Back to Journals » Neuropsychiatric Disease and Treatment » Volume 19
Effective Management of “OFF” Episodes in Parkinson’s Disease: Emerging Treatment Strategies and Unmet Clinical Needs
Authors Masood N, Jimenez-Shahed J ![]()
Received 7 November 2022
Accepted for publication 5 January 2023
Published 25 January 2023 Volume 2023:19 Pages 247—266
DOI https://doi.org/10.2147/NDT.S273121
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Nbaa Masood, Joohi Jimenez-Shahed
Department of Neurology, Icahn School of Medicine at Mount Sinai, Mount Sinai West, New York, NY, USA
Correspondence: Joohi Jimenez-Shahed, Associate Professor of Neurology & Neurosurgery, Icahn School of Medicine at Mount Sinai, Bonnie and Tom Strauss Movement Disorders Center, 1000 10th Avenue, Suite 10C, New York, NY, 10019, USA, Tel +1 212-523-8335, Fax +1 212-523-8342, Email [email protected]
Abstract: Motor complications related to the chronic administration of levodopa and failure to prevent the neurodegenerative disease process counterbalance the pivotal discovery of levodopa as the cornerstone of PD treatment. Excellent motor control is offered early during the course of treatment, but this diminishes as pathological changes in the striatum lead to synaptic dopamine levels becoming completely dependent on exogenous dopamine. This non-physiologic stimulation of dopamine receptors eventually manifests as OFF episodes. As no disease modifying therapy exists for PD that can disrupt these pathological changes, most research and treatment focuses on optimization of dopaminergic stimulation of striatal receptors so that they mimic tonic, physiologic stimulation as closely as possible. Strategies focusing on these challenges have included non-pharmacologic approaches, optimizing levodopa pharmacokinetics, using adjunctive treatments including those with non-dopaminergic mechanisms, and implementing rescue therapies. Device aided therapies, including surgery, are also available. In this review, we will focus on effective management of motor symptoms related to OFF periods, including emerging strategies. Unmet clinical needs will be discussed, including non-motor symptoms, targeted molecular therapies and disease modifying therapy.
Keywords: continuous dopaminergic stimulation, advanced Parkinson’s Disease, motor complications, nonmotor symptoms, dyskinesias, chronic levodopa complications
Introduction
Parkinson’s Disease (PD) is the second most common neurodegenerative disorder, and its prevalence has doubled in the last 25 years.1 It is well established that nigrostriatal dopamine deficiency remains a central factor in the development of the cardinal motor features of PD – tremor, rigidity, bradykinesia, and postural instability. Levodopa, a dopamine precursor, has been the mainstay of PD treatment for the last six decades. While levodopa can substantially ameliorate PD symptoms, it is associated with development of motor complications as the neurodegenerative process continues.1
Normal function of the nigrostriatal pathway relies on tonic dopaminergic stimulation, accomplished by a group of tonically active dopaminergic neurons which maintain a steady flow of dopamine in the synapse, and a quiescent group which can become tonically activated and compensate if there is injury in the system.2,3 There is also a neuronal group which can become phasically activated and release bursts of dopamine, superimposed on background tonic activity.3,4 In PD, there is a loss of presynaptic dopaminergic terminals leading to a reduction in dopamine neurotransmission and resultant dysregulation of movement. Early in the disease, when dopaminergic neuronal loss is mild, there is enough stored dopamine which can function to passively stabilize synaptic dopamine levels by way of endogenous tonic input.2 Thus, despite intermittent oral dosages of exogenous levodopa, there is constant stimulation of the post-synaptic receptors, and patients are generally unaware of increasing symptoms as plasma levodopa concentrations fall. However, as such doses cross the blood brain barrier, they far exceed the typical amount of endogenous dopamine release, and result in non-physiologic, pulsatile stimulation of dopamine receptors.2–4 Over the years as neuronal death becomes more prevalent and dopamine stores are depleted, the endogenous buffering capacity is lost and the striatal dopamine concentrations closely reflect plasma levodopa levels. These plasma levels are prone to large fluctuations, and patients become aware of transitions between periods of good motor response (“ON” state) and periods of poor response/loss of response (“OFF” state).5
Motor complications of PD include motor fluctuations (MF) and dyskinesias, but non-motor fluctuations (NMF) can also occur.6 OFF symptoms can occur under various conditions (Table 1). NMF are defined as the fluctuating presence non-motor symptoms (NMS, neuropsychiatric, autonomic, sensory) in conjunction with the rise/fall of levodopa concentration, and often in association with MF, though non-dopaminergic neurotransmitter systems may also contribute.7 Levodopa-induced dyskinesias (LID) are hyperkinetic movements (eg, chorea, dystonia, ballism or a combination of these) that typically occur during periods of peak levodopa concentrations, and less commonly as a diphasic phenomenon, when levodopa concentrations are either rising or falling.8
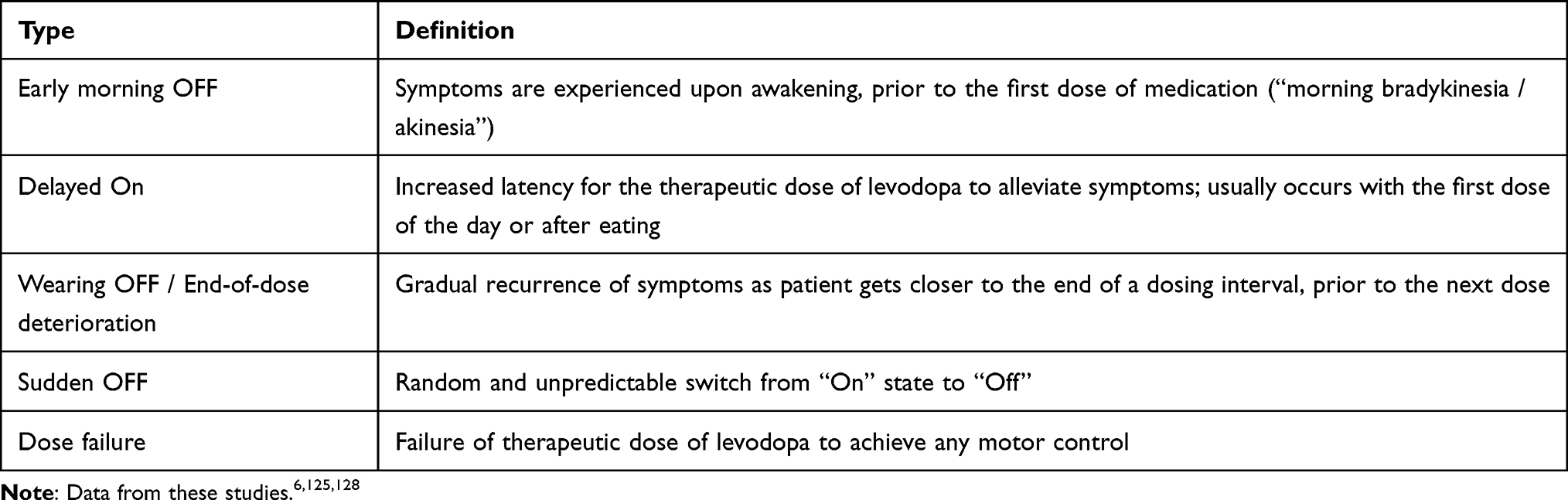 |
Table 1 Types of off Episodes |
It is important to note that pathological changes in the central nervous system (CNS) in PD do not fully account for the development of MF and OFF periods. Several pharmacokinetic properties of levodopa contribute to this, including its short plasma half-life, intestinal absorption and transport via amino acid transporters.9 Levodopa is readily metabolized by DOPA decarboxylase (DDC), catechol-O-methyltransferase (COMT), and monoamine oxidase B (MAO-B). Plasma half-life of levodopa is about 50 minutes; however, this increases to about 1.5 hours with the addition of a DDC inhibitor (eg, carbidopa or benserazide).10 Delayed gastric emptying slows the absorption of levodopa in the proximal small intestine.11 Consumption of high protein meals reduces levodopa absorption as dietary proteins compete for amino acid transporters both in the gastrointestinal tract and the blood brain barrier.2,11,12 Dysphagia and first pass hepatic metabolism further decrease the bioavailability (F) of orally administered levodopa.2 These features cumulatively contribute to fluctuating plasma levodopa levels and result in intermittent dopamine delivery to the CNS. When further combined with loss of the tonic buffer, this creates pulsatile stimulation and desensitization of the postsynaptic dopamine receptors, culminating in motor complications (Figure 1).3
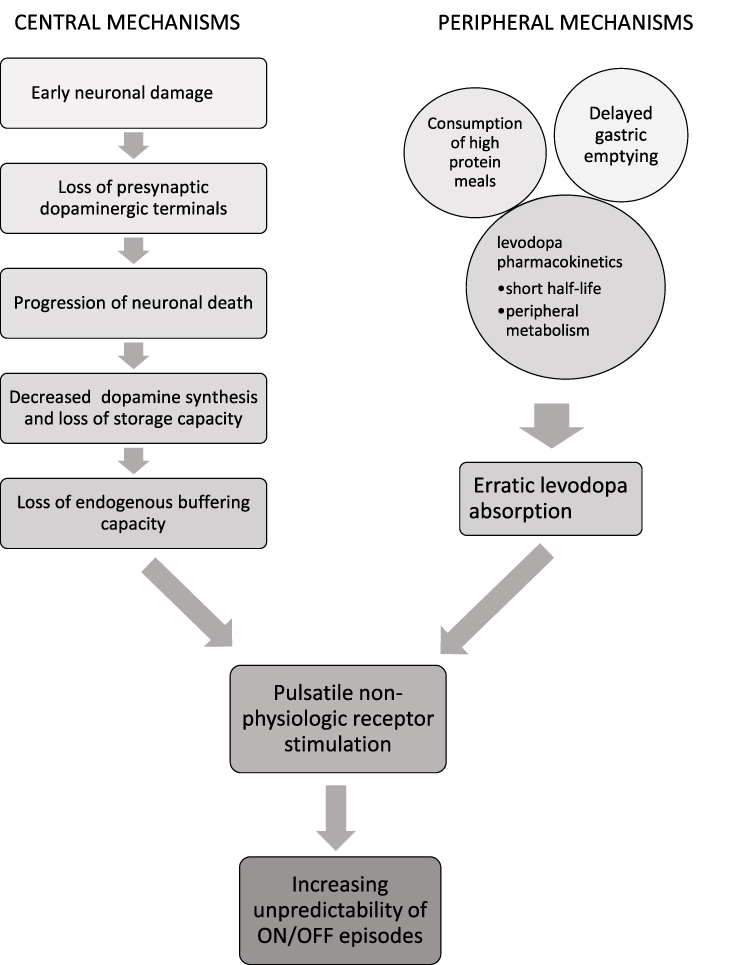 |
Figure 1 Pathogenesis of motor fluctuations in Parkinson’s disease. Both central and peripheral mechanisms contribute to the eventual development of ON/OFF fluctuations by leading to pulsatile non-physiologic dopamine receptor stimulation in the basal ganglia. Successful pharmacologic management of OFF episodes attempts to achieve more continuous drug delivery in order to mitigate the combined effects of these mechanisms. |
Indeed, the ELLDOPA trial showed that high doses of levodopa/DDC inhibitors (DDCI) are a factor in the development of motor complications, which can emerge as early as 5–6 months after treatment with ≥600mg levodopa per day.13,14 As pulsatile stimulation negatively impacts striatal function, the idea of achieving more continuous drug delivery (CDD) in order to better approximate continuous dopamine stimulation (CDS) has become the focus of therapies addressing motor complications (Box 1).
 |
Box 1 Strategies for Management of off Episodes |
In this review we will discuss strategies which can be employed to manage motor complications, including those in the pipeline, and assess what unmet needs still exist in managing this complex and multifactorial phenomenon. While some non-pharmacologic approaches may be helpful, attempts to achieve CDS/CDD are reflected in efforts to increase plasma half-life of levodopa by developing longer acting formulations and preventing enzymatic degradation. There has also been interest in developing alternate administration routes which bypass gastrointestinal absorption and first-pass metabolism. Device aided therapies such as levodopa/carbidopa intestinal gel (LCIG) and continuous subcutaneous apomorphine infusion (CSAI) are also of particular interest as they allow for more steady plasma concentrations of medication. Lastly, surgical approaches and non-dopaminergic agents can also be employed.
