Back to Journals » The Application of Clinical Genetics » Volume 13
Effect of Genetic Testing on Diagnosing Gastrointestinal Pediatric Patients with Previously Undiagnosed Diseases
Authors Altamimi E, Khanfar M ![]() , Rabab'h O
, Rabab'h O ![]() , Dardas Z
, Dardas Z ![]() , Srour L, Mustafa L, Azab B
, Srour L, Mustafa L, Azab B ![]()
Received 9 August 2020
Accepted for publication 10 November 2020
Published 16 December 2020 Volume 2020:13 Pages 221—231
DOI https://doi.org/10.2147/TACG.S275992
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Supplementary video 1 of "Genetic testing on gastrointestinal pediatric patients' [ID 275992].
Views: 238
Eyad Altamimi,1 Mariam Khanfar,2 Omar Rabab’h,3 Zain Dardas,4,5 Luma Srour,4 Lina Mustafa,4 Bilal Azab4,6
1Department of Pediatrics and Neonatology, Jordan University of Science and Technology, Irbid, Jordan; 2Department of Medical Laboratory Sciences, Jordan University of Science and Technology, Irbid, Jordan; 3Center of Cognition and Neuroethics, University of Michigan-Flint, Flint, MI, USA; 4Department of Pathology, Microbiology and Forensic Medicine, School of Medicine, University of Jordan, Amman, Jordan; 5Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA; 6Human and Molecular Genetics, Medical College of Virginia, Virginia Commonwealth University, Richmond, VA, USA
Correspondence: Eyad Altamimi
Department of Pediatrics and Neonatology, Jordan University of Science and Technology, PO Box 3030, Irbid 22110, Jordan
Tel +962 797464254
Email [email protected]
Bilal Azab
Department of Pathology, Microbiology and Forensic Medicine, School of Medicine, University of Jordan, Amman, Jordan
Email [email protected]
Purpose: Four consanguineous Jordanian families with affected members of unknown gastrointestinal related diseases were recruited to assess the utility and efficiency of whole exome sequencing (WES) in reaching the definitive diagnosis.
Patients and Methods: Members from four consanguineous Jordanian families were recruited in this study. Laboratory and imaging tests were used for initial diagnosis, followed by performing WES to test all affected members for the detection of causative variants. Sanger sequencing was used for validation.
Results: We had a 100% success rate identifying each case presented in this study.
Conclusion: This is the first study applying a WES testing approach in the diagnosis of pediatric diseases in Jordan. Our results strongly suggest the need to implement WES as an evident diagnostic tool in the clinical setting, as it will subsequently allow for proper disease management and genetic counseling.
Keywords: pediatric diseases, whole exome sequencing, gastrointestinal diseases, metabolic diseases
Introduction
Pediatric diseases of genetic origin are a group of broad, overlapping conditions that often involve multiple organs. Due to the overlapping symptoms, many children remain undiagnosed and face repeated admissions and prolonged hospital stay.1,2 Although many of those diseases are associated with poor prognosis and even premature deaths, some are compatible with a good quality of life if they are diagnosed early and offered optimal management.3 Diagnostic approaches have often involved standard laboratory, imaging studies, and even more sophisticated interventions that can be invasive, time consuming, and costly. Hence, the need to diagnose such disorders at the molecular level seems most promising. The emergence of whole exome sequencing (WES) gave a momentum chance to look at difficult to diagnose diseases from a different perspective through an individual’s genetic makeup. The first successful use of WES in medical diagnosis was through the identification of the causative variant responsible for a rare form of inflammatory bowel disease in an infant.4 This allowed the extensive use of WES in diagnostic settings where it has proved its success.5–7
In this study, members from four consanguineous Jordanian families were recruited to test the utility of WES in detecting the causative genetic variant responsible for each case.
Patients and Methods
Subject Recruitment and Clinical Evaluation Phase
This study was approved by the institutional review board (IRB) of the Faculty of Medicine and the Research Committee at Jordan University of Science and Technology (JUST), in accordance with the recommendations of the declaration of Helsinki. Written informed consent was obtained from each participant (the legal guardian in the case of the children). Four consanguineous families with children having rare undiagnosed gastrointestinal related diseases (Figure 1) were recruited into this study after rigorous evaluation and consultation between a specialized pediatric gastroenterologist and geneticist. Sample collection took place at the pediatric clinics in King Abdullah University Hospital, Irbid, Jordan. Clinical data and history details were collected, followed by a thorough clinical examination. Standard laboratory testing in addition to any required imaging studies took place for each participant family.
