")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Effect of severe renal impairment on umeclidinium and umeclidinium/vilanterol pharmacokinetics and safety: a single-blind, nonrandomized study
Authors Mehta R, Hardes K, Brealey N, Tombs L, Preece A , Kelleher D
Received 21 May 2014
Accepted for publication 26 August 2014
Published 18 December 2014 Volume 2015:10(1) Pages 15—23
DOI https://doi.org/10.2147/COPD.S68094
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Rashmi Mehta,1 Kelly Hardes,2 Noushin Brealey,3 Lee Tombs,4 Andrew Preece,2 Dennis Kelleher1
1Respiratory Medicines Development Center, GSK, Research Triangle Park, NC, USA; 2Clinical Pharmacology Science and Study Operations, 3Respiratory Medicines Development Centre, GSK, Stockley Park, UK; 4Statistics and Programming, Synergy, Slough, Berkshire, UK
Background: Umeclidinium and vilanterol, long-acting bronchodilators for the treatment of chronic obstructive pulmonary disease, are primarily eliminated via the hepatic route; however, severe renal impairment may adversely affect some elimination pathways other than the kidney.
Objectives: To evaluate the effect of severe renal impairment on the pharmacokinetics of umeclidinium and umeclidinium/vilanterol.
Methods: Nine patients with severe renal impairment (creatinine clearance <30 mL/min) and nine matched healthy volunteers received a single dose of umeclidinium 125 µg; and after a 7- to 14-day washout, a single dose of umeclidinium/vilanterol 125/25 µg.
Results: No clinically relevant increases in plasma umeclidinium or vilanterol systemic exposure (area under the curve or maximum observed plasma concentration) were observed following umeclidinium 125 µg or umeclidinium/vilanterol 125/25 µg administration. On average, the amount of umeclidinium excreted in 24 hours in urine (90% confidence interval) was 88% (81%–93%) and 89% (81%–93%) lower in patients with severe renal impairment compared with healthy volunteers following umeclidinium 125 µg and umeclidinium/vilanterol 125/25 µg administration, respectively. Treatments were well tolerated in both populations.
Conclusion: Umeclidinium 125 µg or umeclidinium/vilanterol 125/25 µg administration to patients with severe renal impairment did not demonstrate clinically relevant increases in systemic exposure compared with healthy volunteers. No dose adjustment for umeclidinium and umeclidinium/vilanterol is warranted in patients with severe renal impairment.
Keywords: chronic obstructive pulmonary disease, exposure, GSK573719, long-acting beta2 agonist, long-acting muscarinic antagonist
Introduction
Umeclidinium (GSK573719) is a long-acting muscarinic antagonist (LAMA) approved in the US, the EU, and several other countries as an inhaled monotherapy1–5 and as a combination therapy with the long-acting beta2 agonist (LABA) vilanterol (GW642444)6,7 for the treatment of chronic obstructive pulmonary disease (COPD). Umeclidinium is generally well tolerated with the overall incidence of adverse events (AEs) similar to that of placebo at doses ≤125 μg once daily.2,4
Umeclidinium and vilanterol are mainly eliminated via the hepatic route: umeclidinium primarily through biliary secretion and metabolism8 and vilanterol primarily through metabolism by cytochrome P450 3A4.9,10 Cytochrome P450 3A4 is highly expressed in the liver and has the capacity to oxidize a wide variety of substrates, thus significantly contributing to drug metabolism.11 Metabolic interactions between substrates and inhibitors of cytochrome P450 3A4 can adversely affect drug efficacy and safety.12
The US Food and Drug Administration Guidance for Industry for studies of patients with renal impairment13 states that pharmacokinetic (PK) characterization should be considered even if the drug and/or its active metabolite(s) are eliminated mainly via the hepatic route, as renal impairment may adversely affect absorption, plasma protein binding, transport, and tissue distribution. These changes may be particularly prominent in patients with severe renal impairment and have been observed even when the renal route is not the primary route of elimination of a drug.
The PK/pharmacodynamic effects of vilanterol have previously been studied in patients with severe renal impairment with the inhaled corticosteroid/LABA combination fluticasone furoate/vilanterol. This 7-day repeat-dose study demonstrated no apparent clinically relevant effects on the vilanterol PK or pharmacodynamic properties or tolerability of the drug combination in patients with severe renal impairment.14 The aim of the current study was to investigate the effect of severe renal impairment (creatinine clearance [Clcr] <30 mL/min) on the plasma PK of inhaled umeclidinium and umeclidinium/vilanterol following single-dose administration. Umeclidinium urine PK as well as safety and tolerability of all treatments were also assessed.
