Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Effects of Autophagy Modulators and Dioxin on the Expression of Epidermal Differentiation Proteins on Psoriasis-Like Keratinocytes in vitro and ex vivo
Authors Kim HR ![]() , Kim HO
, Kim HO ![]() , Kim JC
, Kim JC ![]() , Park CW, Chung BY
, Park CW, Chung BY
Received 26 March 2022
Accepted for publication 1 June 2022
Published 23 June 2022 Volume 2022:15 Pages 1149—1156
DOI https://doi.org/10.2147/CCID.S368105
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Hye Ran Kim, Hye One Kim, Jin Cheol Kim, Chun Wook Park, Bo Young Chung
Department of Dermatology, Hallym University Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul, 07441, Republic of Korea
Correspondence: Bo Young Chung, Department of Dermatology, Hallym University Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul, 07441, Republic of Korea, Tel +82-2-829-9092, Fax +82-2-832-3237, Email [email protected]
Objective: Psoriasis is a chronic inflammatory skin disorder associated with impairment of epidermal differentiation. Many signaling pathways, including those involved in aryl hydrocarbon receptor (AHR) and autophagy dysfunction, are reportedly associated with the pathogenesis of psoriasis. However, the discrete effects of dioxin via AHR activation or autophagy on the epidermal barrier remain unclear. In the current study, we evaluated the effects of autophagy modulators (chloroquine [CQ] and rapamycin) and the AHR agonist TCDD on the expression of epidermal barrier proteins in psoriasis-like keratinocytes and psoriasis lesional skin tissue culture.
Methods: Polycytokine-stimulated human keratinocytes and psoriasis skin biopsies were treated with TCDD, CQ, or rapamycin, and the expression of keratinocyte differentiation-related factors, such as S100A7, S100A8, HRNR, IVL, FLG, and KRT10, was examined by Western blotting or quantitative-polymerase chain reaction.
Results: TCDD upregulated S100A7 and S100A8 expression in polycytokine-stimulated HaCaT cells compared to that in unstimulated cells. CQ decreased HRNR, IVL, and KRT10 mRNA levels, while rapamycin increased HRNR, IVL, and KRT10 mRNA levels in HaCaT cells relative to that in unstimulated cells. Co-treatment with CQ reversed TCDD-induced elevation in FLG, HRNR, and IVL mRNA expression. In psoriasis skin tissue, TCDD induced the upregulation of HRNR, IVL, S100A7, and S100A8 compared with that in normal skin. In ex vivo cultures treated with CQ, IVL expression in psoriasis skin tissue was repressed compared to that in normal skin tissue.
Conclusion: Our data suggest that autophagy modulation or AHR activation affects processes involved in epidermal differentiation and relates to the pathogenesis of chronic inflammatory skin diseases with skin barrier abnormalities such as psoriasis.
Keywords: aryl hydrocarbon receptor, autophagy, keratinocyte differentiation, 2,3,7,8-tetrachlorodibenzo-p-dioxin, chloroquine
Introduction
Psoriasis is a chronic inflammatory skin disorder associated with the impairment of epidermal differentiation.1 In the psoriatic epidermis, the balance between proliferation and differentiation is disrupted.2 The expression levels of several epidermal-differentiation-related markers are altered in psoriasis, with the overexpression of IVL and downregulation of keratin (K)1 and K10.3,4 S100A7 and S100A8 are among epidermal differentiation complex genes and are considerably upregulated in psoriasis.5 Additionally, S100A7 and S100A8 act as antimicrobial peptides that induce T cell and neutrophil chemotaxis.6 Their expression is regulated by pro-inflammatory and Th-derived cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-22, and IL-17.7 Activated keratinocytes in turn produce important pro-inflammatory cytokines and chemokines. These cycles of excessive immune response induce the complex clinical presentation of psoriasis. However, the complicated intracellular epidermal process induced by the immunologic network in psoriasis remains unclear. Many signaling pathways, including those involved in aryl hydrocarbon receptor (AHR) and autophagy dysfunction, are reportedly associated with psoriasis pathogenesis.8,9
