Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Effect of Intestinal Flora on Hyperuricemia-Induced Chronic Kidney Injury in Type 2 Diabetic Patients and the Therapeutic Mechanism of New Anti-Diabetic Prescription Medications
Received 27 July 2023
Accepted for publication 22 September 2023
Published 29 September 2023 Volume 2023:16 Pages 3029—3044
DOI https://doi.org/10.2147/DMSO.S429068
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Wei Yan,1,2 Song Wen,1 Ligang Zhou1,3
1Department of Endocrinology, Shanghai Pudong Hospital, n University, Shanghai, 201399, People’s Republic of China; 2Department of General Practice, Jinshan Hospital, Fudan University, Shanghai, 201508, People’s Republic of China; 3Shanghai Key Laboratory of Vascular Lesions Regulation and Remodeling, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, People’s Republic of China
Correspondence: Ligang Zhou, Department of Endocrinology, Shanghai Pudong Hospital, Fudan University, Shanghai, 201399, People’s Republic of China, Tel +8613611927616, Email [email protected]
Abstract: This article examined the current research on hyperuricemia (HUA) exacerbating diabetic kidney damage and novel anti-diabetic medications for treating these people. Hyperuricemia and type 2 diabetes (T2D), both of which are frequent metabolic disorders, are closely connected. Recent studies have shown that hyperuricemia can increase kidney injury in T2D patients by aggravating insulin resistance, by activating the renin-angiotensin-aldosterone system (RAAS), and by stimulating inflammatory factors, and the diversity, distribution, and metabolites of intestinal flora. Considering this, there are just a few of the research examining the effect of hyperuricemia on diabetic kidney injury via intestinal flora. Through the gut-kidney axis, intestinal flora primarily influences renal function. The primary mechanism is that variations in diversity, distribution, and metabolites of intestinal flora led to alterations in metabolites (such as short-chain fatty acids, Indoxyl sulfate and p-cresol sulfate, Trimethylamine N-oxide TMAO). This article reviewed the research and investigates the association between hyperuricemia and T2D, as well as the influence of hyperuricemia on diabetic kidney injury via intestinal flora. In addition, the current novel antidiabetic drugs are discussed, and their characteristics and mechanisms of action are reviewed. These novel antidiabetic drugs include SGLT2 inhibitors, GLP-1 receptor agonists, DDP-4 inhibitors, glucokinase (GK) enzyme activators (GK agonists), and mineralocorticoid receptor antagonists (MRA). Recent studies suggest that these new anti-diabetic medications may have a therapeutic effect on hyperuricemia-induced kidney impairment in diabetes patients via various mechanisms. Some of these medications may reduce blood uric acid levels, while others may improve kidney function by attenuating the overstimulation of RAAS or by decreasing insulin resistance and inflammation in the kidneys. These novel antidiabetic medicines may have a multifaceted approach to treating hyperuricemia-induced kidney impairment in diabetic patients; nevertheless, additional study is required to establish their efficacy and comprehend their specific mechanisms.
Keywords: hyperuricemia, intestinal flora, type 2 diabetes, T2D, kidney injury, diabetic kidney disease
Introduction
Hyperuricemia and Type 2 diabetes (T2D) are two prevalent metabolic disorders that are increasing globally. Hyperuricemia refers to a blood uric acid content of 420 umol/L or above.1 Diabetic patients with hyperuricemia may be prone to develop gout, renal damage, and other disorders. Poor control of hyperglycemia and hyperglycemia over the long term may increase the risk of cardiovascular events, neurological disorders, kidney disease, and other complications. Hyperuricemia and T2D have a complex interaction, and their coexistence increases the risk of diabetes complications, particularly kidney injury.2
Intestinal flora is a complex microbial system, which is involved in various physiological processes of the body. Studies have shown that intestinal flora is closely related to T2D and hyperuricemia, and there are significant differences in intestinal flora between patients with T2D and hyperuricemia.3,4 Specifically, T2D patients showed an increase in multiple pathogenic bacteria, such as Clostridium hardtii, commensal Clostridium, and Escherichia coli, while healthy controls had a high abundance of butyrate-producing bacteria.5 In patients with hyperuricemia, it is characterized by a decrease in probiotics (eg, Lactobacillus) and an increase in harmful bacteria (eg, Escherichia coli).6 Furthermore, these studies suggest that Intestinal flora is involved in the occurrence and development of T2D and hyperuricemia.
In addition to conventional medications, there are currently many new anti-diabetic therapies that have been approved for clinical application or are on the approach of approval. Compared to conventional anti-diabetic treatments, these novel medications have significant advantages, which is why they have attracted so much interest. For patients with T2D and hyperuricemia, the newer anti-diabetic drugs appear to be a viable option.
For the prevention and treatment of complications associated with related diseases, it is of extremely important to understand the mechanism and diagnostic methods of renal damage in diabetic patients with hyperuricemia. In addition, a comprehensive examination of the association between hyperuricemia and T2D can provide physicians with more effective preventive, diagnosis, and treatment programs to encourage the prevention and treatment of these two conditions. Understanding the mechanism of action of innovative anti-diabetic medicines provides physicians with information and aids in medication selection. In order to serve as a reference for future therapeutic strategies, the purpose of the current review is to demonstrate the research development of hyperuricemia exacerbating renal injury in diabetic patients, with a special emphasis on the research progress of intestinal flora.
Literature Retrieval Strategy
This review was conducted under the standard of PRISMA 2020 procedure. Literature retrieval strategy of this paper is via in PubMed, WanFang Database, VIP database, China hownet (CNKI). The detailed literature inclusion process can be seen in Figure 1. In addition, we also summarized the included epidemiological studies, as shown in Table 1.
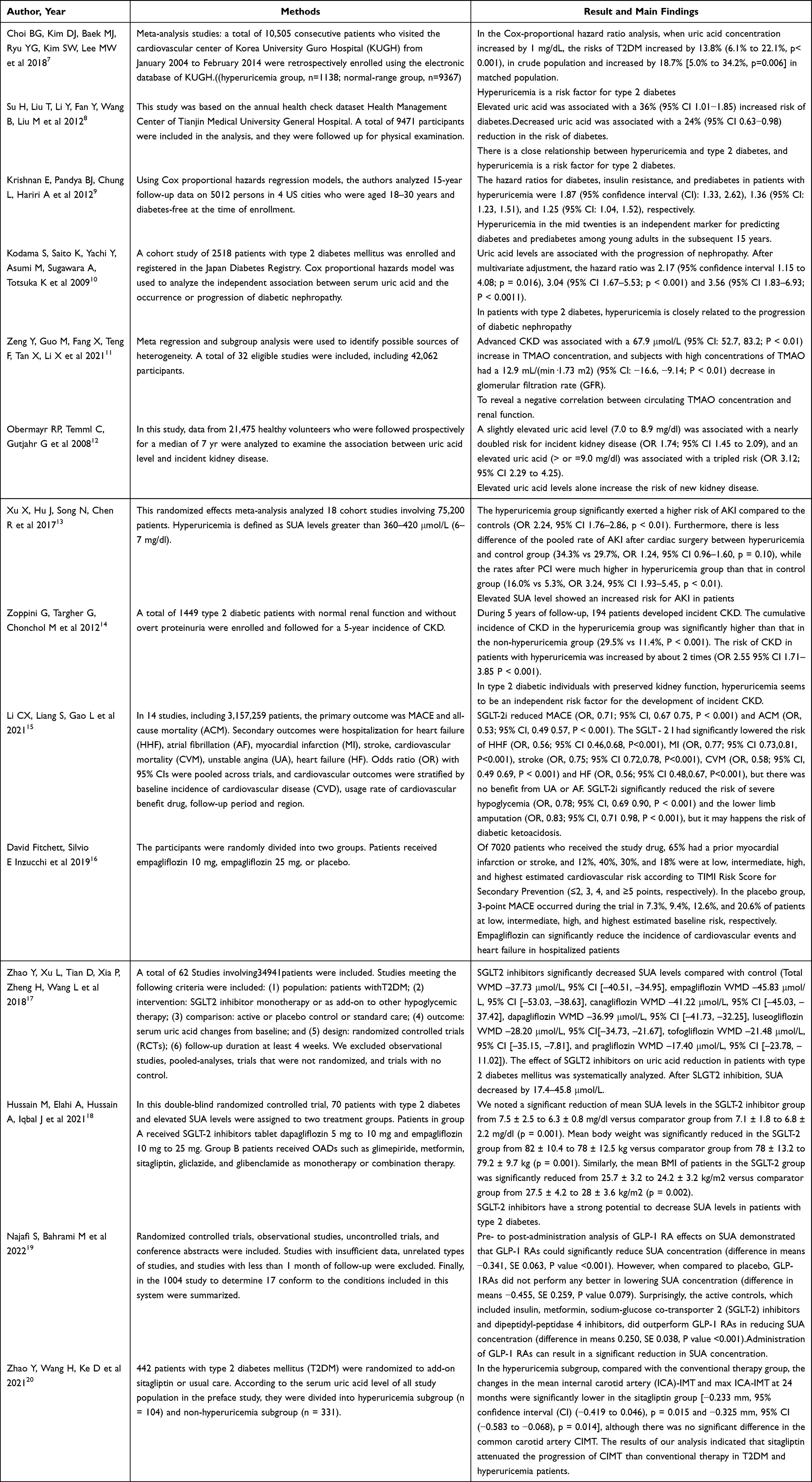 |
Table 1 Summary of Epidemiological Studies |
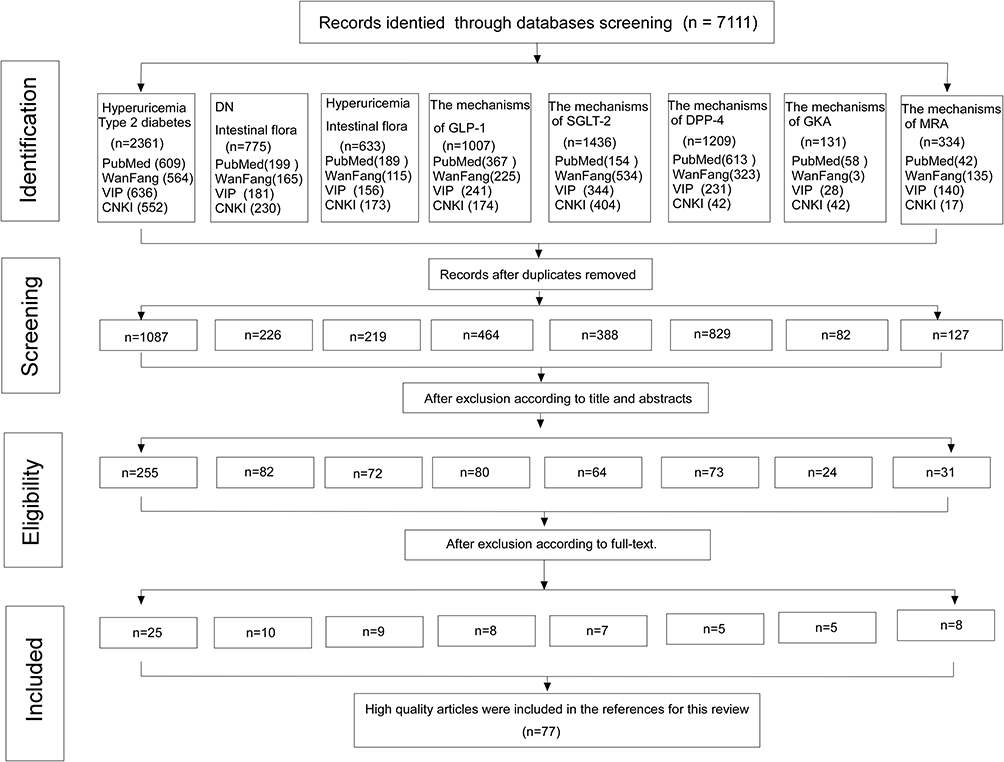 |
Figure 1 Literature inclusion process for the study. Abbreviations: DN, diabetic nephropathy; VIP, vip database; CNKI, China national knowledge infrastructure; SGLT-2, Sodium-glucose cotransporter 2; GLP-1, glucagon-like peptide-1; DPP-4, Dipeptidyl peptidase-4; MRA, mineralocorticoid receptor antagonist; GKA, glucokinase antagonist. |
The Current State of Research on Type 2 Diabetes, Hyperuricemia, Intestinal Flora, and Kidney Injury
The Study Investigated the Relationship Between Hyperuricemia and Type 2 Diabetes
