Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Effects of budesonide on toll-like receptor expression in alveolar macrophages from smokers with and without COPD
Authors Ji J, von Schéele I, Billing B, Dahlén B, Lantz A, Larsson K ![]() , Palmberg L
, Palmberg L
Received 17 December 2015
Accepted for publication 16 February 2016
Published 17 May 2016 Volume 2016:11(1) Pages 1035—1043
DOI https://doi.org/10.2147/COPD.S102668
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Prof. Dr. Richard Russell
Jie Ji,1 Ida von Schéele,1 Bo Billing,2 Barbro Dahlén,2 Ann-Sofie Lantz,2 Kjell Larsson,1 Lena Palmberg1
1Lung and Airway Research, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden; 2Department of Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
Introduction: Alveolar macrophages (AMs) are equipped with innate immune receptors such as toll-like receptor 2 (TLR2) and toll-like receptor 4 (TLR4). In primary bronchial epithelial cells, exposure of toll-like receptor (TLR) ligands or tumor necrosis factor-alpha (TNF-α) increased TLR2 mRNA expression and reduced interleukin-8 (IL-8) release when coincubated with glucocorticosteroids. The aim of this study was to compare TLR2 and TLR4 expression levels and the effect of a glucocorticosteroid after stimulation with TLR ligands on AMs from smokers with and without COPD compared with the healthy controls.
Subjects and methods: Bronchoalveolar lavage was performed, and AMs were isolated from smokers with (n=10) and without COPD (n=11) and healthy controls (n=10) and stimulated ex vivo with peptidoglycan (PGN), lipopolysaccharide (LPS), or TNF-α ± budesonide (Bud). Blocking antibodies to TLR2 or TLR4 were added before stimulation with LPS or PGN ± Bud, respectively. The release of proinflammatory cytokine (TNF-α), chemoattractant (CXCL8), and TLR expression was analyzed by enzyme-linked immunosorbent assay and reverse transcription polymerase chain reaction.
Results: LPS, PGN, and TNF-α induced an increased release of IL-8 and TNF-α in the AMs in all the groups independent of smoking or disease. These responses were inhibited by a glucocorticosteroid (Bud) in all the three groups, except PGN-induced IL-8 secretion in smokers without COPD. Bud increased TLR2 expression in the healthy controls and smokers without COPD. Costimulation of TLR ligands and Bud significantly enhanced TLR2 mRNA expression in both groups of smokers compared with TLR ligands alone. In smokers, costimulation with PGN and Bud significantly increased TLR2 expression when compared with Bud alone. On stimulation with the TLR4 agonist, LPS downregulated TLR4 mRNA expression in all the three groups.
Conclusion: The combination of glucocorticosteroids with TLR ligands can increase TLR2 expression, thereby improving host defense in smokers. Also this combination can decrease the secretion of proinflammatory cytokines and chemokines as an anti-inflammatory response. Our findings indicate that glucocorticosteroid therapy strengthens immune defense pathways, which may have implication during exacerbation caused by microorganisms.
Keywords: smokers, lipopolysaccarid, peptidoglycan, glucocorticosteroid
Introduction
Bacterial colonization of the respiratory tract commonly occurs in people who are continuously exposed to inhaled foreign particles, such as cigarette smoke or dust.1,2 One of the key players of the innate immune defense in the human respiratory tract is the alveolar macrophage (AM).3 The AM orchestrates airway inflammation by secreting and responding to related cytokines and chemokines.4 The number of macrophages in bronchoalveolar lavage (BAL) is usually elevated in smokers with and without COPD compared with nonsmokers.5 The AMs are equipped with different kinds of toll-like receptors (TLRs)6 and, therefore, play a critical role in the continuous recognition of potentially pathogenic organisms entering the lungs.7
TLRs are a well-known family of pattern recognition receptors involved in the initiation of inflammation as well as recognition of pathogen-associated molecular patterns (PAMPs). A wide range of PAMPs, such as peptidoglycans (PGNs) from Gram-positive bacteria and lipopolysaccharides (LPSs) from endotoxin of Gram-negative bacteria, predominantly bind to toll-like receptor 2 (TLR2) and toll-like receptor 4 (TLR4), respectively. It has been previously shown that farmers have a decreased expression of TLR2 on blood monocytes, compared to that of the healthy controls.8 Patients with COPD have a lower expression of TLR2 on sputum neutrophils than the healthy nonsmokers,5,9 and AMs from smokers showed a decreased expression of TLR2.10 Therefore, it could be assumed that the low TLR expression might be a consequence of the continuous exposure to microorganisms due to bacterial colonization in the respiratory tract of both smokers and farmers.
Inhaled corticosteroids are frequently used for the treatment of asthma and, in combination with long-acting β2-agonists, for preventing acute exacerbations in COPD.11 Glucocorticosteroids bind to intracellular glucocorticoid receptors (GRs), which activate a number of anti-inflammatory genes and switch off other genes encoding proinflammatory cytokines or chemokines.12 Organic dust collected in a pig barn, LPS, and tumor necrosis factor-alpha (TNF-α) induced increased interleukin-6 and interleukin-8 (IL-8) protein release and mRNA expression in primary bronchial epithelial cells.13 These effects were reduced by budesonide (Bud) in vitro.13 In parallel, Bud combined with a TLR ligand or TNF-α synergistically enhanced TLR2 expression on bronchial epithelial cells.13 However, this finding has not been well explored in AMs from smokers with and without COPD.