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), an environmental pollutant, is the most potent dioxin and prototype ligand for the AHR, a ligand-activated transcription factor related to sensing environmental stimuli. Binding ligands, including TCDD, induce AHR translocation to the nucleus, heterodimerization with the AHR nuclear translocator, and binding to specific DNA sites to activate target gene transcription, mediating various physiological and pathological effects.10 Exposure to high concentrations of dioxins leads to chloracne in humans.11 Chloracne has been considered to be caused by prolonged AHR activation and excessive hyperkeratinization of pilosebaceous units.10
Chloroquine (CQ), which is used clinically as an antimalarial agent, is a classical autophagy inhibitor that blocks the binding of autophagosomes to lysosomes by altering the acidic environment of lysosomes.12 The mammalian target of rapamycin (mTOR) inhibitor rapamycin upregulates autophagy in primary keratinocytes.13 Autophagy is a critical process for maintaining cell homeostasis by degrading and/or recycling materials via the lysosomes.14 The autophagy pathway includes not only processes of degradation and survival but also regulates various cellular functions, such as, pathogen clearance, antigen presentation, apoptosis, and inflammation.14
We previously found that AHR activation mediated autophagy induces the production of inflammatory cytokines in human keratinocytes via the p65NF-κB/p38MAPK signaling pathways.9 However, the discrete mechanisms associated with autophagy- and AHR-related epidermal differentiation processes remain poorly understood. In this study, we evaluated the effects of autophagy modulators (chloroquine and rapamycin) on TCDD-stimulated expression of skin barrier differentiation proteins using in vitro and ex vivo psoriatic models.
Materials and Methods
Patients and Sample Collection
Skin biopsy tissues were collected from patients with psoriasis (n = 5) and healthy volunteers (n = 5). This study was carried out in accordance with the Declaration of Helsinki. Informed consent was obtained from all subjects before inclusion in the study, and the protocol of the study was approved by the Institutional Review Board of Kangnam Sacred Heart Hospital (IRB no. 2018-05-024; August 31, 2018).
Cell Culture
The immortalized human keratinocyte cell line HaCaT (Welgene, Daegu, South Korea) was maintained in Dulbecco’s Modified Eagle’s medium (DMEM) (Lonza, Walkersville, MD, USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin-streptomycin at 37°C in a humidified CO2 incubator (5% CO2). A mixture of five pro-inflammatory cytokines (10 ng/mL each), namely, TNF-α, IL-17A, IL-22, IL-1, and oncostatin-M (OSM; Prospec, East Brunswick, NJ, USA) designated as M5 cytokines, were used to stimulate the HaCaT cells.15 Upon reaching 70–90% confluence, the cells were separated with 0.25% trypsin/0.01% ethylenediaminetetraacetic acid in HEPES and subcultured.
Ex vivo Culture
Skin biopsy tissues from lesional skin of psoriasis patients and normal skin of healthy controls were prepared for ex vivo culture. Tissues were immersed in a growth medium, skin fragments were prepared, the adipose tissue was removed, and the fragments were washed in serum-free DMEM for 10 min. The whole tissue was then placed in a multi-well plate (Costar, Cambridge, MA, USA) and incubated for 48 h in 1 mL DMEM at 37°C under 5% CO2.
Quantitative PCR
Total RNA was extracted using the RNeasy Plus mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, and cDNA was synthesized from 1 µg of RNA using the Transcriptor first-strand cDNA synthesis kit (Roche Applied Science, Mannheim, Germany). Three rounds of qPCR were performed using TaqMan master mix and a qPCR system (Applied Biosystems, Foster City, CA, USA). mRNA levels of KRT10 (Hs 00166289_m1), S100A7 (Hs 00752780_m1), S100A8 (Hs 00374264_m1), HRNR (Hs 02340614_m1), IVL (Hs 00846307_m1), FLG (Hs 00418578_m1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Hs 02758991_m1) were evaluated. Reactions were performed in triplicate, and fold changes were calculated using the ΔΔCt method.16 Relative quantification was performed using a Light Cycler 96 instrument (Roche Diagnostics, Mannheim, Germany) using the following reaction conditions: initial denaturation at 95°C for 30s; 45 cycles of denaturation at 95°C for 5s, annealing at 60°C for 30s, and extension at 72°C for 10s; final extension at 95°C for 5 s.