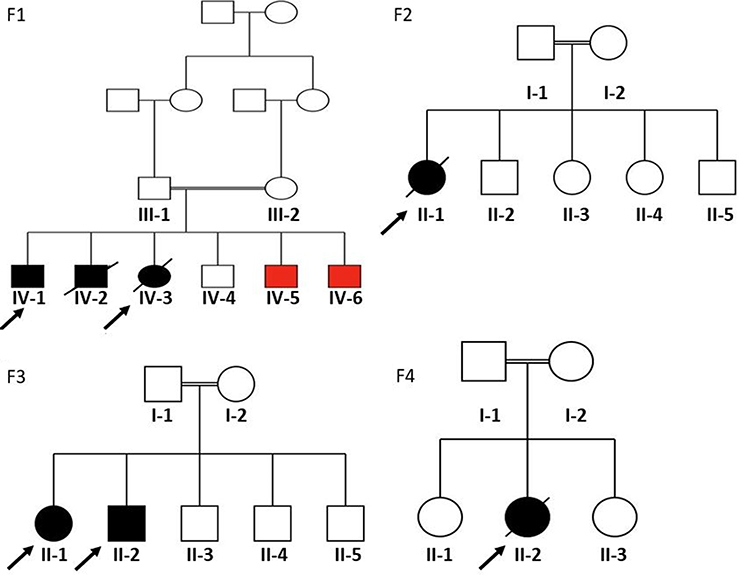 |
Figure 1 Pedigrees of the four participating families. F1 refers to family #1; F2, family #2; F3, family #3, and F4 refers to family #4. Squares and circles indicate male and female members, respectively. Black filled symbols represent affected individuals of relevance in this study, red filled symbols represent affected siblings with different disease, empty symbols for unaffected individuals, crossed symbols for deceased individuals, double lines represent consanguinity, and finally, arrows refer to probands in each case. |
Sample Collection and DNA Testing Phase
Blood samples were collected from affected individuals and their unaffected relatives in the family in EDTA tubes. The DNA was isolated using the QIAprep Spin Mini-prep Kit according to the manufacturer’s instructions. Genomic purity and quality were assessed using nanodrop and gel electrophoresis prior to performing molecular testing.
Whole Exome Sequencing and Analysis
DNA preparation for WES, variant detection, filtration, validation, and segregation analysis were performed as described by Azab et al.8
Results
Available clinical findings of the proband patients recruited in this study are available in Table 1. Identified pathogenic variants for each family can be found in Table 2.
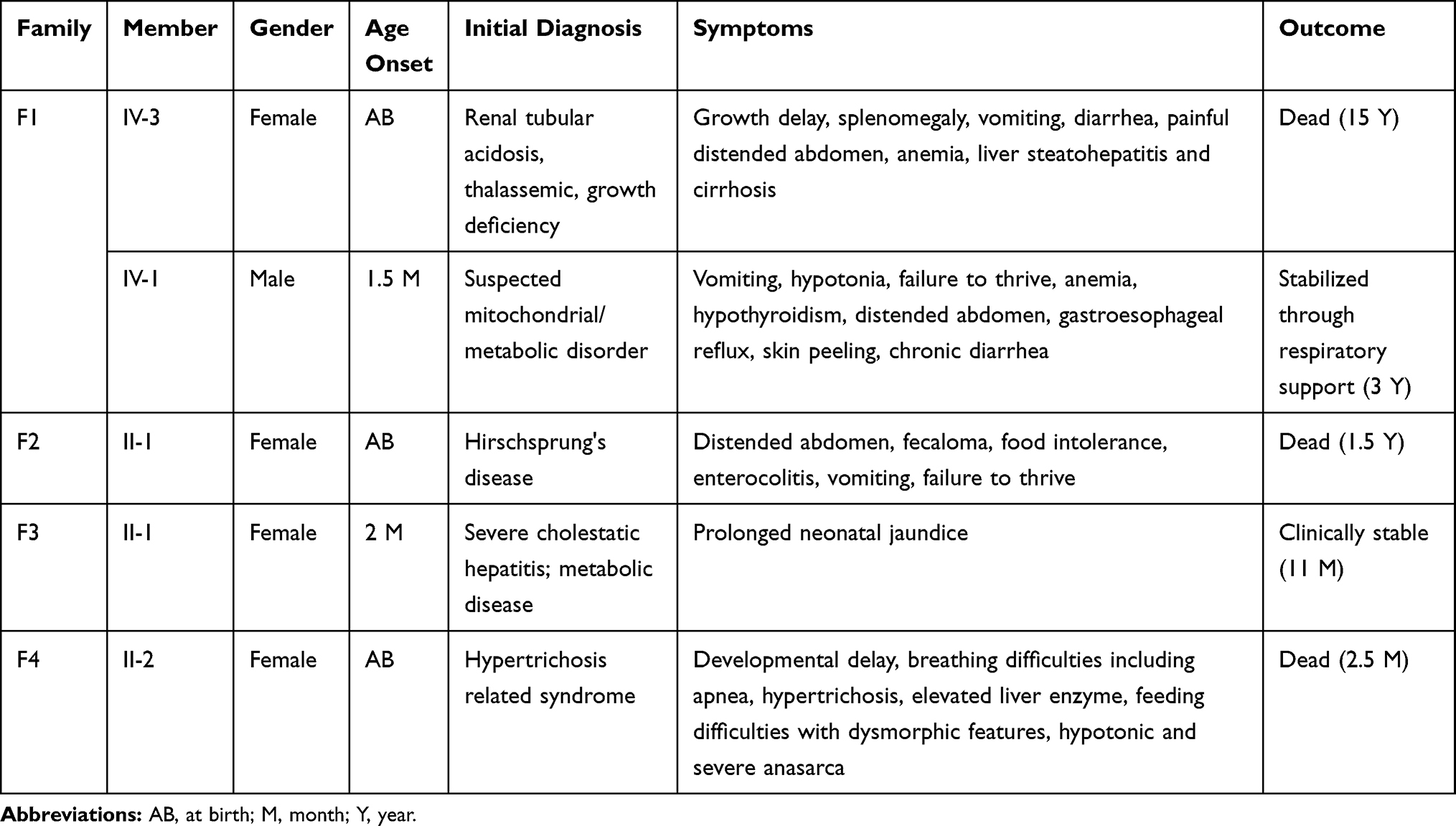 |
Table 1 Clinical Data for Participated Probands in Each Family |
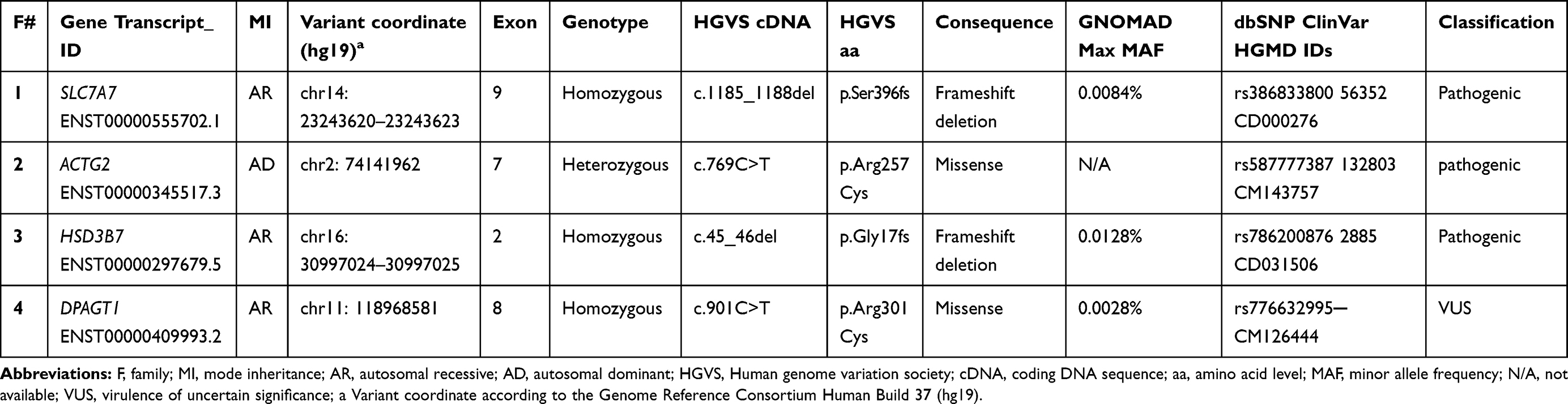 |
Table 2 Identified Variants in Each Participating Family |
Clinical Assessment and Variant Detection in Family F1
Family F1 had a reported family history of neurodevelopmental delay, kidney problems, and early neonatal deaths. The 13-year-old girl (proband IV-3), labeled as a case of renal tubular acidosis and thalassemia intermedia, was referred due to repetitive bouts of vomiting, hematemesis, and