Preliminary results from the current study have been presented previously as an abstract and poster at the European Respiratory Society 2013 meeting.15
Methods
Study design and treatment
This was a single-blind (only volunteers and patients were blinded to study treatment), non-randomized, PK and safety study (http://www.clinicaltrials.gov identifier NCT01571999; GSK study code DB2114636) conducted at two centers (PRA Magyarország Kft., Budapest, Hungary and Pharmaceutical Research Associates CZ, Prague, Czech Republic) between March 29, 2012 and June 22, 2012. The study was approved by local ethics review committees (Medical Research Council, Ethics Committee for Clinical Pharmacology, Budapest, Hungary and Ethics Committee of the Institute for Clinical and Experimental Medicine and Thomayer Hospital, Prague, Czech Republic) and was conducted in accordance with the Declaration of Helsinki 200816 and ICH Good Clinical Practice guidelines.17 All volunteers and patients provided written informed consent prior to any study-specified procedures.
Following screening, volunteers and patients received a single dose of inhaled umeclidinium 125 μg (delivering 113 μg) and a single dose of inhaled umeclidinium/vilanterol 125/25 μg (equivalent to a delivered dose of 113/22 μg), separated by a 7- to 14-day washout period. All doses were administered via the ELLIPTA® dry powder inhaler (GlaxoSmithKline plc, London, UK; ELLIPTA is a trademark of the GSK group of companies). All volunteers and patients were resident in the unit from the evening of day –1 until the morning of day 2 for both treatment periods. Following dose administration on day 1, volunteers and patients were monitored for safety, and plasma and urine PK samples were collected up to 24 hours postdose. Volunteers and patients were discharged once the 24-hour PK sample was collected, and if there were no safety concerns. A follow-up visit was scheduled 7–14 days after the last dose.
Study population
Protocol-specified adult males or females aged 18–70 years with a body weight ≥45 kg and a body mass index between 18 and 33 kg/m2 were eligible for this study. Healthy volunteers had Clcr >80 mL/min as calculated by the Cockcroft–Gault equation using serum creatinine. Patients with severe renal impairment had Clcr <30 mL/min and stable renal function defined as ≤25% difference in Clcr assessed on two occasions separated by ≥4 weeks within the last 3 months. Exclusion criteria for patients with severe renal impairment included a life expectancy of <3 months, hemoglobin <8.5 g/dL (this cut-off point was increased to <11.0 g/dL at the Czech Republic site by a protocol amendment as requested by the Ethics committee), or current hemodialysis treatment. Healthy volunteers were matched to patients with severe renal impairment based on sex, ethnicity, body mass index (±15%), and age (±5 years).
PK collection and analysis
PK blood samples were collected at the following time points: predose, 5 minutes, 15 minutes, 30 minutes, 1, 2, 4, 8, 12, 16, and 24 hours postdose. PK urine samples were collected over the following time intervals: 0–4 hours, 4–8 hours, 8–12 hours, and 12–24 hours postdose.
Plasma samples for umeclidinium and vilanterol and urine samples for umeclidinium were analyzed by a validated analytical method based on solid-phase extraction, followed by high-performance liquid chromatography/tandem mass spectrometry analysis. The lower limit of quantification for umeclidinium and vilanterol in plasma and umeclidinium in urine was 10 pg/mL. However, based on a previous PK study of healthy volunteers that demonstrated renal excretion as a minor route of vilanterol elimination,18 vilanterol urine PK was not assessed in the current study.
The primary endpoints were umeclidinium and vilanterol plasma PK parameters: area under the curve (AUC)(0–0.25h), AUC(0–2h), maximum observed plasma concentration (Cmax), time to Cmax (tmax), AUC(0–24h), AUC(0–∞), time to last quantifiable concentration (tlast), terminal phase half-life (t1/2), and other PK parameters as data permitted. Secondary endpoints were umeclidinium urine PK parameters including urinary recovery of unchanged drug 24 hours postdose, recorded as amount excreted over 24 hours (Ae(0–24h)), and general safety and tolerability assessments.
Safety analysis
AEs were recorded throughout the study and were coded using the Medical Dictionary for Regulatory Activities (MedDRA). Laboratory parameters including clinical chemistry, hematology, urinalysis, vital signs (blood pressure and heart rate), and 12-lead electrocardiogram (ECG) were recorded.
