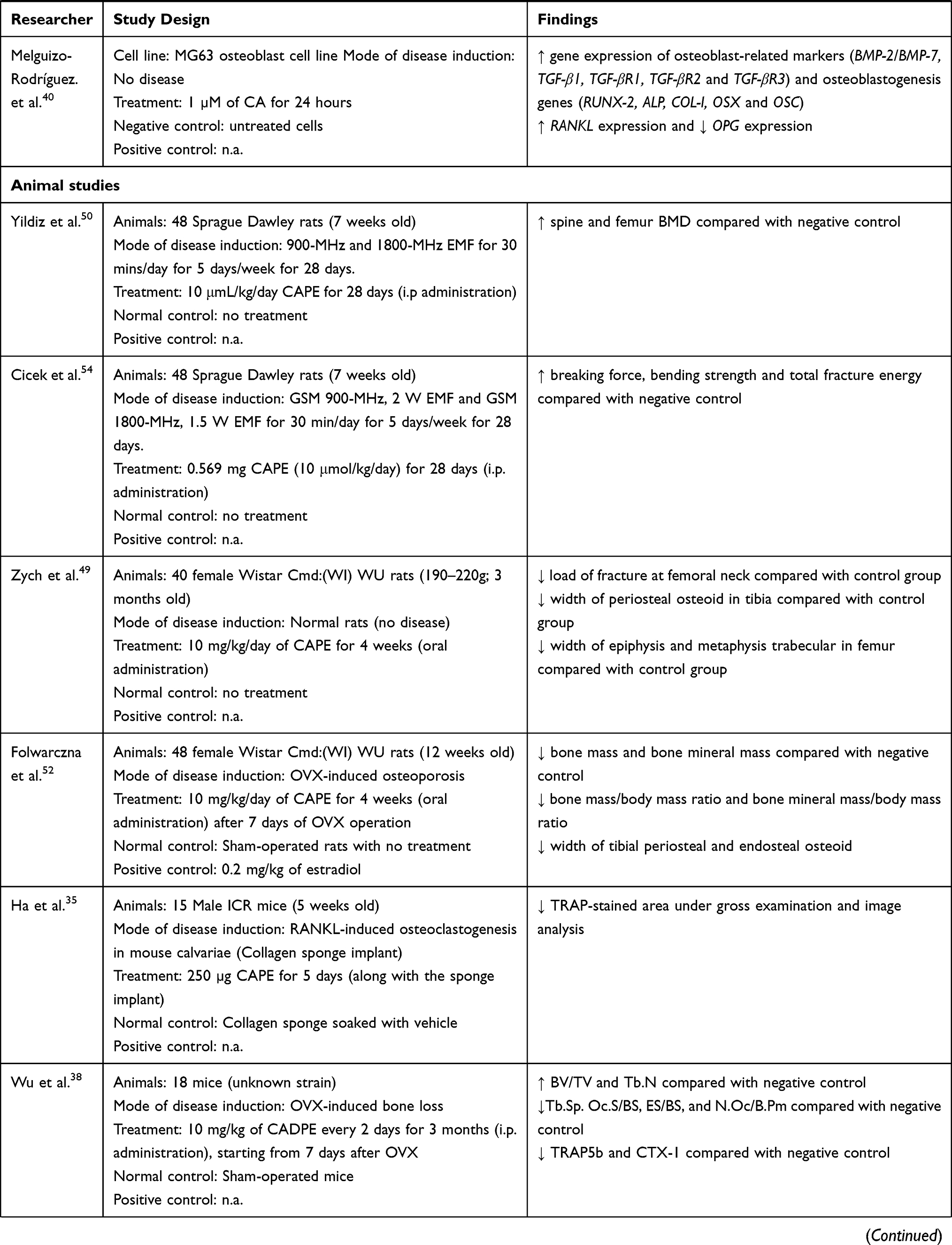
On the other hand, Wu et al. reported that CADPE (0.1–5 µM for 7 days) also concentration-dependently reduced OCL formation in the M-CSF and RANKL-stimulated BMMs and RAW264.7 cells.38 Mechanistic and characterisation examination revealed that CADPE suppressed RANKL-induced tumour necrosis factor receptor-associated factor 6 (TRAF6) activation and protein kinase B (PKB or also known as Akt) and activation of major MAPKs including ERK, JNK and p38.38 Subsequently, CADPE suppressed downstream expression of nuclear factor of activated T-cells cytoplasmic 1 (NFATc1), nuclear translocation of c-Fos protein and expression of osteoclastic markers, such as TRAP and cathepsin K, possibly through the non-receptor tyrosine kinase c-Src signalling.38 Interestingly, CADPE did not significantly affect the NF-kB signalling pathway and M-CSF-induced proliferation and differentiation of BMMs.
Evidence from Animal Studies
Supplementation of CA in animal models of bone loss yielded heterogeneous findings.48,49,52 This observation might be attributable to oral administration. Folwarczna et al. reported that CA (5 and 50 mg/kg, by stomach tube for 4 weeks) improved the bone mechanical properties by increasing the width of the trabecular metaphysis of the femur and decreasing the transverse growth in endosteal of the femur in OVX rats.48 Folwarczna et al. then demonstrated that CA (10 mg/kg/day; oral administration for 4 weeks) could reduce the width of tibial periosteal and endosteal osteoid compared with untreated OVX rats.52 However, CA did not promote or reduce the resorption of compact bone in the tibia of OVX-induced osteoporotic rats as evidenced by negligible changes of bone mass, bone mineral mass, bone mass/body mass ratio and bone mineral mass/body mass ratio.52 On the other hand, Zych et al. reported that CA at a similar dose (10 mg/kg/day; by stomach tube for 4 weeks) worsened the bone mechanical properties of healthy female Wistar Cmd:(WI)WU rats by decreasing the load of fracture at the femoral neck, decreasing the width of periosteal osteoid in the tibia and decreasing the width of the epiphysis and metaphysis trabecular in the femur compared with the negative control group.49
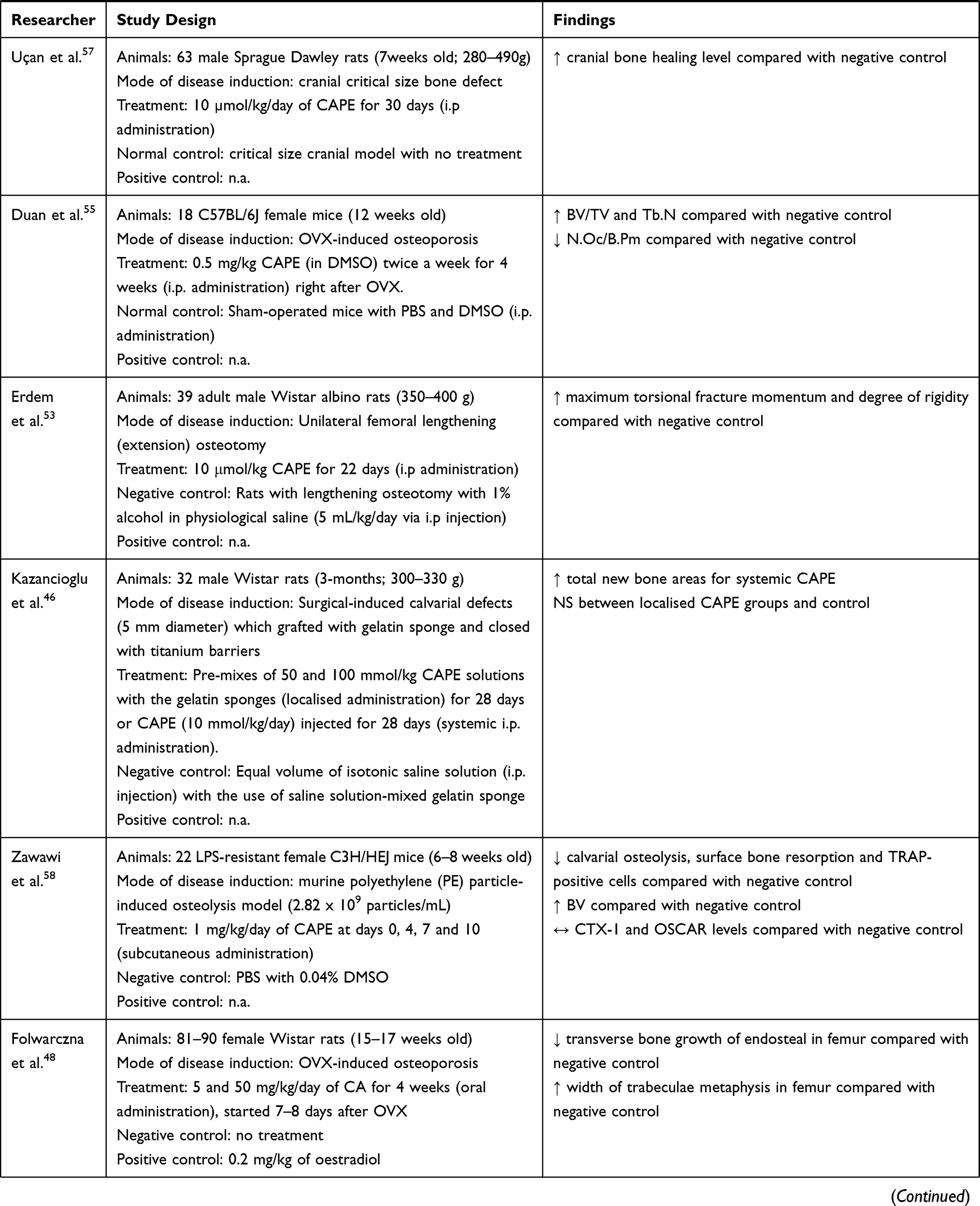
CAPE is the most extensively studied caffeic acid derivative in animal studies. The beneficial effects on new bone formation and healing upon systemic administration of CAPE had been reported.46,47,53,57 Erdem et al. reported that a low dose of CAPE (10 μmol/kg; i.p. injection for 22 days) increased new bone formation and bone strength by increasing maximum torsional fracture momentum and degree of rigidity compared with negative control in rats that underwent unilateral femoral lengthening (osteotomy).53 Similarly, a 30-day i.p. injection of CAPE (10 µmol/kg/day) also increased bone healing level in Sprague Dawley rats with cranial critical size bone defect.57 A higher dose of CAPE (10 mmol/kg/day, i.p. for 20 days) also further promoted the RME procedure-induced new bone formation in midpalatal suture of male Sprague Dawley rats.47 Similarly, a longer treatment period of CAPE (10 mmol/kg/day; i.p. injection for 28 days) also significantly promoted bone healing by increasing the total new bone areas in surgical-induced calvarial defects of male Wistar rats compared with the negative control.46 However, localised administration of CAPE (28 days) on surgical-induced calvarial defects by pre-mixing 50 and 100 mmol/kg CAPE solutions with gelatin sponges did not significantly improve the new bone formation.46
Localised and systemic administration of CAPE was reported to be beneficial in reducing osteolysis and bone loss.35,42–45,50,54–56,58 Ha et al. reported that collagen sponge implant impregnated with 250 µg CAPE and RANKL could reduce osteoclastogenesis with significantly lesser TRAP-stained area in mouse calvariae compared with implants with RANKL only.35 Subcutaneous injection of CAPE (1 mg/kg/day for 10 days) reduced the polyethylene particle-induced calvarial osteolysis, surface bone resorption and TRAP-positive cells formation with an increase of bone volume (BV) on LPS-resistant C3H/HEJ female mice.58 However, no significant changes were observed in carboxy-terminal cross-linked type 1 collagen (CTX-1) and osteoclast-associated receptor levels among untreated and CAPE-treated rats with calvarial osteolysis.58