The aim of this study was to compare TLR expression after costimulation with glucocorticosteroids and TLR ligands on AMs from smokers with and without COPD compared with those from the healthy controls, to find out if the AMs show the same pattern as primary bronchial epithelial cells upon stimulation of TLR ligands and steroids, and to increase knowledge on how this receptor regulation is impaired by the chronic exposure to different inhaled foreign particles, in this case cigarette smoke.
Subjects and methods
Isolation of AM
The AMs were isolated from ten nonallergic, nonasthmatic, nonsmoking healthy controls; eleven smokers without COPD (a postbronchodilator forced expiratory volume in 1 second [FEV1]/forced vital capacity >0.7 and FEV1 >70% of the predicted value); and ten smokers with COPD (postbronchodilator FEV1/forced vital capacity <0.7 and FEV1 40%–70% of the predicted value). The smoker groups included current smokers with a cumulative exposure of >10 pack-years. All subjects gave their written informed consent and the study was approved by the ethics committee at Karolinska Institutet, Stockholm, Sweden. Detailed characterizations of the subjects have previously been published and are summarized in Table 1.3 Bronchoscopy and BAL were performed as previously described.14 After filtering and centrifuging the lavage fluid, the cell pellet was resuspended. Trypan blue exclusion was used to determine total cell number and cell viability. Approximately 0.5 million cells/well were resuspended in 1 mL of Roswell Park Memorial Institute (RPMI) cell medium (RPMI 1640 supplemented with 1% l-glutamine, 1% penicillin-streptomycin, and 5% fetal bovine serum) and put into six-well plates. After 2 hours, the nonadherent cells and supernatants were discarded and the adhered cells (macrophages) were cultured in a 2 mL serum-free RPMI cell culture medium for 20 hours.
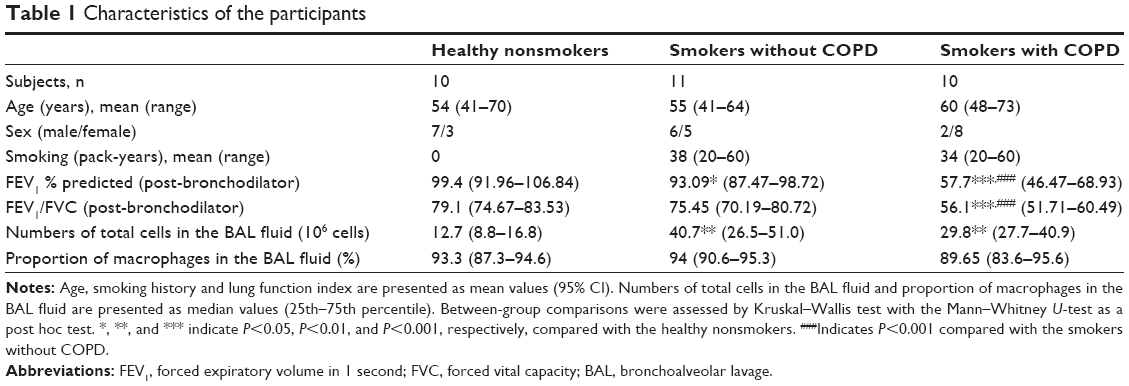 | Table 1 Characteristics of the participants |
Stimulation of AM
The cells were exposed to one of the following stimuli with and without Bud of 10−8 M (Sigma-Aldrich Co., St Louis, MO, USA): LPS (1 μg/mL, Escherichia coli 0111:B4; Sigma-Aldrich Co.), PGN (1 μg/mL; Sigma-Aldrich Co.), and TNF-α (10 ng/mL; R&D Systems, Inc., Minneapolis, MN, USA). The cells were incubated at 37°C in 5% CO2 for 6 hours; supernatants were collected and kept at -70°C until analysis. After stimulation, trypan blue exclusion was used to determine cell viability, and viability >90% was acceptable.
To block TLR2 and TLR4, 2 μL of monoclonal antibody for TLR2 or TLR4 (TLR2, TL2.1, TLR4, HTA 125; eBioscience, San Diego, CA, USA) was added to each well and incubated for 1 hour before exposure. IgG2a was used as an isotype control.
Preparation of mRNA and real-time polymerase chain reaction
Preparation of mRNA from AMs was performed in four healthy controls and five subjects from each of the two smoking groups. After washing the macrophages twice with phosphate-buffered saline, total mRNA was isolated by PureLinkTM Micro-to-Midi Total RNA Purification System (Thermo Fisher Scientific, Waltham, MA, USA). Amplification grade DNase I was used to remove the genomic DNA (Thermo Fisher Scientific). First-stranded cDNA was synthesized from 0.5 μg of total RNA using QuantiTect® Reverse Transcription Kit (Qiagen NV, Venlo, the Netherlands). The Power SYBR Green Master Mix (Thermo Fisher Scientific) was used to perform the reverse transcription polymerase chain reaction (PCR) with TLR2 and TLR4 primers. The primers were designed by software Primer3 and purchased from Thermo Fisher Scientific. For TLR2, the forward primer was 5′-GGCCAGCAAATTACCTGTGT-3′ and the reverse primer was 5′-TTCTCCACCCAGTAGGCATC-3′. For TLR4, the forward primer was 5′-CAACAAAGGTGGGAATGCTT-3′ and the reverse primer was 5′-TGCCATTGAAAGCAACTCTG-3′. Beta-actin was adopted as an internal control gene. For beta-actin, the forward primer was 5′-GAGCACAGAGCCTCGCCTTT-3′ and the reverse primer was 5′-AGAGGCGTACAGGGATAGCA-3′. A total of 50 ng of cDNA was used in each 25 μL PCR reaction volume to identify the products of interest. Data were analyzed using Applied Biosystems ® 7500 Real-Time PCR Systems with 7500 software v.2.0.1 (Thermo Fisher Scientific) and transformed using the ΔCt method. The results were then calculated and expressed semiquantitatively as 2−ΔCt.