chronic non-bloody diarrhea. Upon examination, the petite girl, with growth parameters far below the third centile for age, had hepatosplenomegaly, with significant abdominal distention. Her work-up revealed abnormal non-specific amino acid levels, anemia, elevated ferritin levels, elevated liver enzymes, prolongation of prothrombin time (PT), and international normalized ratio (INR). Her abdominal ultrasound confirmed hepatosplenomegaly with no features of portal hypertension and her upper endoscopy was normal. Her liver biopsy unexpectedly showed steatohepatitis with evolving liver cirrhosis (Figure 2). The patient underwent splenectomy due to hypersplenism, which partially corrected her hematological abnormalities. Her serum ammonia level was elevated, but amino acid chromatography and organic acid abnormalities were thought to reflect renal dysfunction and chronic liver disease. The patient stayed in hospital for almost a year due to recurrent symptoms of diarrhea, vomiting, abdominal pain, respiratory infections, etc., with no settled diagnosis. During her hospitalization, her younger brother (proband IV-1) was admitted to the hospital at the age of 1.5 months with persistent vomiting, hypotonia, respiratory distress, and poor weight gain. Persistent diarrhea developed with no improvement even on amino acid formula. He had anemia, mildly elevated liver enzymes, and thrombocytopenia. His metabolic work-up showed hyperammonemia, significant hyperferritinemia, and non-diagnostic amino and organic profile. WES seemed to be the proper course of action for both siblings. Both probands’ exome sequencing identified a homozygous variant in the SLC7A7 gene (Figure 3, Table 2), which is responsible for lysinuric protein intolerance (LPI). Retrospective analysis of metabolic work-up identified supporting evidence of the LPI diagnosis (Table 3).