Similarly, Duan et al. reported that lower dose and frequency of CAPE injection (0.5 mg/kg twice a week; i.p. injection for 4 weeks) also increased the BV and trabecular number (Tb.N) due to the decrease of bone osteoclast formation (evidenced by decreased osteoclast number/bone perimeter) in OVX mice.55 Tolba et al. also reported that i.p. injection of CAPE (10 and 20 μmol/kg) for 3 weeks increased femur weight and length in rats with dexamethasone-induced bone loss.56 The preservation of skeletal health in their study was associated with an improved antioxidant defence, such as higher levels of glutathione (GSH) and superoxide dismutase (SOD), and the reduction of malondialdehyde (MDA, lipid peroxidation product).56 This event led to an increase of osteoblastogenesis indicated by upregulation of RUNX-2 and ALP (osteoblast marker) levels56 On the other hand, decreased RANKL/osteoprotegerin (OPG) ratio was observed with CAPE treatment, indicating the suppression of osteoclastogenesis, which was further confirmed by lower acid phosphatase level and TRAP activity.56 In another study by Yildiz et al., CAPE (10 μmol/kg/day; i.p. injection for 22 days) also increased the spine and femur BMD in rats with EMF-induced bone loss.50 Similarly, Cicek et al. reported a longer treatment of CAPE (10 μmol/kg/day; i.p. injection for 28 days) also significantly improved the mechanical strength of cortical bone by increasing the breaking force, bending strength and total fracture energy in rats with EMF-induced bone loss compared with negative control.54
Additionally, a study by Wu et al. treated mice with an OVX-induced bone loss with a moderately high dose of CADPE (10 mg/kg; i.p. injection) every 2 days for 3 months.38 Results showed that CADPE could increase the BV fraction (BV/TV) and Tb.N, as well as decreased trabecular spacing (Tb.Sp) compared with the negative control.38 The improvement in the bone structure was contributed by reduced osteoclast number and eroded surface on the bone.38 Assessment of bone remodelling markers also revealed that serum TRAP5b and CTX-1 levels were reduced in CADPE-treated group compared with the negative control.38
On the other hand, CAPE was effective in reducing periodontitis-related bone loss and osteolysis.42–45 CAPE (10 μmol/kg/day, i.p. for 14 days) significantly reduced the subgingival ligature placement-induced periodontitis-mediated articular bone loss, histopathological features and severity of periodontal inflammation with lesser polymorphonuclear cells (PMNLs) infiltration in the junctional epithelium and connective tissues among Wistar albino rats.45 CAPE also suppressed the periodontitis-upregulated interleukin (IL)-1β, IL-6, IL-10, TNFα, MDA levels and the percentage of gingival apoptosis with the parallel restoration of periodontitis-downregulated GSH and glutathione peroxidase (GPx).45 Administration of high-dose CAPE (10 mmol/kg/day; i.p. for 15 days) in streptozotocin (STZ)-induced diabetic male Sprague Dawley rats reduced RANKL-positive osteoclast number, IL-1β levels, oxidative stress index (OSI), alveolar bone loss and histological analysis score in LPS-induced periodontitis. The treated rats also suffered lesser inflammatory reactions, ulcers and hyperemia.42 Similar changes of osteoclast number, IL-1β and OSI were observed in male Sprague Dawley rats with chronic stress and LPS-induced periodontitis treated with CAPE (10 mmol/kg/day, i.p. for 14 days).44 In addition, CAPE also increased the mesial and distal periodontal bone supports (MPBS and DPBS) in these rats.44 The effects of CAPE were sustained with a longer treatment period of CAPE (10 mmol/kg/day, i.p. for 28 days) on male Sprague Dawley rats with LPS-induced periodontitis.43
In contrast to the above findings, Williams et al. reported that subcutaneous injection of CAPE (1 mg/kg; at day 3, 7 and 10) did not reduce paw inflammation or bone loss in CAIA mice.51 Cartilage and bone degradation, as well as TRAP-positive cells on the bone surface and soft tissues, were still apparent in the supplemented CAIA group compared with the normal control.51
Discussion
This systematic review found that although CA and its derivatives is a potential anti-osteoporosis agent by suppressing the formation of osteoclasts and their bone resorption activity, it worsened bone mechanical properties in some cases. The anti-osteoclastogenesis action of CA and its derivatives was mediated by the antioxidant activities, which blocked RANKL-induced TRAF6/Akt and MAPK signalling, as well as M-CSF/c-Src signalling. In animals, CA and its derivatives (mainly CAPE) prevented bone resorption in rodent calvariae when implanted in situ, facilitated the healing of bone defects, preserved bone structure and improved mechanical strength in osteoporosis models induced by OVX, dexamethasone, osteotomy, LPS-mediated periodontitis and EMF. However, CA did not alter bone resorption in OVX-induced osteoporotic rats and worsened the mechanical properties in normal rats. Additionally, CAPE did not suppress bone loss in rats with CAIA-induced bone loss.
