")
Back to Journals » OncoTargets and Therapy » Volume 13
Effects of Atypical Protein Kinase C Inhibitor (DNDA) on Lung Cancer Proliferation and Migration by PKC-ι/FAK Ubiquitination Through the Cbl-b Pathway
Authors BommaReddy RR, Patel R, Smalley T, Acevedo-Duncan M
Received 29 July 2019
Accepted for publication 3 February 2020
Published 24 February 2020 Volume 2020:13 Pages 1661—1676
DOI https://doi.org/10.2147/OTT.S224866
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Raja Reddy BommaReddy, Rekha Patel, Tracess Smalley, Mildred Acevedo-Duncan
Department of Chemistry, University of South Florida, Tampa, FL 33620, USA
Correspondence: Mildred Acevedo-Duncan
University of South Florida, 4202 E Fowler Ave, CHE 205, Tampa, FL 33620, USA
Tel +1 813 748-4715
Email [email protected]
Purpose: The options for treating lung cancers are limited, as diagnosis typically occurs during the late stages of the disease. There is a dire need to develop aPKC (atypical Protein Kinase C) inhibitors due to aPKC overexpression and contributions to lung cancer malignancies. In this study, we investigate the role of atypical PKCs (aPKCs) in cell proliferation and migration in lung cancer cell lines and the effect of the novel aPKC inhibitor DNDA (3,4-amino-2,7 napthalene disulfonic acid).
Methods: The normal and lung cancer cells were treated with various concentrations of DNDA. We used a WST assay to determine lung cell viability, then analyzed cell apoptosis through Annexin V/PI staining and flow cytometry. Immunoprecipitation determined the proteins’ associations, and Western blot allowed testing of the expression of interest proteins. We also employed the UbiTest to identify the ubiquitination of the FAK. The scratch and transwell assays measured cell migration and invasion of lung cancer cells.
Results: Our data from cell viability and flow cytometry showed a significant reduction in cell proliferation and induction of apoptosis with DNDA treatment in lung cancer cells, as well as no toxic effect on normal BEAS-2B lung cells. Western blot results showed that the phosphorylation of PKC-iota and phosphorylation of FAK decreased in A549 lung cancer cells upon DNDA treatment. Immunoprecipitation (IP) data revealed an association of PKC-ι with FAK and FAK with Casitas B-lineage lymphoma proto-oncogene-b (Cbl-b). UbiTest results suggest that PKC-ι regulates FAK cleavage through its ubiquitination by Cbl-b, thereby inhibiting A549 lung cancer cells’ migration. This was evident from scratch, invasion, and migration assays.
Conclusion: Our study data suggest that DNDA inhibits cell proliferation and induces apoptosis in lung cancer cells. Moreover, DNDA inhibit A549 lung cancer cells’ migration by PKC- ι/FAK ubiquitination via Cbl-b.
Keywords: aPKC inhibitor, NSCLC, ubiquitination, FAK, migration
Introduction
On a global scale, lung cancer is one of the major causes of mortality, accounting for more deaths than any other cancer. In 2018, lung cancer estimates from the American Cancer Society described 234,030 new cases (121,680 men and 112,350 women) and 154,050 deaths (83,550 men and 70,500 women) in the United States.1 The largest contributor to lung cancer deaths in the United States is cigarette smoking at 82% of all lung cancer deaths.2 The historical differences in the uptake and reduction in tobacco use drive the worldwide lung cancer burden and trends. Demographic and geographic characteristics such as sex, age, race, ethnicity, and socioeconomic status have also contributed to the dramatic variations in lung cancer rates.3
Lung cancer arises as a result of the progressive appearance of genetic and epigenetic alterations, both in oncogenes and tumor-suppressor genes; ultimately, this leads to deregulated activation of mitogenic and survival signaling pathways. Scientists have extensively studied genetic alterations in non-small cell lung cancer (NSCLC) tumors, including oncogenic mutations in the epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), human epidermal growth factor 2 (HER2), mesenchymal-epithelial transition (MET), phosphatidylinositol-4,5-bisphosphate 3-kinase (PIK3CA), B-Raf proto-oncogene (BRAF), extracellular signal-regulated kinase (ERK) and Kirsten rat sarcoma 2 viral oncogene homolog (KRAS), as well as inactivation of tumor suppressor genes like p53, Phosphatase and Tensin homolog (PTEN), retinoblastoma protein (Rb), and cyclin-dependent kinase inhibitor 2A (p16).4
Protein Kinase C (PKC) is a multigene family composed of serine/threonine protein kinases involved in a variety of fundamental physiological processes that regulate cell growth, differentiation, apoptosis, transformation and tumorigenicity.5 There are 3 different groups in PKC isoenzymes: the classical PKCs (cPKCs), including the α, βI, βII, and γ isoforms stimulated by Ca+2, diacylglycerol (DAG) or phorbol esters; the novel PKCs (nPKCs), including the θ, η, ε, and δ isoforms which are Ca+2 independent and activated by diacylglycerol or phorbol esters; and, finally, the atypical PKCs (aPKCs), including the ζ and ι/λ isoforms that do not respond to Ca+2 and diacylglycerol or phorbol esters. Classifying different isoforms is based on the presence or absence of functional domains that confer specific co-factor and activator requirements.6
There has been considerable interest in aPKCs as drug targets because of the role they play in cancer development and progression. Most types of cells express multiple types of PKC isoform, and each PKC isoform mediates a specific function. Their distribution is variable in different tissues and subcellular locations.7 Among the aPKCs, studies have extensively characterized PKC- ι as an oncogenic kinase.8 PKC- ι is frequently overexpressed at the mRNA and protein levels in cancers of the lung,9 pancreas,10 breast, and many other tumors.11 PKC- ι is crucial in maintaining cell polarity architecture along with other polarity complex components, including the Partitioning-defective Protein 6 (Par 6), Partitioning-defective Protein 3 (Par 3) and Ras-related C3 botulinum toxin substrate 1 (Rac-1), which have been implicated in oncogenesis. The protein-protein interactions of Phox and Bem 1 (PB1) domains between PKC-ι and Par 6 are required for the transformed phenotype and Rac-1 activation in NSCLC cells.12
PKCs has been implicated in tumor cell migration promotion, specifically by the scaffolding of cytoskeletal proteins and phosphorylation of Focal Adhesion Kinase (FAK).13 FAK is a non-receptor tyrosine kinase linked to growth factor receptors and integrin signaling pathways that regulate biological processes like apoptosis, migration, and invasion.14 Studied indicate that Inhibition of FAK expression reduces the migration and invasion of epidermal growth factor (EGF) stimulated human carcinoma cells.15 In addition, FAK inhibition decreases the migratory behavior of endothelial cells and hepatoblastoma cells alike.16 Furthermore, FAK signaling is involved in the process of Transforming Growth Factor-β (TGF-β)-induced epithelial-to-mesenchymal transition (EMT) in hepatocytes.17 Cigarette smoke constituents – NNK (4-(methyl nitrosamino)-1-(3-pyridyl)-1-butanone) – activate the c-Src/FAK/PKC-ι pathway and positively regulate migration and invasion of human lung cancer cells.18
Dephosphorylation and proteolytic cleavage of FAK terminates FAK signaling. Scientists first observed the proteolysis of FAK in chicken embryo fibroblasts transformed by the myc or v-Src oncogene.19 Studies indicate Caspases cleaves FAK during apoptosis,20 Calpain in the v-Src-induced transformation of chicken embryo fibroblasts21 and the E3 ubiquitin ligase, Cbl.22 After integrin stimulation, Cbl recognizes the phosphorylated FAK before it degrades via the Cbl-dependent proteasomal pathway. During this process, Signal-Transducing Adaptor protein-2 (STAP-2) enhances the recruitment of Cbl to FAK. STAP-2 regulates integrin-mediated T cell adhesion by protein degradation of FAK through Cbl.23 Protein degradation controls the turnover rates and steady-state concentrations of all cellular proteins.24
The aPKC inhibitors we are investigating on different cancer cell lines in our laboratory include ICA-1, ζ-stat, ACPD and DNDA. Computational study shows that these small molecule inhibitors bind to the catalytic domain or a potential allosteric pocket in the aPKCs.25 In the present study, we focus on aPKCs’ involvement in cell proliferation, apoptosis, invasion and NSCLC migration. The cells’ lines we used in this study were normal lung cells (BEAS-2B) and metastatic (A549 and H1299) lung cancer cells. A novel aPKC inhibitor, DNDA, showed no toxic effect on normal (BEAS-2B) lung cells, though there was a significant decrease in the cell viability of metastatic (A549 and H1299) lung cancer cells. The treatment with DNDA altered the phosphorylation of aPKCs and FAK. In this study, we also investigate the aPKC role in metastasis by ubiquitination of FAK and its degradation through Cbl-b in NSCLC. Our data supports the notion that DNDA reduces migration of A549 lung cancer cells, specifically by inhibiting PKC-ι and regulating FAK ubiquitination via Cbl-b.