 |
Table 3 Laboratory Tests for Both Patients in Family F1 |
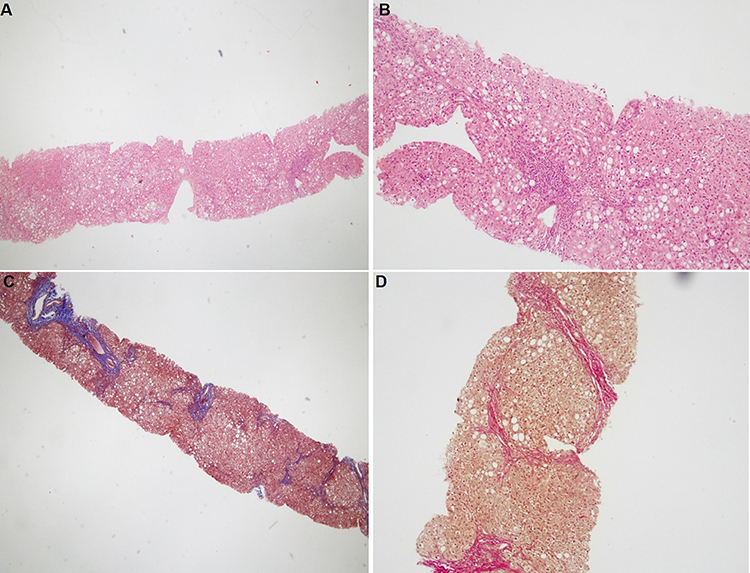 |
Figure 2 Histopathological findings for patient IV-3 in family F1. (A and B) Hematoxylin and eosin staining shows steatosis, hepatocytes ballooning, and portal lymphocytic infiltration. (C and D) Masson’s trichrome and Van Gieson’s stain, respectfully, showing bridging fibrosis partially forming half nodules. Magnification: 40× (A and C) and 100× (B and D). |
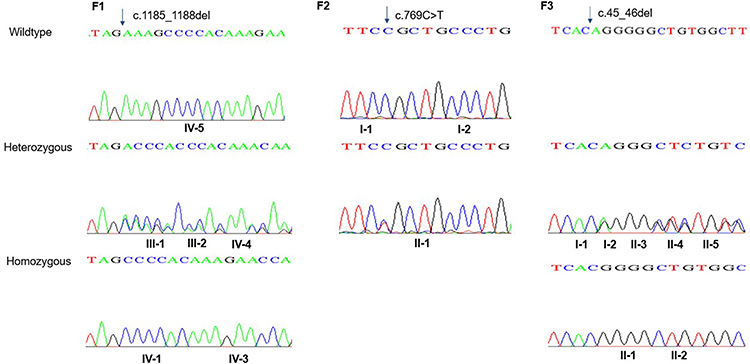 |
Figure 3 Sanger sequence chromatography for family F1 (left panel), family F2 (middle panel), and family F3 (right panel). In F1: the two probands (IV-1 and IV-3) have a pathogenic homozygous variant in SLC7A7 gene. Heterozygous carriers in the family are shown in middle part, while homozygous for the wild type allele is shown at the top. In F2 (middle panel): proband II-1 carries a de novo heterozygous variant in ACTG2 gene; whilst her parents carry the wild type homozygous allele. In the right panel, family F3, the two probands II-1 and II-2 have a homozygous deletion in HSD3B7 gene. While the parents along their children have the heterozygous allele. |
Clinical Assessment and Variant Detection in Family F2
A one-year-old female (proband II-1), born to consanguineous parents, started showing symptoms soon after birth, such as difficulty in passing stool associated with increasing abdominal distention. At five-months-old, she was presented with a picture of intestinal obstruction. Her thyroid function and electrolytes were normal. The patient was labeled as Hirschsprung’s disease and underwent colostomy creation and appendectomy. Soon after surgery her symptoms recurred with more distention and infrequent stooling. The patient underwent diagnostic laparotomy which showed stool impaction in the proximal and distal loop of colostomy and megacolon. The bowel was evacuated, multiple biopsies were taken, and an ileostomy was created following closing colostomy. Her intestinal pathology confirmed the presence of ganglions at the rectum, which excluded Hirschsprung’s disease (Figure 4). However, even with the ileostomy, the patient continued to have feeding intolerance and severe distention. This raised the possibility of dealing with chronic intestinal pseudo-obstruction (CIPO). WES analysis was later performed, and the results showed a heterozygous variant in the ACTG2 gene (Figure 3, Table 2).