Non-Pharmacologic Considerations
Absorption of levodopa is dependent on the functional integrity of the digestive tract that will allow passage through to the small intestine. Constipation due to autonomic dysfunction is highly prevalent in PD, often preceding clinical Parkinsonian symptoms by years. This causes delayed gastric emptying which disrupts levodopa absorption and can lead to a delay in medication effect (delayed ON), no effect of medication (no ON) or unpredictable MF.15 Helicobacter pylori (HP) infection has been repeatedly shown to have higher prevalence in PD patients compared to the general population and has been thought to contribute to worse motor symptom control.16 Hashim et al found that at 12 weeks following successful eradication of HP infection in PD patients with motor fluctuations, there was a significant decrease in time to onset of medication effect, as well as overall improvement in daily ON time.17 More recently, Lolekha et al prospectively evaluated the therapeutic effects of HP eradication on MF. In their cohort of 40 Thai patients with MF, 22 were HP positive and 18 HP negative. At baseline, HP positive patients had longer daily OFF time (4.6 hours vs 3.9 hours). Of the 22 patients who received treatment with triple therapy, 17 had full eradication and they showed a significant decrease in daily OFF time by 0.7 ± 1.3 hours and increase in daily ON time by 0.9 ± 1.2 hours (in contrast to 0.6 hour increase in OFF time and a 1.6 hour decrease in ON time in those who had eradication failure). In total, they found the HP eradication effect to be −1.3 hours of OFF time.18 Small intestine bacterial overgrowth (SIBO) is relatively common in patients with PD, with prevalence in about half of patients with PD compared to about one-quarter of the general population.19 It has been postulated that Enterococcus species which overpopulate the small intestine in patients with SIBO are responsible for metabolism of levodopa, leading to lower absorption.19 Fasano et al were one of the first to establish an association between SIBO and MF (particularly delayed ON and no ON) in a small, unblinded, non-randomized trial.19,20 Additionally, they found that eradication of SIBO with antibiotic treatment had a meaningful clinical effect in improving MF.19,21 Aside from HP and SIBO, there has been growing interest in the gut microbiome of PD patients with ongoing investigation into the role of the microbial environment on levodopa resistance.15
Absorption of levodopa is facilitated via large neutral amino acid (LNAA) transporters which are also needed for absorption of dietary proteins, and it is often recommended that patients avoid taking their medications in close proximity to protein-rich meals. The protein redistribution diet (PRD) is characterized by limitation of protein intake early in the day, with compensation during dinnertime.22 It is important to note that within PRD, patients still consume the recommended daily intake of protein. Compared with those on a high protein diet, patients on PRD had lower motor disability but more dyskinesias, potentially indicating improved levodopa response.22 In a crossover study comparing PRD (15% daily protein before supper) with normal diet (60% of daily protein before supper), patients were randomized to 2 months of normal diet followed by 2 months of PRD. Results showed significantly improved total daily OFF time with PRD (164 minutes) vs normal diet (271 minutes) and significantly improved daily ON time with PRD (852 minutes) vs normal diet (738 minutes).22 While these results are promising, it may be unfeasible for some patients to alter their diets in such a manner. By contrast, low protein diets put patients at risk of weight loss and are not recommended.
Dietary fiber has shown some positive results in regards to optimizing levodopa therapy. Astarola et al found that in those patients with constipation, a diet rich in soluble fiber (28g fiber/day) for 3 months significantly reduced constipation, reduced the levodopa metabolite 3-OMD, and significantly increased levodopa plasma concentration.22 Another trial assessing the water soluble fiber Plantago ovata husk found no significant effect on the pharmacokinetics of levodopa, but did find a lower number of levodopa concentration peaks, potentially suggesting more smoother plasma profile that may be able to avoid peaks and troughs that contribute to MF.22
Strategies to Consider Early After Onset of Motor Fluctuations
Alteration of the levodopa dosing regimen is one of the earliest approaches to consider once MF arise. Doses can either be increased to prolong ON time, or broken down into smaller doses (“fractionating”) which are more frequently administered, to reduce OFF time.23 In 2021, the US Food and Drug Administration (FDA) approved the first functionally scored carbidopa-levodopa tablet that has deep grooves and a drug-free bottom layer that allows for more precise fractionation.24 While dose fractionation may temporarily improve symptoms, it does not prolong levodopa half-life in the plasma and so repeated medication adjustments are often required, which can further contribute to erratic pharmacokinetics.23 Increasing frequency of doses may also indirectly worsen motor symptoms by reducing compliance in some patients.23,25 hi Other limitations include risk of suboptimal control with smaller doses or conversely increased risk of dyskinesia with higher doses.26 Flexilev ® (Sensidose Pharmaceuticals, Uppsala, Sweden) is a rapidly soluble microtablet formulation that has been approved in Sweden since 2014.27 It is unique in that it is delivered via an automatic dose dispenser, My Flexible Individual Dosing (MyFIDTM, Sensidose Pharmaceuticals, Uppsala, Sweden), allowing for more precise and individualized drug delivery.27 Dosing regimens are programmed by a physician and the device is able to collect information on symptoms and medication adherence among other things. It is theorized that this may provide the most optimal mode of oral delivery for achieving CDD.27
Dopamine agonists (DAs) are used in monotherapy as a levodopa-sparing option or as an adjunct to levodopa in order to reduce OFF time (Table 2). Levodopa has a short plasma half-life which increases to approximately 1.5 hours with the addition of a DDCI,10 while DAs have half-lives ranging from 5 to 8 hours depending on the medication.28–30 This allows for a more prolonged motor response due to prolonged stimulation of dopamine receptors, possibly reducing pulsatile stimulation for more tonic stimulation.31 Their use in early PD monotherapy has fallen out of favor due to their less favorable side effect profile and overall lower clinical efficacy than levodopa.32 Pramipexole and ropinirole are DAs available in both conventional immediate release (IR) and once daily extended release (ER) formulations. Addition of either formulation of pramipexole or ropinirole to treatment with levodopa results in reduced OFF time (approximately 1–2 hours) and may allow reduction in levodopa dosing (Table 2). In a study comparing efficacy of pramipexole IR vs pramipexole ER as adjuncts to CD/LD, both were found to be comparable in safety and efficacy, with the advantage of once daily dosing with the ER formulation compared to three times daily dosing.33 Similarly, the PREPARED study compared ropinirole IR and prolonged release (PR, same as ER) as adjuncts to CD/LD in patients experiencing MF and found results comparable, but generally favoring ropinirole PR in regards to reduction in OFF time.34
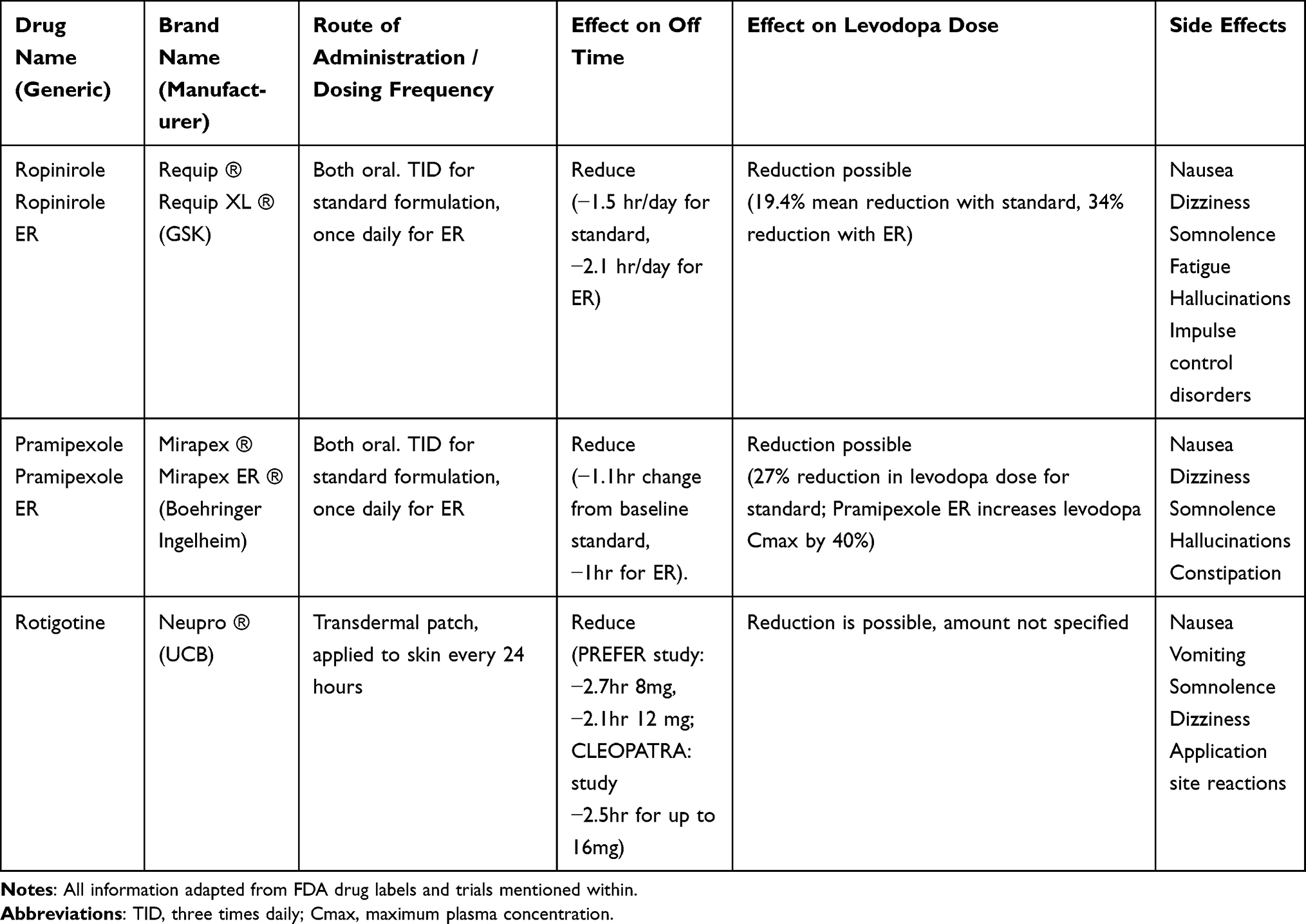 |
Table 2 Effect of Dopamine Agonists When Used as Adjuncts to Levodopa |
Another DA, rotigotine, is the only transdermal patch currently authorized for use as monotherapy and as an adjunct to levodopa in PD. The patch is composed of a thin, matrix-type transdermal system composed of three layers and is available in six doses, which can be combined using the different patches.29 Approximately 45% of rotigotine is released from the patch over 24 hours and this does not differ significantly depending on application sites. Once applied, there is an initial lag of 3 hours (range 1–8 hours) until it can be detected in the serum and time to maximal plasma concentration (Tmax) typically occurs between 15 and 18 hours post-dose, however there is no classic Cmax.29 Steady-state plasma concentrations are achieved within 2–3 days.29