Numerous research have examined the relationship between hyperuricemia and T2D, and the academic consensus today believes that hyperuricemia is an independent risk factor for T2D and its consequences.21 According to the Framingham Heart Study and a recent Korean cohort, each 1 mg/dL increase in uric acid is related with a 20% and 14% increased risk of type 2 diabetes, respectively.7 In prospective Chinese research of 9471 participants, 762 developed type 2 diabetes over a mean follow-up period of 2.9 years. Risk variables include alcohol use, triglycerides, low-density lipoprotein cholesterol, and time-dependent BMI after controlling for age, sex, smoking, family history of diabetes, and current smoking. Across tertiles of baseline blood uric acid, the hazard ratio (HR) and confidence interval (CI) for diabetes were 1.00, 1.18 (95% CI 0.97–1.43), and 1.34 (95% CI1.11–1.66), respectively (P for trend 0.011). Serum uric acid as a continuous variable was found to have a positive linear correlation with the risk of type 2 diabetes, as determined by restricted cubic spline models. Compared to participants whose uric acid levels remained constant (10%), those with the greatest increase in uric acid had a 36% (95%ci 1.01–1.85) greater risk of diabetes.8 Krishnan et al investigated whether hyperuricemia is a marker of diabetes and prediabetes in young adults by including 5012 United States residents who were initially free of T2D.9 Cox proportional hazard regression models revealed in his study that individuals with higher blood uric acid levels also had a greater prevalence of diabetes and prediabetes. Kodama et al conducted a comprehensive meta-analysis of genetic studies examining causal relationships between serum uric acid levels and other metabolic characteristics, including diabetes.10 The study identified several genetic variants associated with uric acid levels and established a causal connection between raised blood uric acid levels and an increased risk of diabetes, suggesting that uric acid control may be a potential therapeutic therapy for the patients with T2D.
The Research into the Connection Between Hyperuricemia and Intestinal Flora
As a massive micro-ecosystem in the human body, intestine micro-ecology is considered an organ that regulates the host’s metabolism. Intestinal flora refers to the vast number of microorganisms found in the digestive tract of humans. In recent years, numerous studies have revealed a close relationship between hyperuricemia and intestinal flora. According to studies, the composition of intestinal flora differs significantly between patients with hyperuricemia and healthy individuals.22 In gout patients, the abundance of Bacteroides faecalis and Bacteroides xylans is greatly increased, whereas faecalibacterium prowazekii and bifidobacterium pseudosmall chain is significantly decreased.6 Similarly, the numbers of Prevotella, Dehalogenobacter, Ruminococcus, and Lactobacillus were reduced in the rat model of hyperuricemia.23 Diet is an important factor affecting the composition of gut microbiota. The composition of Intestinal flora changes in animal models of hyperuricemia caused by high-fructose diet, high-fat diet, high-purpurin diet, and high-oxalate diet.22 Hyperuricemia has an impact on intestinal flora. In addition, intestinal flora can influence the uric acid metabolism and exacerbate hyperuricemia. Through metabolites, such as short chain fatty acid (SCFA) and amino acid metabolites, as well as diverse routes, such as glucagon-like peptide −1 (GLP1), intestinal flora primarily participate in numerous physiological functions of the organism.24 Some intestinal flora can enhance glucose-triggered GLP-1 secretion by upregulating the activity of G-protein-coupled receptor 43/41 (GPR43/41), proglucagon, and proconvertase 1/3, which is the primary mechanism by which intestinal flora regulate GLP-1.25
The Relationship Between Intestinal Flora and Kidney
Intestinal microbiota varies among individuals and is impacted by a range of factors, such as nutrition, age, lifestyle, disease status and medication.26,27 These variables may influence the dynamic composition and diversity of the gut flora, which has an impact on health and disease. Consequently, an increasingly complex and integrated mechanism of the gastrointestinal tract and associated microbiota has been demonstrated. The “intestinal-organ axis” is emerging to be crucial in maintaining the homeostasis of multiple organs, such as the gut-brain axis, gut-kidney axis, gut-liver axis and gut-bone axis.28 Among these cross-talk pathways, intestinal flora has a significant effect on the gut-kidney axis. The intestinal flora is essential to the maintenance of kidney function by synthesizing short-chain fatty acids, p-cresol sulfate, indoxyl sulfate, trimethylamine N-Oxide (TMAO), and other compounds.29 Short-chain fatty acids are necessary for maintaining the integrity of the intestinal epithelium and energy balance. They may attenuate hypoxia damage in renal epithelial cells by promoting mitochondrial biogenesis, whereas para-cresol sulfate and indoxyl sulfate bind to albumin in the blood and are secreted by renal tubules. If uremic retained solute accumulates in the body, the progression of glomerulosclerosis and renal disease will be accelerated. TMAO concentrations were up to 20 times higher in patients with end-stage renal disease than in healthy controls.11 Increased TMAO levels can produce tubulointerstitial fibrosis and contribute to the pathophysiology of atherosclerosis. Thus, TMAO concentration could be used to predict GFR and renal function.11,30
Relationship Between Hyperuricemia and Kidney
Several studies have demonstrated that chronic hyperuricemia can result in kidney injury. Obermayr et al conducted a prospective study on 21,457 healthy participants and followed up for 7 years, they discovered mildly elevated uric acid levels (7.0–8.9 mg/dl) were associated with nearly doubling the risk for new-onset kidney disease (OR=1.74,95% CI 1.45–2.09), whereas elevated uric acid (> OR =9.0 mg/dl) was associated with a threefold increased risk (OR=3.12,95% CI 2.29–4.25).12 This increased risk remained significant even after adjustment for baseline eGFR, sex, age, antihypertensive medications, and metabolic syndrome components (waist circumference, HDL cholesterol, glucose, triglycerides, and blood pressure). A 2017 meta-analysis included 18 studies with a total of 75,200 patients showed that the hyperuricemia group significantly exerted a higher risk of AKI compared to the controls (odds ratio OR 2.24, 95% CI 1.76–2.86, p < 0.01).13 The mechanism by which hyperuricemia affects renal injury may involve vascular endothelial dysfunction, activation of the renin-angiotensin-aldosterone system (RAAS), release of inflammatory factors, and an increase in oxidative stress.31