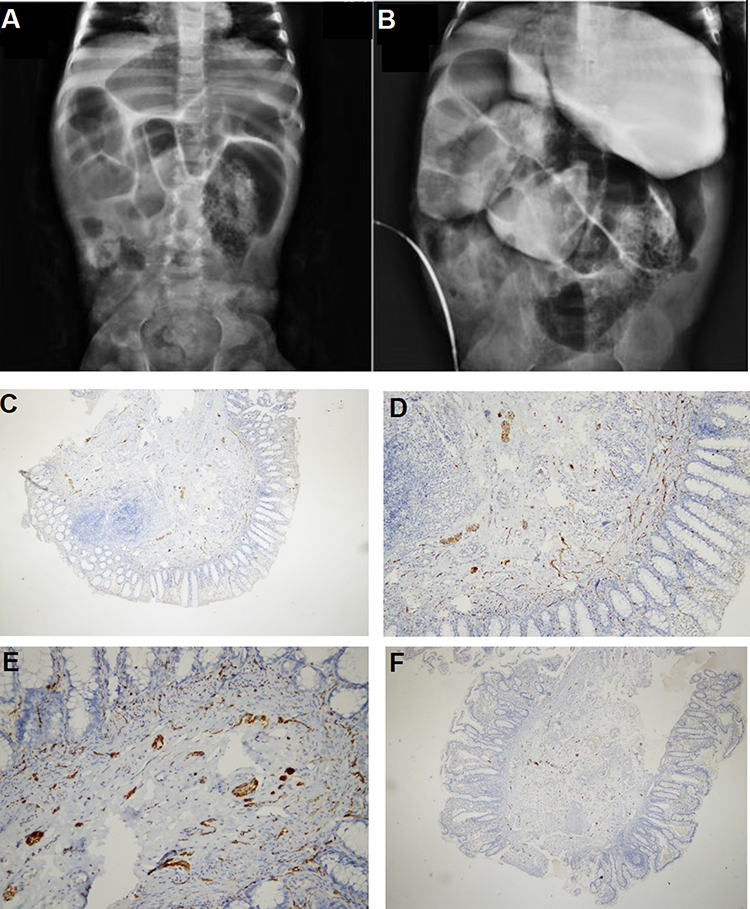 |
Figure 4 Imaging tests for proband II-1 in family F2. (A and B) Abdominal X-ray and barium study showing severe dilated bowel loops. (C and D) Calretinin staining 2 cm away from the anal verge, 4 cm from the anal verge (E), and from the ileostomy site (F). Ganglion cells are present (magnification: 40×, 100×, 200×, and 40× for C, D, E, and F, respectively). |
Clinical Assessment and Variant Detection in Family F3
A two-month-old baby (proband II-1), born to consanguineous parents, suffered from prolonged neonatal jaundice. Her family history showed early neonatal deaths, and her older brother (proband II-2) is diagnosed with cholestatic liver disease, which started in early childhood (now aged 26 years old). Her work-up was normal except for elevated liver enzymes (AST, ALT, ALP, but normal levels of GGT), and prolonged PTT and PT (Table 4). The patient’s liver ultrasound was normal, and her hepatobiliary iminodiacetic acid (HIDA) scan showed no evidence of biliary atresia (Supplementary Material Text S1, Video S1 and Video S2). Thus, a liver biopsy was required for further testing, and the H&E staining revealed severe cholestatic hepatitis (Figure 5). Clinicians then decided to test both probands for urine bile salts through fast atom bombardment mass spectrometry (FAB-MS) (Table 5). Later, the two were recruited for WES analysis. The results showed a homozygous variant in the HSD3B7 gene (Figure 3, Table 2). Subjects with this type of variant fail to synthesize bile acids and end up developing a form of progressive liver disease (cholestatic).
 |
Table 4 Liver Function Tests and Blood Coagulation Tests Conforming Liver Damage in Proband II-1 from Family F3 |
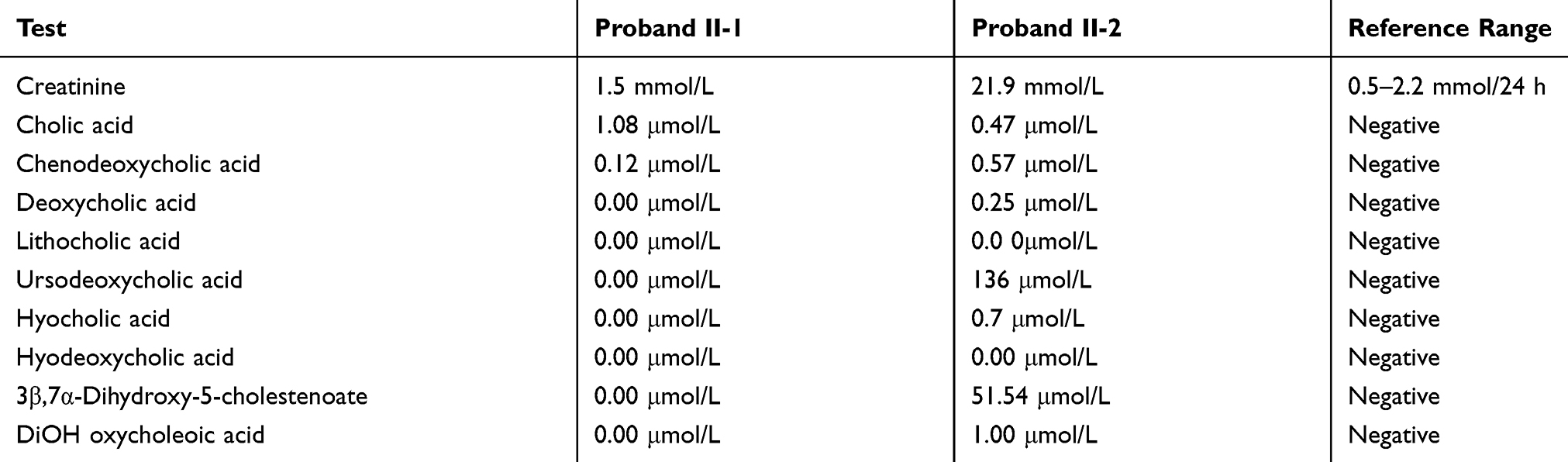 |
Table 5 FAB-MS Test Results for Both Probands in Family F3 Indicate the Presence of Peaks Characteristic of 3-β-HSD Deficiency |
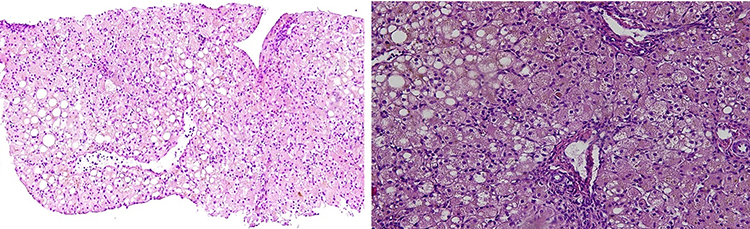 |
Figure 5 Histopathological findings for patient II-1 in family F3. Hematoxylin and eosin staining showing hepatocytes ballooning and feathery changes with marked intrahepatic cholestasis. Mild periportal inflammation and foci of spotty necrosis are also present (magnification: 100× and 200× left to right, respectively). |
Clinical Assessment and Variant Detection in Family F4
A one-month-old female (proband II-2), born to consanguineous parents, was admitted to the NICU for two weeks on respiratory and nutritional support. A week following her discharge, she was presented to the emergency room due to difficulty in breathing and poor feeding, and she had required readmission to the pediatric ICU. Oral feeding could not be established due to poor sucking; therefore, she was fed through a nasogastric tube. The patient had several symptoms that are shown in