Enzyme-linked immunosorbent assay
Detection of interleukin-8/CXCL8 in the supernatant was performed using an in-house enzyme-linked immunosorbent assay (ELISA) method.15 Commercially available antibody pair MAB 208 and BAF 208 (R&D Systems, Inc.) was used to detect IL-8. The detection range for IL-8 was 12.5–6,400 pg/mL. The measurements of TNF-α in the supernatant were performed by using high-sensitive Quantikine ELISA kit (R&D Systems, Inc.). The analyses of TNF-α were performed according to the protocol from the manufacturer. For all the duplicated samples, an intra-assay variation <10% (for TNF-α, <20%) was accepted.
Statistics
Depending on the distribution of the data, the results are presented as mean confidence interval (95% CI) or median values (25th–75th percentile). The percentage of macrophage in the BAL fluid that is presented in Table 1 was used to normalize the release of IL-8 and TNF-α in different donors. Within group comparisons (the stimulation effects of LPS, PGN, and TNF-α; the inhibitory effects of Bud; and the blocking effects of TLR antibodies) were assessed by Wilcoxon signed-rank test. The between-group comparisons were performed by Kruskal–Wallis test with the Mann–Whitney U-test as a post hoc test. A P-value <0.05 was considered significant. All the data were analyzed using STATISTICA 9 (Statsoft Scandinavia AB, Uppsala, Sweden).
Results
The characteristics of the subjects who are a subgroup of a cohort published before3 are shown in Table 1. There are no significant differences among the three groups regarding age and among the smokers regarding smoking history, which is measured as pack years. Although smokers with and without COPD have more total cells in the BAL fluid than the healthy controls, the proportions of BAL macrophages were in the same range with no statistical difference.
In AMs from the healthy controls and smokers with and without COPD, PGN and LPS enhanced the release of IL-8, which could be inhibited by costimulation with Bud (P<0.05; Figure 1A, B, and C) with the exception of PGN-induced IL-8 release in smokers without COPD. TNF-α induced IL-8 secretion in AMs from the non-COPD groups (P<0.05), and Bud only inhibited TNF-α-induced IL-8 release in smokers without COPD (P<0.05; Figure 1D). After both control situation and stimulation with PGN alone, AMs from smokers without COPD released more IL-8 than AMs from the healthy controls (Figure 1A and C).
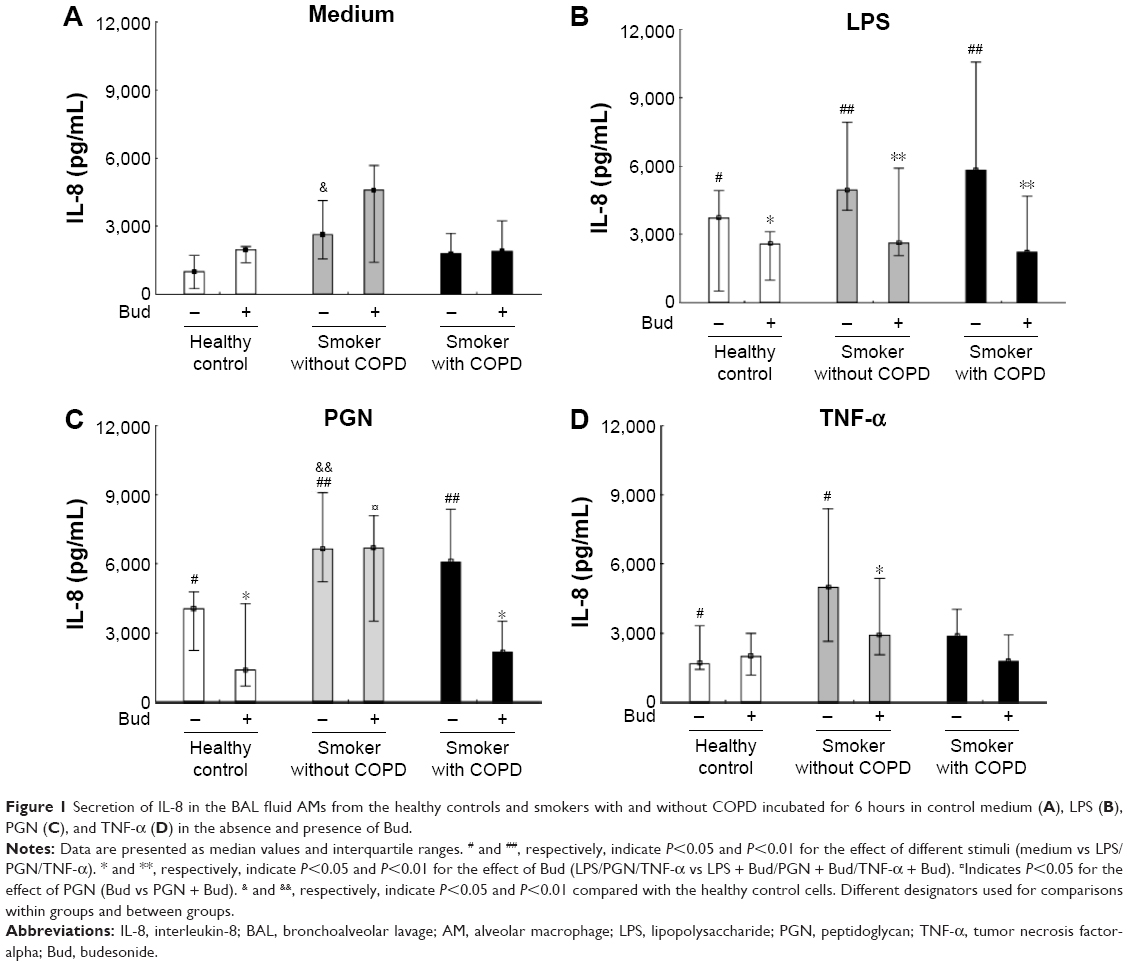 | Figure 1 Secretion of IL-8 in the BAL fluid AMs from the healthy controls and smokers with and without COPD incubated for 6 hours in control medium (A), LPS (B), PGN (C), and TNF-α (D) in the absence and presence of Bud. |
Both LPS and PGN significantly enhanced the release of TNF-α, which could be inhibited by costimulation with Bud in AMs from all the three groups (P<0.05; Figure 2A, B, and C).