One of the most critical mechanisms is that uric acid crystals cause immediate renal injury. In patients with primary hyperuricemia, an excess of uric acid in the blood will precipitate, produce urate crystals, deposit in the renal vascular endothelium, and induce renal vascular lesions. When the PH is 7.1, the majority of uric acid in plasma, glomerular filtration fluid, and renal interstitium will dissociate into urate ions, resulting in the deposition of urate in the tubulointerstitial region of the kidney, which will be surrounded by leukocyte and track macrophage infiltration, increasing in renal tubulointerstitial inflammation and fibrosis, and renal tubular damage.32–34
Another mechanism is that hyperuricemia could have direct effects on the RAAS and indirect effects on angiotensin II and aldosterone via macrophage and T cell polarization.35 In a rat model of hyperuricemia, Stellato D et al36 observed thickening of renal glomerular arterioles, renal tubulointerstitial fibrosis lesions, renal cortical vasoconstriction, and a 35% reduction in single glomerular filtration rate, which may be primarily attributable to the proliferation of vascular smooth muscle cells stimulated by uric acid. In adult adipocytes, soluble uric acid increases NADPH oxidase activity and reactive oxygen species (ROS) generation, which may result in endothelial dysfunction and vasoconstriction.37 In addition, uric acid can raise blood viscosity, decrease nitric oxide levels in the endothelium and macula densus, and promote renal contraction by increasing oxidative stress.38 Long-term contraction of blood arteries results in thickening of the vascular wall, secondary ischemic alterations, and stimulation of tubular interstitial changes, which leads to renal interstitial fibrosis and arterial hypertension. However, uric acid plays a different role according to where it acts. In the cytoplasm or in an atherogenic plaque it plays a pro-oxidant role, promoting oxidative stress and thus contributing to kidney disease and cardiovascular diseases and development.39 In addition, uric acid has antioxidant effects, and studies have shown that in the blood, uric acid may help extend human lifespan by providing protection against oxidative stress-induced aging and cancer, uric acid is an oxidizable substrate of the haem protein/H2O2 systems and is able to prevent oxidative damage by acting as an electron donor.40
Thirdly, hyperuricemia can stimulate the synthesis of certain inflammatory factors. By activating nuclear transcription factor (NF-κB), uric acid can upregulate the production of monocyte chemoattractant protein-1 (MCP-1) in endothelial cells, according to an in vitro cell investigation conducted by Kanellis.41 Under the influence of MCP-1, monocytes accumulate and adhere to endothelial cells, which activates endothelial cells, resulting in a series of alterations to both their function and structure, as well as an increase in the production of harmful cytokines such as tumor necrosis factor-α (TNF-α), intercellular adhesion molecule-1, and interleukin-6 (IL-6), generating an inflammatory cascade. This results in a vicious loop that compromises the endothelial function of the blood vessels. Moreover, high levels of UA significantly upregulated HMGB1 expression by activating Toll-like receptor 4(TLR4) and MEK/ERK pathways.42 HMGB1 amplify inflammatory responses through several pathways, including promoting the secretion of proinflammatory cytokines by monocytes, such as Interleukin—1β (IL-1β) and TNF-α, expression of adhesion molecules, and inflammatory cell infiltration.43 In particular, HMGB1 promotes its own release from endothelial cells through a positive feedback mechanism. After UA-induced HMGB1 binds to the receptor for advanced glycation end products (RAGE), it activates the NF-κB signaling pathway and promotes the production of cytokines such as TNF-α and IL-6, leading to oxidative stress and inflammatory response.44 Therefore, uric acid can act as an inflammatory mediator to trigger the inflammatory response of the endothelium within the cardiovascular system. In a rat model of hyperuricemia,41,45 the renal parenchyma of rats exhibited a substantial increase in macrophage infiltration. It also suggests that hyperuricemia is a promoting factor in the release of inflammatory mediators, which preferably cause renal tubular damage. Therefore, even moderately elevated uric acid could be lethal to the normal function of vascular endothelial as the major consequence of inflammation, which may be at risk of progression to severe kidney injury.
The Kidney Injury Due to the Interactions of Type 2 Diabetes, Hyperuricemia, and Intestinal Flora
Multiple studies have demonstrated that the combination of T2D and hyperuricemia might accelerate the diabetic progression and cause to consequences such as kidney damage. Hyperuricemia and diabetes have a complex relationship, and the presence of both can exacerbate the complications of diabetes, particularly kidney damage.46 Zoppini et al followed 1449 type 2 diabetic patients with normal renal function and no significant proteinuria for 5 years to investigate the onset of diabetic kidney disease (DKD, defined as significant proteinuria or estimated glomerular filtration rate [eGFR] <60 mL/ minute / 1.73 m2). The results showed that 194 patients (13.4%) had new onset CKD. The cumulative incidence of CKD in the hyperuricemia group was significantly higher than that in the non-hyperuricemia group (29.5% vs 11.4%, P < 0.001). Univariate logistic regression analysis showed that the risk of CKD in patients with hyperuricemia was increased by about 2 times [OR=2.55 (95% CI 1.71–3.85), P < 0.001]. After adjustment for age, sex, BMI, smoking status, duration of diabetes, systolic blood pressure, antihypertensive therapy, insulin therapy, hba1c, eGFR, and albuminuria, hyperuricemia was associated with an increased risk of CKD (adjusted OR 2.10 [1.16–3.76], P < 0.01). In continuous analyses, each 1-SD increase in serum uric acid level was significantly associated with a 21% increase in CKD risk.14 It is speculated that hyperuricemia is an independent risk factor for diabetic kidney disease in type 2 diabetic patients with normal renal function.
Currently, the mechanism of renal injury induced or aggravated by hyperuricemia in patients with T2D consists primarily of the three components mentioned below:
- Patients with T2D or diabetic kidney disease (DKD) have a distinct degree of insulin resistance (IR), and hyperuricemia can reduce the level of NO in endothelial cells, which enhances the IR. Long-term hyperuricemia, on the other hand, can result in the deposition of urate crystals in islet B cells, directly impairing insulin secretion, thereby having an effect on glucose metabolism, which is associated with the deterioration of T2D and DKD.47
- Patients with T2D will increase the release of inflammatory factors such as C-reactive protein (CRP), TNF-a, IL-6, etc, due to long-term hyperglycemia and tissue hypoxia, and high uric acid will activate inflammatory cells to release inflammatory mediators such as tumor necrosis factor-α (TNF-a), intercellular adhesion molecule-1 (ICAM-1), interleukin-6 (IL-6), etc. Patients with T2D complicated with hyperuricemia will furtherly the activation of inflammatory factors, adding to the severity of inflammation and oxidative stress, which increase IR, causing glucose metabolism disorders and renal vascular damage, and thereby accelerating the progression of DKD.