Cell Culture and Materials
We obtained the normal lung cell line, BEAS-2B, and metastatic lung cancer cell line, H1299 from American Type Tissue Culture (ATCC). We obtained another lung cancer cell line, A549, from the Moffitt Cancer Centre (Tampa, FL). We subcultured the BEAS-2B, A549, and H1299 cells and maintained them in T75 flasks containing Bronchial Epithelial Cell Growth Media (BEBM) (CC-3171) that we purchased from Lonza (F-12K (10-025-CV)). We purchased Roswell Park Memorial Institute (RPMI) media (10-041-CV) from Corning Life Sciences, and included supplementation of 10% FBS (Fetal Bovine Serum) (S1150) that we purchased from Atlanta Biologics, and finally 1% antibiotics (penicillin 10U/mL and Streptomycin 10 mg/mL) (30-002-CI) that we purchased from Corning Life Sciences. We incubated the cells at 37 ºC and 5% CO2.
We obtained DNDA (3,4-diamino-2,7-naphthalenedisulfonic acid) from the National Institute of Health (NIH; Bethesda, MD, USA). We made the stock solution in Dimethyl sulfoxide (DMSO) and diluted it with sterile distilled water before using it. We purchased the antibodies to anti-Phospho PKC-ζ (T410) (2060), anti-PKC-ζ (SC-216), and anti-PKC-ι (SC-727) from Santa Cruz Biotechnologies; we also purchased Anti-Caspase-3 (9579), anti-Poly adipose ribose polymerase (PARP) (5625), anti- B-cell lymphoma (Bcl-2) (2872) and anti-Bcl-XL (2764) from Cell Signaling Technology. Anti-FAK antibody (AHO0502) was purchased from Invitrogen, anti- Cbl-b antibody (12781-1-AP) was purchased from Protein Tech.
Methods
WST-1 Assay for Cell Viability
This study determined the inhibiting effect of the DNDA on the cell viability of the BEAS-2B normal lung cells and metastatic lung cancer cells (H1299 and A549) by employing a WST-1 assay. We then plated the cells in a 96-well plate at a final density of 2.0 X 103 cells/well. The media was changed after 24 hr of plating, and the cells were treated with different concentrations of DNDA (0.5, 1, 2.5, 5, 10, 20 µM) for 3 consecutive days, as well as incubated at 37ºC. After 24 hrs of each day’s (Day 1, Day 2, Day 3) treatment, we aspirated the media and transferred and pipetted a mixture of 180 μL of fresh media and WST-1 (20 μL) reagent to each well. The Synergy HT BioTek Plate reader acted as a tool to record the absorbance at 450 nm and 630 nm (used as reference absorbance). The measurements represent the average of three independent experiments.
Flow Cytometry
This study plated and treated the cells with DNDA for 3 consecutive days. After the third day of treatment, we aspirated the media and lifted the cells using a Hyclone HyQtase cell detachment solution. Centrifugation (12,000 X g) allowed us to collect the cells before discarding the supernatant. The cells were washed with a 1X binding buffer and resuspended in Annexin V Allophycocyanin (APC)/DAPI (100 μL) staining solution. Subsequently, the cells were mixed and incubated in the dark, at room temperature, for 10 mins, and PBS (200 μL) was added. Samples were analyzed on a BD FacsCanto II cytometer (San Jose, CA) with FacsDiva6.1.3 software. The study included exciting the APC dye using a 633 nm laser, as well as detecting emission at 660/20 nm. The DAPI was excited with a 405 nm laser, and the emission was detected at 450/50 nm.
Protein Isolation and Western Blot Analysis
In this section of the study we seeded the lung cancer cells on 100 mm plates, allowing them to become 50–60% confluent before starting DNDA (10μM) treatment. Following the treatments, we washed the cells with the 1X DBPS and incubated them with trypsin for 10 min. Trypsinized cells were transferred to 1.5 mL Eppendorf tubes before we centrifuged them for 15 min at 1000 X g. From each tube, we decanted the supernatant of the solution, and added a lysis buffer (with the protease and the phosphatase inhibitors) to each tube. We performed the protein assay by using the Bradford method, and we added a Laemelli sample buffer (LSB) to the cell lysate, boiling the mixture for 5 min at 95°C. We also loaded equal amounts (20–40 µg of protein) of samples and separated them using the Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) method. Subsequently, the bands were transblotted to a 0.4-micron nitrocellulose membrane and blocked with 4–5% non-fat dry milk powder in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 hr at room temperature or overnight on a rocker at 4°C. Afterward, we washed the membranes with the TBST and incubated them with primary antibodies at 4°C overnight in a rocker. Subsequently, we washed the membranes and probed them with the corresponding secondary antibodies (mouse or rabbit), finally developing them with the Electro Chemi Luminescence solution and Amersham GE instrument.
ImmunoPrecipitation (IP)
We lysed the cells with an IP lysis buffer (with protease and phosphatase inhibitors), scraped and collected in 1.5 mL tubes. Samples were sonicated for 10 sec on ice, then centrifuged at 4°C for 30 min at 12,000 X g. We collected the supernatant in new 1.5 mL tubes, calculating the protein concentration using a Bradford Assay. We added the lysate and IP lysis buffer to make the final concentration 200 μg/200 μL or 400 μg/400 μL, and incubated the lysates on a rocker at 4°C for 1 hr with 15 μL of magnetic agarose beads to preclear the lysate. We applied a magnetic field to pull beads aside, then added the supernatant to a new tube. Next, 3 μg of antibody was added to the supernatant and incubated at 4°C on a rocker overnight. Subsequently, we added 25 µL of the magnetic agarose beads to every tube and incubated them at 4°C on a rocker for 1 hr at room temperature. After incubation, we applied a magnetic field to pull the beads aside, then discarded the supernatant. The beads were washed with 500 µL 3X of IP lysis buffer by mixing gently before discarding the supernatant. We resuspended the bead pellet in 30 μL of 3X LSB buffer, then heated the samples at 70°C for 5 min and loaded them onto a SDS-PAGE gel.
In-vitro Kinase Assay
This study performed the in-vitro kinase assay by individually re-suspending IP endogenous PKC-ɩ (3 µg) and FAK (3 µg) from the control (untreated) and treated metastatic A549 lung cancer cells with DNDA in kinase buffer (20 mM Tris-HCl (pH 7.5)), 6 mM magnesium acetate, phosphatidylserine (5 μg), and finally adenosine triphosphate (ATP) (0.96 μg) (56). We initiated the reaction by incubating samples in a water bath for 10 min at 30 ºC, eventually terminating the reaction by adding a 3x sample loading buffer on ice. Subsequently, proteins were fractionated by SDS-PAGE – we analyzed these using immunoblotting.
Immuno Fluorescence Microscopy
In this step of the study we seeded the cells at low density in 2 wells chamber slides coated with Poly D-Lysine (1mg/mL) and treated with vehicle (DMSO) and DNDA at 10 µM concentration over the course of three consecutive days. Post-treatment, we fixed the cells in 4% paraformaldehyde for 10 min and permeabilized them with 0.1% Triton-X100 in PBS. After a 1 hr blocking step using 1% Bovine Serum Albumin (BSA) in a TBST (0.1%) solution, we incubated the cells with an anti-FAK primary antibody (1:500) for 1 hr. Subsequently, we used the Alexa 488 conjugated secondary antibody in PBS BSA (0.2%) (1: 200), and we observed fluorescence at 488 nm using a Nikon MICROPHOT-FX microscope with ProgRes®-Capture 2.9.0.1.
Phalloidin Staining of Filamentous (F) Actin
Here, we seeded cells at low density in 2-well chamber slides coated with Poly D-Lysine (1mg/mL), which had also been treated with vehicle (DMSO) and DNDA at 10 µM concentration for 3 consecutive days. We fixed cells in 4% paraformaldehyde and incubated them with Phalloidin-iFluor 594 in a 1% BSA solution for an hour at room temperature. Cells were washed with 1X PBS 3X, and counterstained with DAPI. We captured all images using ProgRes®Capture 2.9.0.1 on a Nikon MICROPHOT-FX fluorescence microscope (Ex/Em = 590/618).
Depletion of PKC-Ɩ and PKC- ζ Protein Expression by RNA Interference via SiRNA
We employed human siRNA for PKC-ɩ and PKC-ζ, which contained 3 unique 27 mer SiRNA duplexes and one universal scrambled negative control siRNA duplex. The Duplex sequences are as follows:
siPRKCZ
SR303747A- 5΄rGrCrArUrGrArUrGrArCrGrArGrGrArUrUrGrArCrUGG3΄
SR303747B-5΄rArGrUrArGrArCrGrArCrArArGrArArCrGrArGrGrArCrGCC3΄
SR303747C-5΄rArGrArArUrGrArCrCrArArArUrUrUrArCrGrCrCrArUrGAA3΄
siPRKCI
SR303741A-5΄rGrGrCrCrUrArCrArGrArUrGrArGrUrArArUrGrArArGrUTA3΄
SR303741B-5΄rGrCrCrArUrArArArCrUrCrGrUrCrArCrArArArUrUrGrArATG3΄
SR303741C-5΄rGrCrCrArUrUrUrArArUrGrCrArUrGrGrArUrArArArCrUTG3΄
We plated the A549 lung cancer cells in 60 mm flasks at 60% confluency, and aspirated the media after 24 hrs of plating before adding back 5 mL of fresh media. The cells were transfected with a 20 nM concentration of siRNA sequences using the transfection reagent siTrans 1.0, then incubated for 24 hrs. We lysed the cells with buffer and collected them in a 1.5 mL tube, and finally subjected the lysates to Western blot analysis.