Table 1. Her work-up included a normal metabolic profile, karyotyping, echocardiogram, and abdominal ultrasound. Her head ultrasound showed a thin corpus callosum and a brain MRI then followed to confirm the finding. The MRI result was normal; hence, congenital brain abnormalities were excluded in the diagnosis. This raised suspicion of dealing with an unknown genetic disease, and thus the patient was a candidate for WES. The analysis showed a missense variant in the DPAGT1 gene (Table 2). The proband’s MRI findings and characteristic features can be seen in Figure 6.
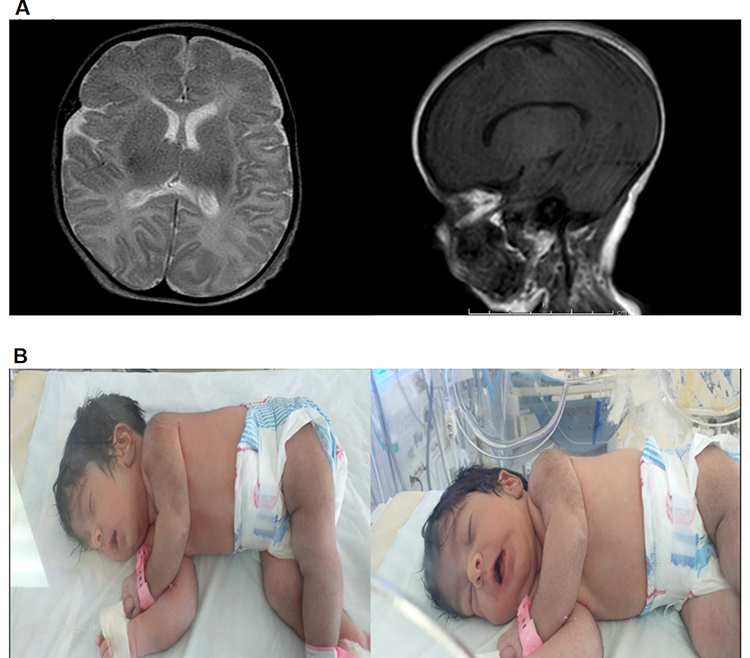 |
Figure 6 MRI and clinical features of proband II-2 in family F4. (A) Brain MRI; myelination is appropriate for proband’s age. Both thalami, both basal ganglia, corpus callosum, brainstem, cerebellum, and ventricular system appear normal. Central midline structures with no mass effect, no hemorrhage or collection seen. (B) Patient’s characteristic features; soft long ears, U-shaped vermilion of the upper lip, thick skin, hypertrichosis, moderate multiple contractures, hypotonia, and severe hypokinesia. |
Discussion
The completion of the human reference genome in 2003, along with the availability of large databases of known single nucleotide polymorphisms (SNPs) and known pathogenic variants, changed our understanding of genetic variations and their relevance to health and disease.9 As a result, the use of next generation sequencing (NGS) platforms, and WES in particular, has been extensively incorporated in the diagnosis of rare, novel, and hard to diagnose diseases.10 Over the past decade, multiple platforms have been introduced to the diagnostic field of genetics, providing increased efficiency and reduced cost. These advances enabled clinicians to widely use WES in order to reach causative variants in multiple diseases, and, consequently, provide proper treatment.9
In 2018, Wojcik et al highlighted the need to improve the genetic diagnostic evaluation for infants suspected of genetic diseases.1 As their study proved that WES or whole genome sequencing (WGS) have only been performed after traditional testing consisting of cytogenetic and molecular tests, biochemical testing, enzyme analysis, and tissue biopsies, all reported as negative. The extremely low usage rate of WES and WGS is an indication of missed opportunities children suffer in their odyssey of getting a proper diagnosis.
Herein, we evaluated the utility of WES in diagnosing challenging cases of five members in four consanguineous Jordanian families, where traditional laboratory and clinical testing failed to accurately reach a definitive diagnosis. We successfully reached a 100% diagnostic rate for each family and were able to detect the causative variant responsible for each case (Table 2).
