Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Effect of simvastatin on MMPs and TIMPs in cigarette smoke-induced rat COPD model
Authors Sun J, Bao J, Shi Y, Zhang B, Yuan L, Li J, Zhang L, Sun M, Zhang L, Sun W ![]()
Received 13 April 2016
Accepted for publication 4 August 2016
Published 22 February 2017 Volume 2017:12 Pages 717—724
DOI https://doi.org/10.2147/COPD.S110520
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Jiawei Sun,1,* Jie Bao,2,* Yanan Shi,3 Bin Zhang,4 Lindong Yuan,5 Junhong Li,1 Lihai Zhang,1 Mo Sun,6 Ling Zhang,2 Wuzhuang Sun1
1Department of Respiratory Medicine, First Hospital of Hebei Medical University, 2Department of Respiratory Medicine, Chest Hospital of Hebei Province, 3Maternal and Child Health Care Center of Hebei Province, 4Department of Emergency, First Hospital of Hebei Medical University, Shijiazhuang, 5Department of Respiratory Medicine, People’s Hospital of Liaocheng, Liaocheng, 6Hebei Medical University, Shijiazhuang, People’s Republic of China
*These authors contributed equally to this work
Background: Proteases may play an important role in the development of chronic obstructive pulmonary disease and emphysema in response to cigarette smoke exposure (CSE). The current study was designed to investigate the expression of matrix metalloproteinase (MMP)-8, MMP-9, MMP-12, tissue inhibitor of MMP (TIMP)-1, and TIMP-4 in rat lung tissues in response to CSE, and assessed the effect of simvastatin in regulating expression of MMPs and TIMPs.
Methods: Thirty normal Sprague Dawley (SD) rats were divided into control (n=10), CSE (n=10), and CSE plus simvastatin (n=10) groups. Animals were whole-body exposed to the cigarette smoke in the box for 1 hour each time, twice a day, 5 days a week for 16 weeks. Animals of CSE + simvastatin group were intra-gastrically administered simvastatin at a dose of 5 mg/kg/day followed by CSE. Bronchoalveolar lavage fluid was harvested for inflammatory cell count and lung tissues were stained for morphologic examination. Expression of mRNA and protein level of MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-4 was assessed by real-time reverse transcription polymerase chain reaction and immunohistochemistry, respectively.
Results: CSE resulted in a significant increase of mean linear intercept (MLI: 34.6±2.0 µm) and bronchial wall thickness and diameter (BWT/D, 0.250±0.062) compared to control (MLI: 24.0±1.7 µm, BWT/D: 0.160±0.034, P<0.01). In contrast, mean alveolar number was significantly decreased in the CSE group than that in the control group (13.5±2.0 of CSE vs 21.5±2.0 N/µm2 of control, P>0.01). Simvastatin slightly but not significantly prevented alteration of MLI, BWT/D, and mean alveolar number (MLI: 33.4±1.4 µm; BWT/D: 0.220±0.052; mean alveolar number: 15.5±2.5 N/µm2, P>0.05). Total white blood cell was significantly increased in the bronchoalveolar lavage fluid of smoking group (3.3±2.5×109 cells/L vs 1.1±1.3×109 cells/L of control, P<0.01), and it was significantly reduced by simvastatin (2.3±2.1×109 cells/L, P<0.01). CSE resulted in significantly increased accumulation of neutrophils and macrophages (neutrophils: 14.5%±1.3% of CSE group vs 9.1%±1.5% of control; macrophage: 91%±3% of CSE group vs 87%±2% of control, P<0.05), and simvastatin significantly reduced neutrophils (12.9%±2.0%, P<0.05) in the bronchoalveolar lavage fluid, but had no effect on macrophage (89%±1.6%, P>0.05). In response to CSE, MMP-8, MMP-9, and MMP-12 mRNA were upregulated more than sevenfold, while TIMP-1 and TIMP-4 increased two- to fivefold. Simvastatin significantly blocked upregulation of MMP-8 and -9 (P<0.01), but had no effect on MMP-12, TIMP-1 and TIMP-4 mRNA (P>0.05). In addition, simvastatin significantly blocked cigarette smoke-induced MMP-8 and -9 protein synthesis, while it had no significant effect on TIMP-1 and -4 protein synthesis even in the presence of cigarette smoke.
Conclusion: CSE resulted in imbalance of MMPs and TIMPs, and by which mechanism, cigarette smoke may lead to insufficient lung tissue repair. Simvastatin partially blocked airway inflammation and MMP production and, thus, statins may modulate composition of the lung extracellular matrix.