Osteoblasts are bone-forming cells derived from bone marrow mesenchymal stem cells and are responsible for the synthesis, secretion and mineralisation of bone matrix.61 The expression of osteoblast markers was increased following CA or CAPE supplementation, an indication that CA and CAPE stimulated osteoblast proliferation, differentiation and maturation.40,56 Osteoblasts and osteocytes regulate the formation of osteoclasts through RANKL/OPG axis. Osteoblasts and osteocytes synthesise RANKL, which binds to RANK to activate the canonical pathway for osteoclastogenesis. They also secrete OPG, which is a decoy receptor for RANKL to suppress osteoclastogenesis. The production of RANKL is stimulated under conditions such as oestrogen deficiency62 and oxidative stress.63 Osteoclastogenesis can also be stimulated via a non-canonical pathway, for instance, through the binding of TNFα with TNF receptor I or II.64 Glucocorticoids are potential modulators of RANKL/OPG axis, whereby dexamethasone is shown to downregulate OPG levels in osteoblasts.65 Tolba et al. showed that the RANKL/OPG level reduced in rats induced with dexamethasone with CAPE treatment.56 Other cellular studies showed that CA and its derivatives suppressed RANKL- and TNFα-induced formation of OCLs from haematopoietic cells,35–39 indicating that CA and its derivatives suppressed both canonical and non-canonical osteoclastogenesis.
The complex formed by the binding of RANKL to RANK causes the recruitment of the adaptor molecule’s tumour necrosis factor receptor-associated factors (TRAFs), including TRAF6.66 This event leads to the activation of several downstream signalling pathways, including c-Src/Akt/phosphatidylinositol 3-kinase and MAPKs (ERK/p38/JNK). CADPE was shown to suppress RANKL-induced activation of TRAF6 activation and the subsequent signalling pathways in multiple osteoclast progenitors, such as BMMs,38 RAW264.738 and RAW D cells.39 Sandra and Ketherin suggested that the downregulation of p38 is the key step of CA-mediated osteoclastogenesis.39 Upon activation, p38 initiates osteoclastogenesis by inducing NF-κB and NFATc1 expression.67,68 Inhibition of p38 MAPK reduces RANKL (canonical) and TNFα-induced (non-canonical) osteoclast formation.69
The NF-κB pathway is another signalling pathway downstream of TRAFs critical for osteoclast differentiation and bone reabsorption activity. Upon activation, IKK (consisting of IKKα, IKKβ and IKKγ) phosphorylates and degrades IκBα, which enables translocation of NF-κB p65/p50 heterodimers into the nucleus to allow transcription of osteoclast-related genes.70 Kwon et al. demonstrated that the anti-osteoclastogenesis effects of CAPE were mediated via the degradation of total IKKβ, thereby preventing the phosphorylation and degradation of IκBα and subsequently suppresses the nuclear translation of p65.37 On the other hand, Wu et al. reported that CADPE did not affect phosphorylation or degradation of IκBα, as well as nuclear translocation, and DNA-binding activity of p65.38 This observation suggests that compared with CAPE, CADPE does not influence the NF-κB signalling pathway.
ROS are one of the important secondary signals in the early stages of osteoclast differentiation.71,72 These ROS are mainly produced as superoxide anions by Nox1.73 Blocking of Nox1 ameliorates ROS production and the downstream MAPKs (JNK, p38 and ERK) and NF-κB activation74 and subsequently suppresses the osteoclast formation.71 The reduction of Nox 1 and Rac1 expression by CAPE is accompanied by RANKL-downstream signalling, denoting that anti-osteoclastogenesis effects of CAPE are dependent on suppression of Nox1-mediated superoxide anion production. Besides, dexamethasone has been reported to increase the expression of oxidative stress-related genes in human osteoblasts.75 Tolba et al. showed that CAPE increased GSH and SOD but reduced MDA in the bone of the rats exposed to dexamethasone, indicating an improvement of redox status in the skeletal environment.56 Additionally, CAPE also reduced the OSI and bone loss with an improvement of bone support in rats with LPS-induced periodontitis.