Rotigotine is unique in that unlike the other non-ergoline DA’s, pramipexole and ropinirole, it has activity at D3/D2/D1 receptors, such that it more closely resembles dopamine.35 The efficacy of adjunctive rotigotine in addressing MF with reduction in OFF time is well established in placebo-controlled clinical trials, all of which show a 1–2 hour advantage over placebo in OFF time reduction.29 Several NMS have also been shown to improve with rotigotine, however most lack high level class I evidence.35 Nocturnal sleep disturbances have been shown to improve as a whole with rotigotine and this was demonstrated in 3 randomized controlled trials (RCTs), thus the Movement Disorder Society Evidence Based Medicine Committee (MDS-EBM) considers it “possibly useful” for the treatment of insomnia and “likely efficacious” in improving sleep.35 The DOLORES study evaluated efficacy of rotigotine for MF-related pain and found insufficient evidence to define it, therefore MDS-EBM considers it as “investigational” for pain related to PD motor complications.35 Given the close resemblance to levodopa and dopamine as well as the potential benefit in NMS, rotigotine offers potential as a strong therapeutic for advanced PD. Other formulations for rotigotine delivery are being investigated including rotigotine loaded nanoparticles for nose-to-brain delivery.36 Alternatively, a ropinirole transdermal patch is being developed in Japan and was found to be non-inferior to ropinirole IR tablets, with maintained efficacy and plasma concentrations for up to 52 weeks.37,38
Prolonging Levodopa Plasma Half-Life
Increasing levodopa plasma half-life can be accomplished by changing the levodopa formulation or inhibiting enzymes which metabolize it. Sustained-release carbidopa-levodopa (CD/LD SR) was the first longer-acting formulation introduced. It is supplied in tablets composed of a polymeric-based drug delivery system that releases the drug in a controlled manner.39 However, clinical experience has revealed increased latency in time to “ON” and dose failures due to greater time to Tmax compared with IR (2 hours vs 0.5 hours for IR).39 Additionally, there is high risk of dyskinesias as the (F) of CD/LD SR is approximately 70% that of IR, and so higher doses are needed, which may accumulate throughout the day and cause worsening of dyskinesias especially in the evening.40 This was improved upon with carbidopa-levodopa extended release (CD/LD ER) capsules (Rytary®, Amneal Pharmaceuticals, Bridgewater, NJ; Numient®, Amneal Pharma Europe Ltd, Dublin, Ireland), a multiparticulate capsule of carbidopa-levodopa composed of both immediate release beads and sustained release beads combined in a specific ratio.41 The idea behind this was for rapid initial increase in plasma concentration with the IR beads, followed by a more sustained plasma concentration from the delayed release beads.3,41,42 As with CD/LD SR, the (F) of levodopa from CD/LD ER capsules relative to CD/LD IR is approximately 70%, but the Tmax is the same as CD/LD IR (1 hour).42 Following the peak, plasma concentrations are maintained for 4–5 hours before declining.42 The quick Tmax is intended to prevent delayed ON or dose failures that are seen with CD/LD SR, while also giving the advantage of a smoother plasma profile that eliminates the steep peaks and troughs which clinically manifest as MF. However, in the practical management of individuals with PD, it has been noted that there was less reduction in OFF time and less reduction of dyskinesia when converting patients from CD/LD IR to CD/LD CR capsules when baseline greater amounts of troublesome dyskinesia are present.43
In order to further improve the pharmacodynamic profile of ER levodopa, IPX203 has been developed (Amneal Pharmaceuticals, Bridgewater, NJ) with the goals of rapidly delivered therapeutic levodopa plasma concentrations, longer duration of such concentrations, minimal peak to trough fluctuations, and greater (F).44 It is designed to be taken every 8 hours. Early data show that it has a rapid initial increase in plasma concentration comparable to CD/LD IR and CD/LD ER capsules, but it is able to sustain the plasma concentration longer (2.6 hours more than CD/LD IR, 0.9 hours more than CD/LD SR).45 In regards to reduction in OFF time, recent data from a phase 3 RCT comparing IPX203 to CD/LD IR revealed a statistically significantly 0.53 hour/day increase in “good on time” as well as significantly less OFF time (−0.48 hour/day) from baseline to the end of study.46 A post-hoc analysis of the data with a modified intent-to-treat population again showed results favoring IPX203 over optimized CD/LD IR regimen in regards to increased “good on time”.47
Enzyme inhibition to prevent metabolism of levodopa has been in use since the early 1970s when carbidopa, a DDCI, was shown to increase the plasma half-life of levodopa from 50 minutes to approximately 1.5 hours.39,48 Benserazide is an alternative DDCI, used more commonly in Europe. Both DDCIs not only increase plasma half-life of levodopa, but also have the added benefit of increasing tolerability of levodopa by allowing lower dosages. COMT and MAO-B are two other major enzymes in the dopamine metabolic pathway. MAO-B inhibitors include selegiline, rasagiline, and safinamide (Table 3). Rasagiline is approved by the FDA for use as monotherapy in early PD while all three are approved for use as adjuncts to levodopa. Selegiline is unique in that it is metabolized into 1-methamphetamine and eventually 1-amphetamine, which may potentially play a role in improving alertness in the elderly, however the concentrations are negligible.49 Rasagiline is more bioavailable than selegiline and may have neuroprotective qualities in vitro.49,50 It has been shown to improve morning OFF as well as wearing OFF, and post-hoc analyses have suggested a possible benefit in measures of depression, cognition and fatigue.51 Safinamide is unique in its mechanism of action by also working at voltage-gated sodium and N-type calcium channels to inhibit release of glutamate.52 This modulation of glutamate is a potentially important feature as excessive glutamate has been implicated in loss of nigral dopaminergic cells, in the development of levodopa-induced dyskinesias and in NMS such as pain.53–55 Furthermore, it has been suggested that it may also modulate metabolism of other neurotransmitters, which could provide benefit for NMS including depression and cognition.52 All three MAO-B inhibitors offer reduced OFF time with the associated possibility of reducing the total levodopa daily dose, and possibly improving NMS (Table 3).
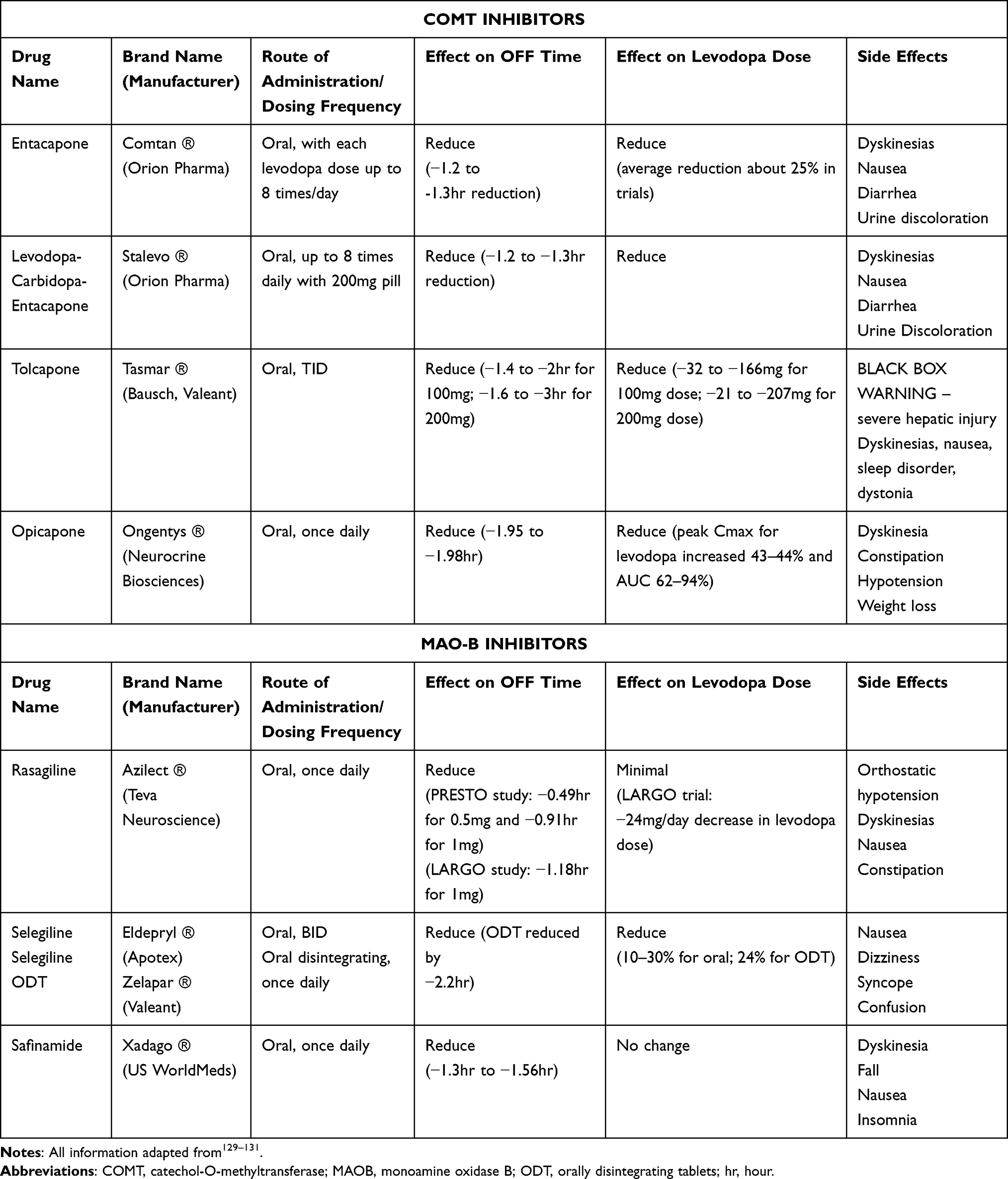 |
Table 3 Effect of Enzyme Inhibitors When Used as Adjuncts to Levodopa |
Entacapone, tolcapone and opicapone are three approved COMT inhibitors (Table 3). Entacapone inhibits peripheral COMT and is available both on its own or in a formulation in combination with CD/LD at set dosage strengths (carbidopa-levodopa-entacapone, CLE, Stalevo®, Orion Pharma, Espoo, Finland). Strong evidence to support the use of entacapone comes from four phase 3 RCTs which showed that the CLE improved pharmacokinetic profile translates clinically to greater efficacy in controlling motor symptoms compared to CD/LD alone, even with a 12% reduction in mean daily levodopa dose.23,56–60 Tolcapone is the most potent of the three COMT inhibitors as it acts both peripherally and centrally, however it is associated with potential for severe hepatic injury and carries a boxed warning from the FDA.61 Opicapone is the newest in the class and contains a pyridine N-oxide residue which gives it high affinity to COMT inhibition.62 Similar to entacapone, it does not cross the blood brain barrier, can only work to inhibit peripheral COMT and the effect on COMT inhibition is dose dependent.62 It has also been found to have the longest half-life, with COMT inhibition detected at 72-hours post-dose.62 This is in contrast to the 8 hour and 18 hour duration of effect found with entacapone and tolcapone, respectively.62 A network meta-analysis comparing the three found that tolcapone was the most efficacious but at the expense of a poor safety profile while entacapone was the least efficacious; opicapone had the best profile in regards to efficacy and safety.63 It should be noted, however, that opicapone had the highest rate of dyskinesias and so may not be suitable for certain patients.63 The addition of all three COMT inhibitors to treatment with carbidopa/levodopa can reduce OFF times from 1.2 to 3 hours, with a variable reduction in the levodopa dose possible (Table 3).
Non-Dopaminergic Adjuncts
Limitations of dopaminergic replacement therapy have led to exploration of alternative drug targets. Medications such as amantadine, zonisamide and istradefylline may still have some indirect dopamine modulating activity, but their primary mechanisms are non-dopaminergic.