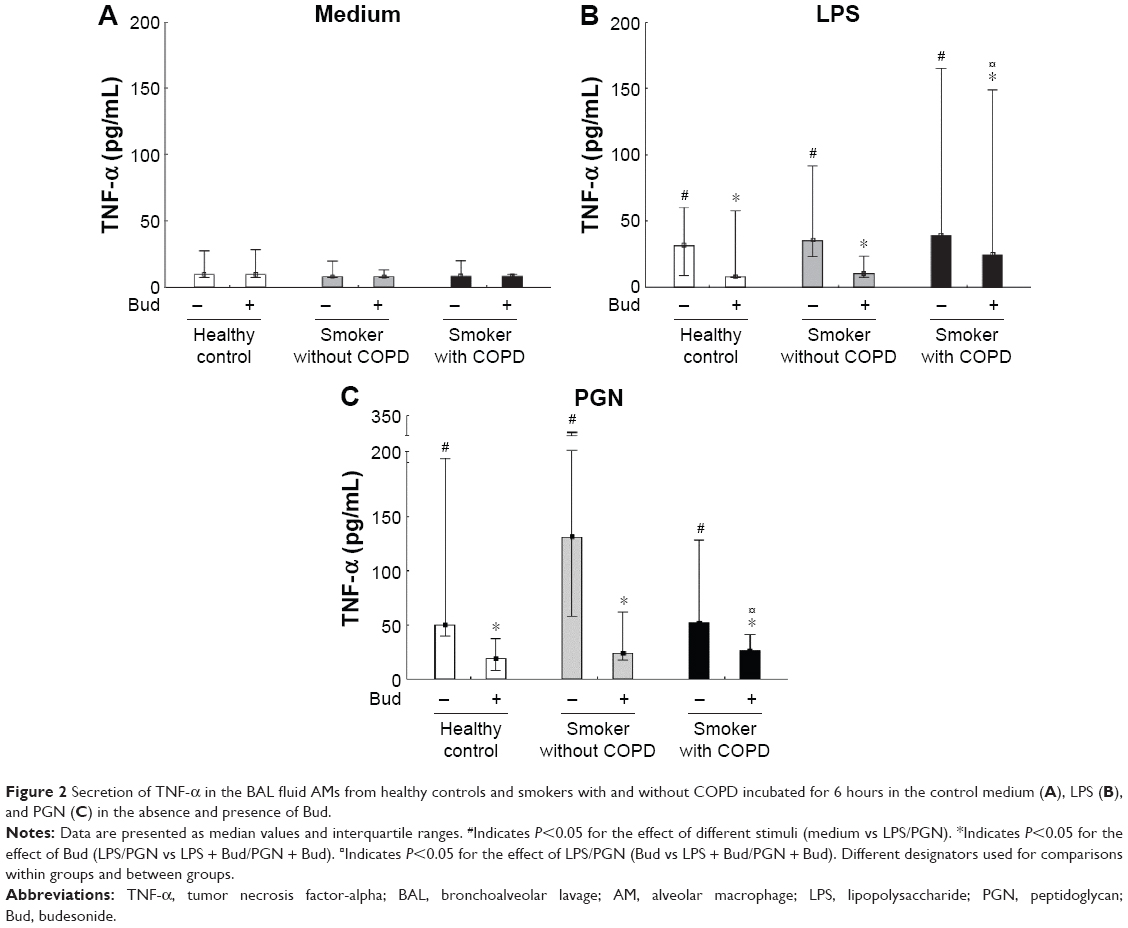 | Figure 2 Secretion of TNF-α in the BAL fluid AMs from healthy controls and smokers with and without COPD incubated for 6 hours in the control medium (A), LPS (B), and PGN (C) in the absence and presence of Bud. |
Bud in itself enhanced TLR2 mRNA expression in AMs from controls and smokers without COPD (P<0.05; Figure 3A). In smokers with and without COPD, the combination of Bud and either LPS or PGN enhanced the expression of TLR2 mRNA (P<0.05; Figure 3A, B and C). Also in smokers without COPD, a significant increased expression of TLR2 was observed between the stimulation combination of Bud and PGN and stimulation with Bud alone (P<0.05; Figure 3C). There is no effect of TNF-α stimulation with or without Bud in any of the groups regarding TLR2 expression (Figure 3D)