UbiTest
We chose to employ the UbiTest kit because it has more advantages than traditional methods for studying ubiquitinated proteins. Enrichment of ubiquitinated proteins by Tandem ubiquitin binding entity (TUBE) pulldown and Deubiquitinating enzymes (DUB) digestion removes all the ubiquitin exposing the epitopes and confirms the substrate ubiquitination. The polyubiquitinated proteins were pulled down from the cell lysates using a non-linkage-selective TUBE. We split the samples in half after eluting the proteins from the TUBE. One half were treated with broad-spectrum DUB, (which hydrolyzes polyubiquitin chains of all linkages), while we left the other half untreated as a control. We compared the DUB treated portion with the DUB untreated portion using immunoblot for the protein of interest.
In the case of the A549 lung cancer cells, we plated them in 100 mm flasks at 50% confluency and treated them with DNDA (10 μM) for 3 consecutive days. In addition, we incubated cells with MG132 (20 μM) for 4 hrs before collecting the lysates with a lysis buffer (containing protease, phosphatase and DUB inhibitors). 20 μL of Ubiquitin Agarose resin was added to 1 mg of cell lysate and incubated for 2 hr on a rocker at room temperature, and beads were further collected at low centrifugation (5000 X g) for 5 min. We then decanted the supernatant and washed the beads twice with 1 mL of TBST before suspending them in 100 μL of elution wash buffer mixed for 5 min at room temperature. The beads were collected by centrifugation (as described above) and resuspended in 50 μL of elution buffer, mixed for 15 min at room temperature and collected once again using centrifugation. We carefully isolated and neutralized the supernatant with 5 μL of 10X neutralization buffer. The sample was divided into two parts. To part 1, we added 2 μL of 10mM DUB; to part 2, we added 2 μL of PBS. The samples were incubated at 30°C for 2 hrs, and adding 6X LSB stopped the reaction.
Scratch Assay
The A549 lung cancer cells were plated in a 6-well plate, and we made a scratch using a 20 μL sterile pipette tip after the cells formed a monolayer. Before beginning the treatment, we serum starved the cells for 24 hrs to eliminate the effect of cell proliferation in determining the wound closure rate. The cells were treated with the DNDA (10 μM) and vehicle (DMSO) for 1, 2 and 3 days and we monitored the closure of the scratch each day using imaging with a Motic iPad at 40X magnification.
Invasion and Migration Assay
This study investigated the migration and invasion of A549 lung cancer cells by plating the cells (cells were serum starved for 24 hrs) in the upper chamber of 96-wells Transwell permeable support (pore size: 0.8 µM) coated with and without 0.4X BME. The lower chamber received the chemoattractant (media with 10% FBS). We treated the A549 cells in the upper chamber with DNDA (10µM) for 3 days and DMSO for the control cells. Post-treatment, the media was aspirated carefully before we washed the cells with PBS. The invasive and migratory cells in the lower chamber were fixed by 4% paraformaldehyde and stained with 0.5% Crystal Violet in 1% ethanol. We washed them with PBS and imaged them using a Motic AE31E iPad at 40X magnification.
Densitometry
We quantified each band intensity using the 1D analysis software Alpha View (Protein Simple, San Jose, California). We subtracted the background intensity from each band to derive the corrected intensity.
Statistical Analysis
A Student’s t-test (two-tailed) tested the statistically significant differences between the DNDA-treated and control (vehicle) groups and a one-way ANOVA with a Tukey’s multiple comparisons test was performed using R Studio software.
Results
aPKCs are Overexpressed in Metastatic Lung Cancer Cells
To identify the role of aPKCs in lung cancer malignancy, this study determined the expression levels of aPKCs in BEAS-2B normal lung cells and metastatic (A549 & H1299) lung cancer cells at 50% and 100% confluences (Figure 1A). We compared the expression levels at 50% confluency between the cell lines. At 50% confluency, cells were rapidly growing owing to the fact that there was more space and replenishing growth factors, including PKC-iota (which we believe plays an important role in proliferation). However, at 100% confluency, cells became contact inhibited, and they were non-proliferating. The inhibitor was used at 50% confluency when the PKC-ɩ levels in the cells were abundant. We applied this method in subsequent experiments to maintain a level of “control” and eliminate any treatment variation. Additionally, we applied this method to decrease treatment bias regarding the 100% confluent cells which could render an alternative effect. PKC- ι was overexpressed in A549 lung cancer cells by approximately 1.53-fold more when compared to those of normal BEAS-2B lung cells. Moreover, PKC-ζ was not overexpressed in A549 and H1299 lung cancer cells. Overexpression of FAK did take place in A549 lung cancer cells by approximately 3.6-fold more and a 2.79-fold increase in H1299 lung cancer cells compared to normal BEAS-2B lung cells (Figure 1B). This data suggests that PKC- ι plays a role in lung cancer proliferation.
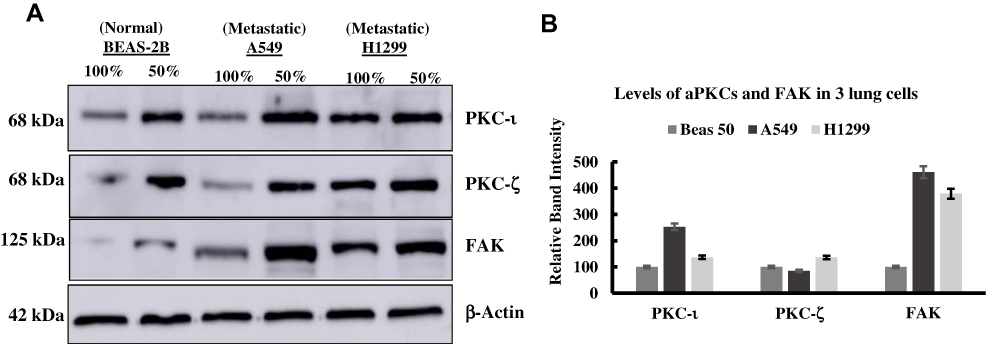 |
Figure 1 (A) Expression profile of PKC-ι, PKC-ζ, FAK proteins in normal (BEAS-2B) and metastatic (H1299, A549) lung cancer cells at different confluences. (B) The bar graph represents the various protein expression levels at 50% confluency. The PKC-ι and FAK were overexpressed in A549 lung cancer cells when compared to normal BEAS-2B lung cells. Equal amounts of protein (30 μg) was loaded in SDS-PAGE as indicated by the loading control β-actin. |
Cell Viability of Normal and Metastatic Lung Cell Lines by the aPKC Inhibitor (DNDA)
To evaluate the effects of DNDA (Figure 2) on cell viability in (BEAS-2B) normal lung cells and metastatic (A549 & H1299) lung cancer cells, we treated cells with varying concentrations of DNDA (0.1, 0.5, 1, 2, 5, 10, 20 µM) for 3 consecutive days. The WST method quantified cell viability by recording the absorbance at 450nm. Resulting data indicated that there was no significant toxicity (p <0.05) for normal lung cells, even at 20 µM (Figure 3A). The lack of toxicity to normal lung cells is crucial because it supports using the aPKC inhibitor as a potential therapeutic agent. The cell viability on H1299 and A549 lung cancer cells showed reduced cell viability in a dose-dependent manner (Figure 3B and C). The results showed that cell viability of H1299 lung cancer cells decreased by approximately 45% (p < 0.001) with a 10 µM DNDA treatment after 3 days (Figure 3D). In A549 lung cancer cells, there was about 39% (p < 0.001) reduction in cell viability using a treatment of 10 µM DNDA over the course of 3 days (Figure 3E). These results illustrate the paramount role that aPKCs play in lung cancer cell proliferation.