NFATc1 is the master regulator of osteoclast-related gene expression, and it is activated by c-Fos and NF-κB.76 Ha et al. observed that CAPE inhibited the recruitment of NF-κB to NFATc1 promoter, and the combined effect of NF-κB inhibition on c-Fos and NFATc1 may have caused CAPE to suppress osteoclastogenesis effectively.35 Holland et al. demonstrated a new fluorinated derivative of CAPE possesses potent anti-osteoclastogenic properties on RAW 264.7 cells by downregulating NFATc1 via suppression of c-Fos and NF-κB signalling pathways.77 Besides, this new fluorinated CAPE also exhibits improved stability with a 2-fold higher potency than CAPE.77 On the other hand, although CADPE did not alter NF-κB signalling, it still could suppress NFATc1 and other osteoclast-related markers, indicating other mechanisms of suppression could be involved, for instance, c-Src and MAPKs signalling pathways.38
Matrix metalloproteinases (MMPs), including gelatinases (MMP-2 and MMP-9) are examples of zinc-dependent extracellular matrix-degrading enzymes, which actively participate in bone resorption.78 MMPs are expressed as inactive proenzymes or zymogens that can be activated by several mediators including AP-1, NF-κB, TNFα and TGFβ.78 Currently, there is no study conducted to investigate the inhibitory effects of CA and CAPE on osteoclastic MMPs activity and its subsequent linkage in bone resorption; interestingly, CA and CAPE were reported to inhibit MMP-9 activity in human hepatocellular carcinoma HEP3B cells.79,80 This observation renders an interesting research gap in osteoclastic MMP inhibition upon CA and its derivatives treatment.
Suppression of osteoclastogenesis by CA or its derivatives have significant therapeutic potential against bone disorders induced by excessive bone resorption. Bone loss after osteotomy is a rapid process that affects both fractured and unfractured bone and may be incompletely reversible.81 CAPE was reported to improve bone formation and mechanical strength of bone in osteotomy.53 Exposure to EMF radiation caused by high-voltage transmission lines and transformers could affect bone health through decreased BMD, serum calcium and ALP level leading to the increase of bone resorption.82 CAPE increased the spine and femur BMD levels50 and increased mechanical strength of bones54 in rats exposed to EMF radiation. Total hip arthroplasty without cement often caused osteolysis induced by polyethylene particles.83 CAPE was shown by Zawawi et al. to prevent calvarial bone resorption in a murine polyethylene particle-induced osteolysis model.58 Therefore, biomaterials impregnated with CA or its derivatives could be adopted to prevent osteolysis in the arthroplasty procedure. CA has been incorporated in chitosan/(3-chloropropyl) trimethoxysilane scaffold for hard-tissue engineering applications and this adopted material exhibits antibacterial and anticancer effects.84 Ucan et al. observed that CAPE increased cranial bone healing in rats with critical size bone defect, suggesting that it could be administered systematically or locally to treat bone fracture/defect healing.57
Similarly, CAPE also effectively reduced the articular bone loss, inflammatory cytokines production and oxidative stress in rats with LPS-mediated periodontitis. Additionally, Wu et al.38 and Duan et al.55 demonstrated that CADPE prevented the ovariectomy-induced bone loss by suppressing osteoclast activity in a mouse model, while Folwarczna et al. showed increased width of trabecular metaphysis in the femur of OVX rats.48 Similarly, Tolba et al. showed improved bone formation and skeletal health in rats with dexamethasone-induced bone loss upon receiving CAPE.56 Additionally, CA and its derivatives may be involved in oestrogen production and signalling. Zych et al. reported that an oral administration of CA (10 mg/kg/day for 4 weeks) significantly restored the serum oestradiol levels in OVX rats.85 Interestingly, CA at 10 and 100 µM did not cause any alteration in calcium content in the femoral-diaphyseal and metaphyseal ex vivo culture, suggesting its bone-protecting effect may not involve calcium metabolism and regulation.86 Additionally, CAPE was reported as a selective human oestrogen receptor β agonist with the EC50 value of 3.72 µM in oestrogen-responsive element transcription.87 A recent in silico study by Zhao et al. suggested potential osteoimmunological effects of CAPE, which may explain its biological activities on both immune and skeletal systems.88 However, the findings from this modelling study requires further validation through in vitro and in vivo models. As oestrogen deficiency due to menopause and glucocorticoids present the most significant cause of primary and secondary osteoporosis globally, CA and its derivatives have the potential to be used as an adjuvant therapy to existing osteoporosis management strategies. The mechanisms of action of CA and its derivatives in osteoclastogenesis have been summarized in Figure 2.
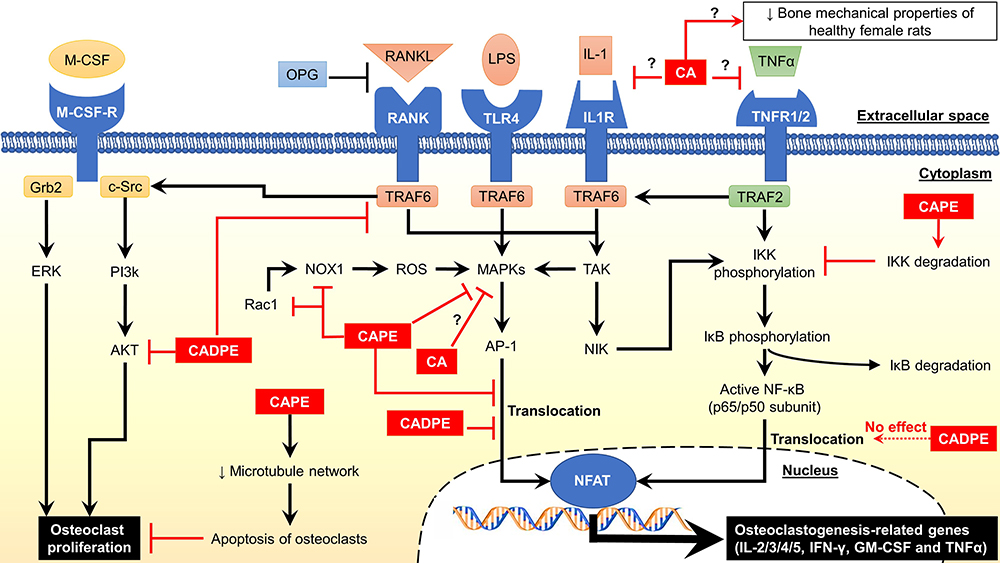 |
Figure 2 Mechanism of action of caffeic acid and its derivatives. Abbreviations: ↓, decrease or downregulate; ?, unknown mechanism; Akt, protein kinase B; AP-1, activator protein 1; CA, caffeic acid; CADPE; caffeic acid 3,4-dihydroxy-phenethyl ester; CAPE, caffeic acid phenethyl ester; c-Src, cellular sarcoma tyrosine kinase; ERK1/2, extracellular signal-regulated kinases 1/2; GM-CSF, granulocyte-macrophage colony-stimulating factor; Grb2, growth factor receptor-bound protein 2; IFN-γ, interferon-gamma; IL, interleukin; IL1R, interleukin-1 receptor; IκB, NF-ĸB inhibitor protein; IKK, IκB kinase; LPS, lipopolysaccharide; M-CSF, macrophage colony-stimulating factor; M-CSF-R, M-CSF receptor; MAPKs, mitogen-activated protein kinases; NFAT, nuclear factor of activated T cells; NF-ĸB, nuclear factor kappa B; NIK, MAPK kinase kinase 14; Nox1, nicotinamide adenine dinucleotide phosphate oxidase 1; OPG, osteoprotegerin; PI3k, phosphoinositide 3-kinase; Rac1, Ras-related C3 botulinum toxin substrate 1; RANK, receptor activator of NF-ĸB; RANKL, receptor activator of NF-ĸB ligand; ROS, reactive oxygen species; TAK, MAPK kinase kinase 7; TLR4, Toll-like receptor 4; TNFα, tumour necrosis factor-alpha; TNFR1/2, TNF receptor 1/2; TRAF2, tumour necrosis factor receptor-associated factor 2; TRAF6; tumour necrosis factor receptor-associated factor 6. |