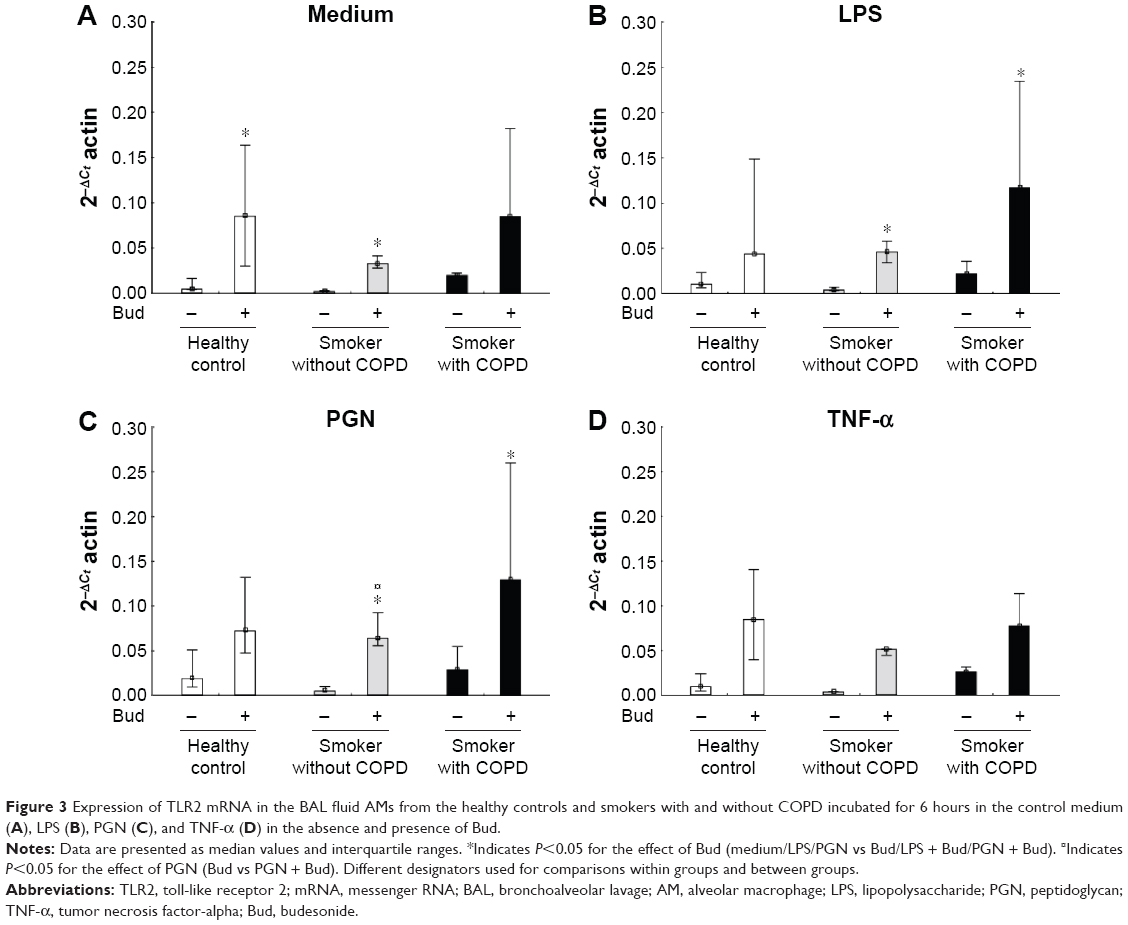 | Figure 3 Expression of TLR2 mRNA in the BAL fluid AMs from the healthy controls and smokers with and without COPD incubated for 6 hours in the control medium (A), LPS (B), PGN (C), and TNF-α (D) in the absence and presence of Bud. |
LPS stimulation alone decreased TLR4 mRNA expression in all the three groups (P<0.05), while there were no significant effects of the combination of LPS and Bud compared with the LPS stimulation alone (Figure 4A and B). There were no effects of PGN or TNF-α stimulation with or without Bud in any of the groups regarding expression of TLR4, with exception of TNF-α stimulation of AMs from smokers with COPD, where a significant reduction was observed (Figure 4C and D). A combined stimulation with PGN and Bud in both smoker groups resulted in significantly decreased TLR4 expression compared with Bud stimulation alone (Figure 4C).