Keywords: tissue injury, tissue repair, smoking
Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide.1 COPD is characterized by chronic airway inflammation and irreversible airflow obstruction, which could result from abnormal airway tissue injury and remodeling. In this regard, the balance between extracellular matrix production and its destruction may be important in determining the outcomes of tissue repair. In particular, balanced and sufficient airway tissue repair is required to restore architectural integrity and normal function of airflow following injury. Insufficient repair may lead to emphysema and, in contrast, excessive repair or remodeling may lead to lung fibrosis.2
Cigarette smoke contains over a 1,000 chemicals. COPD-like airway injury and remodeling are developed in the rodent animals, such as rat and mouse, when they are chronically exposed to cigarette smoke.3,4 One of the potential mechanisms of the cigarette smoke-induced COPD is that cigarette smoke leads to insufficient lung tissue repair through altering production of matrix metalloproteinases (MMPs) and their biological inhibitors, tissue inhibitors of MMPs (TIMPs).5 The MMPs are a large family of proteolytic enzymes6 that are produced by both inflammatory cells and lung structural cells, including epithelial cells and fibroblasts.7,8
Currently, therapy of COPD and emphysema can partially alleviate symptoms, but has relatively little impact on the proteolytic destruction of lung structure.2 Novel therapeutic strategies that potentially block proteolytic tissue destruction, therefore, are of interest as possible approaches to alter the long-term natural history of COPD. In this regard, it has been reported that statins may be associated with reduced acute events and mortality of COPD.9,10
Statins are a class of cholesterol-lowering drugs that inhibit 3-hydroxy-3-methylglutaryl co-enzyme A. Recently, it has been reported that statins have anti-inflammatory and antioxidant effects,11 an inhibitory effect on MMP release from macrophages, lung fibroblasts, and vascular cells,7,12,13 as well as in the animal model of COPD in response to cigarette smoke exposure (CSE).5 Through the effect on MMPs, statins may directly regulate balance of MMPs and their biological inhibitors, TIMPs, and thereby, may modify the alterations in lung structure that compromise lung function.
The current study was designed to extend previous studies of simvastatin regulation on chronic airway inflammation and lung structural alteration in response to CSE. To investigate this, rats were exposed to cigarette smoke for 4 months with or without simvastatin treatment. Animals were sacrificed and alteration of lung structure was assessed by mean linear intercept (MLI), mean alveolar number (MAN), and bronchial wall thickness and diameter (BWT/D). Total and differential inflammatory cell counts in bronchoalveolar lavage fluid (BALF) were performed. Expression of mRNA and protein level of MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-4 were carried out by real-time reverse transcription polymerase chain reaction (RT-PCR) and immunohistochemistry, respectively.
Materials and methods
Animals and CSE
Healthy male Sprague Dawley (SD) rats, 6 weeks old, body weight 110±20 g, were maintained in the animal facilities at the Hebei Medical University. Animals were accommodated for 1 week before the experiment. The study protocol was approved by The Institutional Animal Care and Use Committee, Hebei Medical University. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act. Animals were randomized into the following three groups: control group (ten rats), CSE group (ten rats), and CSE + simvastatin (CSE + S) group (ten rats). Rats in the groups of CSE only or CSE + S were exposed to cigarette smoke (Shijiazhuang Cigarette creating 8 mg tar, 0.8 mg nicotine, and 10 mg carbon monoxide) in a smoking device manufactured by Shijiazhuang Jinyang Science and Technology Inc. (model: JY-01, Shijiazhuang, People’s Republic of China). Briefly, 20 cigarettes were burnt continuously and blown into a box together with oxygen, and animals were passively whole-body exposed to the cigarette smoke in the box for 1 hour each time, twice a day, 5 days a week for 16 weeks. Animals of CSE + S group were intra-gastrically administered simvastatin (MSD, Hangzhou, People’s Republic of China) at a dose of 5 mg/kg/day, each day prior to the smoke exposure.14 The control rats were not treated with any reagent.
BALF collection and analysis
Animals were given an intraperitoneal injection of 10% chloral hydrate (3 mL/kg). After trachea intubation, animals were sacrificed by bleeding at femoral artery. Three lobes of right lung were snap-frozen with liquid nitrogen and one lobe of the right lung was fixed with formaldehyde. Bronchoalveolar lavage was performed on the left lung, using a three-in/three-out pattern of main bronchial instillation and collection 3 mL saline (70%–80% recovery).