Regardless of the positive effects of CA on bone status, some studies have reported negative effects associated with supplementation of CA and its derivatives. CA supplementation did not affect the bone resorption52 and reduced transverse growth of endosteal in femur48 of rats with OVX-induced osteoporosis. In normal rats, CA supplementation even negatively affected their bone mechanical properties.49 Moreover, CAPE supplementation has been reported to stimulate the synthesis of PGE2,89 which mediates osteoclastogenesis through RANKL stimulation and activation of the NF-κB pathway.90 This event will eventually increase TRAP-positive OCLs. Similarly, Williams et al. showed that CAPE did not suppress osteoclastogenesis in rats with CAIA.51
In term of safety, the International Agency for Cancer Research classifies CA as Class 2B (possibly carcinogenic to humans),91 and it was reported to induce renal tubular cell hyperplasia, forestomach hyperplasia, renal cell adenoma and forestomach cancer in rodents.92–94 CA has been reported to be non-mutagenic and non-clastogenic.91 Therefore, its carcinogenicity may involve epigenetic modification. Human toxicity and carcinogenicity of CA and its derivatives remain unknown. CA also showed anti-implantation activity in pregnant mice at a median effective dose of 4.26 mg/kg/day.95 Similarly, 5 mg/kg/day and 150 mg/kg of CA in mice demonstrated anti-implantation activity in early pregnancy.96 On the other hand, 0.15 mg/kg/day, 5 mg/kg/day and 150 mg/kg/day of CA for 21 days in mice showed no maternal toxicity, foetal teratogenesis or post-natal effects on pup development and mortality.96 The same experiment stated that the no-observed-adverse-effect level of CA for pregnant female mice was 0.15 mg/kg/day.96 Therefore, high-dose CA should be cautioned in humans, especially pregnant women.
Several common limitations can be identified from the studies reviewed. Most studies did not adopt a positive control to compare against the anti-osteoclastogenesis or anti-osteoporosis effect of CA. Therefore, the therapeutic effects of CA and currently available anti-resorptive therapy cannot be compared. Although osteoblastogenesis and bone formation are also important in bone remodelling, evidence of CA on these processes is limited in the literature. The actions of CA in humans cannot be confirmed due to the lack of human clinical trials. These aspects can be improved in future studies.
The current review also has several limitations. We only considered articles indexed by PubMed, Scopus, Cochrane Library and Web of Science; therefore, non-indexed articles could be overlooked. We only selected articles studying CA or its derivatives as a single compound to understand its mechanism of action properly without other interference, but not a mixture of compounds or natural products rich in CA. CA are present in foods, and interaction with other compounds in the food matrix might alter its absorption, bioavailability and action on the target tissue. Moreover, the heterogeneous findings of CA in bone loss reduction upon oral administration further emphasise these possibilities.
Conclusions
The current preclinical evidence agrees that CA and its derivatives exert promising skeletal protective effects by inhibiting osteoclastogenesis and bone resorption, but literature on bone formation is limited. Notwithstanding that, the skeletal effects of CA and its derivatives in models of normal bone health should be investigated because the limited studies available show undesirable effects. Human clinical trials to validate the skeletal effects of CA are lacking. Therefore, a well-planned clinical trial should be conducted to confirm the potential of CA as an antiresorptive agent. This information is critical for CA and its derivatives to be incorporated as part of the strategies to prevent bone loss.
Acknowledgments
The researchers are funded by Universiti Kebangsaan Malaysia through Research University Grant (GUP-2020-021). S.O.E. and K.L.P. are post-doctoral researchers funded by Universiti Kebangsaan Malaysia through FPR-1 and RGA-1 grants.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Katsimbri P. The biology of normal bone remodelling. Eur J Cancer Care (Engl). 2017;26(6):e12740. doi:10.1111/ecc.12740
2. Xiao W, Wang Y, Pacios S, et al. Cellular and molecular aspects of bone remodeling. In: Kantarci A, editor. Tooth Movement. Vol. 18. Basel: Karger; 2015:9–16.
3. Andersen TL, Sondergaard TE, Skorzynska KE, et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am J Pathol. 2009;174(1):239–247. doi:10.2353/ajpath.2009.080627
4. Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42(4):606–615. doi:10.1016/j.bone.2007.12.224
5. Jiao H, Xiao E, Graves DT. Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep. 2015;13(5):327–335. doi:10.1007/s11914-015-0286-8
6. Lean JM, Jagger CJ, Kirstein B, et al. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology. 2005;146(2):728–735. doi:10.1210/en.2004-1021
7. Huh YJ, Kim JM, Kim H, et al. Regulation of osteoclast differentiation by the redox-dependent modulation of nuclear import of transcription factors. Cell Death Differ. 2006;13(7):1138–1146. doi:10.1038/sj.cdd.4401793
8. Fontani F, Marcucci G, Iantomasi T, et al. Glutathione, N-acetylcysteine and lipoic acid down-regulate starvation-induced apoptosis, RANKL/OPG ratio and sclerostin in osteocytes: involvement of JNK and ERK1/2 signalling. Calcif Tissue Int. 2015;96(4):335–346. doi:10.1007/s00223-015-9961-0
9. Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–238. doi:10.1002/jbmr.320
10. Jilka RL, Noble B, Weinstein RS. Osteocyte apoptosis. Bone. 2013;54(2):264–271. doi:10.1016/j.bone.2012.11.038
11. Marathe N, Rangaswami H, Zhuang S, et al. Pro-survival effects of 17β-estradiol on osteocytes are mediated by nitric oxide/CGMP via differential actions of CGMP-dependent protein kinases I and II. J Biol Chem. 2012;287(2):978–988. doi:10.1074/jbc.M111.294959
12. Soares DG, Andreazza AC, Salvador M. Avaliação de compostos com atividade antioxidante em células da levedura saccharomyces cerevisiae. Rev Bras Cienc Farm. 2005;41:95–100. doi:10.1590/S1516-93322005000100011
13. Romagnoli C, Marcucci G, Favilli F, et al. Role of GSH/GSSG redox couple in osteogenic activity and osteoclastogenic markers of human osteoblast-like Saos-2 cells. FEBS J. 2013;280(3):867–879.