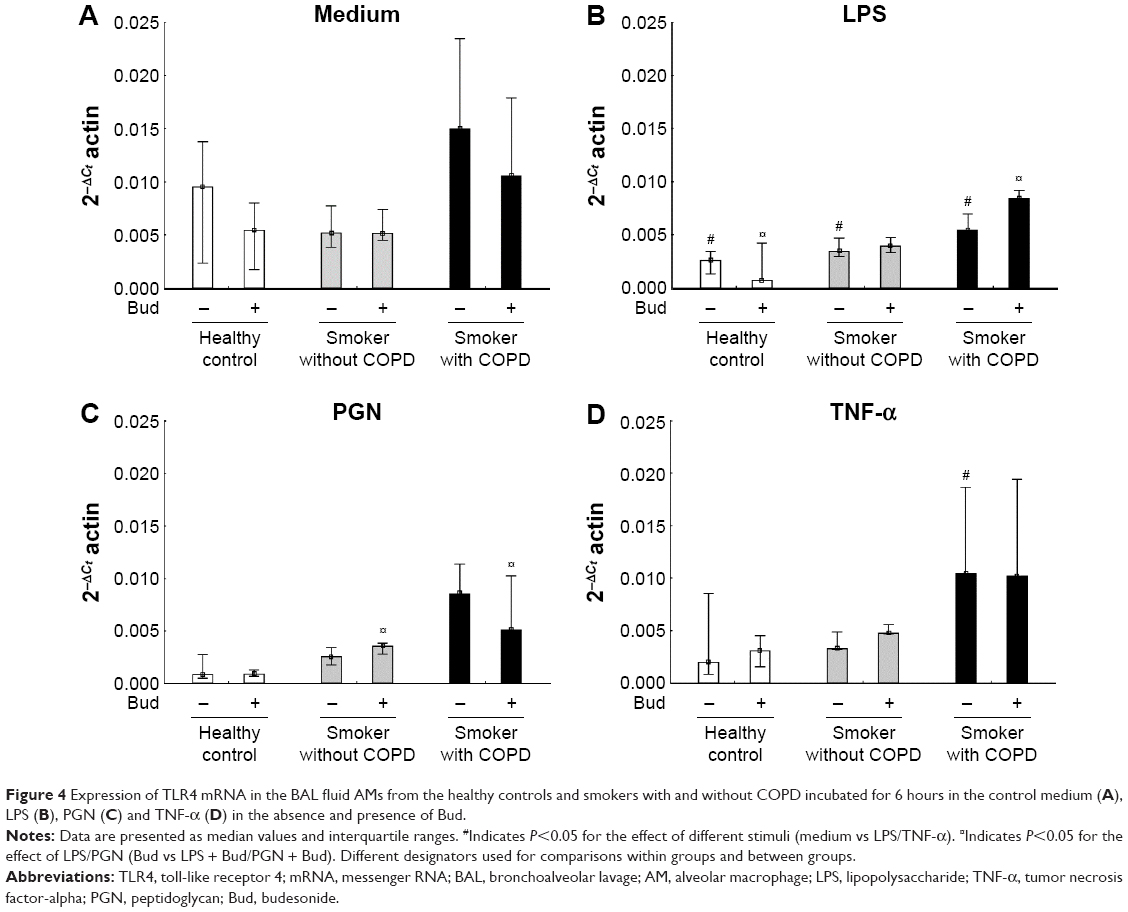 | Figure 4 Expression of TLR4 mRNA in the BAL fluid AMs from the healthy controls and smokers with and without COPD incubated for 6 hours in the control medium (A), LPS (B), PGN (C) and TNF-α (D) in the absence and presence of Bud. |
Blocking TLR2 or TLR4 tended to inhibit PGN- and LPS-induced IL-8 protein secretion in all the three groups (Figure 5A–F). The inhibitory effect of Bud on PGN-induced IL-8 secretion tended to increase further after TLR2 blocking in all the groups (Figure 5A–C). As a control experiment, the appropriate isotype had no significant effect on measured outcomes (data not shown).
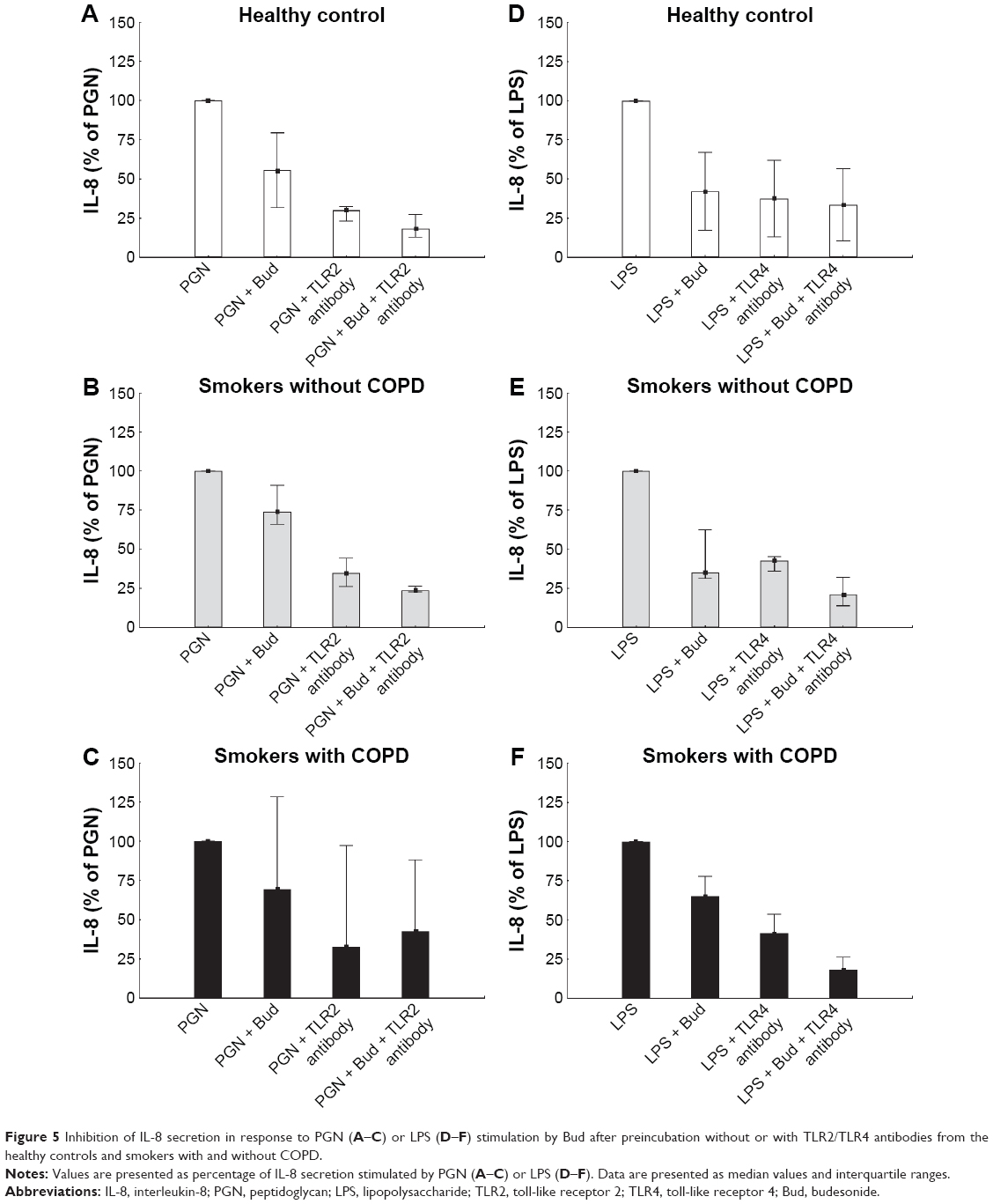 | Figure 5 Inhibition of IL-8 secretion in response to PGN (A–C) or LPS (D–F) stimulation by Bud after preincubation without or with TLR2/TLR4 antibodies from the healthy controls and smokers with and without COPD. |
Discussion
In the present study, we showed that on AMs from the smokers with and without COPD, Bud in itself or the combination of Bud and either PGN or LPS induced increased TLR2 expression. This result is in line with our previous findings on primary human bronchial epithelial cells in which TLR2 expression was enhanced when stimulated with the combination of Bud and a ligand.13 However, in primary bronchial epithelial cells, Bud had no effect in itself. It has been previously shown that COPD patients have a lower expression of TLR2 on sputum neutrophils than that of the healthy nonsmokers,9 and AMs from smokers showed a decreased expression of TLR2.10 However, the expression of TLR2 on AMs in this cohort did not differ between the groups, neither when isolated directly nor after ex vivo incubation in the cell culture medium.3 The discrepancy of TLR2 expression in these studies could be both due to different disease stages in the COPD groups and different methods used to measure protein and mRNA levels. It is reasonable to assume that the increasing TLR2 expression might indicate an enhanced host immune defense against bacteria with beneficial effects by reducing bacterial airway colonization. According to Imasato et al,16 glucocorticosteroids synergistically increase nontypeable Haemophilus influenza-induced TLR2 expression via enhancing the MAPK phosphatase-1 (MKP-1) expression. The upregulation of MKP-1 expression can inhibit the phosphorylation and activation of P38 MAPK, leading to the positive regulation of TLR2 expression. According to Southworth et al,17 dexamethasone alone could increase TLR2 expression in the COPD patients. We found that Bud in itself only increased TLR2 expression in the non-COPD groups. This finding may be due to the large interindividual variance in our COPD patients.