The total cell number in the BALF was counted with an erythrocytometer. The rest of BALF specimens were centrifuged at 1,000 rpm ×10 minutes. Pelleted cells were resuspended with phosphate-buffered saline, and differentially counted by Wright–Giemsa staining. A total of 200 leukocytes was counted in each BALF sample, and the percentage of macrophage, neutrophil, and lymphocyte was calculated.
Histological examination
Air in the alveolar spaces was aspirated with a syringe before injecting 10% formalin in order to avoid alteration of lung structure. The fixed lung tissues were embedded in paraffin wax blocks and cut for hematoxylin and eosin staining. The hematoxylin and eosin stained slides were photographed under a light microscope (Olympus Corporation, Tokyo, Japan). Morphology of the lung tissue was observed and MLI was obtained by the following method. Cross lines were drawn in a field and the total number of alveolar septa at the intersection point of the two lines was counted. The total length of the cross lines divided by the number of intercepts gives the MLI for the region studied, that is, MLI = total length/number of alveolar septa. MLI indicates the average size of alveoli.
MAN was determined by the alveolar number per square millimeter under microscope, that is, MAN = alveolar number/square millimeter. MAN is a parameter for density of alveoli.
BWT/D was obtained by measuring the bronchi with <100 μm circumference in size. At least eight bronchi were measured and the average of the ratio of wall thickness over diameter was calculated.
Immunohistochemistry
Expression of MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-4 in rat lung was assessed by immunohistochemistry. Briefly, paraffin embedded lung tissue slice was deparaffinized and rehydrated. Antigen was retrieved using sodium citrate and with heat-induced retrieval. After blocking with goat serum, antiMMPs or TIMPs antibodies at 1:200 dilution (all antibodies were purchased from Abcam, Shanghai, People’s Republic of China – antiMMP-8 antibody: ab81286; antiMMP-9 antibody: ab76003; antiMMP-12 antibody: ab66157; antiTIMP-1 antibody: ab16412; antiTIMP-4 antibody: ab58425) were applied overnight at 4°C. After horseradish peroxidase-conjugated second antibody was applied, expression of MMPs and TIMPs was visualized using a DAB detection kit. Images of MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-4 were obtained and photographed under microscope (Olympus Corporation).
Real-time RT-PCR
Total RNA was extracted from the snap-frozen rat lung tissues with Trizol reagent (SBS Genetech, Shanghai, People’s Republic of China) following the manufacturer’s instruction. After RNA quantification with ND-1000 spectrophotometer (NanoDrop, USA), 1 μg total RNA was used for reverse transcription using high capacity reverse transcription kit (Baosheng Bio, Dalian, People’s Republic of China). Quantitative real-time PCR was performed using primers/probe and reaction reagents, 7500 Gene Detection System (Applied Biosystem by Life Technology, Grand Island, NY, USA). b-Actin was used as internal control. Data were expressed as fold change versus control, using 2(−ΔΔCt) method.
Statistical analysis
All data were analyzed by SPSS 13.0 statistical software, and presented as mean ± standard deviation. One-way analysis of variance was used for comparison of multiple groups, and Dunnett’s test was used for comparison of paired groups. It was considered statistically significant when P<0.05.
Results
General observation
There were no differences in hair appearance, diet, activity, and reaction to surrounding environment before the experiment. At the early stage of the experiment, animals were in manic anxiety when they were exposed to cigarette smoke. At the late stage of the experiment, animals in the CSE and CSE + S groups were less active, with shriveled hair, yellow teeth, and slow reaction and movement. No animal died during the CSE.
Body weight alteration
There was no difference in baseline body weight among the three groups (P>0.05). After 16 weeks of smoke exposure, however, body weight was significantly reduced in the CSE and CSE + S groups compared to the control rats (445±12 of CSE only, 446±23 of CSE + S group vs 503±15 g of control, P<0.01). Simvastatin could not prevent smoke-induced weight loss (P>0.05 compared to control group).
Histopathological alteration
MLI and BWT/D were significantly increased in the CSE group compared to that in control group (MLI: 34.6±2.0 of CSE vs 24.0±1.7 μm of control; BWT/D: 0.250±0.062 of CSE vs 0.160±0.034 of control, P<0.01). In contrast, MAN was significantly decreased in the CSE group than that in the control group (13.5±2.0 of CSE vs 21.5±2.0 N/μm2 of control, P<0.01). Simvastatin slightly but not significantly blocked alteration of MLI, BWT/D, and MAN in response to cigarette smoke stimulation (MLI: 33.4±1.4 μm; BWT/D: 0.220±0.052; MAN: 15.5±2.5 N/μm2, P>0.05, Figure 1).