Statistical analysis
The sample size calculation was based on feasibility (ie, no hypothesis testing and no formal sample-size calculations). An estimation approach was adopted with approximately 18 volunteers/patients planned for enrollment (nine with severe renal impairment, nine healthy control volunteers). Sample-size sensitivity was performed and showed the effect of variation in the standard deviation and different true ratios on the magnitude of the 90% confidence interval (CI). Estimates of standard deviation were obtained from previous studies.10,14,19
The PK parameters of AUC, Cmax, and Ae(0–24h) of umeclidinium and vilanterol were loge transformed and were analyzed separately. The AUC(0–∞) and AUC(0–24h) were non-calculable due to nonquantifiable (NQ) plasma concentrations in the terminal phase; therefore, AUC(0–0.25h) and AUC(0–2h) for umeclidinium, and AUC(0–0.25h) and AUC(0–1h) for vilanterol were derived and statistically analyzed. The t1/2 could not be calculated for umeclidinium or vilanterol due to NQ plasma concentrations in the terminal phase. For umeclidinium data, a mixed model was fitted with group, treatment, and group*treatment interaction as fixed effects and volunteer/patient as a random effect. For vilanterol data, a fixed-effects model was fitted with group as a fixed effect. Point estimates and their associated 90% CIs were constructed for the difference between severe renal impairment and healthy volunteers for all treatment groups. The point estimates and their associated 90% CIs were then back-transformed to provide point estimates and 90% CIs for the ratio.
Results
Study population disposition and demographics
All 18 enrolled patients and volunteers completed the study. Baseline demographics were matched between the patient and healthy volunteer groups, except for medical history (Table 1). All patients with severe renal impairment were taking prescribed medications prior to and during the study. These medications were representative of medications typically taken in this population to treat related comorbidities20 and were not expected to impact the PK findings in this study.
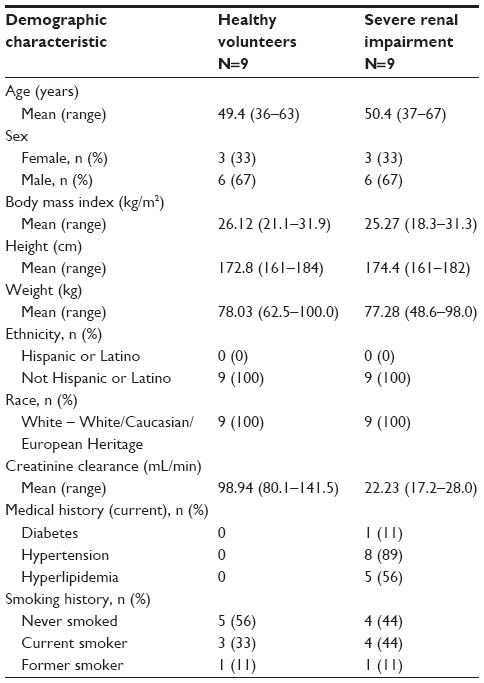 | Table 1 Patient demographics |
PK results
Plasma umeclidinium concentration
Following single-dose administration, 51.8% of postdose samples, overall, were NQ. The percentage of NQ data was similar between treatments and between patients with severe renal impairment and healthy volunteers.
Plasma umeclidinium PK parameters
Summary statistics and the statistical analysis for the plasma umeclidinium PK parameters are shown in Table 2A and B, respectively. Umeclidinium was rapidly absorbed with median tmax of approximately 5 minutes postdose. Median tlast for both umeclidinium 125 μg and umeclidinium/vilanterol 125/25 μg was 2 hours for patients with severe renal impairment and healthy volunteers. Following administration of umeclidinium 125 μg or umeclidinium/vilanterol 125/25 μg, there was no evidence of a clinically relevant increase in umeclidinium plasma exposure, in terms of AUC(0–2h) or Cmax, for patients with severe renal impairment compared with healthy volunteers. For AUC(0–2h), there was an average 10% decrease, with the true mean difference likely to be between 36% lower and 26% higher in patients with severe renal impairment compared with healthy volunteers following umeclidinium 125 μg administration. There was an average 10% increase, with the true mean difference likely to be between 21% lower and 52% higher following umeclidinium/vilanterol 125/25 μg administration. For Cmax, there was an average 11% decrease, with the true mean difference likely to be between 42% lower and 35% higher in patients with severe renal impairment compared with healthy volunteers following umeclidinium 125 μg administration. There was an average 2% decrease, with the true mean difference likely to be between 36% lower and 49% higher following umeclidinium/vilanterol 125/25 μg.