Western Blotting
We conducted the BCA assay (Sigma-Aldrich, St. Louis, MO, USA) to measure protein concentrations. Equal amounts of protein (20 µg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, UK), and blocked with 5% skim milk in TBST for 1 h. The membranes were then incubated overnight at 4°C with rabbit anti-K10 (1:1000; Abcam, Cambridge, UK), rabbit anti-S100A7 (1:1000; Aviva Systems Biology, San Diego, CA, USA), rabbit anti-S100A8 (1:1000; LS Bio, Washington, WA, USA), rabbit anti-FLG (1:1000; LS Bio), and rabbit anti-AHR (1:1000; Abcam). Primary antibodies were detected with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibodies (1:1000; Abcam). We visualized protein bands via enhanced chemiluminescence using a LuminoGraph II (Atto, Tokyo, Japan), and densitometric analysis of the blots was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Small Interfering (si) RNA Transfection
siRNAs targeting AHR (siAHR) and scrambled siRNA sequences (control siRNA) were obtained from Ambion (Thermo Fisher Scientific). HaCaT cells cultured in six-well plates were incubated for 48 h in 0.5 mL of culture medium with a mixture containing 5 nM siRNA and 3 µL of Lipofectamine RNA iMAX (Invitrogen, Carlsbad, CA, USA). After 48 h, transfections were performed using siAHR, or control siRNA
Statistical Analyses
Statistical analyses were performed using GraphPad Prism software (v.5.01; GraphPad Software, La Jolla, CA, USA). Data were analyzed using the one-way analysis of variance (ANOVA) with Tukey’s post-hoc tests. Differences with p < 0.05 were considered statistically significant.
Results
TCDD Profoundly Upregulates S100A7 and S100A8 Expression in Polycytokine (M5)-Stimulated HaCaT Cells
We observed an increase in S100A7 and S100A8 mRNA and protein levels in TCDD-stimulated HaCaT cells compared to those in the control. Similarly, M5 treatment also induced elevation in S100A7 and S100A8 levels in HaCaT cells compared with those in the control. Combination treatment of TCDD and M5 resulted in a more pronounced increase in S100A7 and S100A8 expression compared with those in TCDD- or M5-treated HaCaT cells (Figure 1A–C). In contrast, AHR knockdown resulted in a decrease in the TCDD- and M5-mediated upregulation of S100A7 and S100A8 expression, suggesting the role of AHR in TCDD- and M5-induced elevation of S100A7 and S100A8 expression (Figure 1D–F).
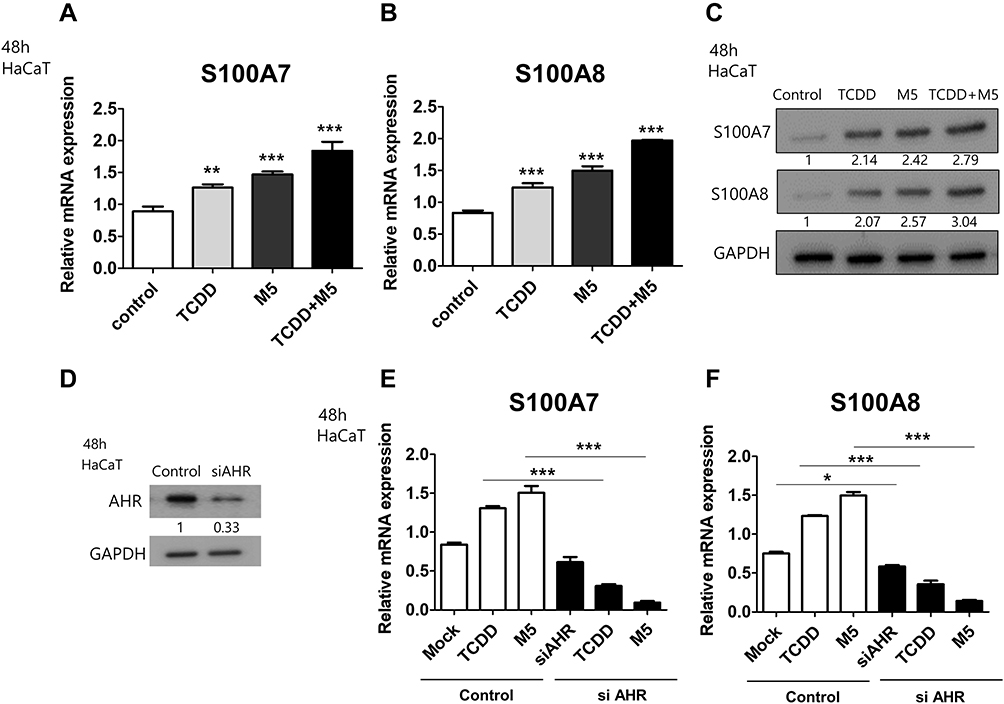 |