- Thirdly, DKD patients are intimately associated to RAAS overstimulation, which plays a crucial role in diabetic vascular disease. Multiple large clinical trials have demonstrated that suppressing RAAS can dramatically decrease the progression of microalbuminuria and macroalbuminuria in patients with DKD. Hyperuricemia can affect the RAAS axis via both direct and indirect pathway.31 Blood uric acid in patients with T2D may contribute to the etiology of DKD by activating the RAAS system, according to studies.48 After T2D complicated by hyperuricemia, it will accelerate glomerular arteriosclerosis and increase the development of T2D and DKD.48
Moreover, in the patients with T2D complicated with hyperuricemia, intestinal flora is also involved in the occurrence and development of DKD under the influence of high uric acid. The composition and diversity of intestinal flora are drastically altered in patients with hyperuricemia, manifested mostly as a decrease in probiotics. A few of studies have demonstrated that the abundance of Lactobacillus and Pseudomonas in the intestine of the patients with hyperuricemia is significantly decreased, whereas Escherichia coli and Proteus are significantly increased,6 resulting in a decrease in short-chain fatty acid(SCFA) production and an increase in p-cresyl sulfate, indoxyl sulfate, and TAMO. The effects of intestinal flora on the kidney are mainly achieved through the gut-kidney axis, which causes injuries to the kidneys via the following three ways:
- Short-chain fatty acids (SCFA) are metabolites of intestinal flora, which play an important role in regulating host immune response and inflammation. Butyric acid in SCFA is the main energy source for intestinal epithelial cells. The absorption of butyrate by intestinal epithelial cells is the main energy source for adenosine phosphate-activated protein kinase (AMPK) activation and glucagon-like peptide- 1(GLP-1) production. SCFA can also regulate the release of peptide YY (PYY) and GLP-1 from L-cells or the regulation of inflammatory factors TNF-α, IL-6 and IL-1β were involved in insulin resistance. Intestinal flora imbalance in patients with hyperuricemia reduces the production SCFA, thereby reducing the secretion of GLP-1 and PYY, leading to increased blood glucose and deterioration of renal injury in diabetic patients, and accelerating the progress of DKD. In addition, the production of SCFA is reduced, which promotes the increase of inflammatory factors such as TNF-α, IL-6 and IL-1β, and aggravates insulin resistance. It further promotes the occurrence and development of diabetic complications.49,50
- Indoxyl sulfate and p-cresol sulfate are also metabolites of intestinal flora, mainly produced by harmful bacteria. The intestinal flora in patients with hyperuricemia is dysregulated, which is characterized by a decrease in beneficial bacteria and an increase in harmful bacteria. When patients with T2D are complicated with hyperuricemia, the production of indoxyl sulfate and p-cresol sulfate will increase. Due to their strong affinity for albumin, they circulation in the blood and are released by renal tubular secretions. If excessive accumulation in the body, it can increase the incidence of glomerulosclerosis and promote the occurrence and development of kidney diseases.51
- Trimethylamine N-oxide (TMAO) is also a metabolite of harmful bacteria in the intestinal flora. In patients with hyperuricemia, the intestinal flora is dysregulated, and TMAO production is increased. Many studies have reported that TMAO concentration levels are positively correlated with the development of a variety of diseases, such as cardiovascular and renal diseases, including atherosclerosis, hypertension, ischemic stroke, atrial fibrillation, heart failure, acute myocardial infarction, and chronic kidney disease.52 It is also positively correlated with the occurrence and development of diabetes, metabolic syndrome, cancer (gastric cancer, colon cancer) and nervous system diseases.52 In recent years, the relationship between TMAO and chronic kidney disease (CKD) has been gradually recognized, the level of TMAO is associated with the occurrence and prognosis of CKD. It can be used as a potential risk factor for the development of CKD and is expected to become a new target for the treatment of CKD.53 TMAO can enter the bloodstream and be excreted by the kidneys. Compared with healthy controls, patients with end-stage renal disease have up to 20-fold higher TMAO concentrations, which directly correlate with the course of CKD.54 TMAO-mediated inflammation is an important mechanism for the occurrence and development of CKD. High concentrations of TMAO promote oxidative stress and inflammation in the kidney.55 Similarly, high concentrations of TMAO can reduce NO production by inducing vascular oxidative stress and inflammation, which triggers CKD complications such as endothelial dysfunction and cardiovascular disease.56 TMAO activates the NLRP3 inflammasome, leading to the release of IL-1β and IL-18, which accelerates renal inflammation.57 In addition, TMAO causes vascular inflammation and myocardial fibrosis to exacerbate cardiovascular disease by activating the NLRP3 inflammasome.58
As described above, hyperuricemia can affect renal function by inducing vascular endothelial dysfunction, stimulating the RAAS, releasing inflammatory substances, elevating oxidative stress, and disrupting intestinal flora (Figure 2). Through the above mechanisms, hyperuricemia leads to glomerular sclerosis and renal tubule fibrosis in patients with type 2 diabetes, resulting in decreased glomerular filtration rate and increased excretion of urinary proteins. Among them, intestinal flora plays a significant role in the process by which hyperuricemia exacerbates kidney injury in individuals with type 2 diabetes, a phenomenon that needs consideration.
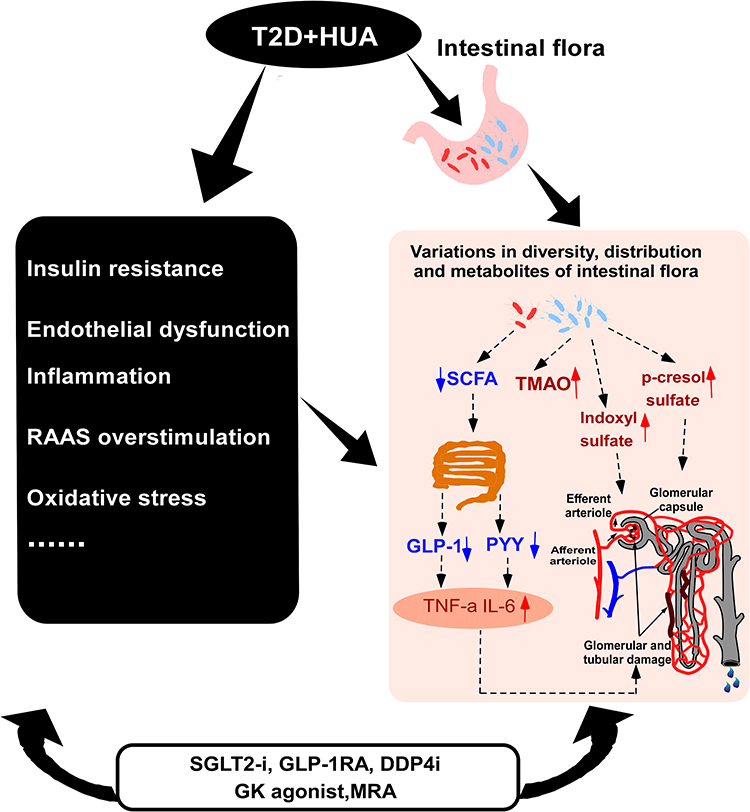 |
Figure 2 The mechanism of aggravated kidney injury in patients with type 2 diabetes mellitus and hyperuricemia. Abbreviations: T2D, type 2 diabetes; HUA, hyperuricemia; PYY, peptide YY; RAAS, renin-angiotensin-aldosterone system; SCFA, Short-chain fatty acids; TMAO, trimethylamine N-oxide; GLP-1, glucagon-like peptide- 1; GK, glucokinase; TNF-a, tumor necrosis factor-α; IL-6, interleukin-6; SGLT-2i, Sodium-glucose cotransporter 2 inhibitor; GLP-1RA, glucagon-like peptide-1 receptor agonists; DPP-4i, Dipeptidyl peptidase-4 inhibitors; MRA, mineralocorticoid receptor antagonist. Notes: These novel antidiabetic medicines, including SGLT2 inhibitors, GLP-1 receptor agonists, DDP-4 inhibitor, Glucokinase enzyme activator (GK agonist), and mineralocorticoid receptor antagonist (MRA), may have a multifaceted approach to treating hyperuricemia-induced kidney impairment in diabetic patients; nevertheless, additional study is required to establish their efficacy and comprehend their specific mechanisms. |
Treatment of Kidney Injury Caused on by Type 2 Diabetes, Hyperuricemia, and Intestinal Flora Metabolites May Benefit from the Recent Innovative Medications
Sodium-glucose cotransporter 2 inhibitor (SGLT-2i) are a relatively recent family of diabetes medications. Because of its good hypoglycemic effect, it has been widely used in clinic in recent years. Sodium-glucose cotransporter 2(SGLT-2) mainly exists on the renal tubules, and its main function is to promote the reabsorption of glucose in the original urine. The mechanism of SGLT-2 inhibitors is to inhibit the activity of SGLT-2, restrict the reabsorption of glucose in the proximal renal tubules, and discharge excess glucose, thereby reducing the blood sugar of patients. And its effect on lowering blood sugar is independent of insulin secretion.59 Numerous studies have demonstrated that SGLT-2 inhibitors have a beneficial effect on cardiovascular and renal protection, and multiple mechanisms have been proposed to explain the cardiovascular and renal benefits of SGLT-2i, including hemodynamic, anti-inflammatory, anti-fibrotic, antioxidant, and metabolic effects.60,61 Studies have shown that SGLT - 2 inhibitors can also reduce the uric acid level, its mechanism has three aspects: 1) Urate reabsorption protein 1 (URAT1) increases uric acid secretion due to glucose reabsorption, and SGLT-2i may increase glucose excretion by blocking renal tubular URAT1, resulting in increased uric acid excretion. In addition, SGLT-2i can also inhibit URAT1 reabsorption of uric acid by reducing serum insulin levels;62 2) SGLT-2i may activate Sirtuin-1 (SIRT1) to inhibit the enzyme xanthine oxidase (OX), leading to a decrease in uric acid levels;63 3) SGLT-2i can down-regulate XO activity and inhibit inflammatory response through RAAS and sympathetic nervous system (SNS), thereby reducing serum uric acid formation.63