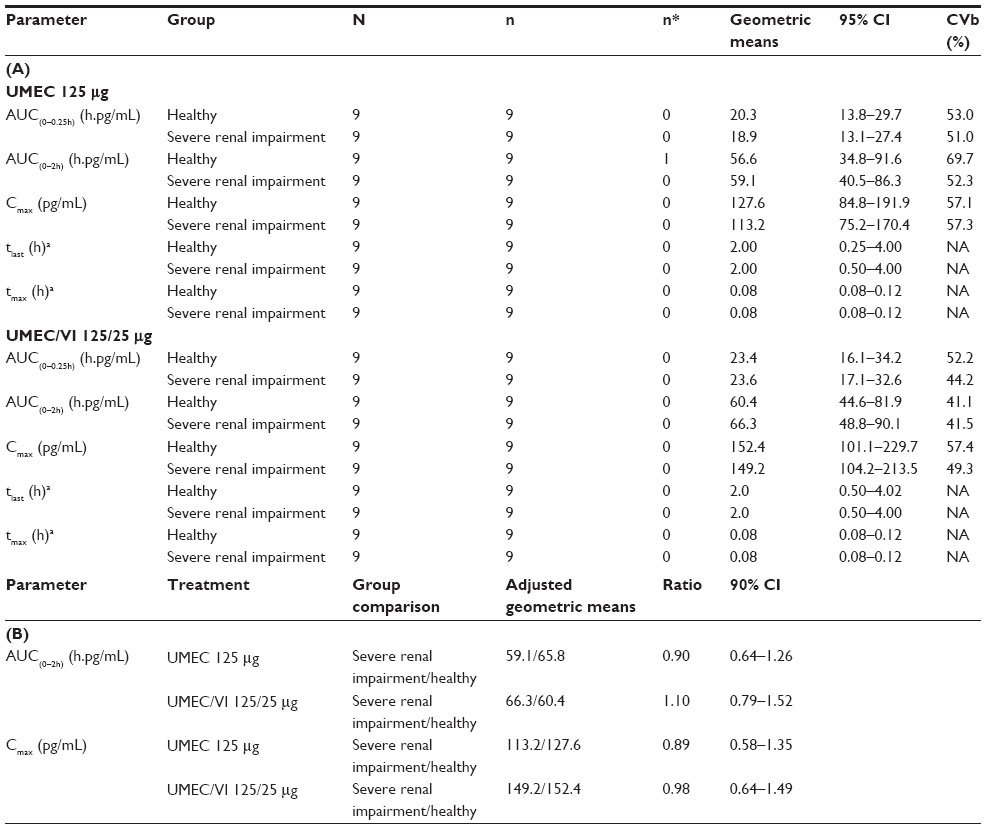 | Table 2 Plasma PK parameters |
Urine umeclidinium PK parameters
Summary statistics and the statistical analysis for the urine umeclidinium PK parameters are shown in Table 3A and B, respectively. On average, umeclidinium urine t1/2 was similar between patients with severe renal impairment and healthy volunteers. The Ae(0–24h) was 88% (90% CI: 81%–93%) and 89% (CI: 81%–93%) lower in patients with severe renal impairment compared with healthy volunteers following umeclidinium 125 μg and umeclidinium/vilanterol 125/25 μg administration, respectively.
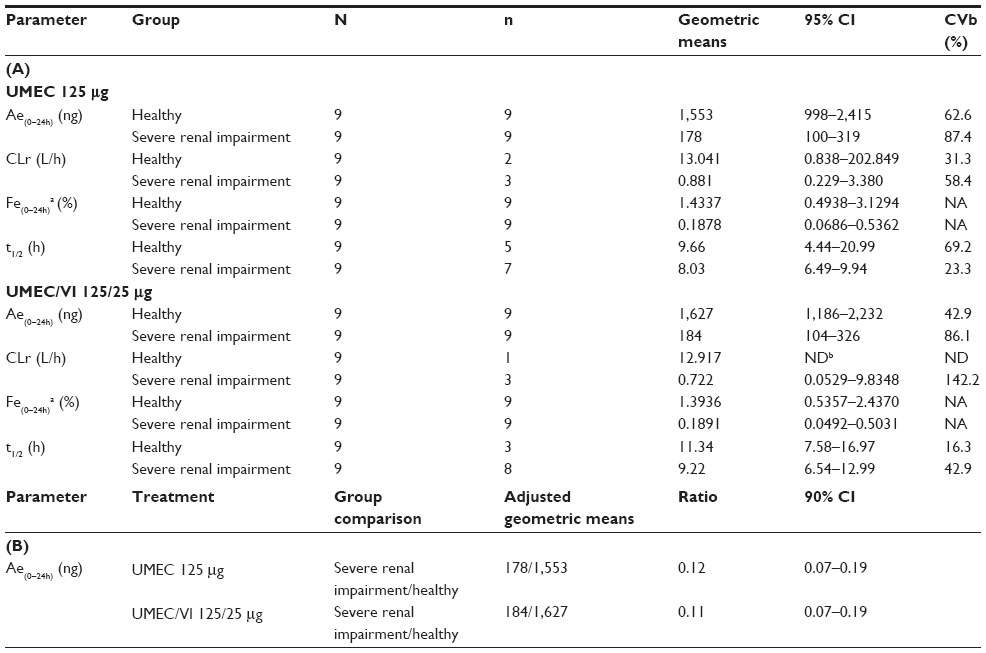 | Table 3 Urine PK parameters |
Plasma vilanterol concentration
Overall, 58.3% of postdose samples were NQ; the percentage of NQ data was 56% in patients with severe renal impairment and 61% in healthy volunteers.