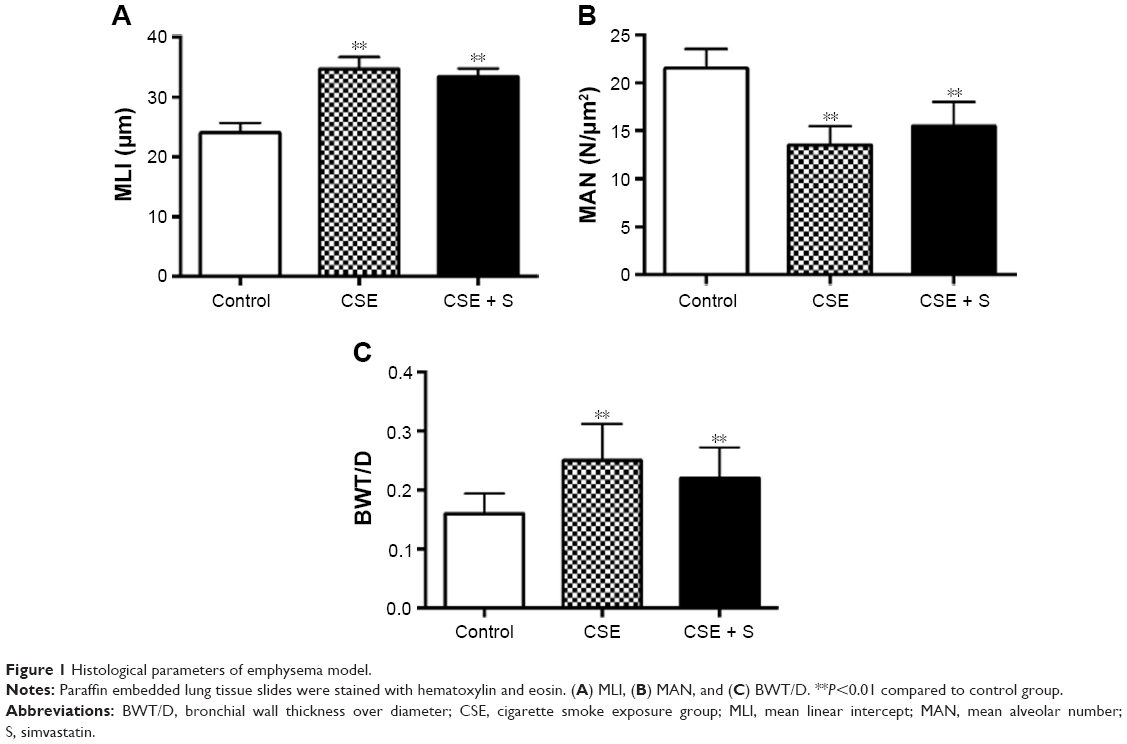 | Figure 1 Histological parameters of emphysema model. |
BALF total and differential cell count
There was no difference in BALF recovery volume among control, CSE, and CSE + S groups (P>0.05). As expected, the number of total white blood cells was significantly increased in the BALF of smoking group (3.3±2.5×109 cells/L vs 1.1±1.3×109 cells/L of control, P<0.01), and it was significantly reduced in the rats treated with simvastatin (2.3±2.1×109 cells/L, P<0.01, Figure 2A). Moreover, differential count of neutrophils, macrophages, and lymphocytes showed that CSE resulted in significantly increased accumulation of neutrophils and macrophages (neutrophils: 14.5%±1.3% of CSE group vs 9.1%±1.5% of control; macrophage: 91%±3% of CSE group vs 87%±2% of control, P<0.05) but with a decrease in lymphocytes in the bronchial alveolar lavage (7.0%±2.9% of CSE group vs 9.3%±1.2% of control, P<0.01). Simvastatin significantly reduced neutrophils (12.9%±2.0%, P<0.05) in the BALF, but had no effect on macrophage (89%±1.6%) or lymphocytes (6.4%±2.3%, Figure 2B).