14. Banfi G, Iorio EL, Corsi MM. Oxidative stress, free radicals and bone remodeling. Clin Chem Lab Med. 2008;46(11):1550–1555. doi:10.1515/CCLM.2008.302
15. Jun JH, Lee S-H, Kwak HB, et al. N-acetylcysteine stimulates osteoblastic differentiation of mouse calvarial cells. J Cell Biochem. 2008;103(4):1246–1255. doi:10.1002/jcb.21508
16. Sendur OF, Turan Y, Tastaban E, et al. Antioxidant status in patients with osteoporosis: a controlled study. Joint Bone Spine. 2009;76(5):514–518. doi:10.1016/j.jbspin.2009.02.005
17. Almeida M, Han L, Martin-Millan M, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282(37):27285–27297. doi:10.1074/jbc.M702810200
18. Maggio D, Barabani M, Pierandrei M, et al. Marked decrease in plasma antioxidants in aged osteoporotic women: results of a cross-sectional study. J Clin Endocrinol Metab. 2003;88(4):1523–1527. doi:10.1210/jc.2002-021496
19. Lean JM, Davies JT, Fuller K, et al. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112(6):915–923. doi:10.1172/JCI200318859
20. Polat B, Halici Z, Cadirci E, et al. The effect of alpha-lipoic acid in ovariectomy and inflammation-mediated osteoporosis on the skeletal status of rat bone. Eur J Pharmacol. 2013;718(1–3):469–474. doi:10.1016/j.ejphar.2013.07.033
21. Mohamad S, Shuid AN, Mohamed N, et al. The effects of alpha-tocopherol supplementation on fracture healing in a postmenopausal osteoporotic rat model. Clinics (Sao Paulo). 2012;67:1077–1085. doi:10.6061/clinics/2012(09)16
22. Hall SL, Greendale GA. The relation of dietary vitamin C intake to bone mineral density: results from the pepi study. Calcif Tissue Int. 1998;63(3):183–189. doi:10.1007/s002239900512
23. Sanders KM, Kotowicz MA, Nicholson GC. Potential role of the antioxidant N-acetylcysteine in slowing bone resorption in early post-menopausal women: a pilot study. Transl Res. 2007;150:215. doi:10.1016/j.trsl.2007.03.012
24. Morton DJ, Barrett-Connor EL, Schneider DL. Vitamin c supplement use and bone mineral density in postmenopausal women. J Bone Miner Res. 2001;16(1):135–140. doi:10.1359/jbmr.2001.16.1.135
25. Mainini G, Rotondi M, Di Nola K, et al. Oral supplementation with antioxidant agents containing alpha lipoic acid: effects on postmenopausal bone mass. Clin Exp Obstet Gynecol. 2012;39(4):489–493.
26. Clifford MN. Chlorogenic acids and other cinnamates – nature, occurrence, dietary burden, absorption and metabolism. J Sci Food Agric. 2000;80(7):1033–1043. doi:10.1002/(SICI)1097-0010(20000515)80:7<1033::AID-JSFA595>3.0.CO;2-T
27. Espindola KMM, Ferreira RG, Narvaez LEM, et al. Chemical and pharmacological aspects of caffeic acid and its activity in hepatocarcinoma. Front Oncol. 2019;9:541. doi:10.3389/fonc.2019.00541
28. Magnani C, Isaac VLB, Correa MA, et al. Caffeic acid: a review of its potential use in medications and cosmetics. Anal Methods. 2014;6(10):3203–3210. doi:10.1039/C3AY41807C
29. Armutcu F, Akyol S, Ustunsoy S, et al. Therapeutic potential of caffeic acid phenethyl ester and its anti-inflammatory and immunomodulatory effects (review). Exp Ther Med. 2015;9(5):1582–1588. doi:10.3892/etm.2015.2346
30. Nardini M, D’Aquino M, Tomassi G, et al. Inhibition of human low-density lipoprotein oxidation by caffeic acid and other hydroxycinnamic acid derivatives. Free Radic Biol Med. 1995;19(5):541–552. doi:10.1016/0891-5849(95)00052-Y
31. Laranjinha J, Vierira O, Almeida L, et al. Inhibition of metmyoglobin/H2O2-dependent low density lipoprotein lipid peroxidation by naturally occurring phenolic acids. Biochem Pharmacol. 1996;51(4):395–402. doi:10.1016/0006-2952(95)02171-X
32. Meyer AS, Donovan JL, Pearson DA, et al. Fruit hydroxycinnamic acids inhibit human low-density lipoprotein oxidation in vitro. J Agric Food Chem. 1998;46(5):1783–1787.
33. Fukumoto LR, Mazza G. Assessing antioxidant and prooxidant activities of phenolic compounds. J Agric Food Chem. 2000;48(8):3597–3604. doi:10.1021/jf000220w
34. Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the prisma statement. PLoS Med. 2009;6(7):e1000097. doi:10.1371/journal.pmed.1000097
35. Ha J, Choi H-S, Lee Y, et al. Caffeic acid phenethyl ester inhibits osteoclastogenesis by suppressing NF kappaB and downregulating NFATc1 and c-Fos. Int Immunopharmacol. 2009;9(6):774–780. doi:10.1016/j.intimp.2009.03.001
36. Ang ES, Pavlos NJ, Chai LY, et al. Caffeic acid phenethyl ester, an active component of honeybee propolis attenuates osteoclastogenesis and bone resorption via the suppression of RANKL-induced NF-kappaB and NFAT activity. J Cell Physiol. 2009;221(3):642–649. doi:10.1002/jcp.21898
37. Kwon YB, Wang FF, Jang HD. Anti-osteoclastic effect of caffeic acid phenethyl ester in murine macrophages depends upon the suppression of superoxide anion production through the prevention of an active-nox1 complex formation. J Nutr Biochem. 2018;58:158–168. doi:10.1016/j.jnutbio.2018.03.023
38. Wu X, Li Z, Yang Z, et al. Caffeic acid 3,4-dihydroxy-phenethyl ester suppresses receptor activator of NF-κB ligand–induced osteoclastogenesis and prevents ovariectomy-induced bone loss through inhibition of mitogen-activated protein kinase/activator protein 1 and Ca2+–nuclear factor of activated T-cells cytoplasmic 1 signaling pathways. J Bone Miner Res. 2012;27(6):1298–1308. doi:10.1002/jbmr.1576
39. Sandra F, Ketherin K. Caffeic acid inhibits RANKL and TNF-α-induced phosphorylation of p38 mitogen-activated protein kinase in RAW-D cells. Indones Biomed J. 2018;10(2):140–143. doi:10.18585/inabj.v10i2.437
40. Melguizo-Rodríguez L, Manzano-Moreno FJ, Illescas-Montes R, et al. Bone protective effect of extra-virgin olive oil phenolic compounds by modulating osteoblast gene expression. Nutrients. 2019;11(8):1722. doi:10.3390/nu11081722
41. Sandra F, Kukita T, Tang QY, et al. Cafeic acid inhibits NFκB activation of osteoclastogenesis signaling pathway. Indones Biomed J. 2011;3(3):216–222. doi:10.18585/inabj.v3i3.153
42. Kızıldağ A, Arabacı T, Albayrak M, et al. A biochemical and immunohistochemical study of the effects of caffeic acid phenethyl ester on alveolar bone loss and oxidative stress in diabetic rats with experimental periodontitis. Biotech Histochem. 2020;95(6):456–463. doi:10.1080/10520295.2020.1718756
43. Kızıldağ A, Arabacı T, Albayrak M, et al. Therapeutic effects of caffeic acid phenethyl ester on alveolar bone loss in rats with endotoxin-induced periodontitis. J Dent Sci. 2019;14(4):339–345. doi:10.1016/j.jds.2019.03.011
44. Kızıldağ A, Arabacı T, Albayrak M, et al. Evaluation of caffeic acid phenethyl ester administration in chronically stressed rats with experimental periodontitis. Cumhur Dent J. 2019;22(1):114–120.