Amantadine
Amantadine is the only drug currently indicated for both reduction in OFF time and treatment of dyskinesias. This unique property is likely due to its effect on multiple neurotransmitter systems. Experimental models show that amantadine enhances dopamine transmission, and increases dopamine biosynthesis, turnover, uptake, and release in rodent models.64 In human models, it is shown to increase striatal L-amino acid decarboxylase activity, which increases dopamine synthesis.64 Antagonism at NMDA receptors is thought to account for its anti-dyskinetic effect.64 It has also been seen to interact with serotonergic, noradrenergic, and cholinergic systems.64 While it is available in both IR and ER formulations, only amantadine ER, Gocovri® (Adamas Pharmaceuticals, Emeryville, California) is indicated for treatment of dyskinesias and reduction in OFF time. Amantadine ER has a long half-life with a median Tmax around 12 hours, which means with bedtime dosing, patients wake up with higher plasma concentrations during the day when benefits are needed.65 In a pivotal phase 3 trial, amantadine was shown to reduce OFF time by −1.2 hour/day.66 The unique duality in improving both dyskinesias and OFF time is an important feature of amantadine ER, as one of the biggest obstacles in treatment of motor complications is the successful management of OFF time without worsening dyskinesias. Aside from motor benefits, amantadine ER was also found to provide statistically significant and clinically meaningful reductions in Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale part II (MDS-UPDRS II) motor experiences of daily living.67
Zonisamide
Zonisamide is an anticonvulsant drug with additional evidence for motor benefit in PD patients. It has been approved as an adjunct to levodopa in Japan since 2003.68 The mechanism of action as an adjunct in PD is unknown, but it has both dopaminergic and non-dopaminergic mechanisms including reversible MAO-B inhibition and modulation of levodopa receptor expression and metabolism.68–72 Zonisamide is taken once daily, therefore improving compliance. The recommended 25mg-50mg dose in PD is markedly lower than that used in epilepsy (200–400mg), which may be more tolerable. Otherwise, the most common side effects of zonisamide amongst PD patients include decreased appetite, weight loss, somnolence, apathy and dizziness.68 Pivotal Phase 2 and 3 trials done in Japan revealed zonisamide 25mg and 50mg to be more effective than placebo in improving UPDRS III (motor) scores, while a post-hoc analysis found that zonisamide 50mg was superior to placebo in improving OFF time (zonisamide 50mg reduced mean daily OFF time by 0.98 hours compared to placebo).68,73–75 An important finding in the post-hoc analysis was that zonisamide 50mg may additionally benefit dyskinesias when compared to placebo, since it was associated with a decrease in disability of dyskinesia score (UPDRS part 4, item 33) from baseline to week 12 by 0.20 points without causing an increase in dyskinesia duration (UPDRS part 4, item 32).75 In regards to NMS, there is conflicting evidence on zonisamide’s role in development of ICD as earlier studies suggested some association between zonisamide and development of these behaviors, however a more recent open label trial found the opposite results. Furthermore, it is suggested that zonismaide may reduce cognitive decline more than the other PD therapies via potential neuroprotective effects such as protection against oxidative stress and potentially against progressive dopaminergic cell loss.76
Istradefylline
Istradefylline is the first adenosine A2A receptor antagonist that is labeled for use in PD.77 Adenosine is a neuromodulator that exhibits action via multiple receptor subtypes in response organ stress or tissue damage and in the CNS is involved in memory, mood, and motor function.78 Adenosine A2A receptors are highly expressed in basal ganglia nuclei in close relation to dopamine D2 and D3 receptors such that activation of A2A receptors inhibits the indirect pathway.78 While istradefylline has not been approved as monotherapy, it exhibits effectiveness in OFF time reduction as an adjunct to conventional PD therapy.78 Four pivotal trials found an overall reduction in OFF time of approximately 1–2 hours, and the mean reduction in daily OFF time relative to placebo was −0.75hr (20mg group) and −0.82hr (40mg group). Open label studies also demonstrated enduring OFF time reduction at 52 weeks and overall positive safety profile.77,79,80 The most common side effects were dyskinesia, dizziness, constipation, decreased weight, hallucinations, and insomnia.77,79 The overall lack of serious side effects associated with istradefylline has been attributed to its lack of direct dopaminergic activity.77,80
In an indirect comparison of istradefylline to other adjunct therapies including dopamine agonists, COMT inhibitors, and MAO-B inhibitors, the mean reduction in OFF time relative to placebo from the 4 pivotal trials of istradefylline [−0.75 hr (20mg dose) and −0.82hr (40mg dose)] was compared to that of other agents. The mean reduction in OFF time relative to placebo was −1.54hr for dopamine agonists, −0.93hr for MAO-B inhibitors, and −0.83hr for COMT inhibitors.77 Taking all this information into account, while istradefylline may be comparable in efficacy to MAO-B and COMT inhibitors, it is less efficacious than dopamine agonists in OFF time reduction.77
Rescue Therapies
Parcopa
An orally disintegrating form of CD/LD (Parcopa®, Schwarz Pharma, Mequon, WI) was FDA approved in 2004 and later by the EMA in 2011. This formulation uses RapiTab™ technology (Schwarz Pharma, Monheim, Germany), which allows the tablet to disintegrate on the tongue within seconds, without the need for water.3,81 An early open label study comparing this orally disintegrating pill (ODT) to conventional CD/LD IR found no statistically significant differences in motor scores in both ON and OFF states between the two groups.82,83 A small pilot study further corroborated this, finding no statistically significant difference between ODT vs conventional CD/LD IR in onset to clinical benefit or overall motor performance, though it should be noted that all trends favored ODT.84 This may be attributed to the fact that while, theoretically, absorption may be quicker due to quick oral disintegration, this formulation still relies on gastrointestinal absorption in the same way as CD/LD IR, while providing no benefit for sustained plasma levels as is seen with CD/LD CR, CD/LD ER, and CLE. While there was no significant difference between UPDRS motor scores in patients receiving ODT vs conventional oral therapy, Nausieda et al found a patient preference for ODT, citing accessibility of the medication to treat OFF times, reduced concerns about swallowing, its use for nighttime dosing and the fact that it is less noticeable thus less stigmatizing when taking the medication in front of others.82 CD/LD ODT most commonly addresses early morning OFF and delayed ON features of PD (Table 1).
Inbrija
Levodopa inhalation powder, Inbrija® (Acorda Therapeutics, New York, NY) is delivered via Arcus® technology (Acorda Therapeutics, New York, NY) to the pulmonary system. It consists of capsules which are broken, releasing the powder which is then orally inhaled by the patient. Single dose pharmacokinetics show a median Tmax of 30 minutes which clinically translates to a quick onset of action.85 Clinical improvement is seen within 5–10 minutes, which makes it a faster rescue therapy than the alternative apomorphine subcutaneous injection. The dose-normalized Cmax of levodopa from Inbrija is approximately 50% of that from IR.85 Placebo-controlled studies show superiority in reduction of UPDRS part III (motor scores) from pre-dose to 30 minutes post-dose, with benefit evident up to 60 minutes after inhalation.86 While respiratory side effects are the most common (eg, mild cough), no significant differences were seen in spirometry measures between Inbrija and placebo at initiation of treatment, at 4 weeks, 12 weeks, and even at 12 months.87–89 Some patients may prefer administering medication via an inhaler compared to a more invasive injection. CD/LD inhalation powder may also be particularly useful in patients with difficulty swallowing pills and those with severely delayed gastric emptying due to its unique property in bypassing gastrointestinal absorption.
INP-107 (Impel Neuropharma, Seattle, WA) is an inhalation powder aimed for intranasal delivery via POD® devices (Impel Neuropharma, Seattle, WA). Similar to the already available CD/LD inhalation powder, it bypasses gastrointestinal absorption and allows for rapid relief of symptoms.90 Unlike the oral inhalation route implicated with use of Inbrija, intranasal delivery is more natural and convenient as it avoids side effects such as dry mouth.91 INP107 is currently in development with phase 2 trials complete.90
Apomorphine – Sublingual and Subcutaneous
Apomorphine is an alkaloid derived from morphine which has structural similarity to dopamine and functions as a DA but with key differences in its dopamine receptor affinity.92 Similar to rotigotine, apomorphine has a high affinity for D1 receptors, thus making it closer in action to dopamine.92 Apomorphine is available in the form of a sublingual film (Kynmobi®, Sunovion Pharmaceuticals, Marlborough, MA), a subcutaneous injection (Apokyn®, Supernus Pharmaceuticals, Rockville, MD; APO-Go®, Britannia Pharmaceuticals, Reading, UK), and a subcutaneous infusion. The sublingual film and subcutaneous injection are indicated as rescue therapies; apomorphine subcutaneous infusion will be discussed separately. Both rescue formulations bypass first pass metabolism and are efficacious in reducing OFF time more rapidly than orally ingested pills. In a direct comparison of the two, apomorphine injection had a higher maximum plasma concentration (Cmax), a shorter Tmax but overall similar systemic exposure (AUC).93 So while Cmax was rapidly achieved (<1 hour) for both, the injection was about 15 minutes faster than the sublingual film.93 While it may be important for some patients to achieve a quicker response, patient preference surveys have found them to generally prefer oral disintegrating formulations as opposed to injection or inhalation.93 Regardless of formulation, a disadvantage of apomorphine is the severe nausea which requires additional treatment with trimethobenzamide or domperidone, neither of which medications are available in the US and so alternative treatments may be required.94 Clinically, because of their short duration of onset, apomorphine sublingual and subcutaneous formulations can be used to address any of the OFF types that are commonly encountered in PD (Table 1). However, attempts to further improve the latency to ON with apomorphine have led to the development of AZ-009, an inhaled formulation that is rapidly systemically absorbed with a median Tmax of 1–2 minutes, and with early evidence for clinical efficacy in successfully treating morning OFF amongst PD patients.95,96
Infusion Therapies
Levodopa-Carbidopa Intestinal Gel (LCIG)
Levodopa-carbidopa intestinal gel (LCIG, trade name Duodopa® / Duopa®, AbbVie LTD, Maidenhead, UK) is a stable gel suspension designed for continuous delivery of levodopa, developed in Sweden in the 1990s and approved in Europe in 2004 and US in 2015.97 The levodopa containing gel is delivered straight to the proximal small intestine via a percutaneous endoscopic gastrostomy with jejunal (PEG-J) tube that is connected to a portable infusion pump, which is carried by the patient.98 The gel is infused for 16 hours/day during waking hours, though some patients may use it for longer.97
LCIG allows for a two-pronged approach in addressing OFF episodes in PD. Firstly, continuous infusion allows for a steady stream of levodopa circulating to the CNS that is a contrast from the peaks and troughs with repeated dosing with conventional oral therapy. Second, the PEG-J tube allows for direct delivery to the primary area of levodopa absorption, thereby bypassing gastric absorption which can be delayed and lead to erratic absorption in advanced PD.99 A pivotal RCT comparing LCIG to CD/LD CR found lower mean plasma concentration variation, lower average change of variation of plasma concentration and lower mean levodopa exposure with LCIG as compared to CR.100 These findings underlie the clinical effects of increased ON time (associated with reduced OFF time) and reduced dyskinesia. Other trials after this have confirmed reduction in time spent in the OFF state in patients receiving LCIG vs other conventional therapies including optimizing CD/LD IR and CD/LD ER.100,101 In a placebo controlled, double blinded study comparing LCIG with optimized medical therapy, LCIG provided a −4.04 hour/day decrease in OFF time vs −2.14 hour/day difference seen with optimized medical therapy.101 A qualitative review of long-term effects of LCIG found that in a majority of the studies reviewed, there was a statistically significant reduction in mean OFF time by at least 2 hours at a minimum of 12 months, with the longest follow-up being 120 months.99