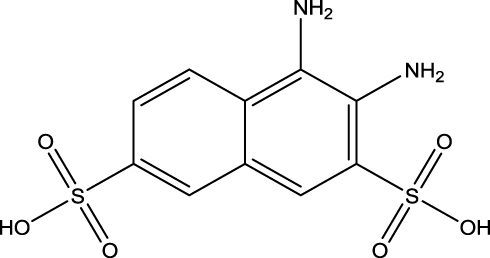 |
Figure 2 Chemical Structure of DNDA (3,4- diamino-2,7-napthalene disulfonic acid). |
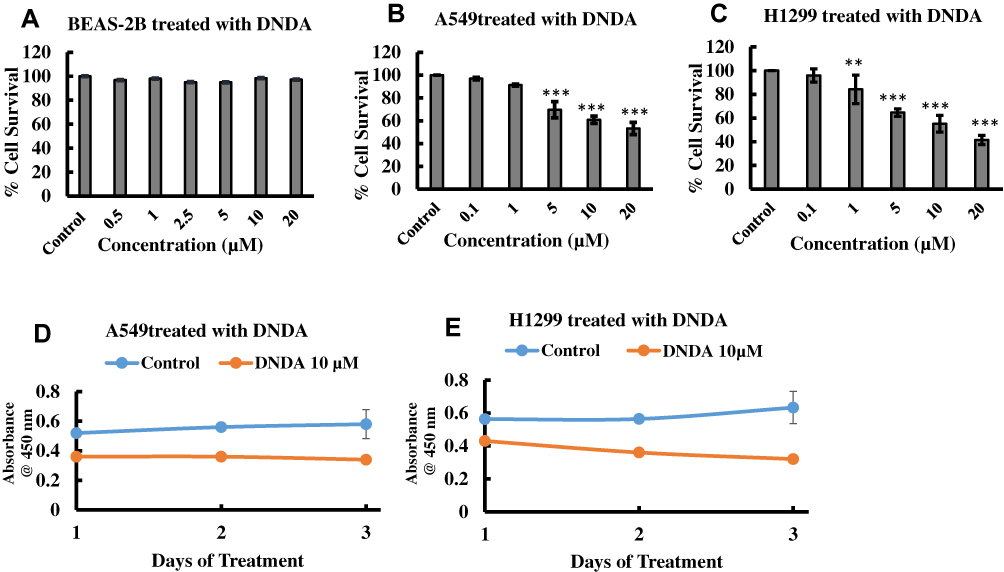 |
Figure 3 (A–C) Dose Response curve of DNDA on BEAS-2B (normal lung cells) and metastatic (A549 & H1299) lung cancer cells. The cells were treated for 3 consecutive days with the vehicle (DMSO), 0.5, 1, 2.5, 5, 10, 20 μM of DNDA and the cells were quantified using WST-1 assay by recording the absorbance at 450 nm after third day treatment. The results indicate DNDA had no toxic effect on normal lung cells and cell viability was reduced in a dose dependent manner in metastatic A549 and H1299 lung cancer cells. (D) Effect of DNDA 10 μM on cell viability of H1299 lung cancer cells treated for 1,2,3 days. Cells were treated for 3 consecutive days and absorbance of WST-1 at 450 nm was recorded for each day by using BioTek Plate reader. DNDA reduced cell viability of H1299 lung cancer cells by 45% and (E) DNDA 10 μM reduced cell viability of A549 lung cancer cells by 39%. The data represents three independent experiments, Mean ± S.E.M. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc test. Statistical significance is represented by p value where ** < 0.01, *** < 0.001. |
Induction of Apoptosis in Metastatic Lung Cancer Cells
Since DNDA treatment of metastatic (A549 & H1299) lung cancer cells significantly reduced cell proliferation, we further used Western blot analysis and flow cytometry methods (Figure 4C–H) to investigate whether knocking down aPKCs could induce apoptosis by determining the expression levels of various apoptotic and anti-apoptotic proteins (Figure 4A and B). Our data showed a decrease in amounts of survival proteins like Bcl-2 by 5% and 53% (p < 0.001), Bcl-XL by 22% and 44% (p < 0.01), and Survivin by 10% and 51% (p < 0.001) in H1299 and A549 cells, respectively. There was a decrease in Caspase-3 by 4.5% and 44% (p < 0.001) and an increase in cleaved Caspase-3 by 3% and 49%, as well as a decrease in PARP by 9% and 15% (p < 0.05) in H1299 and A549 lung cancer cells, respectively (Figure 4A and B). Additionally, we performed flow cytometry to analyze the apoptotic events that DNDA treatment induced after 3 days. There was no significant effect on the early apoptosis in both metastatic cell lines. The late apoptotic event results showed an increase of 0.8% (p <0.05) and 10.8% (p < 0.001) in H1299 (Figure 4C–F) and A549 lung cancer cells respectively. The Western blot data of apoptotic markers and flow cytometry analysis results suggest that inhibition of aPKCs by DNDA in metastatic lung cancer cells induced apoptosis in the current study.
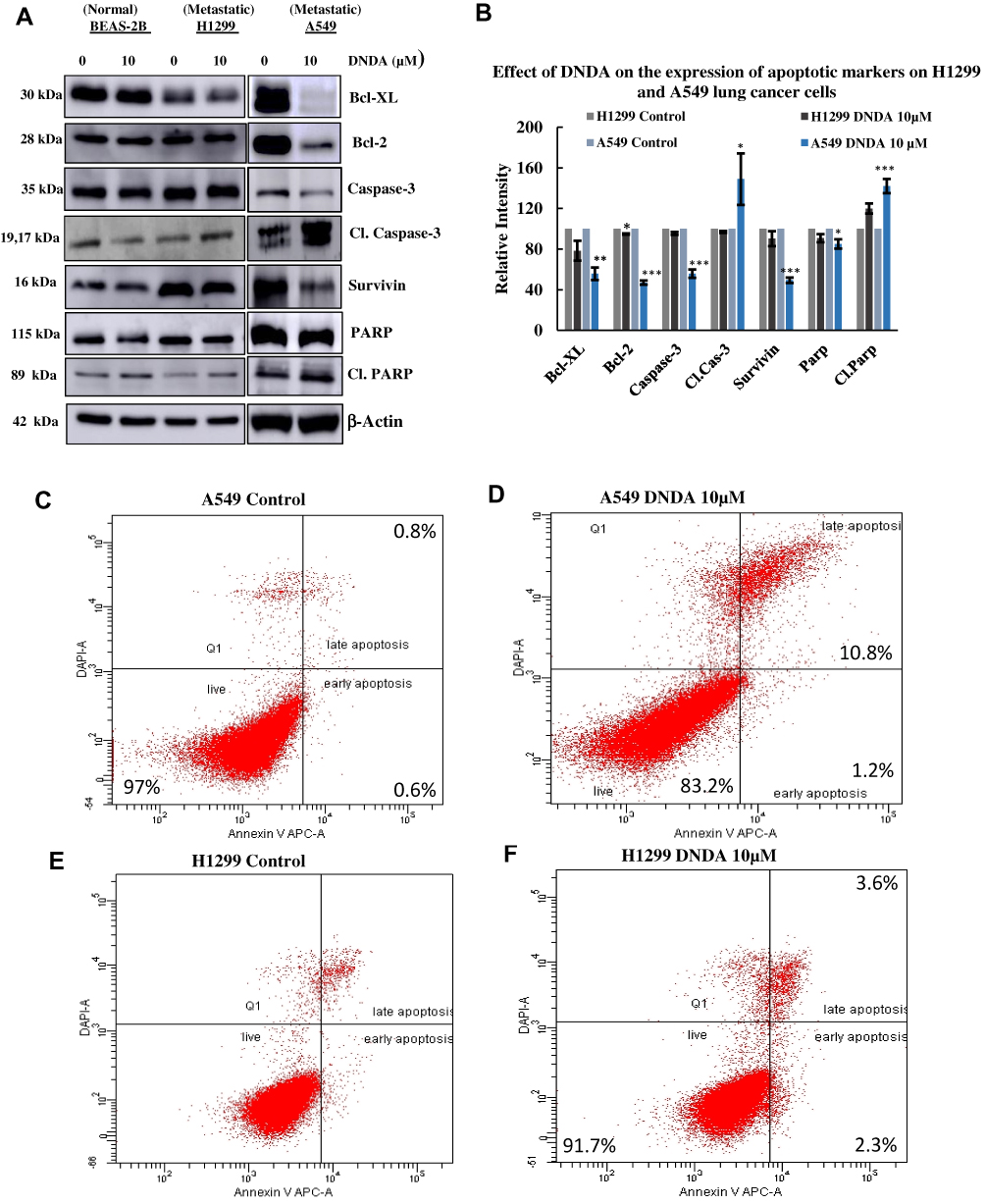 |
Figure 4 (A, B) Induction of apoptosis in metastatic lung cancer cells. The metastatic lung cancer cells were treated with DNDA 10 μM for 3 days and an equal amount of protein (30 μg) from cellular extracts were subjected to SDS-PAGE, followed by Western blot analysis. Expression levels of apoptotic markers from Western blot data revealed that treatment with DNDA promotes apoptosis by significant reduction of anti-apoptotic markers – Bcl-xL, BCL-2, Caspase-3, PARP, Survivin and increase in cleaved caspase −3, Cl.Parp fragments in A549 lung cancer cells and a decrease of Bcl-xL, BCL-2 in H1299 lung cancer cells. β-actin was analyzed as a loading control. (C–H) Flow cytometry analysis of apoptotic events on metastatic lung cancer cells. Density plots of various events in apoptosis with the treatment of DNDA 10 μM on A549 (C, D) and H1299 (E, F) lung cancer cells from flow cytometry. The cells were treated for 3 days with the vehicle (DMSO), 10 μM of DNDA and the cells were subjected to analysis on a BD FacsCanto II cytometer. The late apoptosis of DNDA treated cells (A549) were significantly higher than the control. The data represents n =3 independent experiments, Mean ± S.D. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc test. Statistical significance is represented by p value where * < 0.05, ** < 0.01, *** < 0.001. |
Inhibitory Effect of DNDA on aPKCs in Lung Cancer Cells
DNDA is an inhibitor of both aPKCs, so we first determined the inhibition of DNDA in both aPKCs and FAK in lung cancer cells. We analyzed the phospho and total levels of aPKCs and FAK in both metastatic cells using Western blot analysis (Figure 5A). There was a significant reduction in the phosphor levels of PKC- ι (T555) by 46% (p = 0.0002) and FAK (Y397) in A549 cells upon treatment with DNDA. In comparison, there was no significant reduction in phospho PKC-ζ T410 (12%) in A549 lung cancer cells. There was a significant decrease in phospho-PKC- by 18% (p = 0.004), and there was no significant decrease in phospho-PKC-ζ (7.5%) or phospho- FAK (4%) in H1299 lung cancer cells; likewise, we observed no significant changes in the expression of total PKC-ι and FAK in both A549 & H1299 lung cancer cells with DNDA treatment for 3 days (Figure 5B and C). These results suggest that targeting PKC-ι inhibition will decrease the FAK activity in lung cancer cells, making the DNDA an excellent candidate for lung cancer treatment.