45. Yiğit U, Kırzıoğlu FY, Uğuz AC, et al. Is caffeic acid phenethyl ester more protective than doxycycline in experimental periodontitis? Arch Oral Biol. 2017;81:61–68. doi:10.1016/j.archoralbio.2017.04.017
46. Kazancioglu HO, Bereket MC, Ezirganli S, et al. Effects of caffeic acid phenethyl ester on wound healing in calvarial defects. Acta Odontol Scand. 2015;73(1):21–27. doi:10.3109/00016357.2014.942876
47. Kazancioglu HO, Aksakalli S, Ezirganli S, et al. Effect of caffeic acid phenethyl ester on bone formation in the expanded inter-premaxillary suture. Drug Des Devel Ther. 2015;9:6483–6488. doi:10.2147/DDDT.S97797
48. Folwarczna J, Pytlik M, Zych M, et al. Effects of caffeic and chlorogenic acids on the rat skeletal system. Eur Rev Med Pharmacol Sci. 2015;19(4):682–693.
49. Zych M, Folwarczna J, Pytlik M, et al. Administration of caffeic acid worsened bone mechanical properties in female rats. Planta Med. 2010;76(5):407–411. doi:10.1055/s-0029-1240603
50. Yildiz M, Cicek E, Cerci SS, et al. Influence of electromagnetic fields and protective effect of cape on bone mineral density in rats. Arch Med Res. 2006;37(7):818–821. doi:10.1016/j.arcmed.2006.03.006
51. Williams B, Tsangari E, Stansborough R, et al. Mixed effects of caffeic acid phenethyl ester (CAPE) on joint inflammation, bone loss and gastrointestinal inflammation in a murine model of collagen antibody-induced arthritis. Inflammopharmacology. 2017;25(1):55–68. doi:10.1007/s10787-016-0306-z
52. Folwarczna J, Zych M, Burczyk J, et al. Effects of natural phenolic acids on the skeletal system of ovariectomized rats. Planta Med. 2009;75(15):1567–1572. doi:10.1055/s-0029-1185904
53. Erdem M, Gulabi D, Sen C, et al. Effects of caffeic acid phenethyl ester and melatonin on distraction osteogenesis: an experimental study. SpringerPlus. 2014;3:8. doi:10.1186/2193-1801-3-8
54. Cicek E, Gokalp O, Varol R, et al. Influence of electromagnetic fields on bone fracture in rats: role of cape. Biomed Environ Sci. 2009;22(2):157–160. doi:10.1016/S0895-3988(09)60039-8
55. Duan W, Wang Q, Li F, et al. Anti-catabolic effect of caffeic acid phenethyl ester, an active component of honeybee propolis on bone loss in ovariectomized mice: a micro-computed tomography study and histological analysis. Chin Med J (Engl). 2014;127(22):3932–3936.
56. Tolba MF, El-Serafi AT, Omar HA. Caffeic acid phenethyl ester protects against glucocorticoid-induced osteoporosis in vivo: impact on oxidative stress and RANKL/OPG signals. Toxicol Appl Pharmacol. 2017;324:26–35. doi:10.1016/j.taap.2017.03.021
57. Uçan MC, Koparal M, Ağaçayak S, et al. Influence of caffeic acid phenethyl ester on bone healing in a rat model. J Int Med Res. 2013;41(5):1648–1654. doi:10.1177/0300060513490613
58. Zawawi MS, Perilli E, Stansborough RL, et al. Caffeic acid phenethyl ester abrogates bone resorption in a murine calvarial model of polyethylene particle-induced osteolysis. Calcif Tissue Int. 2015;96(6):565–574. doi:10.1007/s00223-015-9982-8
59. Renwick A. First-pass metabolism within the lumen of the gastrointestinal tract. Presystem Drug Eliminat. 2013;1:1.
60. Al Shoyaib A, Archie SR, Karamyan VT. Intraperitoneal route of drug administration: should it be used in experimental animal studies? Pharm Res. 2019;37(1):12. doi:10.1007/s11095-019-2745-x
61. Fakhry M, Hamade E, Badran B, et al. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J Stem Cells. 2013;5(4):136–148. doi:10.4252/wjsc.v5.i4.136
62. Beekman KM, Zwaagstra M, Veldhuis-Vlug AG, et al. Ovariectomy increases RANKL protein expression in bone marrow adipocytes of C3H/HEJ mice. Am J Physiol Endocrinol Metab. 2019;317(6):E1050–E1054. doi:10.1152/ajpendo.00142.2019
63. Mazière C, Salle V, Gomila C, et al. Oxidized low density lipoprotein enhanced RANKL expression in human osteoblast-like cells. Involvement of ERK, NFkappaB and NFAT. Biochim Biophys Acta Mol Basis Dis. 2013;1832(10):1756–1764. doi:10.1016/j.bbadis.2013.05.033
64. Feng W, Guo J, Li M. RANKL-independent modulation of osteoclastogenesis. J Oral Biosci. 2019;61(1):16–21. doi:10.1016/j.job.2019.01.001
65. Humphrey EL, Williams JHH, Davie MWJ, et al. Effects of dissociated glucocorticoids on OPG and RANKL in osteoblastic cells. Bone. 2006;38(5):652–661. doi:10.1016/j.bone.2005.10.004
66. Negishi-Koga T, Takayanagi H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231(1):241–256. doi:10.1111/j.1600-065X.2009.00821.x
67. Cong Q, Jia H, Li P, et al. P38α MAPK regulates proliferation and differentiation of osteoclast progenitors and bone remodeling in an aging-dependent manner. Sci Rep. 2017;7(1):45964. doi:10.1038/srep45964
68. Kang J-H, Lim H, Jeong J-E, et al. Attenuation of RANKL-induced osteoclast formation via p38-mediated NFATc1 signaling pathways by extract of euphorbia lathyris l. J Bone Metab. 2016;23(4):207–214. doi:10.11005/jbm.2016.23.4.207
69. Thouverey C, Caverzasio J. Focus on the p38 MAPK signaling pathway in bone development and maintenance. Bonekey Rep. 2015;4:711. doi:10.1038/bonekey.2015.80
70. Boyce BF, Xiu Y, Li J, et al. NF-κB-mediated regulation of osteoclastogenesis. Endocrinol Metab (Seoul). 2015;30(1):35–44. doi:10.3803/EnM.2015.30.1.35
71. Lee NK, Choi YG, Baik JY, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106(3):852–859. doi:10.1182/blood-2004-09-3662
72. Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279(6):L1005–1028. doi:10.1152/ajplung.2000.279.6.L1005