With LCIG, there is a unique set of adverse effects that is attributable to the invasive nature of the delivery system. The most frequent adverse effects are related to the surgical procedure including wound infection, tube dislocation, and abdominal pain.99 Peripheral neuropathy as a result of levodopa-based therapy has been observed more frequently in patients on LCIG versus oral formulations.102 While the exact mechanism of the development of neuropathy is unclear, vitamin B deficiency and hyperhomocysteinemia have been cited, and thus vitamin B supplementation has been employed to manage this.99 Diphasic dyskinesias may develop in up to 15% of patients treated with LCIG and can be addressed by increasing the infusion time to 24 hours.103
LCIG is considered one of the advanced options in the treatment of PD, alongside deep brain stimulation (DBS) and continuous apomorphine infusion (CSAI). While there have been trials that extensively compare levodopa infusion to conventional oral therapies, there are less head-to-head data thus far regarding surgery vs infusion options. A review comparing available data on LCIG and subthalamic nucleus (STN) DBS found that while the two were comparable in the motor control domain, LCIG offers some cognitive benefit.104 This can be helpful when considering advanced therapeutics as those patients with dementia may potentially be better suited to LCIG vs DBS.104 Other important aspects to consider include economic (LCIG is the more costly than DBS and CSAI in many places), patients’ willingness to carry a harness with the pump, and other co-morbid conditions.103
As a result of its design, LCIG is the closest to physiological delivery of levodopa to the CNS.3 Other pros of this include the ability to individualize treatment by adjusting flow rates and administering boluses, which can be more precisely targeted than by splitting conventional pills.99 This is an effective treatment option for patients with severe motor complications in which OFF episodes are not treated by conventional oral therapy, however its costs and efficacy in comparison to other advanced therapeutics needs to be considered closely.103
A newer device aided therapy with a lighter, more practical pump system is Levodopa-entacapone-carbidopa intestinal gel (LECIG) infusion (Lecigon®/Lecigimon®; Lobsor Pharmaceuticals AB, Uppsala, Sweden).105 This was developed based on data showing that the addition of oral entacapone to LCIG allowed for a dose reduction in levodopa whilst maintaining clinical efficacy.105 Senek et al conducted a randomized, open-label, cross-over study to measure systemic levodopa exposure with LECIG infusion using a 20% reduced dose vs patients’ usual dose of LCIG.106 Despite the reduced dosage with LECIG, systemic levodopa exposure did not differ between the two treatments, the dose-adjusted levodopa exposure was significantly higher during LECIG administration, and the treatment response scale (TRS) did not differ between the two.105,106 Patients on LECIG also had lower plasma concentrations of the levodopa metabolite 3-OMD, the formation of which is hypothesized to play a role in development of neuropathy.106 It is therefore speculated that rates of neuropathy may be lower with LECIG than with LCIG.106 All adverse effects were mild, including headache and nausea.106 While these results are promising, larger comparative studies are needed to assess safety, efficacy and long-term outcomes.105
Continuous Subcutaneous Apomorphine Infusion (CSAI)
CSAI is approved in Europe and under review by the FDA.107 The infusion is delivered via a battery-driven, portable pump system that delivers a continuous dose of drug, although bolusing is an option.92 Similar to LCIG, the infusion is typically 12–16 hours during the waking day, but can be extended to 24 hours in certain cases.92 Its efficacy was established in the TOLEDO trial which found that from baseline to 12 weeks, the CSAI arm had an absolute difference of reduction in OFF time of −1.9 hours when compared to placebo and a 2 hour increase in ON time without troublesome dyskinesias.108 CSAI offers a smooth continuous pharmacokinetic profile that is comparable to LCIG without the need for a surgical procedure. The most common adverse events are skin nodules at the injection site. A special consideration for both the apomorphine subcutaneous injection and apomorphine infusion is that a challenge test needs to be done in a supervised setting by a trained health-care professional to avoid the risk of hypotension.94 A disadvantage in some patients may be the severe nausea caused by apomorphine (all forms), thus patients are routinely administered anti-emetics.109
Under Investigation
Several levodopa subcutaneous infusions are under development to address persistent challenges in levodopa administration in order to better achieve CDD. These include ND0612 (NeuroDerm, Rehovot, Israel), ABBV-951 (Abbvie Inc, Chicago, IL), and Infudopa® (Dizlin Pharmaceuticals, Gothenburg, Sweden). These aim to address problems associated with levodopa solutions, including high instability at room temperature and limited solubility in water requiring large volumes.110 Successful subcutaneous solutions require high aqueous solubility and excellent chemical stability.111 While ND0612 achieves a highly concentrated CD/LD solution, its alkaline pH causes a high rate of skin nodules – around 95% of people receiving ND0612.112 ABBV-951 is a phosphate prodrug of CD/LD that appears more stable and is at a near neutral pH, thus avoiding excessive skin irritation.113 Lastly, Dizlin pharmaceuticals is currently developing DZ102, which consists of a stock solution (CD/LD dissolved at a low pH) and a buffer that brings the solution to a physiologically favorable pH when mixed.111 The stock solution is stable at room temperature for >3 months and the buffered mixture allows for better skin tolerability.111
In a phase 2 place-controlled study, patients receiving levodopa standard of care (SoC) and ND0612 had a −2.1 hour/day reduction in OFF time vs a −1.4 hour/day reduction for patients receiving levodopa SoC and placebo.112 A new drug application has been submitted to the FDA for ABBV-951 based on results of an open-label phase 3 study which demonstrated superiority of ABBV-951 vs placebo in reduction in off time (−2.75 hours/day ABBV-951 arm, −0.96 placebo arm) and increase in on time without troublesome dyskinesias (+2.72 hours/day ABBV-951 arm vs +0.97 hours/day in placebo arm).114 DIZ102 was found to be non-inferior to LCIG in regards to fluctuations of levodopa plasma levels, a property which is essential for infusions.111 If approved, these investigational products may provide a less invasive and better tolerated alternative to infusion therapies, especially in older patients in whom DBS is not an option and a PEG-J procedure may be less desired.
Deep Brain Stimulation (DBS)
The current FDA-approved indication for DBS in PD includes patients who have had PD for at least 4 years, with at least 4 months of motor complications. DBS is the most frequently performed surgical procedure for PD, with two main target sites – subthalamic nucleus (STN) and globus pallidus interna (GPi).115 In a seminal trial, Deuschl et al demonstrated that STN stimulation was superior to medical management alone as evidenced by greater improvements from baseline to 6 months in UPDRS III (motor) and PDQ-39 (mobility, ADLs, emotional wellbeing, bodily discomfort).116 Numerous trials have supported this, such as the Veterans Affairs NIH trial (VA-NIH) that showed bilateral DBS (either STN or GPi) was superior to best medical therapy (BMT) as seen by the 4.6 hour increase in ON time without troublesome dyskinesia in the group treated with DBS (pooled for both targets) vs 0 hour/day improvement in the group assigned to continued BMT at 6 months, based on patient motor diaries.117 There was an associated 2.4 hour/day decrease in OFF time from baseline to 6 months in patients with DBS.117 Another study found a statistically significant increase in the percent of patients experiencing no OFF time during waking hours (from 3% at baseline to 29% at 1 year in the DBS + BMT group vs 2% increase in BMT alone group) as well as an increase in the percent of patients experiencing no dyskinesias during waking hours (37% increase in the DBS+BMT group vs −1% in the BMT alone group).118 It is important to note that while results from the DBS + BMT arm of the study were pooled for both STN and GPi, the majority of DBS patients in the study underwent bilateral STN electrode placement.118 In further randomized studies, one reporting effects from a multiple independent constant current-controlled device found bilateral STN stimulation provided a 3.74 hour increase in good ON time without troublesome dyskinesias at 3 months, while another one evaluating bilateral STN with constant-current stimulation found it to improve ON time without troublesome dyskinesias by 4.27 hours/day at 3 months.119,120 These differences likely relate to the baseline characteristics of the enrolled cohorts and methods of assessment. Regardless, multiple studies have confirmed the major benefits of DBS in PD, regardless of target, to be reduced OFF time, increased ON time, and reduced dyskinesias (Table 4).
 |
Table 4 Comparison of DBS Target Sites |
One of the challenges in the device-aided treatment of PD with MF is choosing the best device (LCIG, CSAI, or DBS) for an individual patient. Currently, there are no trials directly comparing all three; however, recent meta-analyses have provided some guidance. A network meta-analysis done by Antonini et al demonstrated that DBS and LCIG had greater improvements in OFF time than CSAI with DBS resulting in greatest OFF time reduction.121 The change in OFF time was −1.45 hours/day for DBS and −1.35 hours/day for LCIG.121 However, LCIG had the highest ranking in QoL improvement.121 Marsilli et al also found DBS and LCIG to be superior to CSAI and BMT in improving motor function, however at the expense of higher lifetime costs (CSAI had the lowest lifetime costs and LCIG had the highest).122 An advantage of CSAI is that no surgical procedure is required, however it is not available in all countries. LCIG may be an option for patients with dementia or cognitive impairment in whom DBS is not an option.122
Unmet Needs / Future Perspectives
Motor OFF times in PD are responsible for a substantial portion of the social and economic impact of PD on individuals, their caregivers and on health-care systems. One study assessing data from the “Financial and Social Impact of PD Survey” (commissioned by MJ Fox Foundation and Parkinson’s Foundation) found that compared to those patients experiencing no OFF, patients experiencing OFF episodes were less likely to work full-time, more likely to have days with lower productivity, and more likely to miss work due to PD.123 Additionally, they found that those providing care for patients with OFF episodes were more likely to stop working their other jobs due to PD and were more likely to have annual income losses when compared to caregivers of patients who were not experiencing OFF episodes.123 Patients experiencing OFF were also more likely to have more neurologist consultations, more hospital and ICU admissions, and more ER visits as a result of their PD.124
Several factors play into this, including the heterogeneity of these episodes between patients with varying frequencies and severities of each fluctuation even within the same patient. This is further complicated by misunderstanding of symptoms by patients which results in underreporting and thus under-recognition by clinicians.125 Some patients may also see these complications as an inevitable part of their disease and thus do not mention them.125 Better tools for clinicians to assess motor complications are needed, since clinical scales that are commonly used in practice do not adequately assess these symptoms.126 A survey done by Armstrong et al, in which PD patients and their caregivers were administered the Wearing off Questionnaire 19 (WOQ 19), found that while most fluctuating motor symptoms were assessed in some way with the WOQ 19, there were some motor, but numerous NMS which were reported but not included in WOQ 19, including fatigue and irritability.125 Furthermore, a cross-sectional study focusing on the frequency and severity of NMS in patients with advanced PD who were experiencing MF showed that all patients experienced at least 2 NMS and all reported worsening of at least 2 of their NMS during their motor OFF episodes.125 The Neuropsychiatric Fluctuations Scale (NFS) was recently developed but does not address sensory or autonomic fluctuations.126 The Nonmotor Fluctuation Assessment Scale (NoMoFa) is a scale that was recently developed to address neuropsychiatric, autonomic, and sensory fluctuations and represents an important step in the ability to quantify NMF alongside MF and any improvements that may occur with intervention. Thus, both the recognition and treatment of NMF still represent a major unmet area in the management of PD.
Aside from scales to assess symptoms, physicians rely on home diaries and patient self-report to assess for MF, NMS and MMF, but it remains imperative that patients are educated on the nature of these symptoms, why they occur, the relation to their medications, and the availability of treatment options. Wearables and sensors may be better able to identify symptom dynamics (for both MF and NMF), and importantly may be able to provide objective data that can not only signal the need for treatment of OFF episodes, but also support the development of more targeted therapies or facilitate a holistic approach to managing PD symptomatology, beyond motor manifestations.