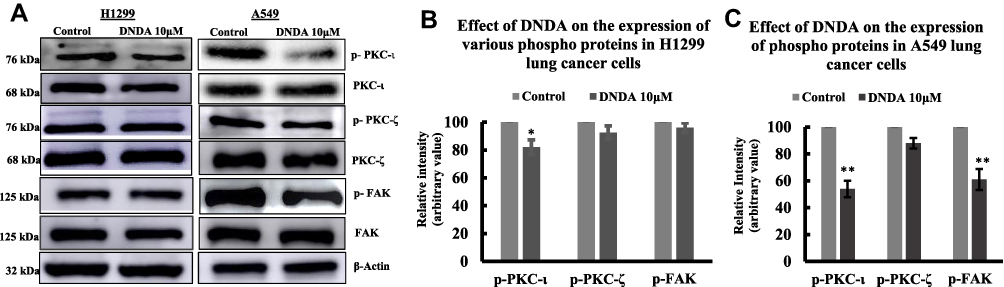 |
Figure 5 Effect of DNDA on the expression of phospho and total aPKCs, and FAK in metastatic (H1299, A549) lung cancer cells. (A) Western blot analysis for the effect of DNDA in A549 and H1299 lung cancer cells following 3 days of treatment with DNDA 10μM on the expression of phospho and total PKC-ι, PKC-ζ, and FAK. Equal amounts of protein (30 μg) were loaded in SDS-PAGE as normalized by loading control β-actin. (B) The bar graph depicts the densitometry of phospho PKC-ι, PKC-ζ, and FAK levels upon DNDA 10μM treatment in H1299 lung cancer cells and (C) A549 lung cancer cells. The data represents three independent experiments, Mean ± S.D. * represents p value where * < 0.05, ** < 0.001. |
Small-Interfering RNA (siRNA)
To establish a proof of concept that aPKCs were regulating the phosphorylation of FAK, we treated cells with siRNA against PKC-ι and PKC–ζ. The A549 lung cancer cells were transfected with either one of the three siRNA sequences for both PKC-ι and PKC–ζ, while we used a scrambled RNA sequence as a control. By running the Western blot analyses, we analyzed the effectiveness of each sequence as it pertains to knocking down the target protein expression. Results showed that siRNA sequence B significantly reduced the PKC-ι expression by 53% (p < 0.01) compared to other sequences (Figure 6A and E). Moreover, siRNA sequence A significantly decreased the PKC-ζ expression by 40% (p < 0.01) compared to other sequences (Figure 6B and E). We used siPRKCI sequence B and siPRKCZ sequence A to establish the mechanism of aPKCs in regulating FAK activity. Interestingly, knocking down PKC-ι significantly reduced FAK expression by a figure of 34% (p < 0.01) (Figure 6C and F). On the contrary, knocking down PKC-ζ did not change the expression of FAK (Figure 6D and F).
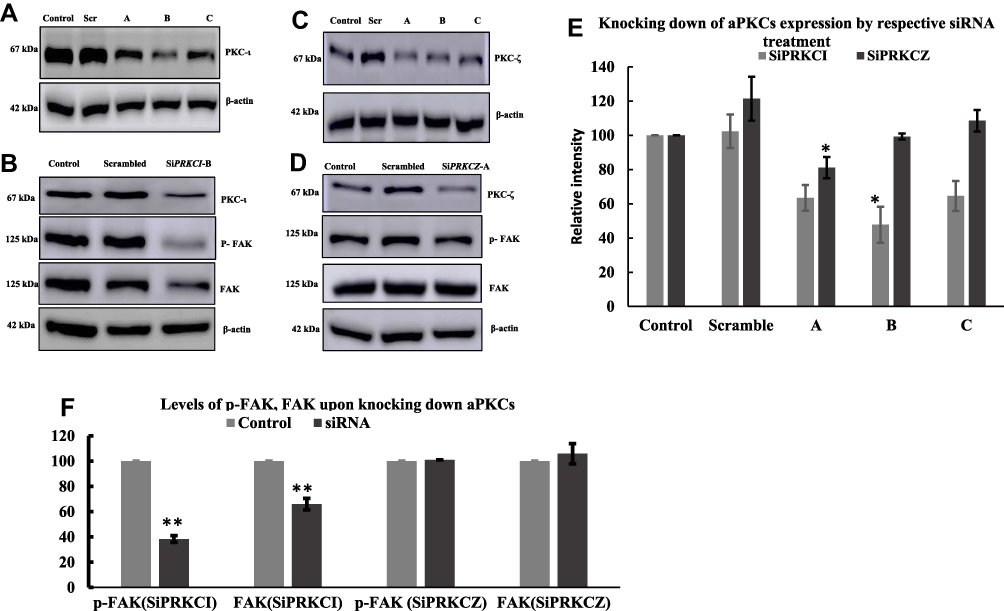 |
Figure 6 siRNA treatment of A549 cells with SiPRKCI (A, B) and SiPRKCZ (C, D). Cells were plated in 60 mm flask and at 60% confluency, media was replaced with fresh media. The siRNA concentration used was 20 nM and SiTrans1.0 was used to transfect the cells. One negative scrambled control sequence was used to ensure the targeted gene silencing was not from the reagents. The cells were collected after 24 hrs of transfection and subjected to Western blotting to determine the expression of proteins PKC-ι, PKC-ζ and β-actin. (E, F) Results showed that for PRKCI, the Sequence B knocked down more PKC-ι when compared to other sequences and Sequence A worked for PKC-ζ. Interestingly, knocking down PKC-ι reduced the protein level of p-FAK and FAK. In Contrast, knocking down PKC-ζ did not reduce the expression level of p-FAK and FAK. The data represents three independent experiments, Mean ± S.E.M. * represents p value where * < 0.05, ** < 0.005. |
ImmunoPrecipitation (IP)
To investigate the association of aPKCs with FAK and Cbl-b, we individually considered IP PKC-ι, PKC-ζ, FAK, Cbl-b and checked for the associated proteins by performing Western blot analyses. Our results showed there was an association of PKC-ι with FAK and the reverse IP also showed an association of FAK with PKC-ι (Figure 7A and B). The PKC- ζ showed no observable association with FAK (Figure 7C). Cbl-b did not associate with either PKC-ι or PKC- ζ, (Figure 7A, C, D) but there was an association between Cbl-b and FAK (Figure 7B and D). The FAK IP sample treated with DNDA 10 μM showed increased FAK degradation when we detected cleaved FAK fragment at approximately 100 kDa (Figure 7B). The extent of association of various proteins between the control and DNDA treated groups were quantified in the bar graph (Figure 7E–G). From the immunofluorescence images (Figure 7H), it is evident the DNDA treated cells had cleaved FAK when compared to control cells.
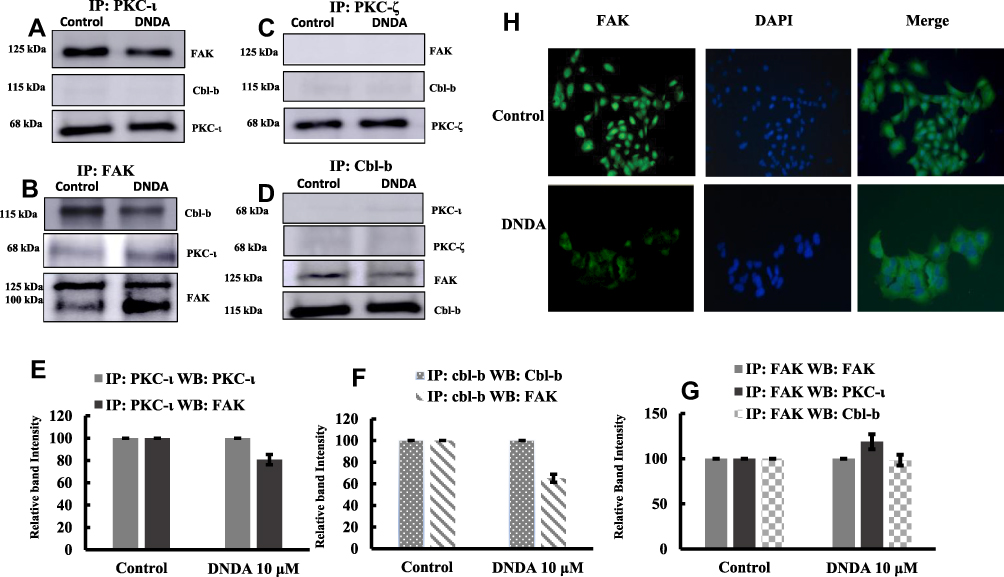 |
Figure 7 (A-D) Immunoprecipitation results showing the association of PKC-ι with FAK and FAK with Cbl-b. Initially, 3 μg of PKC-ι, PKC-ζ, FAK and Cbl-b were IP separately from whole cell lysates of control and DNDA treated A549 NSCLC cells and probed with various antibodies to determine the association between PKC-ι, PKC-ζ, FAK and Cbl-b. Immunoblots showed the association of PKC-ι with FAK, Cbl-b with FAK. In FAK IP samples, cleaved fragments of FAK were observed with the treatment of DNDA. There was no association found between PKC-ζ with FAK or Cbl-b. (E-G) Bar graphs show the densitometry of the associated proteins between control and DNDA treated cells (H) Immunofluorescence of FAK also complements FAK cleavage with treatment of DNDA when compared to the cells treated with vehicle (DMSO). |
Kinase Assay
Considering the association of PKC-ι and FAK in the A549 lung cancer cells, we wanted to determine whether FAK was the direct substrate of PKC-ι. Toward this goal, we performed an in-vitro kinase activity assay. Derived from the co-IP of PKC-ι and FAK, the data revealed that the levels of p-FAK and FAK had noticeably increased compared to those of individual IP samples. We did not observe any association of p-PKC-ι with individual IP FAK, indicating that the FAK is a possible downstream substrate of PKC-ι in A549 lung cancer cells. Reduction of PKC-ι expression by the DNDA also caused a marked reduction in the levels of p-PKC-ι, p-FAK and FAK (Figure 8A–E). Such data suggests that FAK is downstream of PKC- ι.