73. Bedard K, Krause K-H. The Nox family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313.
74. Sedeek M, Nasrallah R, Touyz RM, et al. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. Clin J Am Soc Nephrol. 2013;24(10):1512LP–1518. doi:10.1681/ASN.2012111112
75. Hurson CJ, Butler JS, Keating DT, et al. Gene expression analysis in human osteoblasts exposed to dexamethasone identifies altered developmental pathways as putative drivers of osteoporosis. BMC Musculoskelet Disord. 2007;8:12. doi:10.1186/1471-2474-8-12
76. Kim JH, Kim N. Regulation of NFATc1 in osteoclast differentiation. J Bone Metab. 2014;21(4):233–241. doi:10.11005/jbm.2014.21.4.233
77. Holland L, Mula RVR, Machiah D, et al. Fluorinated caffeic acid phenethyl ester: a novel anti-osteogenic molecule to attenuate excessive bone damage during autoimmune arthritis. FASEB J. 2016;30:lb451.
78. Liang H, Xu J, Xue M, et al. Matrix metalloproteinases in bone development and pathology: current knowledge and potential clinical utility. Metalloproteinases Med. 2016;3:93–102. doi:10.2147/MNM.S92187
79. Park WH, Kim SH, Kim CH. A new matrix metalloproteinase-9 inhibitor 3,4-dihydroxycinnamic acid (caffeic acid) from methanol extract of Euonymus alatus: isolation and structure determination. Toxicology. 2005;207(3):383–390. doi:10.1016/j.tox.2004.10.008
80. Jin UH, Chung TW, Kang SK, et al. Caffeic acid phenyl ester in propolis is a strong inhibitor of matrix metalloproteinase-9 and invasion inhibitor: isolation and identification. Clin Chim Acta. 2005;362(1–2):57–64. doi:10.1016/j.cccn.2005.05.009
81. Karlsson MK, Josefsson PO, Nordkvist A, et al. Bone loss following tibial osteotomy: a model for evaluating post-traumatic osteopenia. Osteoporos Int. 2000;11(3):261–264. doi:10.1007/s001980050290
82. Kunt H, Senturk I, Gonul Y, et al. Effects of electromagnetic radiation exposure on bone mineral density, thyroid, and oxidative stress index in electrical workers. Onco Targets Ther. 2016;9:745–754. doi:10.2147/OTT.S94374
83. Orishimo KF, Claus AM, Sychterz CJ, et al. Relationship between polyethylene wear and osteolysis in hips with a second-generation porous-coated cementless cup after seven years of follow-up. J Bone Joint Surg Am. 2003;85(6):1095–1099. doi:10.2106/00004623-200306000-00018
84. Shiu J, Ho M-H, Yu S-H, et al. Preparation and characterization of caffeic acid grafted chitosan/CPTMS hybrid scaffolds. Carbohydr Polym. 2010;79(3):724–730. doi:10.1016/j.carbpol.2009.09.025
85. Zych M, Folwarczna J, Trzeciak HI. Natural phenolic acids may increase serum estradiol level in ovariectomized rats. Acta Biochimica Polonica. 2009;56(3):503–507. doi:10.18388/abp.2009_2486
86. Lai YL, Yamaguchi M. Phytocomponent p-hydroxycinnamic acid stimulates bone formation and inhibits bone resorption in rat femoral tissues in vitro. Mol Cell Biochem. 2006;292(1–2):45–52. doi:10.1007/s11010-006-9175-x
87. Jung BI, Kim MS, Kim HA, et al. Caffeic acid phenethyl ester, a component of beehive propolis, is a novel selective estrogen receptor modulator. Phytother Res. 2010;24(2):295–300. doi:10.1002/ptr.2966
88. Zhao YH, Pang XK, Nepal A, et al. Caffeic acid phenethyl ester effects: in silico study of its osteoimmunological mechanisms. Lett Drug Des Discov. 2020;17(5):556–562. doi:10.2174/1570180815666180803111902
89. Larki-Harchegani A, Hemmati AA, Arzi A, et al. Evaluation of the effects of caffeic acid phenethyl ester on prostaglandin E2 and two key cytokines involved in bleomycin-induced pulmonary fibrosis. Iran J Basic Med Sci. 2013;16(7):850–857.
90. Saegusa M, Murakami M, Nakatani Y, et al. Contribution of membrane-associated prostaglandin E2 synthase to bone resorption. J Cell Physiol. 2003;197(3):348–356. doi:10.1002/jcp.10356
91. lnternational Agency for Research on Cancer. Caffeic acid. IARC monographs on the evaluation of carcinogenic risks to humans. Some naturally occurring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins. Vol 56. Lyon, France: lnternational Agency for Research on Cancer, World Health Organization; 1993:115–134.
92. Hagiwara A, Hirose M, Takahashi S, et al. Forestomach and kidney carcinogenicity of caffeic acid in F344 rats and C57BL/6N x C3H/HEN F1 mice. Cancer Res. 1991;51:5655–5660.
93. Hirose M, Fukushima S, Shirai T, et al. Stomach carcinogenicity of caffeic acid, sesamol and catechol in rats and mice. Jpn J Cancer Res. 1990;81:207–212. doi:10.1111/j.1349-7006.1990.tb02550.x
94. Hirose M, Takesada Y, Tanaka H, et al. Carcinogenicity of antioxidants BHA, caffeic acid, sesamol, 4-methoxyphenol and catechol at low doses, either alone or in combination, and modulation of their effects in a rat medium-term multi-organ carcinogenesis model. Carcinogenesis. 1997;19(1):207–212. doi:10.1093/carcin/19.1.207
95. Zheng YQ, Xu FB, Zhou S, et al. [abortifacient activity of caffeic acid and its antiprogestational action in early pregnant mice]. Acta Pharmacol Sin. 1987;8(3):250–254.
96. Liu Y, Qiu S, Wang L, et al. Reproductive and developmental toxicity study of caffeic acid in mice. Food Chem Toxicol. 2019;123:106–112. doi:10.1016/j.fct.2018.10.040
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.