Figure 1 Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the expression of S100A7 and S100A8 in M5-stimulated HaCaT cells. (A, B) Levels of S100A7 and S100A8 mRNA normalized against that of GAPDH, in HaCaT cells treated with 10 nM TCDD or M5 (TNF-α, IL-17A, IL-22, IL-1, and OSM; 10 ng/mL each) for 48 h. Data are presented as mean ± SD of at least three independent experiments (each performed in duplicate). **p < 0.01, ***p < 0.001 compared with unstimulated cells. (C) Western blotting of HaCaT cells stimulated with 10 nM TCDD or M5 or 10 nM TCDD + M5 for 48 h using antibodies against S100A7 and S100A8. Relative expression was normalized to GAPDH expression. Results are representative of three independent experiments. The densitometry value of each independent band is compared with the control. (D) HaCaT cells were transiently transfected with small interfering(si)-aryl hydrocarbon receptor (AHR) followed by Western blotting targeting AHR. Relative expression was normalized to GAPDH expression. Results are representative of three independent experiments. The densitometry value of each independent band is compared with the control. (E, F) HaCaT cells were transiently transfected with siAHR and stimulated with 10 nM TCDD or M5 for 48 h, followed by qPCR targeting S100A7 and S100A8. Data are presented as mean ± standard deviation (SD) of three independent experiments (each performed in duplicate). *p < 0.05, **p < 0.01, ***p< 0.001 compared with unstimulated cells. |
Effects of CQ Treatment on TCDD-Induced Expression of Epidermal Barrier-Related Markers in HaCaT Cells
We then examined the effects of the autophagy inhibitor CQ and autophagy inducer rapamycin on epidermal barrier differentiation-related marker levels in HaCaT cells. CQ decreased HRNR, IVL, and KRT10 mRNA levels compared with that in the control. In contrast, rapamycin treatment increased HRNR and IVL mRNA levels in HaCaT cells compared with that in the control (Figure 2A–C).
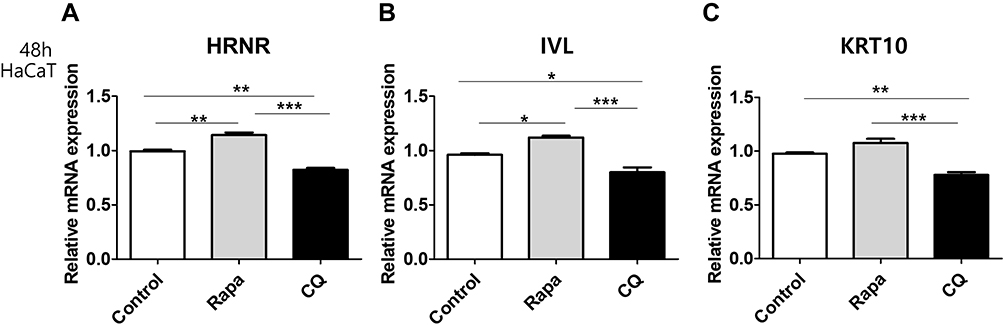 |
Figure 2 Effects of chloroquine (CQ) and rapamycin on the expression of HRNR, IVL, and KRT10 mRNA in HaCaT cells. qRT-PCR analysis of (A) HRNR, (B) IVL, and (C) KRT10 mRNA from HaCaT cells treated with 20 µg/mL CQ and 50 nM rapamycin for 48 h. Data are presented as mean ± SD of three independent experiments (each performed in duplicate). *p < 0.05, **p < 0.01, ***p < 0.001 compared with unstimulated cells. |
Following TCDD stimulation, FLG, HRNR, and IVL expression increased in HaCaT cells compared to that in the control (Figure 3A–C). Additionally, we observed that CQ repressed TCDD-induced upregulation of FLG, HRNR, and IVL expression (Figure 3A–C).