A considerable number of current medical investigations have demonstrated that SGLT-2 inhibitors have a certain cardiovascular protective effect. Li et al conducted a meta-analysis in 2021 on cardiovascular outcomes associated with SGLT-2 inhibitors and other hypoglycemic medications in T2D patients. A total of 14 studies including 3,157,259 patients were included. The findings demonstrated that SGLT-2 inhibitors can decrease the risk of cardiovascular disease.15 There is currently inadequate information to clarify the relationship between heart failure and SGLT-2 inhibitor benefits. It is believed to be associated with weight loss, improvement in blood lipids, influence on endothelial function and vascular sclerosis, oxidative stress, and inflammatory cytokines. SGLT-2 inhibitors can successfully lower the incidence of a renal composite endpoint and protect the kidneys. The systematic review/meta-analysis by Neuen et al,16 which included four randomized controlled trials (ie, EMPAREG OUTCOME, CANVAS Program, CREDENCE, and DECLARE-TIMI 58), demonstrated that SGLT-2 is capable of inhibiting the progression of chronic kidney disease (CKD) in patients with type 2 diabetes.64 It may protect the kidney by reducing glucose and uric acid levels, alleviating renal hyperfiltration, reducing proteinuria, oxidative stress, and chronic inflammation.
SGLT2 Inhibitors may also protect the kidney by decreasing hyperuricemia (HUA). Zhao et al conducted a meta-analysis comprising 62 research on the effect of SGLT-2 inhibitors on serum uric acid levels.17 Patients with T2D had significantly reduced serum uric acid levels when administering SGLT-2 inhibitors, according to a study involving 34,941 participants. Patients with diabetes and hyperuricemia may obtain additional benefits from SGLT-2 inhibitors. Alicia and colleagues did a meta-analysis on the effects of SGLT2 inhibitors on serum uric acid levels in diabetic and non-diabetic patients in 2022.18 There were a total of 43 randomized controlled studies with 31,921 patients enrolled. The research shown that SGLT-2 inhibitors decreased serum uric acid levels in both diabetic and non-diabetic individuals. Therefore, SGLT-2 inhibitors may assist to the management of hyperuricemia in patients with and without diabetes. It is unknown how SGLT2 inhibitors lower serum uric acid, however it is considered to be associated to the renal SLC2A9 and GLUT9 transporter.62
GLP-1 receptor agonists are also emerging as novel antidiabetic agents that have received great attention in the treatment of obesity and diabetes due to their potent incretin effects, which are incretin hormones secreted by L cells in the distal small intestine and colon. Animal studies have shown that administration of native GLP-1 reduces blood glucose levels in a glucose-dependent manner, improves the function and quality of B cells,65 increases insulin sensitivity and has direct cardioprotective effects.66 Numerous studies have shown that GLP-1 receptor agonists have potential renal protective effects. The beneficial effects of GLP-1 receptor agonists on the kidney are generally believed to be related to lowering blood glucose and blood pressure (BP), lowering insulin levels, and leading to weight loss.67 Emerging evidence suggests that the potential kidney-protective effects of GLP-1 receptor agonists are independent of their glucose-lowering effects, and that some of them may play a role in inhibiting the development and progression of DKD.68 Studies have shown that GLP-1RA can inhibits the RAAS, which may imply additional potential renal protective mechanisms of GLP-1 receptor agonists in DKD.69 In addition, the potential renal protection of GLP-1 receptor agonists may also be related to the improvement of glomerular atherosclerosis, renal vasodilatation, renal hypoxia, improvement of insulin sensitivity, weight loss, and reduction of intestinal lipid uptake.19 There are also reports of a presumed association with intestinal microbiota, but more studies are needed to confirm this.25 In addition, investigations have indicated that GLP-1 receptor agonists have a substantial effect on serum uric acid levels, but their effect on uric acid reduction is smaller than that of SGLT-2 inhibitors.19 The mechanism by which GLP-1 receptor agonists reduce uric acid is still unclear, and it is speculated that GLP-1RA improves renal tubules and glomeruli to promote uric acid excretion.
For more than a decade, Dipeptidyl peptidase-4 (DPP-4) inhibitors have been used as second-line treatment for patients with T2D, and they are now routinely utilized in clinical practice. The primary mechanism of DPP-4 inhibitors is to inhibit the activity of dipeptidyl peptidase IV, hence preventing the degradation of incretin, GLP-1, and glucose-dependent insulinotropic peptide (GIP), resulting in increased endogenous levels of GLP-1 and GIP. At the same time, it also leads to the inhibition of glucagon secretion.70 DPP-4 inhibitors may improve the two main risk factors of DKD, thus producing potential renal benefits in addition to blood glucose control. DPP-4 mainly degrades GLP-1 and other incretin, so DPP-4 inhibitors can restore GLP-1 signaling pathway.71 GLP-1 receptor mediated cAMP-PKA pathway signaling in the kidney, which is important for renal homeostasis,72 and GLP-1 receptor stimulation can regulate cardiac natriuretic hormone (ANP) and renin-angiotensin system (RAS), which may be GLP-1-mediated renal protection.73 DPP-4 can also degrade many peptide substrates in addition to GLP-1, and DPP-4 inhibitors may affect different pathways of diabetic and non-diabetic CKD.74 Recent studies, including CARMELINA (NCT01897532) have shown that DPP-4 inhibitors have a kidney protective effect in patients with DKD, and in an animal experiment, knocking out the GLP-1R gene in rats and removing 5/6 of the rats’ kidneys showed delayed progression of chronic kidney disease (CKD). These results suggest that DPP-4 inhibitors have protective effects on GLP-1R independent kidneys. Proteomic analysis showed that collagen I homeostasis, heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), Y box binding protein-1 (YB-1), thymosin β4 and transforming growth factor-β1 may potentially contribute to the protection of kidneys by DPP-4 inhibition.75 A recent study indicated that DDP-4i could contribute to the reduction of uric acid.20
In recent years, glucokinase (GK) has become a target for anti-diabetes treatment because of its glucose sensor and hyperglycemia control function. Glucokinase (GK) is mainly distributed in the liver and pancreas. GK acts as a “glucose sensor” in pancreatic cells, mainly triggering glucose-stimulated insulin secretion, and promoting hepatic glycogen synthesis in the liver. Therefore, glucokinase (GK) can promote insulin secretion and hepatic glycogen synthesis. Studies have shown that impaired islet function, resulting in impaired glucokinase (GK) activity, leads to abnormal insulin secretion and impaired liver glycogen synthesis in T2D, and up-regulation of GK activity by drugs can solve the above pathophysiological disorders.76 The main mechanism of glucokinase activator (GKA) is to activate glucokinase (GK), thereby promoting insulin secretion, while inhibiting glucagon release, and promoting liver glycogen synthesis, and at the same time, it has the effect of promoting GLP-1 secretion.