Effective management of OFF episodes in PD involves the manipulation of levodopa pharmacokinetics using non-pharmacologic approaches, adjunctive medications or alternate routes of delivery, the use of rescue therapies, or device-assisted therapies. While some of them are efficacious alone, a combination of these approaches is generally needed.125 Efficacy is generally defined as reduction in absolute time spent in the OFF state (as defined by motor symptoms) without worsening of dyskinesia, and with evidence of improved QoL. None of these strategies, however, can completely ameliorate MF and dyskinesias. More notable effects can be seen with device aided therapies such as infusion therapies and DBS, which have been shown to deliver sustained reduction in OFF times due to their mechanisms, offer better control of dyskinesias, and have greater amounts of data exploring their effects on NMS and NMF. However, adequate guidelines for more timely intervention against MF and NMF with device-assisted therapies are needed.
Finally, due to the progressive nature of neurodegenerative disease, the risk of developing MF only increases as the disease advances and another major unmet need is for medications which can prevent disease progression. This approach will require better understanding of PD pathophysiology. While some therapies for monogenetic forms of PD are in development, it is not yet known whether these approaches will impact the evolution of MF and/or NMS/NMF. Alternative therapeutic targets will also need to be considered, as is currently being done with different neurotransmitter systems and identifying potential biomarkers.127
Conclusion
While levodopa has been the cornerstone of PD treatment, adding or switching to different therapeutic strategies is required to mitigate the effects of MF. A working knowledge of the differences between these therapeutic strategies, their expected benefits, and risk profiles is required in order to make effective recommendations for management. Early on, it may be possible to adjust doses of medications patients are already taking, such as with dose fractionation or with non-medical approaches such as dietary changes to restrict eating within a certain time frame of taking medication. These benefits are likely short-lived and eventually other approaches are needed to try and achieve CDS/CDD in an effort to minimize OFF times.
As we have reviewed here, these approaches include the use of longer acting levodopa formulations, the use of enzyme inhibitors to prevent metabolic degradation of levodopa, and the use of non-dopaminergic medications, often in combination. The selection of specific strategies for an individual patient may be dictated by cost, patient preference, impact on associated levodopa dosing, risk of worsening dyskinesia, or other side effects. For example, a patient with mainly MF and no dyskinesia may benefit from a dosing schedule change, or adjunctive dopaminergic or non-dopaminergic medications, while a patient with both MF and LID may benefit more from a rescue therapy, adjunctive non-dopaminergic medication or device assisted therapy. In fact, device aided therapies such as infusions and DBS are highly effective options that may provide more sustained improvement in MF, however more definitive guidelines regarding which patients to refer for these advanced therapies and when are needed. The resulting under-utilization of these options is further complicated by the fact that they are not universally available in all countries or may be subject to regulatory policies.
Emerging therapeutic targets for management of OFF episodes take advantage of the pitfalls of currently approved treatments, optimizing aspects such as absorption, drug half-life, and ease of administration. Newer agents such as IPX203 and new inhaled levodopa aim to improve upon pre-existing formulations, and will likely result in clinically relevant, albeit incremental, improvements in patient’s motor control. Newer therapies also aim to increase ease of administration, eg, adapting once daily formulations, which may help improve compliance. Levodopa infusion therapies currently in development may advance our ability to achieve CDS/CDD, which could potentially be seen as “disease modifying” if physiologic stimulation can be achieved for a greater portion of the disease course.
While all these improvements are essential in the effort to improve the daily lives of PD patients, they still represent tools which clinicians must learn to use in combination, and strategically to mitigate risk of current and future functional impairments amongst patients with PD. Patients must receive adequate education about these treatments, their expected effects, and any adverse effects or consequences of their use. Clinician knowledge balanced with consideration of patient preferences as well as psychosocial and economic factors are therefore important requisites of true shared decision-making in the effective management of OFF periods in PD.
Disclosure
Dr. Joohi Jimenez-Shahed reports that outside of the submitted work, she has received grants and personal fees from Medtronic and Amneal, and personal fees from St. Jude Medical, Abbvie, Alpha Omega, Bracket, Teva, Photopharmcis, and BlueRock. She is a member of the data safety monitoring commitee for BlueRock Therapeutics. The author reports no other conflicts of interest in this work.
References
1. Dorsey ER, Sherer T, Okun MS, Bloem BR. The emerging evidence of the Parkinson pandemic. J Parkinsons Dis. 2018;8(s1):S3–S8. doi:10.3233/JPD-181474
2. Muller T, Mohr JD. Long-term management of Parkinson’s disease using levodopa combinations. Expert Opin Pharmacother. 2018;19(9):1003–1011. doi:10.1080/14656566.2018.1484108
3. Jimenez-Shahed J. A review of current and novel levodopa formulations for the treatment of Parkinson’s disease. Ther Deliv. 2016;7(3):179–191. doi:10.4155/tde.15.96
4. Goto Y, Otani S, Grace AA. The Yin and Yang of dopamine release: a new perspective. Neuropharmacology. 2007;53(5):583–587. doi:10.1016/j.neuropharm.2007.07.007
5. Wright BA, Waters CH. Continuous dopaminergic delivery to minimize motor complications in Parkinson’s disease. Expert Rev Neurother. 2013;13(6):719–729. doi:10.1586/ern.13.47
6. Vijiaratnam N, Foltynie T. Therapeutic strategies to treat or prevent off episodes in adults with Parkinson’s disease. Drugs. 2020;80(8):775–796. doi:10.1007/s40265-020-01310-2
7. Martinez-Fernandez R, Schmitt E, Martinez-Martin P, Krack P. The hidden sister of motor fluctuations in Parkinson’s disease: a review on nonmotor fluctuations. Mov Disord. 2016;31(8):1080–1094. doi:10.1002/mds.26731
8. Pandey S, Srivanitchapoom P. Levodopa-induced dyskinesia: clinical features, pathophysiology, and medical management. Ann Indian Acad Neurol. 2017;20(3):190–198. doi:10.4103/aian.AIAN_239_17
9. Stocchi F, Bonamartini A, Vacca L, Ruggieri S. Motor fluctuations in levodopa treatment: clinical pharmacology. Eur Neurol. 1996;36(Suppl 1):38–42. doi:10.1159/000118882
10. Sinemet(R) [package insert]. Whitehouse Station. NJ: Merck & Co., INC; 2020
11. Freitas ME, Hess CW, Fox SH. Motor complications of dopaminergic medications in Parkinson’s disease. Semin Neurol. 2017;37(2):147–157. doi:10.1055/s-0037-1602423
12. LeWitt PA. Levodopa therapy for Parkinson’s disease: pharmacokinetics and pharmacodynamics. Mov Disord. 2015;30(1):64–72. doi:10.1002/mds.26082
13. Aradi SD, Hauser RA. Medical management and prevention of motor complications in Parkinson’s disease. Neurotherapeutics. 2020;17(4):1339–1365. doi:10.1007/s13311-020-00889-4
14. Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–2508. doi:10.1056/NEJMoa033447
15. Beckers M, Bloem BR, Verbeek MM. Mechanisms of peripheral levodopa resistance in Parkinson’s disease. NPJ Parkinsons Dis. 2022;8(1):56. doi:10.1038/s41531-022-00321-y
16. Nyholm D, Hellstrom PM. Effects of helicobacter pylori on levodopa pharmacokinetics. J Parkinsons Dis. 2021;11(1):61–69. doi:10.3233/JPD-202298
17. Hashim H, Azmin S, Razlan H, et al. Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS One. 2014;9(11):e112330. doi:10.1371/journal.pone.0112330
18. Lolekha P, Sriphanom T, Vilaichone RK. Helicobacter pylori eradication improves motor fluctuations in advanced Parkinson’s disease patients: a prospective cohort study (HP-PD trial). PLoS One. 2021;16(5):e0251042. doi:10.1371/journal.pone.0251042
19. Danau A, Dumitrescu L, Lefter A, Tulba D, Popescu BO. Small intestinal bacterial overgrowth as potential therapeutic target in Parkinson’s disease. Int J Mol Sci. 2021;22(21). doi:10.3390/ijms222111663
20. Vizcarra JA, Wilson-Perez HE, Fasano A, Espay AJ. Small intestinal bacterial overgrowth in Parkinson’s disease: tribulations of a trial. Parkinsonism Relat Disord. 2018;54:110–112. doi:10.1016/j.parkreldis.2018.04.003
21. Fasano A, Bove F, Gabrielli M, et al. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord. 2013;28(9):1241–1249. doi:10.1002/mds.25522
22. Boelens Keun JT, Arnoldussen IA, Vriend C. Dietary approaches to improve efficacy and control side effects of levodopa therapy in Parkinson’s disease: a systematic review. Adv Nutr. 2021;12(6):2265–2287. doi:10.1093/advances/nmab060
23. Brooks DJ. Optimizing levodopa therapy for Parkinson’s disease with levodopa/carbidopa/entacapone: implications from a clinical and patient perspective. Neuropsychiatr Dis Treat. 2008;4(1):39–47. doi:10.2147/ndt.s1660
24. Avion Pharmaceuticals. Dhivy [package insert]. U.S. Food and Drug Administration website; 2022. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214869s000lbl.pdf.
25. Grosset KA, Bone I, Grosset DG. Suboptimal medication adherence in Parkinson’s disease. Mov Disord. 2005;20(11):1502–1507. doi:10.1002/mds.20602
26. Stocchi F. The levodopa wearing-off phenomenon in Parkinson’s disease: pharmacokinetic considerations. Expert Opin Pharmacother. 2006;7(10):1399–1407. doi:10.1517/14656566.7.10.1399
27. Aquilonius SM, Nyholm D. Development of new levodopa treatment strategies in Parkinson’s disease-from bedside to bench to bedside. Ups J Med Sci. 2017;122(2):71–77. doi:10.1080/03009734.2017.1285374
28. GSK. Requip (R) (ropinirole hydrochloride) tablets [package insert]. U.S. Food and Drug Administration; 2007. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020658s018s020s021lbl.pdf.
29. UCB. Neupro (R) (Rotigotine transdermal system) [package insert]. U.S. Food and Drug Administration; 2022. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021829s001lbl.pdf.
30. Boehringer Ingelheim Pharmaceuticals, Inc. Mirapex (R) (pramipexole dihydrochloride) [package insert]. U.S. Food and Drug Administration; 2022. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020667s014s017s018lbl.pdf.
31. Kondo T. Initial therapy for Parkinson’s disease: levodopa vs. dopamine receptor agonists. J Neurol. 2002;249(Suppl 2):II25–II29. doi:10.1007/s00415-002-1205-3
32. Pringsheim T, Day GS, Smith DB, et al. Dopaminergic therapy for motor symptoms in early Parkinson disease practice guideline summary: a report of the AAN guideline subcommittee. Neurology. 2021;97(20):942–957. doi:10.1212/WNL.0000000000012868
33. Ji N, Meng P, Xu B, Zhou X. Efficacy and safety of pramipexole in Parkinson’s disease with anxiety or depression: a meta-analysis of randomized clinical trials. Am J Transl Res. 2022;14(3):1757–1764.
34. Stocchi F, Giorgi L, Hunter B, Schapira AH. PREPARED: comparison of prolonged and immediate release ropinirole in advanced Parkinson’s disease. Mov Disord. 2011;26(7):1259–1265. doi:10.1002/mds.23498
35. Raeder V, Boura I, Leta V, et al. Rotigotine transdermal patch for motor and non-motor Parkinson’s disease: a review of 12 years’ clinical experience. CNS Drugs. 2021;35(2):215–231. doi:10.1007/s40263-020-00788-4
36. Bhattamisra SK, Shak AT, Xi LW, et al. Nose to brain delivery of rotigotine loaded chitosan nanoparticles in human SH-SY5Y neuroblastoma cells and animal model of Parkinson’s disease. Int J Pharm. 2020;579:119148. doi:10.1016/j.ijpharm.2020.119148
37. Hattori N, Mochizuki H, Hasegawa K, et al. Ropinirole patch versus placebo, ropinirole extended-release tablet in advanced Parkinson’s disease. Mov Disord. 2020;35(9):1565–1573. doi:10.1002/mds.28071
38. Mochizuki H, Hattori N, Hasegawa K, et al. Long-term study of ropinirole patch in Parkinson’s disease patients with/without basal l-dopa. Parkinsonism Relat Disord. 2021;83:105–109. doi:10.1016/j.parkreldis.2020.12.023
39. Mylan Pharmaceuticals, Inc. Sinemet (R) CR (carbidopa levodopa) sustained-release tablets [package insert]. U.S. Food and Drug Administration. 2022. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/019856s016s024s028lbl.pdf.