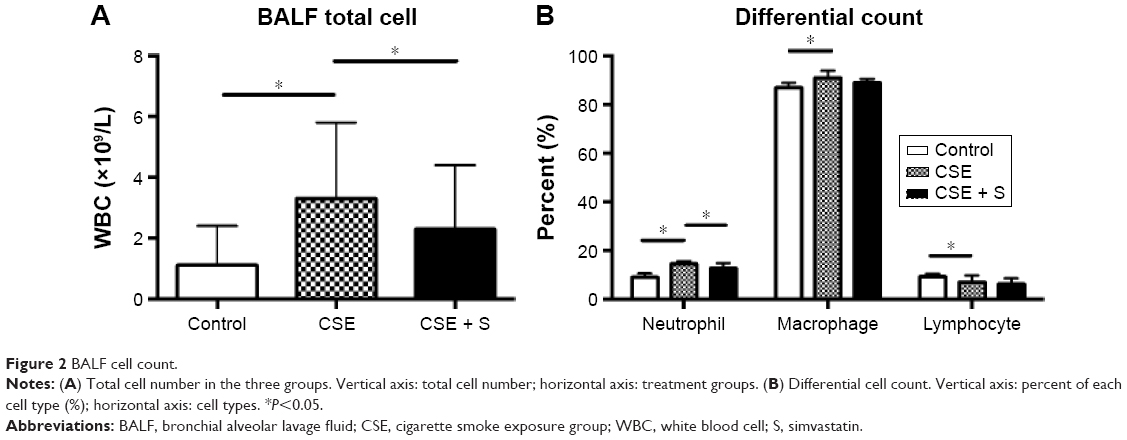 | Figure 2 BALF cell count. |
Comparison of mRNA and protein of MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-4
Expression of MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-4 mRNA was significantly upregulated in the CSE group compared to that in control group (P<0.01). Simvastatin significantly blocked upregulation of MMP-8 and −9 in response to CSE (P<0.01), but had no effect on MMP-12, TIMP-1, and TIMP-4 mRNA (P>0.05, Figure 3). Consistent with the effect on mRNA expression of MMPs and TIMPs, CSE resulted in a significant stimulation of MMP-8 and −9 protein synthesis (Figure 4B and E), but had no significant effect on MMP-12 (Figure 4H), TIMP-1 (Figure 4K), and TIMP-4 (Figure 4N) protein release. Simvastatin significantly blocked cigarette smoke-induced protein synthesis of MMP-8 (Figure 4C) and −9 (Figure 4F), but had no significant effect on MMP-12 (Figure 4I). Moreover, simvastatin significantly stimulated TIMP-1 (Figure 4L) and −4 (Figure 4O) protein synthesis even in the presence of cigarette smoke.