Bud with LPS or PGN significantly decreased LPS- or PGN-induced release of IL-8 and TNF-α in all the groups (except in smokers without COPD), similar to what has been demonstrated in the previous studies.17–21 The lack of the effect of Bud regarding PGN stimulation and IL-8 secretion in smokers without COPD could at least in part be due to higher secretion in both medium control situation and after PGN stimulation. Neither of these findings were seen regarding TNF-α secretion. Glucocorticosteroids effectively control the inflammation by binding to their receptors (GRs)’ and switching off multiple activated inflammatory genes that encode for cytokines or chemokine production.12 Therefore, the downregulation of LPS- or PGN-induced IL-8 and TNF-α production might be a consequence of inhibited activation of transcription factors, such as nuclear factor κB (NF-κB) or activator protein 1 (AP-1), directly or indirectly (by binding to GRs) induced by the glucocorticosteroid (Bud). These anti-inflammatory properties of Bud (inhibition of IL-8 and TNF-α release) appear paradoxical in the context of its increasing effect on TLR2 expression, which would likely increase the binding of PAMPs, leading to further activation of NF-κB and a subsequent release of proinflammatory cytokines or chemokines. Our finding that AMs from smokers without COPD expressed TLR2 significantly more after costimulation with PGN and Bud, compared with both stimulation with Bud or PGN separately, together with the lack of inhibitory effect of Bud on IL-8 secretion after costimulation, might illustrate this paradox. Both nontreated and PGN-treated AMs from smokers without COPD released more IL-8 than AMs from the healthy controls. This may indicate that AMs from smokers without COPD are more active and less sensitive to glucocorticosteroids regarding IL-8 release than the healthy controls.
The lack of increase in the cytokine release might be explained by the downstream blockage of the TLR2 receptor signaling by a glucocorticosteroid, and a mere increase in the amount of TLR2 does not relieve this blocking effect.22 Besides, the changes in the cytokine levels may take place at a later stage, which is beyond the stimulation time in this experiment (6 hours). To identify the role of TLR signaling in IL-8 secretion and whether IL-8 secretions are driven by the TLR pathways, we blocked TLR2 and TLR4 with the corresponding antibodies. After blocking, there was a tendency of decreased LPS- or PGN-induced-IL-8 secretion. The inhibitory effect of Bud on PGN-induced IL-8 secretion tended to increase further after TLR2 blocking in all the groups. These results might imply that TLR2 and/or TLR4 may play a role in AM activation as a response to LPS/PGN stimulation. Also, the inhibitory effect of Bud is partly mediated through the TLR2 and/or TLR4 pathway.