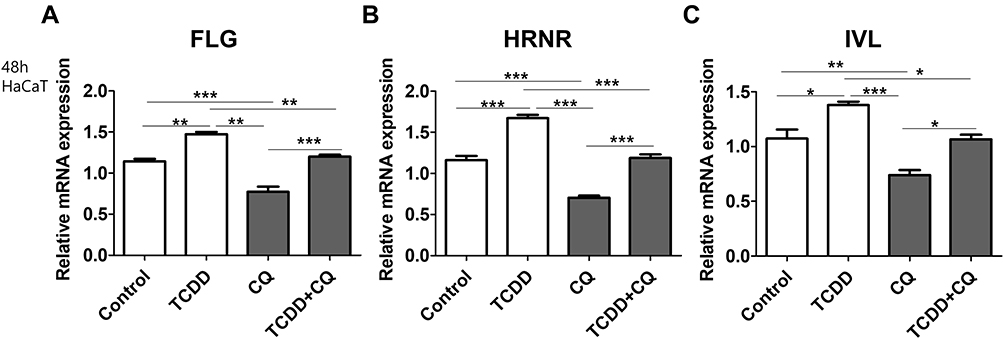 |
Figure 3 Effects of TCDD and CQ co-treatment on the expression of epidermal barrier-related markers in HaCaTcells. (A) FLG, (B) HRNR, and (C) IVL mRNA levels in HaCaT cells treated with 10 nM TCDD, 20 µg/mL CQ, or 10 nM TCDD + 20 µg/mL CQ for 48 h, relative to the expression of GAPDH. Data are presented as mean ± SD of at least three independent experiments (each performed in duplicate). *p < 0.05, **p < 0.01, ***p < 0.001 compared with unstimulated cells. |
Effects of TCDD or Autophagy Modulation on the Expression of Skin Barrier-Related Markers in ex vivo Cultures of Human Psoriasis Skin Biopsies
To confirm the effects of dioxin and autophagy regulation on psoriatic pathology, we investigated the effects of TCDD, autophagy inhibition by CQ, and autophagy stimulation by rapamycin on the production of skin barrier-related markers in an ex vivo culture of human psoriasis lesional skin tissue. Full-thickness skin biopsies were obtained from patients with psoriasis (n = 5) and healthy human volunteers (n = 5), and human skin explants were treated with TCDD (10 nM), CQ (20 µg/mL), or rapamycin (50 nM), and cultured for 48 h before mRNA quantification. HRNR, IVL, S100A7, and S100A8 expression significantly increased in TCDD-treated psoriatic tissues compared with that in TCDD-treated normal skin tissues (Figure 4A–D). IVL expression significantly decreased in CQ-treated psoriatic tissues compared with that in CQ-treated normal skin tissues. In contrast, IVL mRNA expression was significantly upregulated in rapamycin-treated psoriatic skin tissues compared with those in rapamycin-treated normal skin tissues (Figure 4B).
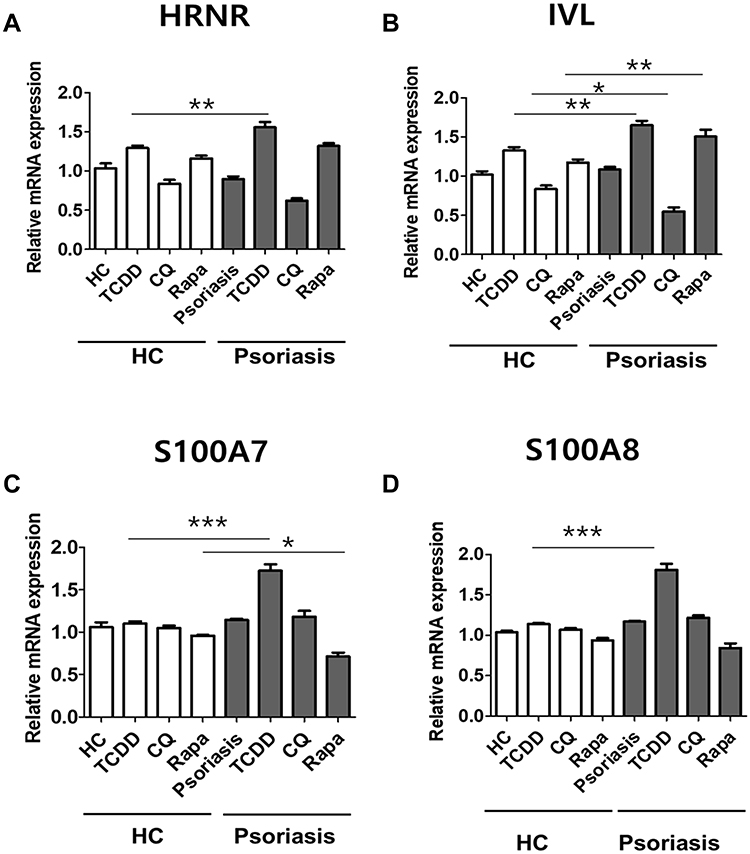 |
Figure 4 Effects of AHR or autophagy modulation on the expression of skin barrier markers in human psoriasis skin biopsies. (A) HRNR, (B) IVL, (C) S100A7, and (D) S100A8 expression in human psoriatic (n = 5) and healthy (n = 5) skin explants following treatment with 10 nM TCDD, 20 µg/mL CQ, or 10 nM rapamycin. Data are presented as mean ± SD of three independent experiments (each performed in duplicate). *p < 0.05, **p < 0.01, ***p < 0.001 compared with unstimulated cells. |
Discussion
Previous studies have shown that exposure of human keratinocytes and three-dimensional skin-equivalent models to TCDD significantly increases the expression of genes encoding barrier proteins, such as FLG, HRNR, IVL, S100A7, and S100A8.17,18 Our finding shows that TCDD upregulated FLG, HRNR, IVL, S100A7, and S100A8 expression in HaCaT cells is consistent with that of previous studies.17,18 It is suggested that toxic AHR ligands, such as TCDD, persist in the human body because they are not readily metabolized by CYP1A1, unlike physiological AHR ligands. Thus, exaggerated AHR activation triggers pathologic keratinocyte differentiation.19 Notably, in the present study, the epidermal differentiation protein expression upregulation by TCDD was pronounced in polycytokine-stimulated in vitro and ex vivo psoriatic models. These results suggest that pathologic epidermal proliferation induced by TCDD is exaggerated in psoriasis.