Mineralocorticoid receptor antagonist (MRA) is primarily used to treat chronic kidney disease in Type 2 Diabetes (T2D) patients. MRA has no influence on blood sugar regulation. Its primary mechanism is to inhibit MR overactivation, reduce inflammatory factors, decrease the reduction of fibrosis medium, inhibit the proliferation of fibroblasts, inhibit the apoptosis of podocytes, delay myocardial hypertrophy and myocardial cell apoptosis, inhibit vascular contraction, reduce vascular damage, and prevent vascular sclerosis, thereby improving renal fibrosis and glomerulosclerosis, and enhancing kidney function.77 Future research will determine whether or not MRA improves renal function via intestinal flora (Figure 3).
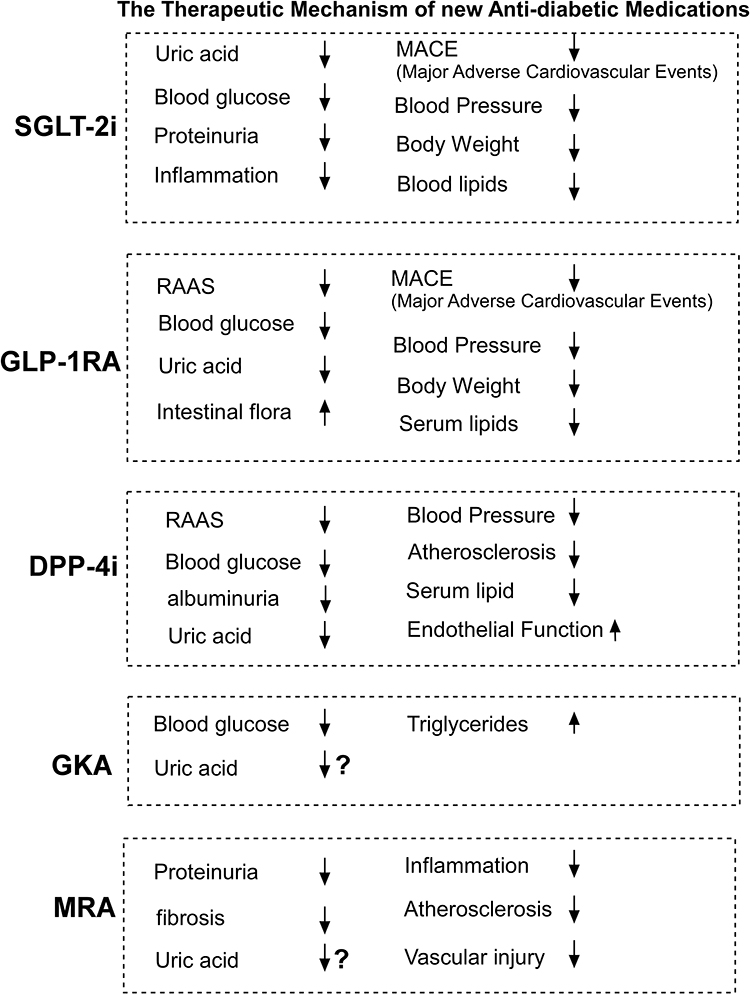 |
Figure 3 The therapeutic mechanism of new antidiabetic medicines on hyperuricemia-induced chronic kidney injury in the patients with T2D. Abbreviations: SGLT-2i, Sodium-glucose cotransporter 2 inhibitor; GLP-1RA, glucagon-like peptide-1 receptor agonists; DPP-4i, Dipeptidyl peptidase-4 inhibitors; MRA, mineralocorticoid receptor antagonist; GKA, glucokinase antagonist. Notes: It is unclear whether the GKA and MRA could contribute to the reduction of uric acid. SGLT2i, sodium-glucose cotransporter 2 inhibitors, GLP-1RA, glucagon like peptide-1 receptor agonist, DDP-4i, DDP-4 inhibitor, GKA, glucokinase activator, MRA, mineralocorticoid antagonist. |
In conclusion, hyperuricemia and T2D are two prevalent metabolic conditions whose prevalence has increased in recent years. More and more research indicated that hyperuricemia and T2D are tightly connected and mutually affect each other. At present, hyperuricemia is considered to be an independent risk factor for T2D, and its presence significantly increases the probability of T2D. In addition, patients with T2D are prone to hyperuricemia, with elevated levels of uric acid in the serum. The coexistence of these two diseases can easily lead to serious complications such as nervous system diseases and cardiovascular diseases, especially aggravating kidney diseases in diabetic patients. The main pathways by which hyperuricemia aggravates renal damage in T2D are the aggravation of insulin resistance, the activation of RAAS, the activation of inflammatory factors, and the change of intestinal flora, while the latter of which might play an essential role. Therefore, when T2D patients with hyperuricemia have renal injury, the role of intestinal flora should be considered, and the intestinal flora should be adjusted if necessary. Although the relationship between hyperuricemia and T2D has been widely studied, the mechanism of hyperuricemia through intestinal flora to aggravate T2D renal injury is not completely clear, and it can only make inferences based on clinical research and theory. Therefore, this article has certain limitations. In the future, animal experiments can be done to study the specific mechanism of hyperuricemia aggravating T2D renal injury through intestinal flora.
For patients with T2D and hyperuricemia, innovative diabetic medications, such as SGLT2 inhibitors, GLP-1 receptor agonists, and DDP-4 inhibitor, can be selected to regulate blood glucose. Not only can these new anti-diabetes medications regulate glucose levels, but they can also preserve heart and kidney function. To determine whether GK agonist is beneficial for DKD, additional clinical studies are required. When hyperuricemia exacerbates DKD in type 2 diabetic individuals, mineralocorticoid receptor antagonists appear to be an alternative therapy option. Because of SGLT-2i inhibitors, GLP-1 agonists, DDP-4 inhibitors such as innovation diabetes drugs for cardiovascular and renal protection concrete mechanism is not entirely clear, so there are some limitations in this article, you need to do in the future more animal experiments to clarify specific mechanisms of cardiovascular and renal, It is expected that intestinal flora can be used as a new target for the treatment of diabetic nephropathy in the future, and continue to actively explore, develop new anti-diabetic drugs, and benefit human health.
Funding
This work was supported by Youth Research Foundation of Jinshan Hospital of Fudan University(JYQN-LC-202212), the Project of Key Medical Discipline of Pudong Hospital of Fudan University (Zdxk2020-11), Project of Key Medical Specialty and Treatment Center of Pudong Hospital of Fudan University (Zdzk2020-24), Integrative Medicine special fund of Shanghai Municipal Health Planning Committee (ZHYY- ZXYJHZX-2-201712), Special Department Fund of the Pudong New Area Health Planning Commission (PWZzk2017-03), Outstanding Leaders Training Program of Pudong Health Bureau of Shanghai (PWR12014-06), Pudong New Area Clinical Plateau Discipline Project (PWYgy-2021-03), the Natural Science Foundation of China (21675034), National Natural Science Foundation of China (81370932), Shanghai Natural Science Foundation (19ZR1447500), Fudan Zhangjiang Clinical Medicine Innovation Fund Project (KP0202118), Pudong New Area Clinical Characteristic Discipline Project (PWYts2021-11), Pudong New Area Clinical Characteristic Discipline Project (PWYts2021-01), Wenzhou Medical University Education Grant (JG2021197).
Disclosure
The authors declare there is no conflict of interest.
References
1. Lin S. Interpretation of Chinese guidelines for the diagnosis and treatment of hyperuricemia and gout (2019). J Clin Med. 2020;37(06):460–462.