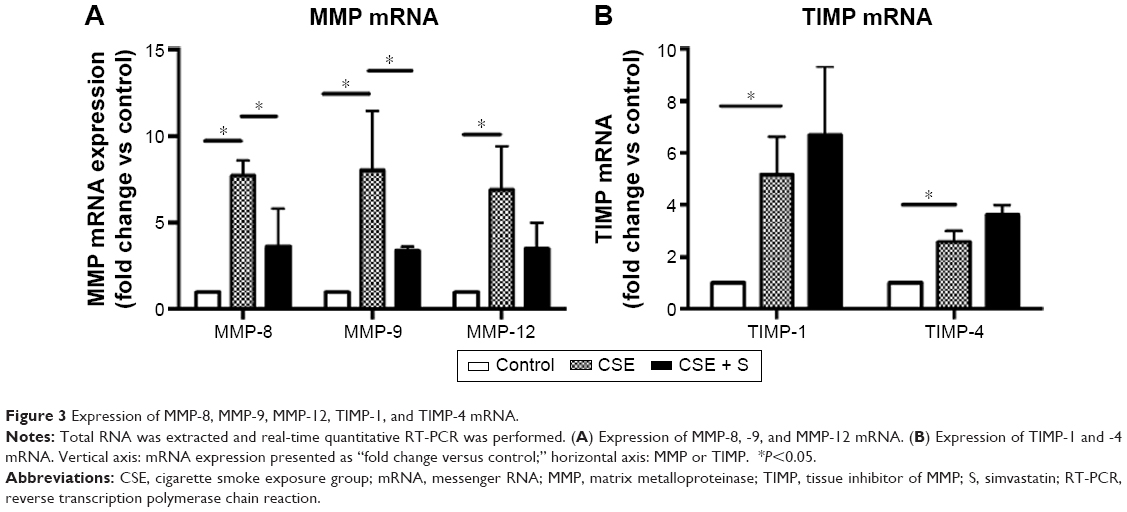 | Figure 3 Expression of MMP-8, MMP-9, MMP-12, TIMP-1, and TIMP-4 mRNA. |
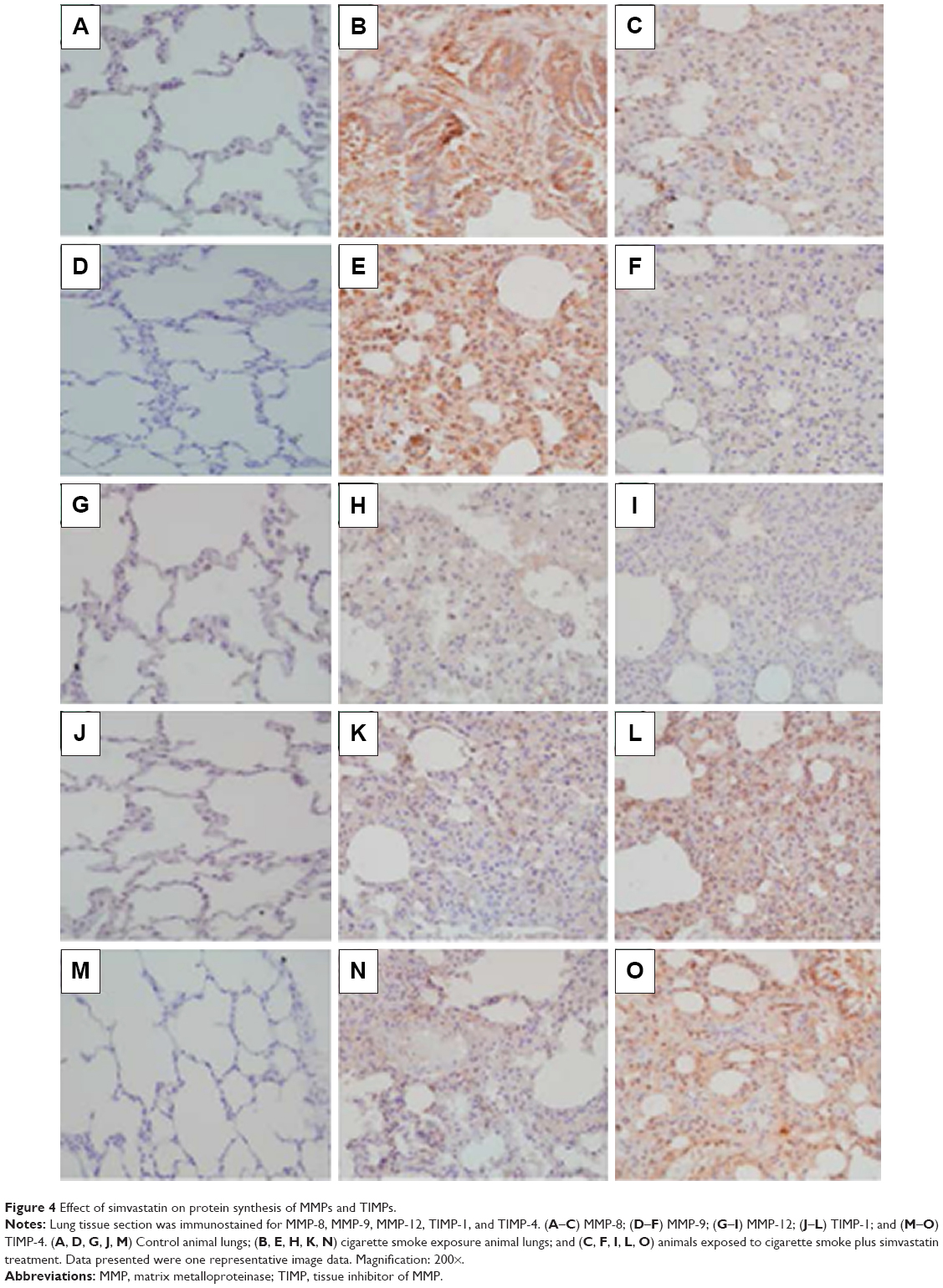 | Figure 4 Effect of simvastatin on protein synthesis of MMPs and TIMPs. |
Discussion
COPD is characterized by chronic airway inflammation and irreversible airway obstruction in response to irritant gas or particulates exposure.15 The current study demonstrated that simvastatin decreased accumulation of inflammatory cells in the airways of cigarette smoke-induced COPD rat models. Furthermore, simvastatin decreased upregulation of MMP-8 and −9, but had no effect on MMP-12, TIMP-1, and TIMP-4 mRNA and protein in the lung tissues of the rats exposed to cigarette smoke. However, simvastatin could not reverse or block the alteration of MLI, MAN, or BWT/D in response to cigarette smoke stimulation. These findings suggested that simvastatin may modulate airway inflammation and part of MMP (MMP-8 and −9) release.