In the present study, autophagy inhibitor CQ suppressed the expression of epidermal-differentiation-related markers, such as FLG, HRNR, IVL, and K10, whereas autophagy inducer rapamycin upregulated their expression. Our results demonstrate that autophagy affects the regulation of epidermal differentiation. Cumulative evidence suggests that autophagy is essential for physiological epidermal differentiation and dysregulation of autophagy is closely associated with barrier-disrupted inflammatory skin diseases, such as psoriasis. Akinduro et al reported that autophagy is constitutively active in normal epidermal differentiation in mice skin and that impaired autophagy leads to abnormal keratinocyte differentiation. In the psoriatic epidermis, the expression of autophagy-related markers was different from that in normal skin.13 Similarly, another study reported that BNIP3-induced autophagy plays a crucial role in epidermal differentiation.20
In the present study, we used M5-stimulated keratinocytes as an in vitro model of psoriasis. A mixture of five pro-inflammatory cytokines, TNF-α, IL-17A, IL-22, IL-1, and oncostatin-M designated as M5 cytokines, were used to stimulate HaCaT cells and induce inflammatory chemokines and cytokines expression observed in keratinocytes in psoriasis.16 M5-stimulated keratinocytes showed increased S100A7 (psoriasin) and S100A8 expression compared to that in the control. S100A7 and S100A8 were also considerably upregulated in psoriasis lesional skin.21 S100A7 and S100A8 are epidermal differentiation complex genes,22 and also act as antimicrobial peptides.14 The overexpression of S100A7 and S100A8 is associated with dysregulated keratinocyte differentiation.23,24 The expression of S100A7 is stimulated by key cytokines involved in the pathogenesis of psoriasis, IL-22 and the combination of IL-17 and TNF-α.7 Our results in the in vitro psoriatic model confirmed that excessive immune response induces changes in the expression of epidermal differentiation markers. Combination treatment of TCDD and M5 induced a more profound increase in S100A7 and S100A8 expression compared with that in TCDD-treated HaCaT cells. A previous study demonstrated that TCDD + M5 treatment in HaCaT cells increased AHR expression compared to that in only TCDD-treated HaCaT cells.9 Furthermore, AHR knockdown resulted in a decrease in TCDD- or M5-mediated upregulation in S100A7 and S100A8 expression, suggesting that both TCDD- or M5-induced upregulation is mediated by AHR. Ex vivo experiments using psoriasis skin biopsy tissue also showed pronounced elevation of IVL, HRNR, S100A7, and S100A8 mRNA levels following TCDD treatment compared to that in normal skin tissues following TCDD treatment. These results led us to speculate that psoriatic inflammatory skin might be more vulnerable to environmental toxicants, such as TCDD.