2. Bartakova V, Kuricova K, Pacal L, et al. Hyperuricemia contributes to the faster progression of diabetic kidney disease in type 2 diabetes mellitus. J Diabetes Complications. 2016;30(7):1300–1307. doi:10.1016/j.jdiacomp.2016.06.002
3. Gurung M, Li Z, You H, et al. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine. 2020;51:102590. doi:10.1016/j.ebiom.2019.11.051
4. Yang HT, Xiu WJ, Liu JK, et al. Gut Microbiota Characterization in Patients with Asymptomatic Hyperuricemia: probiotics increased. Bioengineered. 2021;12(1):7263–7275. doi:10.1080/21655979.2021.1976897
5. Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi:10.1038/nature11450
6. Guo Z, Zhang J, Wang Z, et al. Intestinal Microbiota Distinguish Gout Patients from Healthy Humans. Sci Rep. 2016;6:20602. doi:10.1038/srep20602
7. Choi BG, Kim DJ, Baek MJ, et al. Hyperuricaemia and development of type 2 diabetes mellitus in Asian population. Clin Exp Pharmacol Physiol. 2018;45(6):499–506. doi:10.1111/1440-1681.12911
8. Su H, Liu T, Li Y, et al. Serum uric acid and its change with the risk of type 2 diabetes: a prospective study in China. Prim Care Diabetes. 2021;15(6):1002–1006. doi:10.1016/j.pcd.2021.06.010
9. Krishnan E, Pandya BJ, Chung L, Hariri A, Dabbous O. Hyperuricemia in young adults and risk of insulin resistance, prediabetes, and diabetes: a 15-year follow-up study. Am J Epidemiol. 2012;176(2):108–116. doi:10.1093/aje/kws002
10. Kodama S, Saito K, Yachi Y, et al. Association between serum uric acid and development of type 2 diabetes. Diabetes Care. 2009;32(9):1737–1742. doi:10.2337/dc09-0288
11. Zeng Y, Guo M, Fang X, et al. Gut Microbiota-Derived Trimethylamine N-Oxide and Kidney Function: a Systematic Review and Meta-Analysis. Adv Nutr. 2021;12(4):1286–1304. doi:10.1093/advances/nmab010
12. Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol. 2008;19(12):2407–2413. doi:10.1681/ASN.2008010080
13. Xu X, Hu J, Song N, Chen R, Zhang T, Ding X. Hyperuricemia increases the risk of acute kidney injury: a systematic review and meta-analysis. BMC Nephrol. 2017;18(1):27. doi:10.1186/s12882-016-0433-1
14. Zoppini G, Targher G, Chonchol M, et al. Serum uric acid levels and incident chronic kidney disease in patients with type 2 diabetes and preserved kidney function. Diabetes Care. 2012;35(1):99–104. doi:10.2337/dc11-1346
15. Li CX, Liang S, Gao L, Liu H. Cardiovascular outcomes associated with SGLT-2 inhibitors versus other glucose-lowering drugs in patients with type 2 diabetes: a real-world systematic review and meta-analysis. PLoS One. 2021;16(2):e0244689. doi:10.1371/journal.pone.0244689
16. Fitchett D, Inzucchi SE, Cannon CP, et al. Empagliflozin Reduced Mortality and Hospitalization for Heart Failure Across the Spectrum of Cardiovascular Risk in the EMPA-REG OUTCOME Trial. Circulation. 2019;139(11):1384–1395. doi:10.1161/CIRCULATIONAHA.118.037778
17. Zhao Y, Xu L, Tian D, et al. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: a meta-analysis of randomized controlled trials. Diabetes Obes Metab. 2018;20(2):458–462. doi:10.1111/dom.13101
18. Hussain M, Elahi A, Hussain A, Iqbal J, Akhtar L, Majid A. Sodium-Glucose Cotransporter-2 (SGLT-2) Attenuates Serum Uric Acid (SUA) Level in Patients with Type 2 Diabetes. J Diabetes Res. 2021;2021:9973862. doi:10.1155/2021/9973862
19. Najafi S, Bahrami M, Butler AE, Sahebkar A. The effect of glucagon-like peptide-1 receptor agonists on serum uric acid concentration: a systematic review and meta-analysis. Br J Clin Pharmacol. 2022;88(8):3627–3637. doi:10.1111/bcp.15344
20. Zhao Y, Wang H, Ke D, et al. Sitagliptin on carotid intima-media thickness in type 2 diabetes and hyperuricemia patients: a subgroup analysis of the PROLOGUE study. Ther Adv Chronic Dis. 2021;12:20406223211026993. doi:10.1177/20406223211026993
21. Li C, Hsieh MC, Chang SJ. Metabolic syndrome, diabetes, and hyperuricemia. Curr Opin Rheumatol. 2013;25(2):210–216. doi:10.1097/BOR.0b013e32835d951e
22. Chen R, Liu Y. Research progress on the relationship between intestinal flora and hyperuricemia and gout. J Nanjing Med University. 2020;40(10):1560–1564.
23. Yu Y, Liu Q, Li H, Wen C, He Z. Alterations of the Gut Microbiome Associated With the Treatment of Hyperuricaemia in Male Rats. Front Microbiol. 2018;9:2233. doi:10.3389/fmicb.2018.02233
24. Cani PD, Jordan BF. Gut microbiota-mediated inflammation in obesity: a link with gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2018;15(11):671–682. doi:10.1038/s41575-018-0025-6
25. Wang Y, Dilidaxi D, Wu Y, Sailike J, Sun X, Nabi XH. Composite probiotics alleviate type 2 diabetes by regulating intestinal microbiota and inducing GLP-1 secretion in db/db mice. Biomed Pharmacother. 2020;125:109914. doi:10.1016/j.biopha.2020.109914
26. Wolter M, Grant ET, Boudaud M, et al. Leveraging diet to engineer the gut microbiome. Nat Rev Gastroenterol Hepatol. 2021;18(12):885–902. doi:10.1038/s41575-021-00512-7
27. Weersma RK, Zhernakova A, Fu J. Interaction between drugs and the gut microbiome. Gut. 2020;69(8):1510–1519. doi:10.1136/gutjnl-2019-320204
28. Ahlawat S. Gut-organ axis: a microbial outreach and networking. Lett Appl Microbiol. 2021;72(6):636–668. doi:10.1111/lam.13333
29. Zhang Q, Zhang Y, Zeng L, et al. The Role of Gut Microbiota and Microbiota-Related Serum Metabolites in the Progression of Diabetic Kidney Disease. Front Pharmacol. 2021;12:757508. doi:10.3389/fphar.2021.757508
30. Zixin Y, Lulu C, Xiangchang Z, et al. TMAO as a potential biomarker and therapeutic target for chronic kidney disease: a review. Front Pharmacol. 2022;13:929262. doi:10.3389/fphar.2022.929262
31. Su HY, Yang C, Liang D, Liu HF. Research Advances in the Mechanisms of Hyperuricemia-Induced Renal Injury. Biomed Res Int. 2020;2020:5817348. doi:10.1155/2020/5817348
32. Marangella M. Uric acid elimination in the urine. Pathophysiological implications. Contrib Nephrol. 2005;147:132–148. doi:10.1159/000082551
33. Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32(17):2336–2347. doi:10.1038/emboj.2013.171
34. Hasegawa J, Maejima I, Iwamoto R, Yoshimori T. Selective autophagy: lysophagy. Methods. 2015;75:128–132. doi:10.1016/j.ymeth.2014.12.014
35. Chaudhary K, Malhotra K, Sowers J, Aroor A. Uric Acid - key ingredient in the recipe for cardiorenal metabolic syndrome. Cardiorenal Med. 2013;3(3):208–220. doi:10.1159/000355405
36. Stellato D, Morrone LF, Di Giorgio C, Gesualdo L. Uric acid: a starring role in the intricate scenario of metabolic syndrome with cardio-renal damage? Intern Emerg Med. 2012;7(1):5–8. doi:10.1007/s11739-011-0642-3
37. Cristobal-Garcia M, Garcia-Arroyo FE, Tapia E, et al. Renal oxidative stress induced by long-term hyperuricemia alters mitochondrial function and maintains systemic hypertension. Oxid Med Cell Longev. 2015;2015:535686. doi:10.1155/2015/535686
38. Song X, Sun Z, Chen G, et al. Matrix stiffening induces endothelial dysfunction via the TRPV4/microRNA-6740/endothelin-1 mechanotransduction pathway. Acta Biomater. 2019;100:52–60. doi:10.1016/j.actbio.2019.10.013
39. Ndrepepa G. Uric acid and cardiovascular disease. Clin Chim Acta. 2018;484:150–163. doi:10.1016/j.cca.2018.05.046
40. Glantzounis GK, Tsimoyiannis EC, Kappas AM, Galaris DA. Uric acid and oxidative stress. Curr Pharm Des. 2005;11(32):4145–4151. doi:10.2174/138161205774913255
41. Kanellis J, Watanabe S, Li JH, et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension. 2003;41(6):1287–1293. doi:10.1161/01.HYP.0000072820.07472.3B
42. Rabadi MM, Kuo MC, Ghaly T, et al. Interaction between uric acid and HMGB1 translocation and release from endothelial cells. Am J Physiol Renal Physiol. 2012;302(6):F730–41. doi:10.1152/ajprenal.00520.2011
43. Choe JY, Choi CH, Park KY, Kim SK. High-mobility group box 1 is responsible for monosodium urate crystal-induced inflammation in human U937 macrophages. Biochem Biophys Res Commun. 2018;503(4):3248–3255. doi:10.1016/j.bbrc.2018.08.139
44. Cai W, Duan XM, Liu Y, et al. Uric Acid Induces Endothelial Dysfunction by Activating the HMGB1/RAGE Signaling Pathway. Biomed Res Int. 2017;2017:4391920. doi:10.1155/2017/4391920
45. Khosla UM, Zharikov S, Finch JL, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005;67(5):1739–1742. doi:10.1111/j.1523-1755.2005.00273.x
46. Shi L, Wang Q, Zhang L. Effects of hyperuricemia on early diabetic nephropathy in elderly patients. Chine J Gerontol. 2009;29(22):2862–2864.