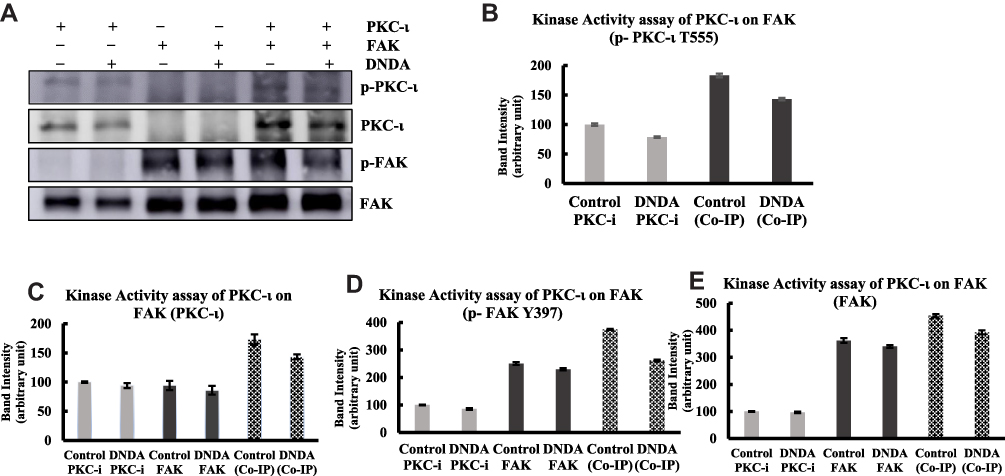 |
Figure 8 Kinase activity assay of PKC-ι on FAK phosphorylation. (A) A549 cells were treated with either DMSO or DNDA 10μM for 3 days. PKC-ι and FAK were IP (3 μg) separately and co-IP from whole cell extracts using specific antibodies. The proteins were then separated by SDS-PAGE, followed by immunoblotting to determine the p- PKC-ι at T555, PKC-ɩ, p-FAK (Y-397) and FAK. The inhibition of PKC-ι activity by DNDA, inhibits the phosphorylation of FAK. (B-E) Densitometry was used to quantify the intensity of respective bands. The data represents three independent experiments; Mean ± S.D. |
Ubiquitination
We investigated the ubiquitination of FAK by employing three different methods. First, we applied a conventional method in which the FAK was IP and probed with the ubiquitin antibody. The immunoblot showed the smear of bands when developed for the ubiquitin, and a FAK band (125 kDa) increased by 40% in the DNDA-treated lane (Figure 9A). Second, we performed the UbiTest, which offered a greater number of advantages compared to the conventional methods in terms of finding the FAK ubiquitination. Figure 9B shows that lanes (1–4) remained unincubated with MG132, and lanes (5–8) were incubated with MG132 for 4 hrs prior to collecting the lysate (Figure 9B). The PBS-treated (non-DUB-treated) lane (e.g., lane 5) contains all the ubiquitylated proteins from the lysate. These proteins travel at higher molecular weights and are very difficult to visualize. However, in the corresponding DUB-treated lane (e.g., lane 6), ubiquitin removal means that any ubiquitylated proteins travel at their unmodified molecular weight. If the protein of interest is ubiquitylated, we therefore expect to see the increase of a band at the unmodified molecular weight of that protein in the DUB-treated lane of that sample. When we probed the blot for the ubiquitin, it became evident that the DUB-treated lanes had a less intense smear of bands compared to those of the non-DUB-treated lanes. When we probed the blot for FAK (lane 5), FAK and its cleaved products were visually predominant compared to DUB-treated (lane 6). Lane 8 (DNDA- and DUB-treated) had 66% more FAK and cleaved FAK than lane 7 (DNDA- and non-DUB-treated) (Figure 9B and C)
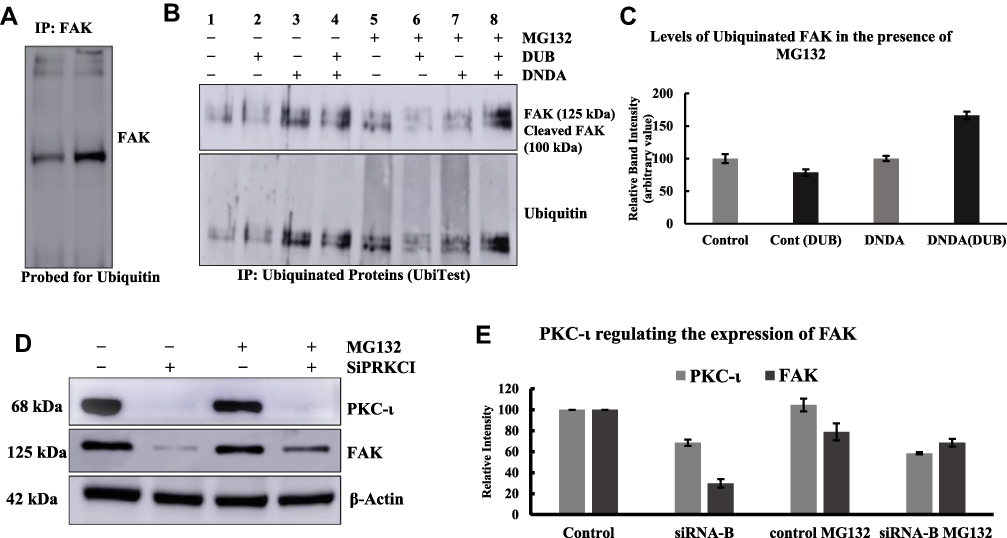 |
Figure 9 Determination of ubiquitination of FAK in A549 lung cancer cells by the treatment with DNDA 10μM for 3 days. (A) The IP of FAK (3 μg) was immunoblotted for ubiquitin and found that there was an increase in the FAK (125 kDa) band in the DNDA treated. (B, C) UbiTest was employed to determine the ubiquitination of FAK. The results showed the FAK (125 kDa) and cleaved FAK (100 kDa) were ubiquitinated with the DNDA treatment. (D) To explore the PKC-ι involvement in the ubiquitination of FAK, after transient transfection, cells were treated with 20μM proteasomal inhibitor MG-132 or DMSO for 4 hrs before collecting the lysates. The FAK protein expression was concurrently decreased when PKC-ι was repressed in the absence of MG132. However, FAK expression was increased when PKC-ι was repressed in the presence of MG132. (E) The intensity of respective bands were quantified by using densitometry. This result indicate that the ubiquitinated FAK undergoes proteasomal degradation and PKC-ι regulates this process. |
Last, we employed the SiRNA treatment with siPRKCI in the presence of MG132 to check whether the proteasomal inhibitor treatment would bring back the FAK expression. In theory, if the proteasome is inhibited, ubiquitinated proteins cannot be degraded. The Western blots data showed that PKC- ι knockdown decreased the expression of FAK by 70% in the absence of MG132. On the contrary, FAK expression returned with siPRKCI in the presence of MG132 by 40% (Figure 9D and E). This data implies that FAK undergoes ubiquitination and the presence of MG132 inhibited its subsequent degradation.
Scratch Assay
To examine the anti-migratory effect of DNDA in the A549 lung cancer cells, we employed a scratch wound healing assay. We treated cells either with vehicle control or DNDA for 24, 48 and 72 hr. Control cells demonstrated their migration potential consider the results of 62% wound repair in 72 hr, and DNDA suppressed the wound healing by 21%. This implies that DNDA treatment inhibits A549 lung cancer cell migration (Figure 10A and B).