In the present study, the upregulation in FLG, HRNR, and IVL mRNA expression via AHR activation through TCDD was suppressed by CQ in keratinocytes. Notably, we found that the AHR agonist TCDD and autophagy inhibitor CQ showed the opposite effect on epidermal barrier-related factor expression in keratinocytes. Therefore, our results suggest that autophagy modulation is a potential therapeutic approach for reversing epidermal differentiation pathology. However, with respect to the relationship between autophagy regulators and AHR activation, which of them is upstream/downstream is not clear based on our data. Thus, further experiments for confirming the relationship between autophagy regulators and AHR activation will be needed.
This study had some limitations. Our study was conducted using HaCaT cells and skin tissue cultures. Therefore, experiments with normal human epidermal keratinocytes are needed to provide more accurate confirmation. The experiments performed in monolayer keratinocyte cultures might not fully represent the stratified epidermis in vivo, and the effects of AHR activation or autophagy modulation on actual skin barrier functions, such as transepidermal water loss or skin hydration, could not be checked owing to limitations with in vitro conditions.
Conclusions
The present study reports that autophagy modulation or AHR activation affects the expression of epidermal differentiation-related markers in both in vitro and ex vivo psoriatic models. In summary, autophagy inhibitor CQ suppressed TCDD-induced upregulation of epidermal barrier protein levels. Our data suggest that autophagy modulation or AHR activation relates to the pathogenesis of chronic inflammatory skin disease with epidermal barrier abnormalities such as psoriasis. However, further experiments will be needed to confirm the relationship between autophagy regulators and AHR activation in epidermal differentiation.
Abbreviations
AHR, aryl hydrocarbon receptor; CQ, chloroquine; IL, interleukin; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; TNF-α, tumor necrosis factor-α.
Data Sharing Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the 2018 Amorepacific Grant. The funding source had no involvement in study design; data collection, analysis, and interpretation, manuscript writing, and the decision to submit the article for publication.
Disclosure
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
1. Raychaudhuri SP. A cutting edge overview: psoriatic disease. Clin Rev Allergy Immunol. 2013;44(2):109–113. doi:10.1007/s12016-012-8309-z
2. Segre JA. Epidermal barrier formation and recovery in skin disorders. J Clin Invest. 2006;116(5):1150–1158. doi:10.1172/JCI28521
3. Ishida-Yamamoto A, Iizuka H. Differences in involucrin immunolabeling within cornified cell envelopes in normal and psoriatic epidermis. J Invest Dermatol. 1995;104(3):391–395. doi:10.1111/1523-1747.ep12665870
4. Piruzian E, Bruskin S, Ishkin A, et al. Integrated network analysis of transcriptomic and proteomic data in psoriasis. BMC Syst Biol. 2010;4:41. doi:10.1186/1752-0509-4-41
5. Eckert RL, Broome AM, Ruse M, et al. S100 proteins in the epidermis. J Invest Dermatol. 2004;123(1):23–33. doi:10.1111/j.0022-202X.2004.22719.x
6. Jinquan T, Vorum H, Larsen CG, et al. Psoriasin: a novel chemotactic protein. J Invest Dermatol. 1996;107(1):5–10. doi:10.1111/1523-1747.ep12294284
7. Hegyi Z, Zwicker S, Bureik D, et al. Vitamin D analog calcipotriol suppresses the Th17 cytokine-induced proinflammatory S100 “alarmins” psoriasin (S100A7) and koebnerisin (S100A15) in psoriasis. J Invest Dermatol. 2012;132(5):1416–1424. doi:10.1038/jid.2011.486
8. Varshney P, Saini N. PI3K/AKT/mTOR activation and autophagy inhibition plays a key role in increased cholesterol during IL-17A mediated inflammatory response in psoriasis. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5):1795–1803. doi:10.1016/j.bbadis.2018.02.003