A limitation of this study is the small study sample and the great interindividual variance, which diminishes the statistical power. In addition, we stimulated AMs for 6 hours and we only made our analysis at one single time point. It cannot be denied that the stimulated effects may take place after a longer stimulation. Despite this, we found significant effects of TLR ligands or endogenous ligand stimulation in the absence or presence of a glucocorticosteroid. Although significant effects were demonstrated in smokers but not in controls, there were no statistically significant differences between nonsmokers and smokers. We thus found consistently higher expression of TLR2 on AMs from smokers with and without COPD than from controls when AMs were cocultured with Bud and LPS/PGN, while there was no significant increase in TLR2 expression in the healthy controls. It is therefore reasonable to believe that there is a difference between nonsmoking controls and smokers with and without COPD. Our results indicate that smokers who are more likely to have bacterial colonizations than the healthy individuals will benefit from a glucocorticosteroid.
Conclusion
By costimulating AMs with LPS/PGN and a glucocorticosteroid or stimulation with a glucocorticosteroid itself, we found that on the one hand glucocorticosteroids increased TLR2 expression in smokers that might have beneficial effects associated with an improved host defense and on the other hand they attenuated the release of proinflammatory cytokines (TNF-α) and chemokines (IL-8) in both the healthy controls and smokers with and without COPD as an expression of an anti-inflammatory response. Therefore, our findings might contribute to the understanding of why glucocorticosteroid therapy, which strengthens our immune defense pathway, has implication during exacerbation caused by microorganisms.
Acknowledgments
The authors would like to thank, Dr Maria Skedinger, Dr Anita Simhag Britt-Marie Sundblad and Kristin Blidberg for valuable technical assistance. This study was funded by grants from Swedish Heart and Lung Foundation, Karolinska Institutet, AstraZeneca Sweden, Swedish research council and King Gustaf V’s and Queen Victoria’s Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Palmberg L, Larsson BM, Malmberg P, Larsson K. Induction of IL-8 production in human alveolar macrophages and human bronchial epithelial cells in vitro by swine dust. Thorax. 1998;53(4):260–264. | ||
Knobloch J, Hag H, Jungck D, Urban K, Koch A. Resveratrol impairs the release of steroid-resistant cytokines from bacterial endotoxin-exposed alveolar macrophages in chronic obstructive pulmonary disease. Basic Clin Pharmacol Toxicol. 2011;109(2):138–143. | ||
Pons AR, Sauleda J, Noguera A, et al. Decreased macrophage release of TGF-beta and TIMP-1 in chronic obstructive pulmonary disease. Eur Respir J. 2005;26(1):60–66. | ||
Culpitt SV, Rogers DF, Shah P, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167(1):24–31. | ||
von Scheele I, Larsson K, Dahlen B, et al. Toll-like receptor expression in smokers with and without COPD. Respir Med. 2011;105(8):1222–1230. | ||
Maris NA, Dessing MC, de Vos AF, et al. Toll-like receptor mRNA levels in alveolar macrophages after inhalation of endotoxin. Eur Respir J. 2006;28(3):622–626. | ||
Twigg HL 3rd. Macrophages in innate and acquired immunity. Semin Respir Crit Care Med. 2004;25(1):21–31. | ||
Sahlander K, Larsson K, Palmberg L. Altered innate immune response in farmers and smokers. Innate Immun. 2010;16(1):27–38. | ||
von Schéele I, Larsson K, Palmberg L. Interactions between alveolar epithelial cells and neutrophils under pro-inflammatory conditions. Eur Clin Respir J. 2014;1:24545. | ||
Droemann D, Goldmann T, Tiedje T, Zabel P, Dalhoff K, Schaaf B. Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir Res. 2005;6:68. | ||
Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol. 2011;163(1):29–43. | ||
Barnes PJ. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br J Pharmacol. 2006;148(3):245–254. | ||
von Scheele I, Larsson K, Palmberg L. Budesonide enhances toll-like receptor 2 expression in activated bronchial epithelial cells. Inhal Toxicol. 2010;22(6):493–499. | ||
Ji J, von Scheele I, Bergstrom J, et al. Compartment differences of inflammatory activity in chronic obstructive pulmonary disease. Respir Res. 2014;15:104. | ||
Larsson K, Tornling G, Gavhed D, Muller-Suur C, Palmberg L. Inhalation of cold air increases the number of inflammatory cells in the lungs in healthy subjects. Eur Respir J. 1998;12(4):825–830. | ||
Imasato A, Desbois-Mouthon C, Han J, et al. Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of toll-like receptor 2. J Biol Chem. 2002;277(49):47444–47450. | ||
Southworth T, Metryka A, Lea S, Farrow S, Plumb J, Singh D. IFN-gamma synergistically enhances LPS signalling in alveolar macrophages from COPD patients and controls by corticosteroid-resistant STAT1 activation. Br J Pharmacol. 2012;166(7):2070–2083. | ||
Armstrong J, Sargent C, Singh D. Glucocorticoid sensitivity of lipopolysaccharide-stimulated chronic obstructive pulmonary disease alveolar macrophages. Clin Exp Immunol. 2009;158(1):74–83. | ||
Lea S, Plumb J, Metcalfe H, et al. The effect of peroxisome proliferator-activated receptor-gamma ligands on in vitro and in vivo models of COPD. Eur Respir J. 2014;43(2):409–420. | ||
Higham A, Booth G, Lea S, Southworth T, Plumb J, Singh D. The effects of corticosteroids on COPD lung macrophages: a pooled analysis. Respir Res. 2015;16:98. | ||
Homma T, Kato A, Hashimoto N, et al. Corticosteroid and cytokines synergistically enhance toll-like receptor 2 expression in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2004;31(4):463–469. | ||
Chinenov Y, Rogatsky I. Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Mol Cell Endocrinol. 2007;275(1–2):30–42. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.