Plasma vilanterol PK parameters
Summary statistics and the statistical analysis for the plasma vilanterol PK parameters are shown in Table 4A and B, respectively. Vilanterol was rapidly absorbed with a median tmax occurring at approximately 5 minutes postdose, following which plasma concentrations declined rapidly for both patients with severe renal impairment and healthy volunteers. Median tlast was 1 hour for both study populations. Following dosing with umeclidinium/vilanterol 125/25 μg, there was no evidence of a clinically relevant increase in vilanterol plasma exposure, in terms of AUC(0–1h) or Cmax, for patients with severe renal impairment compared with healthy volunteers. For AUC(0–1h), there was an average 21% increase, with the true mean increase likely to be between 13% lower and 70% higher in patients with severe renal impairment compared with healthy volunteers. For Cmax, there was an average 3% increase, with the true mean increase likely to be between 27% lower and 46% higher in patients with severe renal impairment compared with healthy volunteers.
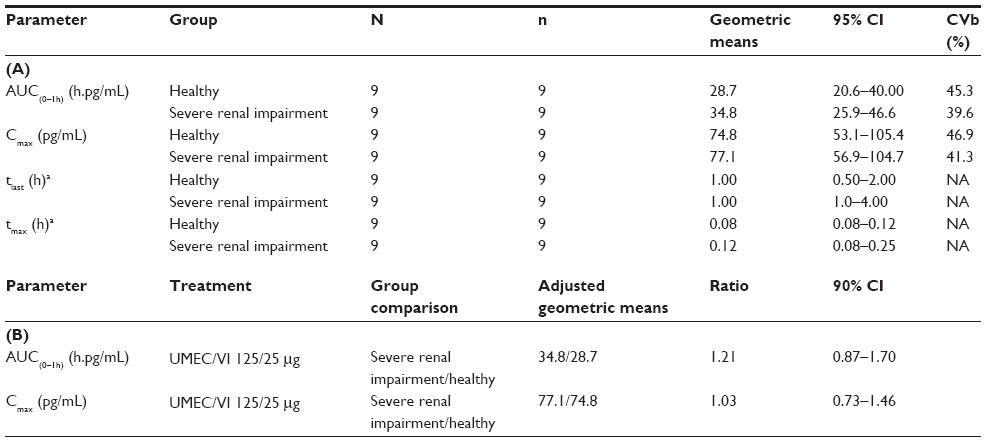 | Table 4 Plasma PK parameters |
Safety
One healthy volunteer experienced two AEs (moderate headache and intermittent mild vomiting) during the umeclidinium/vilanterol 125/25 μg treatment period; both AEs were considered possibly related to the study medication by the investigator. Neither AE led to study withdrawal. No AEs occurred in patients with severe renal impairment and no serious AEs were reported during the study.
There were no clinically relevant changes in mean laboratory or vital-sign parameters, and no laboratory values were considered clinically significant by the investigators. There were no clinically relevant differences between patients with severe renal impairment and healthy volunteers in mean ECG interval changes from baseline following administration of treatment. No clinically significant abnormalities were recorded on the ECG traces.
Discussion
The primary goal of this study was to evaluate the effect of severe renal impairment (Clcr <30 mL/min) on the plasma PK of inhaled umeclidinium 125 μg and umeclidinium/vilanterol 125/25 μg following single-dose administration. Patients with severe renal impairment had a lower mean Clcr (22.2 mL/min) at baseline compared with healthy volunteers (98.9 mL/min), as specified by the predefined study criteria.