9. Kim HR, Kang SY, Kim HO, Park CW, Chung BY. Role of aryl hydrocarbon receptor activation and autophagy in psoriasis-related inflammation. Int J Mol Sci. 2020;21(6):2195. doi:10.3390/ijms21062195
10. Furue M, Takahara M, Nakahara T, Uchi H. Role of AhR/ARNT system in skin homeostasis. Arch Dermatol Res. 2014;306(9):769–779. doi:10.1007/s00403-014-1481-7
11. Caputo R, Monti M, Ermacora E, et al. Cutaneous manifestations of tetrachlorodibenzo-p-dioxin in children and adolescents. Follow-up 10 years after the Seveso, Italy, accident. J Am Acad Dermatol. 1988;19(5 Pt 1):812–819. doi:10.1016/s0190-9622(88)70238-8
12. Mushtaque MD. Reemergence of chloroquine (CQ) analogs as multi-targeting antimalarial agents: a review. Eur J Med Chem. 2015;90:280–295. doi:10.1016/j.ejmech.2014.11.022
13. Akinduro O, Sully K, Patel A, et al. Constitutive autophagy and nucleophagy during epidermal differentiation. J Invest Dermatol. 2016;136(7):1460–1470. doi:10.1016/j.jid.2016.03.016
14. Sil P, Muse G, Martinez J. A ravenous defense: canonical and non-canonical autophagy in immunity. Curr Opin Immunol. 2018;50:21–31. doi:10.1016/j.coi.2017.10.004
15. Guilloteau K, Paris I, Pedretti N, et al. Skin inflammation induced by the synergistic action of IL-17A, IL-22, oncostatin M, IL-1α, and TNF-α recapitulates some features of psoriasis. J Immunol. 2010;184(9):5263–5270. doi:10.4049/jimmunol.0902464
16. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
17. Kennedy LH, Sutter CH, Leon Carrion SL, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol Sci. 2013;132(1):235–249. doi:10.1093/toxsci/kfs325
18. Sutter CH, Bodreddigari S, Campion C, Wible RS, Sutter TR. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol Sci. 2011;124(1):128–137. doi:10.1093/toxsci/kfr205
19. Loertscher JA, Sattler CA, Allen-Hoffmann BL. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation pattern of human keratinocytes in organotypic culture. Toxicol Appl Pharmacol. 2001;175(2):121–129. doi:10.1006/taap.2001.9202
20. Moriyama M, Moriyama H, Uda J, Matsuyama A, Osawa M, Hayakawa T. BNIP3 plays crucial roles in the differentiation and maintenance of epidermal keratinocytes. J Invest Dermatol. 2014;134(6):1627–1635. doi:10.1038/jid.2014.11
21. Schonthaler HB, Guinea-Viniegra J, Wculek SK, et al. S100A8-S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity. 2013;39(6):1171–1181. doi:10.1016/j.immuni.2013.11.011
22. Kypriotou M, Huber M, Hohl D. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the “fused genes” family. Exp Dermatol. 2012;21(9):643–649. doi:10.1111/j.1600-0625.2012.01472.x
23. Voss A, Bode G, Sopalla C, et al. Expression of S100A8/A9 in HaCaT keratinocytes alters the rate of cell proliferation and differentiation. FEBS Lett. 2011;585(2):440–446. doi:10.1016/j.febslet.2010.12.037
24. Ekman AK, Vegfors J, Eding CB, Enerbäck C. Overexpression of psoriasin (S100A7) contributes to dysregulated differentiation in psoriasis. Acta Derm Venereol. 2017;97(4):441–448. doi:10.2340/00015555-2596
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Metformin Inhibits HaCaT Cell Proliferation Under Hyperlipidemia Through Reducing Reactive Oxygen Species via FOXO3 Activation
Zhang L, Liu X, Huang M, Wang R, Zhu W, Li Y, Shen L, Li C
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1403-1413
Published Date: 22 July 2022
Adipose-Derived Stem Cell Exosomes Alleviate Psoriasis Serum Exosomes-Induced Inflammation by Regulating Autophagy and Redox Status in Keratinocytes
Kim HR, Lee SY, You GE, Kim HO, Park CW, Chung BY
Clinical, Cosmetic and Investigational Dermatology 2023, 16:3699-3711
Published Date: 23 December 2023
Indole-3-Lactic Acid Attenuates IMQ-Induced Psoriasiform Dermatitis in Mice via AhR-Dependent Suppression of IL-17A
Meng Z, Ren J, Lai Y, Lu S, Wu H, Jiang Y, Zhang Z, Tan G, Shi Z
Psoriasis: Targets and Therapy 2026, 16:585731
Published Date: 27 March 2026