40. Contin M, Riva R, Albani F, Baruzzi A. Pharmacokinetic optimisation in the treatment of Parkinson’s disease. Clin Pharmacokinet. 1996;30(6):463–481. doi:10.2165/00003088-199630060-00004
41. Hsu A, Yao HM, Gupta S, Modi NB. Comparison of the pharmacokinetics of an oral extended-release capsule formulation of carbidopa-levodopa (IPX066) with immediate-release carbidopa-levodopa (Sinemet((R))), sustained-release carbidopa-levodopa (Sinemet((R)) CR), and carbidopa-levodopa-entacapone (Stalevo((R))). J Clin Pharmacol. 2015;55(9):995–1003. doi:10.1002/jcph.514
42. Impax Laboratories. Rytary (R) (carbidopa and levodopa) extended-release capsules [package insert]. U.S. Food and Drug Administration; 2015.
43. Hauser RA, Zeitlin L, Fisher S, D’Souza R. Carbidopa and levodopa extended release capsules in patients with and without troublesome and non-troublesome dyskinesia. J Parkinsons Dis. 2020;10(3):915–925. doi:10.3233/JPD-202010
44. Modi NB, Mittur A, Dinh P, Rubens R, Gupta S. Pharmacodynamics, efficacy, and safety of IPX203 in Parkinson disease patients with motor fluctuations. Clin Neuropharmacol. 2019;42(5):149–156. doi:10.1097/WNF.0000000000000354
45. Modi NB, Mittur A, Rubens R, Khanna S, Gupta S. Single-dose pharmacokinetics and pharmacodynamics of IPX203 in patients with advanced Parkinson disease: a comparison with immediate-release carbidopa-levodopa and with extended-release carbidopa-levodopa capsules. Clin Neuropharmacol. 2019;42(1):4–8. doi:10.1097/WNF.0000000000000314
46. Hauser RA, Espay AJ, LeWitt P, et al. A phase 3 trial of IPX203 vs CD-LD IR in Parkinson’s disease patients with motor fluctuations (RISE-PD) (S16.010). Neurology. 2022;98(18 Supplement):1225.
47. Hauser RA, Fernandez HH, Klos K, et al. Duration of benefit per dose: post hoc analysis of “good on” time per dose for IPX203 vs CD-LD IR in the RISE-PD phase 3 trial.
48. Tolosa E, Marti MJ, Valldeoriola F, Molinuevo JL. History of levodopa and dopamine agonists in Parkinson’s disease treatment. Neurology. 1998;50(6Suppl 6):S2–S10. doi:10.1212/wnl.50.6_suppl_6.s2
49. Somerset Pharmaceuticals, Inc. Eldepryl (R) (Selegiline Hydrochloride) [package insert]. U.S. Food and Drug Administration; 2008. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020647s006s007lbl.pdf.
50. Jost WH. A critical appraisal of MAO-B inhibitors in the treatment of Parkinson’s disease. J Neural Transm. 2022;129(5–6):723–736. doi:10.1007/s00702-022-02465-w
51. Stocchi F, Fossati C, Torti M. Rasagiline for the treatment of Parkinson’s disease: an update. Expert Opin Pharmacother. 2015;16(14):2231–2241. doi:10.1517/14656566.2015.1086748
52. Bette S, Shpiner DS, Singer C, Moore H. Safinamide in the management of patients with Parkinson’s disease not stabilized on levodopa: a review of the current clinical evidence. Ther Clin Risk Manag. 2018;14:1737–1745. doi:10.2147/TCRM.S139545
53. Blandini F, Greenamyre JT, Nappi G. The role of glutamate in the pathophysiology of Parkinson’s disease. Funct Neurol. 1996;11(1):3–15.
54. Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol. 2012;96(1):69–86. doi:10.1016/j.pneurobio.2011.10.005
55. Watson CJ. Insular balance of glutamatergic and GABAergic signaling modulates pain processing. Pain. 2016;157(10):2194–2207. doi:10.1097/j.pain.0000000000000615
56. Brooks DJ, Sagar H; Group UK-IES. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind, six month study. J Neurol Neurosurg Psychiatry. 2003;74(8):1071–1079. doi:10.1136/jnnp.74.8.1071
57. Group PS. Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol. 1997;42(5):747–755. doi:10.1002/ana.410420511
58. Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in Parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology. 1998;51(5):1309–1314. doi:10.1212/wnl.51.5.1309
59. Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M, Celomen Study G. Efficacy and safety of entacapone in Parkinson’s disease patients with suboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand. 2002;105(4):245–255. doi:10.1034/j.1600-0404.2002.1o174.x
60. Larsen JP, Worm-Petersen J, Siden A, et al. The tolerability and efficacy of entacapone over 3 years in patients with Parkinson’s disease. Eur J Neurol. 2003;10(2):137–146. doi:10.1046/j.1468-1331.2003.00559.x
61. Bausch. Tasmar (R) (tolcapone) [package insert]. U.S. Food and Drug Administration; 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/020697s004lbl.pdf.
62. Greenwood J, Pham H, Rey J. Opicapone: a third generation COMT inhibitor. Clin Park Relat Disord. 2021;4:100083. doi:10.1016/j.prdoa.2020.100083
63. Song Z, Zhang J, Xue T, et al. Different Catechol-O-Methyl transferase inhibitors in Parkinson’s disease: a bayesian network meta-analysis. Front Neurol. 2021;12:707723. doi:10.3389/fneur.2021.707723
64. Rascol O, Fabbri M, Poewe W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 2021;20(12):1048–1056. doi:10.1016/S1474-4422(21)00249-0
65. Adamas Pharma, LLC. Gocovri TM (amantadine) extended-release capsules [package insert]. U.S. Food and Drug Administration. 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208944lbl.pdf.
66. Hauser R, Chernick D, Formella A. Gocovri reduces disruptive motor episodes and improves function in Parkinson’s disease patients with OFF episodes and dyskinesia: analysis of phase 3 trial data (1882). Neurology. 2020;94(15 Supplement):1882.
67. Hauser RA, Lytle J, Formella AE, Tanner CM. Amantadine delayed release/extended release capsules significantly reduce OFF time in Parkinson’s disease. NPJ Parkinsons Dis. 2022;8(1):29. doi:10.1038/s41531-022-00291-1
68. Li C, Xue L, Liu Y, Yang Z, Chi S, Xie A. Zonisamide for the treatment of Parkinson disease: a current update. Front Neurosci. 2020;14:574652. doi:10.3389/fnins.2020.574652
69. Uemura MT, Asano T, Hikawa R, Yamakado H, Takahashi R. Zonisamide inhibits monoamine oxidase and enhances motor performance and social activity. Neurosci Res. 2017;124:25–32. doi:10.1016/j.neures.2017.05.008
70. Willmore LJ. Antiepileptic drugs and neuroprotection: current status and future roles. Epilepsy Behav. 2005;7(Suppl 3):S25–8. doi:10.1016/j.yebeh.2005.08.006
71. Rosler TW, Arias-Carrion O, Hoglinger GU. Zonisamide: aspects in neuroprotection. Exp Neurol. 2010;224(2):336–339. doi:10.1016/j.expneurol.2010.04.017
72. Ikeda K, Yanagihashi M, Miura K, et al. Zonisamide cotreatment delays striatal dopamine transporter reduction in Parkinson disease: a retrospective, observational cohort study. J Neurol Sci. 2018;391:5–9. doi:10.1016/j.jns.2018.05.013
73. Murata M, Hasegawa K, Kanazawa I, et al. Zonisamide improves wearing-off in Parkinson’s disease: a randomized, double-blind study. Mov Disord. 2015;30(10):1343–1350. doi:10.1002/mds.26286
74. Saenz-Farret M, Tijssen MAJ, Eliashiv D, Fisher RS, Sethi K, Fasano A. Antiseizure drugs and movement disorders. CNS Drugs. 2022;36(8):859–876. doi:10.1007/s40263-022-00937-x
75. Tsuboi Y, Nakamura M, Maruyama H, Matsumoto Y. Zonisamide improves wearing off in Parkinson’s disease without exacerbating dyskinesia: post hoc analysis of phase 2 and phase 3 clinical trials. J Neurol Sci. 2021;430:120026. doi:10.1016/j.jns.2021.120026
76. Asanuma M, Miyazaki I, Diaz-Corrales FJ, et al. Neuroprotective effects of zonisamide target astrocyte. Ann Neurol. 2010;67(2):239–249. doi:10.1002/ana.21885
77. Cummins L, Cates ME. Istradefylline: a novel agent in the treatment of “off” episodes associated with levodopa/carbidopa use in Parkinson disease. Ment Health Clin. 2022;12(1):32–36. doi:10.9740/mhc.2022.01.032
78. Zheng J, Zhang X, Zhen X. Development of Adenosine A2A receptor antagonists for the treatment of Parkinson’s disease: a recent update and challenge. ACS Chem Neurosci. 2019;10(2):783–791. doi:10.1021/acschemneuro.8b00313
79. Kyowa Kirin, Inc. Nourianz TM (istradefylline) tablets [package insert]. U.S. Food and Drug Administration. 2019. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/022075s000lbl.pdf.
80. Kondo T, Mizuno Y; Japanese Istradefylline Study G. A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin Neuropharmacol. 2015;38(2):41–46. doi:10.1097/WNF.0000000000000073
81. Patel AB, Jimenez-Shahed J. Profile of inhaled levodopa and its potential in the treatment of Parkinson’s disease: evidence to date. Neuropsychiatr Dis Treat. 2018;14:2955–2964. doi:10.2147/NDT.S147633
82. Nausieda PA, Pfeiffer RF, Tagliati M, Kastenholz KV, DeRoche C, Slevin JT. A multicenter, open-label, sequential study comparing preferences for carbidopa-levodopa orally disintegrating tablets and conventional tablets in subjects with Parkinson’s disease. Clin Ther. 2005;27(1):58–63. doi:10.1016/j.clinthera.2005.01.004
83. Gupta HV, Lyons KE, Pahwa R. Old drugs, new delivery systems in Parkinson’s disease. Drugs Aging. 2019;36(9):807–821. doi:10.1007/s40266-019-00682-9
84. Ondo WG, Shinawi L, Moore S. Comparison of orally dissolving carbidopa/levodopa (Parcopa) to conventional oral carbidopa/levodopa: a single-dose, double-blind, double-dummy, placebo-controlled, crossover trial. Mov Disord. 2010;25(16):2724–2727. doi:10.1002/mds.23158
85. Acorda Therapeutics, Inc. Inbrija TM (levodopa inhalation powder) [package insert]. U.S. Food and Drug Administration; 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/209184s000lbl.pdf.