In patients with severe renal impairment, excretion of unchanged umeclidinium into the urine was approximately ninefold lower when compared with healthy volunteers. This is concordant with the lower Clcr baseline values observed in patients with severe renal impairment. However, no differences in systemic exposure (plasma AUC, Cmax) considered clinically relevant (ie, necessitating umeclidinium dose adjustment) were observed between these patients with severe renal impairment and healthy volunteers. Moreover, tmax, tlast, and urine t1/2 values were similar between both study populations. These data support the conclusion that pathways other than the kidney have sufficient capacity to accommodate the impaired renal function, consistent with previous findings showing that umeclidinium is primarily metabolized via hepatic pathways and excreted via biliary secretion.8,21 Vilanterol systemic exposure, in terms of AUC(0–1h), Cmax, and tlast, was also similar between patients with severe renal impairment and healthy volunteers. It has previously been reported that vilanterol undergoes rapid and extensive first-pass metabolism in the liver, primarily via O-dealkylation.22 The O-dealkylated metabolites, which exhibit negligible pharmacological activity, are subsequently excreted in the urine.22 Additionally, there were no differences in in vitro plasma protein binding following umeclidinium and umeclidinium/vilanterol administration (GSK, data on file, GSK study numbers QBR106268 February 16, 2012, and QBR113236 September 13, 2012). which is another common concern for patients with renal impairment as plasma protein binding is often disrupted in this patient population,13 and the unbound concentrations of systemically active drugs and metabolites are commonly believed to determine the rate and extent of delivery to the sites of action.13 The low or NQ plasma concentrations observed in this study were likely due to the rapid hepatic clearance of the single dose of medication administered; this may partially explain the lack of observed difference in PK parameters between patients with renal impairment and healthy subjects.
Following umeclidinium 125 μg and umeclidinium/vilanterol 125/25 μg administration in healthy volunteers, umeclidinium and vilanterol were rapidly absorbed (tmax ~5 minutes) and eliminated (tlast 2 hours for umeclidinium, 1 hour for vilanterol). These data are consistent with previous study data of umeclidinium 250–1,000 μg and umeclidinium/vilanterol 500/50 μg in healthy volunteers (tmax 5–15 minutes).7,23 Urinary excretion of umeclidinium following umeclidinium 125 μg administration was similar to that following umeclidinium/vilanterol 125/25 μg administration in healthy volunteers and was on average 1.4% of the total dose, which is similar to previously reported data (1%–1.5% of the total dose).23 The lack of effect of combining vilanterol with umeclidinium on umeclidinium and vilanterol PK parameters in healthy volunteers has also been observed in previous studies involving patients with COPD.6,10,19,24 One of these studies was a population PK analysis that included patients with renal impairment. No clinically significant association was found between levels of systemic exposure and different degrees of renal impairment.24
Results from renal impairment studies for other LAMA therapies are consistent in the sense that significant effects of renal impairment were seen only if the primary route of excretion is via the kidneys, which has been observed for tiotropium25 and glycopyrronium,26 but not for aclidinium bromide.27,28 The potential risks associated with the former treatments should therefore be weighed carefully against the expected benefits in patients with severe renal impairment.
The effects of vilanterol have previously been studied following inhaled administration of the corticosteroid/LABA combination fluticasone furoate/vilanterol (200/25 μg or 100/25 μg once daily for 7 days) in patients with severe renal impairment and varying degrees of hepatic impairment. Results indicate that severe renal impairment had no clinically relevant effects on the PK parameters of fluticasone furoate/vilanterol,14 and it has been shown that both of these agents are primarily metabolized in the liver.14,22
No safety concerns were identified during the current study. Umeclidinium 125 μg and umeclidinium/vilanterol 125/25 μg were well tolerated in patients with severe renal impairment and healthy volunteers. These findings are consistent with previous safety findings for umeclidinium and umeclidinium/vilanterol in healthy volunteers,7,10 and in patients with COPD.1,2,4,6
This study had strengths and limitations. The number of patients with severe renal impairment and healthy volunteers included in this study (9 per arm) may be considered small; however, the study size was in line with US Food and Drug Administration guidance.13 The study focused on patients with severe renal impairment, but specifically did not include dialysis patients.
Conclusion
In conclusion, administration of umeclidinium 125 μg or umeclidinium/vilanterol 125/25 μg to patients with severe renal impairment did not result in clinically significant increases in umeclidinium or vilanterol exposure compared with healthy control volunteers. The data suggest that no dose adjustment for umeclidinium or umeclidinium/vilanterol is warranted in patients with severe renal impairment and there is no requirement for specific monitoring of renal function for use of umeclidinium or umeclidinium/vilanterol. Overall, umeclidinium 125 μg and umeclidinium/vilanterol 125/25 μg were well tolerated.