Chronic airway inflammation is believed to play a major role in the development of COPD. Cigarette smoke contains more than a 1,000 chemicals that stimulate the airway cells to release inflammatory cytokines, such as interleukin (IL)-1β, −6 and −8,16–18 and cause airway injury.19 Under the effect of these inflammatory cytokines, neutrophils and macrophages migrate to the site of injury and result in further tissue injury and remodeling. Consistent with this concept, the current study demonstrated that CSE in the rats resulted in accumulation of neutrophils and macrophages and that there is significant damage of alveolar structure (MLI increase and MAN decrease) and airway tissue remodeling (altered BWT/D).
Proteases have been believed to play a major role in the pathogenesis of COPD and emphysema. In this context, mice deficient in the macrophage elastase MMP-12 have been reported to be resistant to the development of cigarette smoke-induced emphysema;20 upregulation of MMP-9 in the lungs has also been reported to be associated with emphysema not only in experimental animal models21,22 but also clinical patients.23
However, expression of MMPs in normal lung is very low or undetectable. In the presence of inflammation or irritants, including cigarette smoke, however, expression of MMPs is dramatically increased and readily detectable in a variety of lung diseases, including COPD and emphysema. In the cigarette smoke-induced COPD animal model, cigarette smoke may stimulate MMPs directly or indirectly through inflammatory cytokines, such as IL-1β and tumor necrosis factor-α. In this context, MMP-1 (collagenase 1) and −8 (collagenase 2) have been reported to be upregulated in COPD lungs and epithelial cells.24 Consistent with previous reports, the current study demonstrated that expression of MMP-8, −9, and −12 mRNA was significantly upregulated in the model of cigarette smoke-induced COPD. In addition, expression of TIMP-1 and −4 mRNA was also increased in the COPD model. However, the fold of TIMP-1 and −4 increase (two- to fivefold) was less than that of MMP-8, −9, and −12 (>7-fold). Furthermore, simvastatin not only significantly blocked cigarette smoke-induced MMP-8 and −9 protein synthesis, but also significantly stimulated TIMP-1 and −4 protein synthesis in the presence of cigarette smoke, suggesting that imbalance of MMPs versus TIMPs may play a role in the pathogenesis of COPD and emphysema in response to CSE, and that simvastatin blocks development of COPD and emphysema, at least partially, through modulating protein synthesis of MMP-8, MMP-9, TIMP-1, and TIMP-4.
Statins were originally developed for their cholesterol-lowering properties and efficacy in the cardiovascular disease. However, previous studies indicated that statins not only have an effect of immunomodulation, antioxidant, antithormbogenic and vascular actions,25,26 but also decrease the expression of MMPs in vascular cells and macrophages in vitro.13,27 Consistent with this effect on MMPs, statins have been reported to inhibit the development of emphysema in experimental animal models. In this regard, Lee et al28 found that simvastatin inhibited lung parenchymal destruction and MMP-9 expression in a rat model of cigarette smoking-induced emphysema. Moreover, in a mouse model of elastase-induced emphysema, it had been reported that simvastatin reduced mRNA expression for tumor necrosis factor-α and MMP-12 in the whole lung.29 The current study extends these observations and further demonstrated that simvastatin significantly decreased alveolar accumulation of neutrophils and expression of MMP-8 and −9 in the whole lung. However, cigarette smoke had no significant effect on MMP-12 protein synthesis and simvastatin could not significantly decrease MMP-12 expression in response to CSE. Moreover, simvastatin had no effect on alteration of MLI, MAN, and BWT/D, suggesting simvastatin could significantly inhibit airway inflammation and MMP-8 and −9 release, but may not block proteinase-induced alteration of lung structures, and, thus, may have no direct effect on tissue repair and remodeling.
Taken together, the current study demonstrates that simvastatin can inhibit accumulation of neutrophils in the airways, and decrease mRNA expression and protein synthesis of MMP-8 and −9 in rat lungs in response to CSE. Simvastatin, however, has no significant effect on lung tissue repair and remodeling.
Disclosure
The authors report no conflicts of interest in this work.