In family F1, two siblings of consanguineous parents displaying complex, multisystem manifestations raised the suspicion of inborn error of metabolism (IEM).11 Diagnosis of IEM represents a real challenge considering the overlapping clinical presentations of multisystem disorders and limitations of specific biomarkers. In some cases, the metabolic changes may be transient and affected by substrate intake.12 In family F1, this was further complicated by other comorbidities; such as being a thalassemia carrier, where organomegaly and hematological abnormalities were thought to be secondary, and renal tubular defect, which was blamed for the aminoaciduria proband IV-3 manifested. Both siblings’ exome sequencing identified a homozygous variant in the SLC7A7 gene, and this variant is one of the 60 already reported variants in this gene causing LPI, an inherited genetic metabolic disorder caused by defective cationic amino acid (CAA) transport at the basolateral membrane of epithelial cells in the kidney and intestines.13,14 Functional studies on this variant confirmed failing to induce the CAA transport.15 Considering the non-diagnostic biochemical findings in our patients, WES settled the diagnosis for this family.
The delayed diagnosis and utility of WES in diagnosing LPI was previously reported; Posey et al reported a five-year-old boy with osteoporosis and recurrent fracture that followed for three years before the diagnosis of LPI was confirmed by WES.16 Another report by Cimbalistienè et al described a 17-year delay in diagnosing LPI in a child with hepatosplenomegaly, recurrent episodes of drowsiness, and osteoporosis. WES confirmed the diagnosis.17
We believe that without WES the two patients would have continued to be misdiagnosed. Following WES results, better management was provided to the two patients, and they were put on L-carnitine and special formula. Unfortunately, at the age of 15 years, proband IV-3 developed a picture of intestinal obstruction and passed away as a complication of her illness. Meanwhile, her brother is stabilized on oxygen support and regular citrulline supplement.
In family F2 (proband II-1), the infantile presentation of progressive intestinal obstruction suggested congenital ganglionic colon. Presence of colonic ganglions and the small intestinal nature of involvement suggested CIPO. Although isolated hypoganglionosis has been reported to cause CIPO, however, the diagnosis of hypoganglionosis can be misleading. Moreover, no unbiased genetic study was used in these diseases to determine if there is an underlying genetic cause. Many cases of CIPO are caused by well characterized variants, so we decided to perform WES. The analysis matched what Ravenscroft et al have previously reported in their study; a de novo heterozygous variant in the ACTG2 gene that is associated with CIPO.18 ACTG2 dysfunction is associated with visceral myopathy with bladder and intestinal involvement.19 Our patient manifested only intestinal involvement by recurrent intestinal obstruction in the absence of an organic blockage. Unfortunately, the proband’s status worsened, as she suffered multiple septic episodes related to her central line and died aged 16.5 months.
In the case of family F3, cholestasis is marked as defective bile secretion with build-up levels of bile salts in the body. A significant portion of infantile cholestasis disorders are secondary to genetic defects,20 with limited biomarkers and non-diagnostic histological features. The availability of WES can test rapidly, accurately, and cost-effectively for a large number of genetic causes.21
Furthermore, WES is now proposed in diagnostic algorithms of neonatal cholestasis. Nicastro et al reported a genetic testing detection rate of 60% in their infantile cholestatic cohort.22,23
Considering that infantile cholestasis represents a heterogeneous hereditary group and owing to the history of hepatic diseases and early neonatal deaths in family F3, the use of WES analysis was prompted to identify the genetic cause. WES results showed a homozygous variant in the HSD3B7 gene in both siblings. Subjects with this type of variant produce abnormal bile acids that fail to leave the liver and end up developing progressive liver disease and even liver cirrhosis and failure.24,25 Untreated patients eventually require liver transplant.25 Proband II-1 is now 11 months old, maintained on the second-choice medication (ursodeoxycholic acid), waiting for the first-choice drug (cholic acid) to be available. It is worthy to mention that her successful diagnosis could have been a blessing had her older brother, proband II-2, been properly diagnosed at the beginning of his onset. Even though he is alive now, he suffers from liver cirrhosis which could have been preventable with the use of proper medication earlier.
In family F4, whole exome analysis of proband II-2 showed a missense variant in the DPAGT1 gene. Pathogenic variants in DPAGT1 are known to manifest two distinct phenotypes, either limb-girdle congenital myasthenic syndrome (CMS) with tubular aggregates, or congenital disorder of glycosylation (DPAGT1-CDG).26 Interestingly, in this study, our proband shared the same homozygous missense variant and diagnosis that has been reported only once before; where a Spanish non-consanguineous family had a baby boy with similar clinical features and prognosis as proband II-2.27 Although this variant has only been reported once before, we believe it harbors a deleterious outcome. Imtiaz et al previously reported a novel missense variant in the same amino acid position (Arg 301), this residue is highly conserved among 42 different species, and no change over this residue has ever been found in healthy controls.28 Moreover, prediction tools such as SIFT, Polyphen 2, and MutationTaster gave predicted outcome of deleterious, probably damaging, and disease causing, respectively, as this variant seems to likely affect the protein misfolding. We believe that our patient presents further clinical evidence that this homozygous variant is the candidate disease causing variant. Both infants in the Spanish case report and in our study passed away aged 1.5 months and 2.5 months, respectfully, due to multiorgan failure.