Acknowledgments
This study was funded by GSK (http://www.clinicaltrials.gov identifier NCT01571999 GSK study code DB2114636). The authors would like to acknowledge the Principal Investigators of the study, István Udvaros, MD, MSc (Budapest, Hungary) and Petr Sramek (Prague, Czech Republic). The authors would also like to acknowledge: 1) Pharmaceutical Research Associates (Zuidlaren, the Netherlands) for their role in project management, clinical conduct, and safety laboratory analyses; 2) York Bioanalytical Solutions Ltd (York, UK) for their assistance with pharmacokinetic sample analysis; 3) Phast Clinical Data, Inc. (Raleigh, NC, USA) for assistance with noncompartmental pharmacokinetic analysis; 4) Synergy (Berks, UK) for their statistical input into protocol and study set up, reporting analysis plan production, and production of the analysis; reporting datasets,tables, figures, and listings; and statistical contribution to the final study report; and 5) Niche Science and Technology (Surrey, UK) for their editorial support in the form of development of a draft outline for the Clinical Pharmacology Study report (CPSR) in consultation with the authors; development of the first draft in consultation with the authors; editorial suggestions to draft versions of the CPSR; assembling tables; and collating author comments. Editorial support in the form of development of a draft outline in consultation with the authors; development of a manuscript first draft in consultation with the authors; editorial suggestions to draft versions of this paper; assembling tables; collating author comments; copyediting; fact checking; and referencing were provided by Tara N Miller, PhD (employed by Gardiner-Caldwell Communications, Lyndhurst, NJ, USA, until November 2013) and Jackie Phillipson, PhD, at Gardiner-Caldwell Communications (Macclesfield, UK), and was funded by GSK.
Author contributions
All authors participated in the study conception and design, data analysis, and interpretation of data. All authors met the criteria for authorship as defined by the International Committee of Medical Journal Editors.
Disclosure
The study was sponsored by GSK. LT is a consultant for Synergy. All other authors are employees of, and own stock in, GSK.
References
Church A, Beerahee M, Brooks J, Mehta R, Shah P. Dose response of umeclidinium administered once or twice daily in patients with COPD: a randomised cross-over study. BMC Pulm Med. 2014;14:2. | |
Donohue JF, Anzueto A, Brooks J, Mehta R, Kalberg C, Crater G. A randomized, double-blind, dose-ranging study of the novel LAMA GSK573719 in patients with COPD. Respir Med. 2012;106:970–979. | |
Cahn A, Tal-Singer R, Pouliquen IJ, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single and repeat inhaled doses of umeclidinium in healthy subjects: two randomized studies. Clin Drug Investig. 2013;33:477–488. | |
Decramer M, Maltais F, Feldman G, et al. Bronchodilation of umeclidinium, a new long-acting muscarinic antagonist, in COPD patients. Respir Physiol Neurobiol. 2013;185:393–399. | |
Tal-Singer R, Cahn A, Mehta R, et al. Initial assessment of single and repeat doses of inhaled umeclidinium in patients with chronic obstructive pulmonary disease: two randomised studies. Eur J Pharmacol. 2013;701:40–48. | |
Feldman G, Walker RR, Brooks J, Mehta R, Crater G. 28-Day safety and tolerability of umeclidinium in combination with vilanterol in COPD: a randomized placebo-controlled trial. Pulm Pharmacol Ther. 2012;25:465–471. | |
Kelleher DL, Mehta RS, Jean-Francois BM, et al. Safety, tolerability, pharmacodynamics and pharmacokinetics of umeclidinium and vilanterol alone and in combination: a randomized crossover trial. PLoS One. 2012;7:e50716. | |
Kelleher D, Hughes S, Mehta R, Tombs L, Kelly K, Church A. Absorption, distribution, metabolism, and elimination (ADME) of umeclidinium (UMEC) in healthy adults [abstract]. Eur Respir J. 2012; 40:P2153. | |
Kempsford R, Allen A, Bal J, Rubin D, Tombs L. The effect of ketoconazole on the pharmacokinetics and pharmacodynamics of inhaled fluticasone furoate and vilanterol trifenatate in healthy subjects. Br J Clin Pharmacol. 2013;75:1478–1487. | |
Mehta R, Kelleher D, Preece A, Hughes S, Crater G. Effect of verapamil on systemic exposure and safety of umeclidinium and vilanterol: a randomized and open-label study. Int J Chron Obstruct Pulmon Dis. 2013;8:159–167. | |