References
Rennard SI, Drummond MB. Early chronic obstructive pulmonary disease: definition, assessment, and prevention. Lancet. 2015;385(9979):1778–1788. | ||
Rennard SI. Overview of causes of COPD. New understanding of pathogenesis and mechanisms can guide future therapy. Postgrad Med. 2002;111(6):28–30, 33–34, 37–38. | ||
Heulens N, Korf H, Cielen N. Vitamin D deficiency exacerbates COPD-like characteristics in the lungs of cigarette smoke-exposed mice. Respir Res. 2015;16:110. | ||
Kamiide Y, Inomata N, Furuya M, Yada T. Ghrelin ameliorates catabolic conditions and respiratory dysfunction in a chronic obstructive pulmonary disease model of chronic cigarette smoke-exposed rats. Eur J Pharmacol. 2015;755:88–94. | ||
Wang Y, Jiang X, Zhang L, Wang L, Li Z, Sun W. Simvastatin mitigates functional and structural impairment of lung and right ventricle in a rat model of cigarette smoke-induced COPD. Int J Clin Exp Pathol. 2014;7(12):8553–8562. | ||
Parks WC, Shapiro SD. Matrix metalloproteinases in lung biology. Respir Res. 2001;2(1):10–19. | ||
Kamio K, Liu XD, Sugiura H. Statins inhibit matrix metalloproteinase release from human lung fibroblasts. Eur Respir J. 2010;35(3):637–646. | ||
Jouneau S, Khorasani N, De Souza P, et al. EMMPRIN (CD147) regulation of MMP-9 in bronchial epithelial cells in COPD. Respirology. 2011;16(4):705–712. | ||
Ishida W, Kajiwara T, Ishii M. Decrease in mortality rate of chronic obstructive pulmonary disease (COPD) with statin use: a population-based analysis in Japan. Tohoku J Exp Med. 2007;212(3):265–273. | ||
Søyseth V, Brekke PH, Smith P, Omland T. Statin use is associated with reduced mortality in COPD. Eur Respir J. 2007;29(2):279–283. | ||
Hothersall E, McSharry C, Thomson NC. Potential therapeutic role for statins in respiratory disease. Thorax. 2006;61(8):729–734. | ||
Bellosta S, Via D, Canavesi M, et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol. 1998;18(11):1671–1678. | ||
Ikeda U, Shimpo M, Ohki R, et al. Fluvastatin inhibits matrix metalloproteinase-1 expression in human vascular endothelial cells. Hypertension. 2000;36(3):325–329. | ||
Menard G, Turmel V, Bissonnette EY. Serotonin modulates the cytokine network in the lung: involvement of prostaglandin E2. Clin Exp Immunol. 2007;150(2):340–348. | ||
Marin L, Colombo P, Bebawy M, Young PM, Traini D. Chronic obstructive pulmonary disease: patho-physiology, current methods of treatment and the potential for simvastatin in disease management. Expert Opin Drug Deliv. 2011;8(9):1205–1220. | ||
Moon HG, Zheng Y, An CH, Kim YK, Jin Y. CCN1 secretion induced by cigarette smoking extracts augments IL-8 release from bronchial epithelial cells. PLoS One. 2013;8(7):e68199. | ||
Wang H, Yang T, Shen Y, et al. Ghrelin inhibits interleukin-6 production induced by cigarette smoke extract in the bronchial epithelial cell via NF-kappaB pathway. Inflammation. 2016;39(1):190–198. | ||
Rusznak C, Mills PR, Devalia JL, Sapsford RJ, Davies RJ, Lozewicz S. Effect of cigarette smoke on the permeability and IL-1beta and sICAM-1 release from cultured human bronchial epithelial cells of never-smokers, smokers, and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2000;23(4):530–536. | ||
Goldkorn T, Filosto S, Chung S. Lung injury and lung cancer caused by cigarette smoke-induced oxidative stress: molecular mechanisms and therapeutic opportunities involving the ceramide-generating machinery and epidermal growth factor receptor. Antioxid Redox Signal. 2014;21(15):2149–2174. | ||
Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–2004. | ||
Selman M, Cisneros-Lira J, Gaxiola M, et al. Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in Guinea pigs. Chest. 2003;123(5):1633–1641. | ||
Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32(4):311–318. | ||
Russell RE, Culpitt SV, DeMatos C, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26(5):602–609. | ||
Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117(3):684–694. | ||
Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effects beyond lipid lowering – are they clinically relevant? Eur Heart J. 2003;24(3):225–248. | ||
Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol. 2006;6(5):358–370. | ||
Luan Z, Chase AJ, Newby AC. Statins inhibit secretion of metalloproteinases-1, −2, −3, and −9 from vascular smooth muscle cells and macrophages. Arterioscler Thromb Vasc Biol. 2003;23(5):769–775. | ||
Lee JH, Lee DS, Kim EK. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172(8):987–993. | ||
Takahashi S, Nakamura H, Seki M. Reversal of elastase-induced pulmonary emphysema and promotion of alveolar epithelial cell proliferation by simvastatin in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L882–L890. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.