86. LeWitt PA, Hauser RA, Grosset DG, et al. A randomized trial of inhaled levodopa (CVT-301) for motor fluctuations in Parkinson’s disease. Mov Disord. 2016;31(9):1356–1365. doi:10.1002/mds.26611
87. LeWitt PA, Hauser RA, Pahwa R, et al. Safety and efficacy of CVT-301 (levodopa inhalation powder) on motor function during off periods in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Neurol. 2019;18(2):145–154. doi:10.1016/S1474-4422(18)30405-8
88. LeWitt PA, Pahwa R, Sedkov A, Corbin A, Batycky R, Murck H. Pulmonary safety and tolerability of inhaled levodopa (CVT-301) administered to patients with Parkinson’s disease. J Aerosol Med Pulm Drug Deliv. 2018;31(3):155–161. doi:10.1089/jamp.2016.1354
89. Grosset DG, Dhall R, Gurevich T, et al. Inhaled levodopa in Parkinson’s disease patients with OFF periods: a randomized 12-month pulmonary safety study. Parkinsonism Relat Disord. 2020;71:4–10. doi:10.1016/j.parkreldis.2019.12.012
90. Pharmaceuticals I. Pipeline impel pharmaceuticals 2022; 2022. Available from: https://impelpharma.com/treatments/.
91. Vass G, Huszar E, Barat E, et al. Comparison of nasal and oral inhalation during exhaled breath condensate collection. Am J Respir Crit Care Med. 2003;167(6):850–855. doi:10.1164/rccm.200207-716BC
92. Carbone F, Djamshidian A, Seppi K, Poewe W. Apomorphine for Parkinson’s disease: efficacy and safety of current and new formulations. CNS Drugs. 2019;33(9):905–918. doi:10.1007/s40263-019-00661-z
93. Agbo F, Isaacson SH, Gil R, et al. Pharmacokinetics and comparative bioavailability of apomorphine sublingual film and subcutaneous apomorphine formulations in patients with Parkinson’s disease and “OFF” episodes: results of a randomized, three-way crossover, open-label study. Neurol Ther. 2021;10(2):693–709. doi:10.1007/s40120-021-00251-6
94. Britannia Pharmaceuticals. Apokyn (R) (apomorphine hydrochloride injection) [package insert]. U.S. Food and Drug Administration; 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021264s014lbl.pdf.
95. Thijssen E, den Heijer JM, Puibert D, van Brummelen EMJ, Naranda T, Groeneveld GJ. Safety and pharmacokinetics of multiple dosing with inhalable apomorphine (AZ-009), and its efficacy in a randomized crossover study in Parkinson’s disease patients. Parkinsonism Relat Disord. 2022;97:84–90. doi:10.1016/j.parkreldis.2022.02.014
96. Thijssen E, den Heijer J, Puibert D, et al. A randomized trial assessing the safety, pharmacokinetics, and efficacy during morning Off of AZ-009. Mov Disord. 2022;37(4):790–798. doi:10.1002/mds.28926
97. Virhammar J, Nyholm D. Levodopa-carbidopa enteral suspension in advanced Parkinson’s disease: clinical evidence and experience. Ther Adv Neurol Disord. 2017;10(3):171–187. doi:10.1177/1756285616681280
98. Inc. A. Duopa website AbbVie Inc; 2022. Available from: https://www.duopa.com/.
99. Antonini A, Odin P, Pahwa R, et al. The long-term impact of levodopa/carbidopa intestinal gel on ‘Off’-time in patients with advanced Parkinson’s disease: a systematic review. Adv Ther. 2021;38(6):2854–2890. doi:10.1007/s12325-021-01747-1
100. Nyholm D, Nilsson Remahl AI, Dizdar N, et al. Duodenal levodopa infusion monotherapy vs oral polypharmacy in advanced Parkinson disease. Neurology. 2005;64(2):216–223. doi:10.1212/01.WNL.0000149637.70961.4C
101. Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014;13(2):141–149. doi:10.1016/S1474-4422(13)70293-X
102. Romagnolo A, Merola A, Artusi CA, Rizzone MG, Zibetti M. Levodopa-induced neuropathy: a systematic review. Mov Disord Clin Pract. 2019;6(2):96–103. doi:10.1002/mdc3.12688
103. Dijk JM, Espay AJ, Katzenschlager R, de Bie RMA. The choice between advanced therapies for Parkinson’s disease patients: why, what, and when? J Parkinsons Dis. 2020;10(s1):S65–S73. doi:10.3233/JPD-202104
104. Liu XD, Bao Y, Liu GJ. Comparison between levodopa-carbidopa intestinal gel infusion and subthalamic nucleus deep-brain stimulation for advanced Parkinson’s disease: a systematic review and meta-analysis. Front Neurol. 2019;10:934. doi:10.3389/fneur.2019.00934
105. Nyholm D, Jost WH. Levodopa-entacapone-carbidopa intestinal gel infusion in advanced Parkinson’s disease: real-world experience and practical guidance. Ther Adv Neurol Disord. 2022;15:17562864221108018. doi:10.1177/17562864221108018
106. Senek M, Nielsen EI, Nyholm D. Levodopa-entacapone-carbidopa intestinal gel in Parkinson’s disease: a randomized crossover study. Mov Disord. 2017;32(2):283–286. doi:10.1002/mds.26855
107. Dhamija RK, Srivastava AK, Garg D. Apomorphine in Parkinson’s disease-the questions raised. Ann Indian Acad Neurol. 2020;23(3):377–378. doi:10.4103/aian.AIAN_11_20
108. Katzenschlager R, Poewe W, Rascol O, et al. Apomorphine subcutaneous infusion in patients with Parkinson’s disease with persistent motor fluctuations (Toledo): a multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2018;17(9):749–759. doi:10.1016/S1474-4422(18)30239-4
109. Gaire S, Kafle S, Bastakoti S, Paudel A, Karki K. Continuous subcutaneous apomorphine infusion in advanced Parkinson’s disease: a systematic review. Cureus. 2021;13(9):e17949. doi:10.7759/cureus.17949
110. Nyholm D, Askmark H, Gomes-Trolin C, et al. Optimizing levodopa pharmacokinetics: intestinal infusion versus oral sustained-release tablets. Clin Neuropharmacol. 2003;26(3):156–163. doi:10.1097/00002826-200305000-00010
111. Bergquist F, Ehrnebo M, Nyholm D, et al. Pharmacokinetics of intravenously (DIZ101), subcutaneously (DIZ102), and intestinally (LCIG) infused levodopa in advanced Parkinson disease. Neurology. 2022. doi:10.1212/WNL.0000000000200804
112. Giladi N, Gurevich T, Djaldetti R, et al. ND0612 (levodopa/carbidopa for subcutaneous infusion) in patients with Parkinson’s disease and motor response fluctuations: a randomized, placebo-controlled phase 2 study. Parkinsonism Relat Disord. 2021;91:139–145. doi:10.1016/j.parkreldis.2021.09.024
113. Rosebraugh M, Liu W, Neenan M, Facheris MF. Foslevodopa/foscarbidopa is well tolerated and maintains stable levodopa and carbidopa exposure following subcutaneous infusion. J Parkinsons Dis. 2021;11(4):1695–1702. doi:10.3233/JPD-212813
114. ClinicalTrials.gov. Study comparing continuous subcutaneous infusion of ABBV-951 with oral carbidopa/levodopa tablets for treatment of motor fluctuations in adult participants with advanced Parkinson’s disease; 2021. Available from: https://clinicaltrials.gov.
115. Fasano A, Daniele A, Albanese A. Treatment of motor and non-motor features of Parkinson’s disease with deep brain stimulation. Lancet Neurol. 2012;11(5):429–442. doi:10.1016/S1474-4422(12)70049-2
116. Deuschl G, Schade-Brittinger C, Krack P, et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2006;355(9):896–908. doi:10.1056/NEJMoa060281
117. Weaver FM, Follett K, Stern M, et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63–73. doi:10.1001/jama.2008.929
118. Williams A, Gill S, Varma T, et al. Deep brain stimulation plus best medical therapy versus best medical therapy alone for advanced Parkinson’s disease (PD SURG trial): a randomised, open-label trial. Lancet Neurol. 2010;9(6):581–591. doi:10.1016/S1474-4422(10)70093-4
119. Vitek JL, Jain R, Chen L, et al. Subthalamic nucleus deep brain stimulation with a multiple independent constant current-controlled device in Parkinson’s disease (INTREPID): a multicentre, double-blind, randomised, sham-controlled study. Lancet Neurol. 2020;19(6):491–501. doi:10.1016/S1474-4422(20)30108-3
120. Okun MS, Gallo BV, Mandybur G, et al. Subthalamic deep brain stimulation with a constant-current device in Parkinson’s disease: an open-label randomised controlled trial. Lancet Neurol. 2012;11(2):140–149. doi:10.1016/S1474-4422(11)70308-8
121. Antonini A, Pahwa, R, Odin P, et al. Impact of device-aided therapies on QoL and Off-time improvement in advanced Parkinson’s Disease patients: comparative effectiveness results from a Bayesian network meta-analysis; 2021. Available from: https://www.mdsabstracts.org/abstract/impact-of-device-aided-therapies-on-qol-and-off-time-improvement-in-advanced-parkinsons-disease-patients-comparative-effectiveness-results-from-a-bayesian-network-meta-analysis/.
122. Marsili L, Bologna M, Miyasaki JM, Colosimo C. Parkinson’s disease advanced therapies - A systematic review: more unanswered questions than guidance. Parkinsonism Relat Disord. 2021;83:132–139. doi:10.1016/j.parkreldis.2020.10.042
123. Abeynayake I, Tanner CM. The economic impact of OFF periods in Parkinson disease. Am J Manag Care. 2020;26(12 Suppl):S265–S269. doi:10.37765/ajmc.2020.88518
124. Thach A, Jones E, Pappert E, Pike J, Wright J, Gillespie A. Real-world assessment of “OFF” episode-related healthcare resource utilization among patients with Parkinson’s disease in the United States. J Med Econ. 2021;24(1):540–549. doi:10.1080/13696998.2021.1913009
125. Tanner CM. Exploring the clinical burden of OFF periods in Parkinson disease. Am J Manag Care. 2020;26(12 Suppl):S255–S264. doi:10.37765/ajmc.2020.88517
126. Del Prete E, Schmitt E, Meoni S, et al. Do neuropsychiatric fluctuations temporally match motor fluctuations in Parkinson’s disease? Neurol Sci. 2022;43(6):3641–3647. doi:10.1007/s10072-021-05833-8
127. Gouda NA, Elkamhawy A, Cho J. Emerging therapeutic strategies for Parkinson’s disease and future prospects: a 2021 update. Biomedicines. 2022;10(2):5. doi:10.3390/biomedicines10020371
128. Chou KL, Stacy M, Simuni T, et al. The spectrum of “off” in Parkinson’s disease: what have we learned over 40 years? Parkinsonism Relat Disord. 2018;51:9–16. doi:10.1016/j.parkreldis.2018.02.001
129. Borgohain R, Szasz J, Stanzione P, et al. Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Mov Disord. 2014;29(2):229–237. doi:10.1002/mds.25751
130. Schapira AH, Fox SH, Hauser RA, et al. Assessment of safety and efficacy of safinamide as a levodopa adjunct in patients with Parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol. 2017;74(2):216–224. doi:10.1001/jamaneurol.2016.4467
131. Waters CH, Sethi KD, Hauser RA, Molho E, Bertoni JM; Zydis Selegiline Study G. Zydis selegiline reduces off time in Parkinson’s disease patients with motor fluctuations: a 3-month, randomized, placebo-controlled study. Mov Disord. 2004;19(4):426–432. doi:10.1002/mds.20036
132. Follett KA, Weaver FM, Stern M, et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2010;362(22):2077–2091. doi:10.1056/NEJMoa0907083
133. Odekerken VJ, van Laar T, Staal MJ, et al. Subthalamic nucleus versus globus pallidus bilateral deep brain stimulation for advanced Parkinson’s disease (NSTAPS study): a randomised controlled trial. Lancet Neurol. 2013;12(1):37–44. doi:10.1016/S1474-4422(12)70264-8
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.