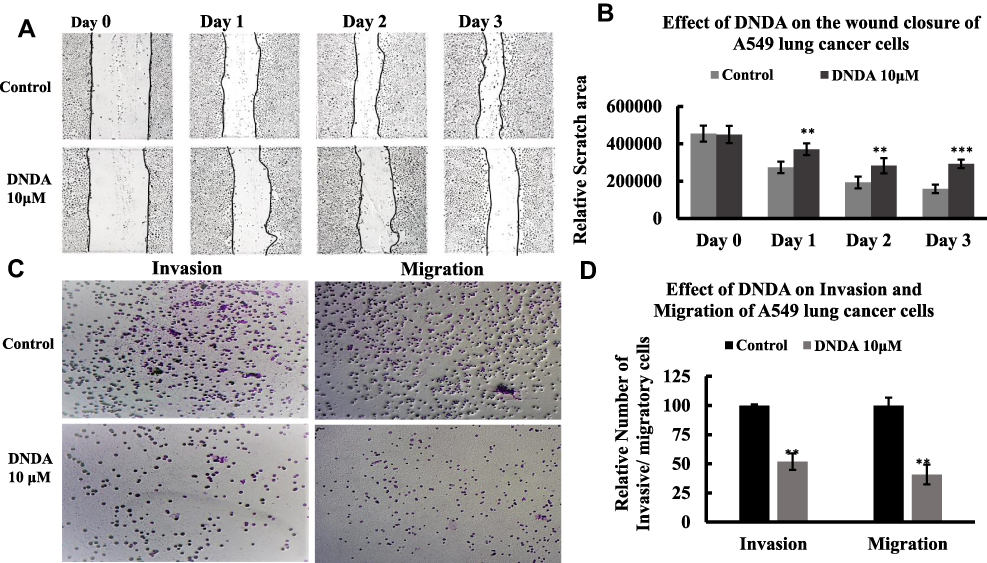 |
Figure 10 (A, B) DNDA impairs the in-vitro migration potential of A549 lung cancer cells independent of cell proliferation. (A) The A549 lung cancer cells were grown in 6- well plates and after the monolayer was formed, scratch was made using sterile 20 μL pipette tip, cells were washed with PBS and the treated with DNDA 10μM each day and photographs were captured for three days. The cells were starved for 24 hrs before the scratch was made to avoid the wound healing from the cell proliferation. (B) the bar graph represents the wound closure rate of DNDA treated cells compared to those of Control cells (untreated cells). (C, D) Crystal violet staining was performed to show the inhibition of invasion and migration of the A549 lung cancer cells by DNDA treatment. The cells were platted in upper chamber of 96-well Transwell permeable support (pore size: 0.8 μm) coated with BME and without coating to study the invasive and migration ability of A549 lung cancer cells. The 10% FBS was added to the lower chamber. The cells were treated for 3 days and cells that are invaded and migrated to the lower chamber were stained with crystal violet. The number of invasive and migratory cells were counted by using the ImageJ software. The DNDA treatment for 3 days reduced the invasion and migration of A549 lung cancer cells. Data represents the n=3 independent experiments, mean ± S.E.M. * represents p value where ** < 0.005, *** <0.0005. |
Invasion and Migration
We plated the A549 cells in the upper chamber of 96-well Transwell permeable support coated with and without BME, which allowed us to study the invasive and migratory abilities of lung cancer cells. The lower chamber was supplied with media that possessed 10% FBS, which attracted the cells to migrate. We treated the cells with DNDA for 3 days, fixing cells in the lower chamber with 4% paraformaldehyde and staining them with 0.5% Crystal Violet in 1% ethanol. Cells were washed with PBS and counted using the ImageJ software FIJI bundle. DNDA treatment reduced the invasive behavior of A549 cells by 50% (p = 0.002) and significantly decreased migration potential by 60% (p = 0.005) (Figure 10C and D). These results suggest that DNDA reduces the invasion and migration of A549 lung cancer cells.
Discussion
Remarkable advances in molecular pathology have enhanced understanding of the underlying heterogeneity of NSCLC. Studies have clearly identified specific oncogenic mutations and multiple signaling pathways that lead to malignant transformations in lung cancer.26 Atypical PKCs are the most common dysregulated kinases in breast, prostate, colon, pancreatic, kidney and liver cancers.27 PKC-ι is overexpressed in NSCLC cell lines and primary tumors, and there is compelling evidence that the PKC-ι isozyme is an oncogene in NSCLC.28
Recently, our laboratory published a study on the effect of DNDA on cell proliferation, apoptosis and its role in the EMT pathway of metastatic melanoma cells.29 The nature of aPKCs functionality and involvement with major cancer-causing pathways such as AKT, NFκB and WNT drove us to focus on a detailed investigation into lung cancer. Our collaborator docked more than 300,000 compounds against the two isoforms of aPKCs. Out of this in-silico experiment, we identified the DNDA small molecule as one of the best hits of aPKCs. Our recently published study showed that DNDA interacted with the catalytic domains of PKC- ι (amino acid residues of Asp 339, Asp 382, Leu 385 and Thr 395) and PKC-ζ (at Asp 337, Asp 380, Leu 383 and Thr 393). Moreover, DNDA showed selective inhibition of PKC-ι and PKC-ζ from the kinase activity data.29 We investigated the role of aPKCs in NSCLC carcinogenesis. After examining the levels of aPKCs, we found PKC-ι protein expression to be significantly higher in both metastatic A549 and H1299 lung cancer cells, whereas there was no increase in the expression of PKC-ζ in metastatic A549 and H1299 lung cancer cells; these results support other findings.30 Normal lung cells showed low levels of aPKCs (Figure 1A and B). Such findings suggest a relationship between cell growth and aPKCs, which agrees with data from other studies.9 The cell viability data showed that DNDA treatment resulted in a significant decrease in the cell viability of metastatic lung cancer cells (Figure 3). The apoptotic markers determined by Western blot demonstrated that DNDA treatment also induced apoptosis in metastatic lung cancer cells (Figure 4A and B). Furthermore, flow cytometry data complemented these findings (Figure 4C–F).
To investigate the role of aPKCs in the carcinogenesis of NSCLC, we examined the phospho levels of PKC-ι, PKC-ζ and FAK in metastatic A549 and H1299 lung cancer cells (Figure 5). Data revealed that, when treated with DNDA, there was a significant reduction in the phosphorylation of PKC-ι and FAK in A549 lung cancer cells and no significant reduction in the phosphorylation of FAK in H1299 lung cancer cells. Furthermore, when we immunoprecipitated FAK after DNDA treatment, we found there to be an increase of protein cleavage of FAK in comparison to the control. This infers that FAK is being degraded upon treatment of DNDA.
To elucidate how the aPKCs regulate the FAK protein expression, we performed siRNA experiments for aPKCs. The resulting data showed that the knock down of PKC-ι resulted in a significant decrease in FAK expression. On the contrary, the knock down of PKC-ζ had no effect in terms of FAK expression (Figure 6). This data suggests that PKC-ι is involved in regulating FAK but not PKC-ζ in A549 lung cancer cells.
Additionally, in this study we explored the association of aPKCs with FAK and Cbl-b. Our resulting data indicated an association of PKC-ι with FAK (Figure 7A and B). Our results agreed with Jie Shen et al18 in which NNK promotes migration and invasion of lung cancer cells through activation of a c-Src/PKC-ι/FAK loop. Moreover, we observed cleaved fragments of FAK in the DNDA-treated IP (FAK) sample, suggesting that DNDA treatment degrades FAK. This finding was also complemented by immunofluorescence data (Figure 7H), where FAK cleavage was also observed with the DNDA treatment. Moreover, an in-vitro kinase assay revealed that PKC-ι and FAK were associated, but it also revealed that DNDA treatment decreased the FAK phosphorylation and dissociation (Figure 8).
In previous studies, the ubiquitination of FAK via Cbl-b resulted in cell detachment in gastric cancer MGC803 cells.31 However, scientists have not yet investigated FAK ubiquitination via the Cbl-b pathway with PKC-ι involvement in NSCLC. In accordance with the IP data, we observed an association between FAK with Cbl-b (Figure 7B and D). To explore the ubiquitination of FAK, we probed the IP (FAK) for ubiquitin and showed an increase in the FAK band at 125 kDa with DNDA treatment (Figure 9A). Data from the UbiTest also demonstrated that the DNDA treatment caused higher ubiquitinated FAK and its cleaved products (Figure 9B). Furthermore, we examined the involvement of PKC-ι in FAK degradation. We treated the A549 lung cancer cells with siPRKCI as well as the proteasomal inhibitor (MG132), then applied a Western blot analysis. The data showed that the knock down of PKC-ι significantly reduced the expression of FAK in the absence of MG132. On the contrary, there was a significant increase in FAK levels in the presence of MG132, indicating the role of PKC-ι in FAK protein stability (Figure 9D and E).
The most common feature of cell motility is the formation of the F-actin rich protrusions by which cells extend forward to adhere to their surroundings. A recent study found that, in lung cancer adenocarcinoma cells, the downregulation of PKCζ/Pard3/Pard6 mediated EMT and invasion.32 The migration of A549 lung cancer cells from the scratch assay showed that DNDA treatment for 3 days had decreased the migratory ability of lung cancer cells (Figure 10A and B). The invasion and migration analysis of A549 cells demonstrated that DNDA treatment also caused a significant reduction of the invasion of cells and a dramatic decrease in migratory behavior (Figure 10C and D). Jie Shen et al demonstrated that specific inhibition of c-Src, PKC-ι or FAK expression by RNA interference significantly reduced NNK-induced cell migration and invasion.18 Our data suggests the DNDA treatment reduces A549 lung cancer cell invasion and migration. Moreover, the phalloidin F-actin staining revealed that the DNDA treatment reduced the actin polymerization (Figure 11). The pathway by which aPKC regulates F-actin expression is still elusive, and further efforts are required to understand the mechanisms underlying NSCLC metastasis.