47. Chen X, Luo S, Liao S. Study on the relationship between reduced glucose tolerance and blood lipid and uric acid levels. Chine J Med Med. 2006;8(005):696–697.
48. Mauer M, Doria A. Uric acid and risk of diabetic kidney disease. J Nephrol. 2020;33(5):995–999. doi:10.1007/s40620-020-00796-z
49. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7(3):189–200. doi:10.1080/19490976.2015.1134082
50. He J, Zhang P, Shen L, et al. Short-Chain Fatty Acids and Their Association with Signalling Pathways in Inflammation, Glucose and Lipid Metabolism. Int J Mol Sci. 2020;21(17). doi:10.3390/ijms21176356
51. Croci S, D’Apolito LI, Gasperi V, Catani MV, Savini I. Dietary Strategies for Management of Metabolic Syndrome: role of Gut Microbiota Metabolites. Nutrients. 2021;13(5). doi:10.3390/nu13051389
52. Gatarek P, Kaluzna-Czaplinska J. Trimethylamine N-oxide (TMAO) in human health. EXCLI J. 2021;20:301–319. doi:10.17179/excli2020-3239
53. Cho CE, Caudill MA. Trimethylamine-N-Oxide: friend, Foe, or Simply Caught in the Cross-Fire? Trends Endocrinol Metab. 2017;28(2):121–130. doi:10.1016/j.tem.2016.10.005
54. Tang WH, Wang Z, Kennedy DJ, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116(3):448–455. doi:10.1161/CIRCRESAHA.116.305360
55. Sun G, Yin Z, Liu N, et al. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem Biophys Res Commun. 2017;493(2):964–970. doi:10.1016/j.bbrc.2017.09.108
56. Li T, Gua C, Wu B, Chen Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem Biophys Res Commun. 2018;495(2):2071–2077. doi:10.1016/j.bbrc.2017.12.069
57. Fang Q, Zheng B, Liu N, et al. Trimethylamine N-Oxide Exacerbates Renal Inflammation and Fibrosis in Rats With Diabetic Kidney Disease. Front Physiol. 2021;12:682482. doi:10.3389/fphys.2021.682482
58. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J Am Heart Assoc. 2017;6(9). doi:10.1161/JAHA.117.006347
59 Caruso I, Giorgino F. SGLT-2 inhibitors as cardio-renal protective agents. Metabolism. 2022;127:154937. doi:10.1016/j.metabol.2021.154937.
60. Garla VV, Butler J, Lien LF. SGLT-2 Inhibitors in heart failure: guide for prescribing and future perspectives. Curr Cardiol Rep. 2021;23(6):59. doi:10.1007/s11886-021-01486-3
61. Grubic Rotkvic P, Cigrovski Berkovic M, Bulj N, Rotkvic L, Celap I. Sodium-glucose cotransporter 2 inhibitors’ mechanisms of action in heart failure. World J Diabetes. 2020;11(7):269–279. doi:10.4239/wjd.v11.i7.269
62. Novikov A, Fu Y, Huang W, et al. SGLT2 inhibition and renal urate excretion: role of luminal glucose, GLUT9, and URAT1. Am J Physiol Renal Physiol. 2019;316(1):F173–F85. doi:10.1152/ajprenal.00462.2018
63. Ahmed MI, Gladden JD, Litovsky SH, et al. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction >60%. J Am Coll Cardiol. 2010;55(7):671–679. doi:10.1016/j.jacc.2009.08.074
64. Neuen BL, Young T, Heerspink HJL, et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019;7(11):845–854. doi:10.1016/S2213-8587(19)30256-6
65. Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117(18):2340–2350. doi:10.1161/CIRCULATIONAHA.107.739938
66. Verge D, Lopez X. Impact of GLP-1 and GLP-1 receptor agonists on cardiovascular risk factors in type 2 diabetes. Curr Diabetes Rev. 2010;6(4):191–200. doi:10.2174/157339910791658853
67. Thomas MC. The potential and pitfalls of GLP-1 receptor agonists for renal protection in type 2 diabetes. Diabetes Metab. 2017;43(Suppl 1):2S20–2S7. doi:10.1016/S1262-3636(17)30069-1
68. Roscioni SS, Heerspink HJ, de Zeeuw D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nat Rev Nephrol. 2014;10(2):77–87. doi:10.1038/nrneph.2013.251
69. Greco EV, Russo G, Giandalia A, Viazzi F, Pontremoli R, De Cosmo S. GLP-1 Receptor Agonists and Kidney Protection. Medicina. 2019;55(6). doi:10.3390/medicina55060233
70. Thornberry NA, Gallwitz B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best Pract Res Clin Endocrinol Metab. 2009;23(4):479–486. doi:10.1016/j.beem.2009.03.004
71. Hasan AA, Hocher B. Role of soluble and membrane-bound dipeptidyl peptidase-4 in diabetic nephropathy. J Mol Endocrinol. 2017;59(1):R1–R10. doi:10.1530/JME-17-0005
72. von Websky K, Reichetzeder C, Hocher B. Physiology and pathophysiology of incretins in the kidney. Curr Opin Nephrol Hypertens. 2014;23(1):54–60. doi:10.1097/01.mnh.0000437542.77175.a0
73. Muskiet MHA, Tonneijck L, Smits MM, et al. GLP-1 and the kidney: from physiology to pharmacology and outcomes in diabetes. Nat Rev Nephrol. 2017;13(10):605–628. doi:10.1038/nrneph.2017.123
74. Schernthaner G, Mogensen CE, Schernthaner GH. The effects of GLP-1 analogues, DPP-4 inhibitors and SGLT2 inhibitors on the renal system. Diab Vasc Dis Res. 2014;11(5):306–323. doi:10.1177/1479164114542802
75. Hasan AA, von Websky K, Reichetzeder C, et al. Mechanisms of GLP-1 receptor-independent renoprotective effects of the dipeptidyl peptidase type 4 inhibitor linagliptin in GLP-1 receptor knockout mice with 5/6 nephrectomy. Kidney Int. 2019;95(6):1373–1388. doi:10.1016/j.kint.2019.01.010
76. Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414(1):1–18. doi:10.1042/BJ20080595
77. Kolkhof P, Joseph A, Kintscher U. Nonsteroidal mineralocorticoid receptor antagonism for cardiovascular and renal disorders - New perspectives for combination therapy. Pharmacol Res. 2021;172:105859. doi:10.1016/j.phrs.2021.105859
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Plasma Asprosin Concentrations are Associated with Progression of Diabetic Kidney Disease
Xu M, Zhang C, Zhang L, Qu H, Wang Y
Diabetes, Metabolic Syndrome and Obesity 2024, 17:2235-2242
Published Date: 5 June 2024
Integrated Data Mining and Animal Experiments to Investigate the Efficacy and Potential Pharmacological Mechanism of a Traditional Tibetan Functional Food Terminalia chebula Retz. in Hyperuricemia

Liu W, Zhang M, Tan J, Liu H, Wang L, Liao J, Huang D, Jie W, Jin X
Journal of Inflammation Research 2024, 17:11111-11128
Published Date: 17 December 2024
Neutrophil Extracellular Traps in Diabetic Kidney Disease: Mechanisms of Pathogenesis and Emerging Therapeutic Strategies
Wang B, Zhang R, Liu X, Shang Y, Jin T, Gao C, Yang N, Jin J, He Q
Drug Design, Development and Therapy 2026, 20:583077
Published Date: 21 February 2026
Traditional Chinese Medicine in Hyperuricemia: Current Advances, Mechanistic Insights, and Clinical Challenges
Lei S, Gong X, Du Y, Liu R, Liu Y
International Journal of General Medicine 2026, 19:619201
Published Date: 14 July 2026