Ingelman-Sundberg M. Human drug metabolizing cytochrome P450 enzymes: properties and polymorphisms. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:89–104. | |
Zhou SF, Xue CC, Yu XQ, Li C, Wang G. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3A4 and the role of therapeutic drug monitoring. Ther Drug Monit. 2007;29:687–710. | |
US Department of Food and Health Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research. Guidance for Industry, Pharmacokinetics in Patients with Impaired Renal Function – Study Design, Data Analysis, and Impact on Dosing and Labeling. Rockville: Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research; 1998. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072127.pdf. Accessed October 10, 2014. | |
Allen A, Davis A, Hardes K, Tombs L, Kempsford R. Influence of renal and hepatic impairment on the pharmacokinetic and pharmacodynamic properties and tolerability of fluticasone furoate and vilanterol in combination. Clin Ther. 2012;34:2316–2332. | |
Kelleher D, Hardes K, Brealey N, Tombs L, Preece A, Mehta R. Effect of severe renal impairment (SRI) on umeclidinium (UMEC) and vilanterol (VI) pharmacokinetics (PK) [abstract]. Eur Respir J. 2013;42:4148. | |
WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964 and amended (latest) by the 59th WMA General Assembly, Seoul, Korea, October 2008 [webpage on the Internet]. Ferney-Voltaire: WMA The World Medical Association [updated Oct 2008]. Available from: http://www.wma.net/en/30publications/10policies/b3/index.html. Accessed October 10, 2014. | |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice E6(R1). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed October 10, 2014. | |
Harrell AW, Siederer SK, Bal J, et al. Metabolism and disposition of vilanterol, a long-acting β(2)-adrenoreceptor agonist for inhalation use in humans. Drug Metab Dispos. 2013;41:89–100. | |
Cahn A, Mehta R, Preece A, Blowers J, Donald A. Safety, tolerability and pharmacokinetics and pharmacodynamics of inhaled once-daily umeclidinium in healthy adults deficient in CYP2D6 activity: a double-blind, randomized clinical trial. Clin Drug Invest. 2013;33:653–664. | |
Coyle DW. Management of Chronic Kidney Disease Comorbidities. Medscape education. Available from: http://www.medscape.org/viewarticle/736181. Accessed October 10, 2014. | |
Mehta R, Hardes K, Kelleher D, Preece A, Tombs L, Brealey N. Effects of moderate hepatic impairment on the pharmacokinetic properties and tolerability of umeclidinium and vilanterol in inhalational umeclidinium monotherapy and umeclindinum/vilanterol combination therapy: an open-label, non-randomised study. Clin Ther. 2014;36:1016–1027. | |
Harrell AW, Siederer SK, Bal J, et al. Metabolism and disposition of vilanterol, a long-acting β(2) adrenoreceptor agonist, for inhalation use in humans. Drug Metab Dispos. 2013;41:89–100. | |
Mehta R, Hardes K, Cahn A, et al. Safety, tolerability and pharmacokinetics (PK) of repeated doses of GSK573719 inhalation powder, a new long-acting muscarinic antagonist, in healthy adults [abstract]. Eur Respir J. 2011;38(Suppl 55):3972. | |
Goyal N, Beerahee M, Kalberg C, Church A, Kilbride S, Mehta R. Population pharmacokinetics of inhaled umeclidinium and vilanterol in patients with chronic obstructive pulmonary disease. Clin Pharmacokinet. 2014;53:637–648. | |
Spiriva® HandiHaler® (tiotropium bromide inhalation powder) capsules for respiratory inhalation [prescribing information]. Ridgefield: Boehringer Ingelheim Pharmaceuticals, Inc.; 2014. Available from: http://bidocs.boehringer-ingelheim.com/BIWebAccess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing+Information/PIs/Spiriva/Spiriva.pdf. Accessed October 10, 2014. | |
European Medicines Agency. Annex I: Summary of Product Characteristics: Glycopyrronium Bromide. London: European Medicines Agency; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002430/WC500133769.pdf. Accessed October 10, 2014. | |
Tudorza™ Pressair™ (aclidinium bromide inhalation powder) [prescribing information update]. St Louis: Forest Laboratories, Inc.; 2014. Available from: http://www.frx.com/pi/tudorza_pi.pdf. Accessed October 10, 2014. | |
Schmid K, Pascual S, Gil EG, Ortiz S, Jansat JM. Pharmacokinetics and safety of aclidinium bromide, a muscarinic antagonist, in adults with normal or impaired renal function: A phase I, open-label, single-dose clinical trial. Clin Ther. 2010;32:1798–1812. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.