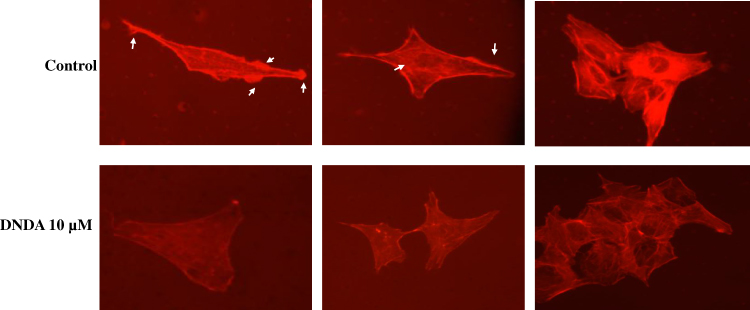 |
Figure 11 Effect of DNDA on the F-actin organization in A549 lung cancer cells. F-actin reorganization was modulated by DNDA 10μM treatment for 3 days in A549 cells. Actin organization was visualized by performing the F-actin staining with Phalloidin TxRed. White arrows, in the vehicle control cells, indicate the membrane protrusions enriched in actin meshwork, which is characteristic for motile cells. In contrast, cells treated with DNDA were flattened with visible, stretched actin stress fibers. |
In conclusion, the sequence of events following the treatment of DNDA of lung cancer cells is illustrated by the mechanism described in Figure 12. DNDA treatment disrupts the association of PKC-ɩ and FAK, leading to the ubiquitination of FAK by Cbl-b (an E3-ligase). Subsequently, the ubiquitinated FAK is recognized and degraded by the 26S proteasome system; such degradation affected the downstream signaling pathways of migration and invasion.
 |
Figure 12 Proposed pathway by which DNDA inhibits the migration of A549 cells by PKC-ι/FAK ubiquitination via Cbl-b. |
Acknowledgments
We are grateful for the support for this work by grants from the Daniel Tanner Foundation, Frederick H. Leonhardt Foundation, H.O. West Foundation, Kyrias Foundation, Jin and Joo Lee Family Foundation, Miami Foundation for Cancer Research, Inc., Yolanda and Salvatore Gigante Charitable Foundation Trust, Bennack-Polan Foundation, Michael Carlisle Charitable Trust 2, Gackstatter Foundation, Arthur T. Cantwell Charitable Trust, and the Marion E. Jerome Foundation, Inc. The abstract of this paper was presented at the American Association for Cancer Research Annual Meeting 2019 as an abstract presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29-Apr 3; Atlanta, GA. Philadelphia (PA): AACR; Cancer Res. 2019;79(13 Suppl): Abstract nr 4437.DOI: https://doi.10.1158/1538-7445.AM2019-4437
Disclosure
Raja Reddy BommaReddy reports a Provisinal patent application. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Jerome A. Smoking and tobacco use; 50th anniversary surgeon general’s report. Centers Dis Control Prev. 2018. doi:10.1111/j.1440-1754.2007.01157.x
3. Thun M, Peto R, Boreham J, Lopez AD. Stages of the cigarette epidemic on entering its second century. Tob Control. 2012;21(2):96–101. doi:10.1136/tobaccocontrol-2011-050294
4. Shames DS, Wistuba II. The evolving genomic classification of lung cancer. J Pathol. 2014;232:121–133. doi:10.1002/path.4275
5. Meier R, Hemmings BA. Regulation of protein kinase B. J Recept Signal Transduct. 1999;19(1–4):121–128. doi:10.3109/10799899909036639
6. Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270(48):28495–28498. doi:10.1074/JBC.270.48.28495
7. Lin H-L, Lai -C-C, Yang L-Y. Critical care nurses’ knowledge of measures to prevent ventilator-associated pneumonia. Am J Infect Control. 2014;42(8):923–925. doi:10.1016/j.ajic.2014.05.012
8. Fields AP, Regala RP. Protein kinase C iota: human oncogene, prognostic marker and therapeutic target. Pharmacol Res. 2007;55(6):487–497. doi:10.1016/J.PHRS.2007.04.015
9. Regala RP, Weems C, Jamieson L, et al. Atypical protein kinase Cι is an oncogene in human non-small cell lung cancer. Cancer Res. 2005;65(19):8905–8911. doi:10.1158/0008-5472.CAN-05-2372
10. Scotti C, Tonnarelli B, Papadimitropoulos A, et al. Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc Natl Acad Sci. 2010;107(16):7251–7256. doi:10.1073/pnas.1000302107
11. Murray NR, Kalari KR, Fields AP. Protein kinase Cι expression and oncogenic signaling mechanisms in cancer. J Cell Physiol. 2011;226(4):879–887. doi:10.1002/jcp.22463
12. Gunaratne A, Thai BL, Di Guglielmo GM. Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor β-induced epithelial-to-mesenchymal transition. Mol Cell Biol. 2013;33(5):874–886. doi:10.1128/MCB.00837-12
13. Fogh BS, Multhaupt HAB, Couchman JR. Protein kinase C, focal adhesions and the regulation of cell migration. J Histochem Cytochem. 2014;62(3):172–184. doi:10.1369/0022155413517701
14. Carelli S, Zadra G, Vaira V, et al. Up-regulation of focal adhesion kinase in non-small cell lung cancer. Lung Cancer. 2006;53(3):263–271. doi:10.1016/j.lungcan.2006.06.001
15. Hauck CR, Sieg DJ, Hsia DA, et al. Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor-stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res. 2001;61(19):7079–7090.
16. Yu H, Gao M, Ma Y, Wang L, Shen Y, Liu X. Inhibition of cell migration by focal adhesion kinase: time-dependent difference in integrin-induced signaling between endothelial and hepatoblastoma cells. Int J Mol Med. 2018;41(5):2573–2588. doi:10.3892/ijmm.2018.3512
17. Cicchini C, Laudadio I, Citarella F, et al. TGFβ-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314(1):143–152. doi:10.1016/j.yexcr.2007.09.005
18. Shen J, Xu L, Owonikoko TK, et al. NNK promotes migration and invasion of lung cancer cells through activation of c-Src/PKCι/FAK loop. Cancer Lett. 2012;318(1):106–113. doi:10.1016/j.canlet.2011.12.008
19. Datta A, Shi Q, Boettiger DE. Transformation of chicken embryo fibroblasts by v-src uncouples 1 integrin-mediated outside-in but not inside-out s ignaling. Mol Cell Biol. 2001;21(21):7295–7306. doi:10.1128/MCB.21.21.7295-7306.2001
20. Carragher NO, Fincham VJ, Riley D, Frame MC. Cleavage of focal adhesion kinase by different proteases during SRC-regulated transformation and apoptosis. distinct roles for calpain and caspases. J Biol Chem. 2001;276(6):4270–4275. doi:10.1074/jbc.M008972200
21. Carragher NO, Westhoff MA, Riley D, et al. v-Src-induced modulation of the calpain-calpastatin proteolytic system regulates transformation. Mol Cell Biol. 2002;22(1):257–269. doi:10.1128/MCB.22.1.257-269.2002
22. Huang C, Roles of E3 ubiquitin ligases in cell adhesion and migration. Cell Adh Migr. 2010;4(1):10–18. doi:10.4161/cam.4.1.9834
23. Sekine Y, Tsuji S, Ikeda O, et al. Signal-transducing adaptor protein-2 regulates integrin-mediated T cell adhesion through protein degradation of focal adhesion kinase. J Immunol. 2007;179(4):2397–2407.
24. Lundin VF, Leroux MR, Stirling PC. Quality control of cytoskeletal proteins and human disease. Trends Biochem Sci. 2010;35(5):288–297. doi:10.1016/j.tibs.2009.12.007
25. Pillai P, Desai S, Patel R, et al. A novel PKC-ι inhibitor abrogates cell proliferation and induces apoptosis in neuroblastoma. Int J Biochem Cell Biol. 2011;43(5):784–794. doi:10.1016/j.biocel.2011.02.002
26. Brambilla E, Gazdar A. Pathogenesis of lung cancer signalling pathways: roadmap for therapies. Eur Respir J. 2009;33(6):1485–1497. doi:10.1183/09031936.00014009
27. Parker PJ, Justilien V, Riou P, Linch M, Fields AP. Atypical protein kinase Cι as a human oncogene and therapeutic target. Biochem Pharmacol. 2014;88(1):1–11. doi:10.1016/j.bcp.2013.10.023
28. Osada H, Takahashi T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene. 2002;21(48):7421–7434. doi:10.1038/sj.onc.1205802
29. Ratnayake WS, Apostolatos AH, Ostrov DA, Acevedo-Duncan M. Two novel atypical PKC inhibitors; ACPD and DNDA effectively mitigate cell proliferation and epithelial to mesenchymal transition of metastatic melanoma while inducing apoptosis. Int J Oncol. 2017;51(5):1370–1382. doi:10.3892/ijo.2017.4131
30. Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein kinase C and cancer: what we know and what we do not. Oncogene. 2014;33(45):5225–5237. doi:10.1038/onc.2013.524
31. Fan Y, Qu X, Ma Y, et al. Cbl-b promotes cell detachment via ubiquitination of focal adhesion kinase. Oncol Lett. 2016;12(2):1113–1118. doi:10.3892/ol.2016.4730
32. Zhou Q, Dai J, Chen T, et al. Downregulation of PKCζ/Pard3/Pard6b is responsible for lung adenocarcinoma cell EMT and invasion. Cell Signal. 2017;38:49–59. doi:10.1016/j.cellsig.2017.06.016
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.