In this study, all patients were successfully diagnosed following failed attempts due to the nonspecific and overlapping symptoms in each case. Of worthy notice, those patients were brought to our attention due to their gastrointestinal related problems, which led to primary assumption of dealing with GI diseases. However, through WES we managed to identify two families suffering from metabolic disorders with multisystem involvement. This is an achievement that would not have been possible without the use of WES. Furthermore, we managed to provide better management of disease through the right administration of proper medication in proband II-1 of family F3. And although three of the patients passed away while waiting for the WES results, better awareness and genetic counseling have been provided to their families.29
Conclusion
Children with suspected genetic conditions have a great chance of getting properly diagnosed and managed through WES, as it has proved its efficiency and great diagnostic yield. Moreover, WES cost-effectiveness is maximized by early application in the diagnostic process. Therefore, pediatricians are highly encouraged to consider early referral of children with undiagnosed syndromes to clinical geneticists.30
Abbreviations
CAA, cationic amino acid; CDG, congenital disorders of glycosylation; CIPO, chronic intestinal pseudo-obstruction; IEM, inborn error of metabolism; IRB, Institutional Review Boards; LPI, lysinuric protein intolerance; NGS, next generation sequencing; SNP, single nucleotide polymorphisms; WES, whole exome sequencing; WGS, whole genome sequencing.
Data Sharing Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material. For additional inquiries, please refer to the corresponding author.
Ethics Approval and Informed Consent
The study was approved by the institutional review board of the Faculty of Medicine and the Research Committee at Jordan University of Science and Technology (JUST). Written informed consent to participate in this study and to publish each case's details was provided by the participants (the legal guardian in case of the children). Additionally, the parents of the proband in family F4 have provided their written informed consent to publish the patient’s images and videos.
Consent for Publication
Consent for publishing data in this study was obtained from each participating family.
Acknowledgments
The authors thank the patients’ families for participating in this project.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by a grant from the Deanship of Scientific Research, Jordan University of Science and Technology (Number 20180083).
Disclosure
The authors report no conflicts of interest for this work and declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wojcik MH, Schwartz TS, Yamin I, et al. Genetic disorders and mortality in infancy and early childhood: delayed diagnoses and missed opportunities HHS public access author manuscript. Gene Med. 2018;20. 254. doi:10.1038/gim.2018.17
2. Yoon PW. Contribution of birth defects and genetic diseases to pediatric hospitalizations: A population-based study. Arch Pediatr Adolesc Med. 1997;151(11):1096–1103. doi:10.1001/archpedi.1997.02170480026004
3. Tan TY, Dillon OJ, Stark Z, et al. Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA Pediatrics. 2017;171(9):855. doi:10.1001/jamapediatrics.2017.1755
4. Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255–262. doi:10.1097/GIM.0b013e3182088158
5. Goh G, Choi M. Application of Whole Exome Sequencing to Identify Disease-Causing Variants in Inherited Human Diseases. Genomics & Informatics. 2012;10(4):214. doi:10.5808/gi.2012.10.4.214
6. Bras J, Singleton A. Exome sequencing in Parkinson’s disease. Clin Genet. 2011;80(2):104–109. doi:10.1111/j.1399-0004.2011.01722.x
7. Sassi C, Guerreiro R, Gibbs R, et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p.L166V and p.S230R) in British early-onset Alzheimer’s disease. Neurobiol Aging. 2014;35(10):
8. Azab B, Dardas Z, Rabab’h O, et al. Enteric anendocrinosis attributable to a novel Neurogenin-3 variant. European Journal of Medical Genetics. 2020;63(9):103981. doi:10.1016/j.ejmg.2020.103981
9. Warr A, Robert C, Hume D, Archibald A, Deeb N, Watson M. Exome sequencing: current and future perspectives. G3 Genes Genomes Genet. 2015;5(8):1543–1550. doi:10.1534/g3.115.018564
10. Sawyer SL, Hartley T, Dyment DA, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89(3):275–284. doi:10.1111/cge.12654
11. Mussap M, Zaffanello M, Fanos V. Metabolomics: a challenge for detecting and monitoring inborn errors of metabolism. Ann Transl Med. 2018;6(17):338. doi:10.21037/atm.2018.09.18
12. Champion MP. An approach to the diagnosis of inherited metabolic disease. Arch Dis Child Educ Pract Ed. 2010;95(2):40–46. doi:10.1136/adc.2008.151183
13. Sebastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet Part C Semin Med Genet. 2011;157(1):54–62. doi:10.1002/ajmg.c.30287
14. Sperandeo MP, Andria G, Sebastio G. Lysinuric protein intolerance: update and extended mutation analysis of theSLC7A7 gene. Hum Mutat. 2008;29(1):14–21. doi:10.1002/humu.20589
15. Sperandeo MP, Paladino S, Maiuri L, et al. A y+LAT-1 mutant protein interferes with y+LAT-2 activity: implications for the molecular pathogenesis of lysinuric protein intolerance. Eur J Hum Genet. 2005;13(5):628–634. doi:10.1038/sj.ejhg.5201376
16. Posey JE, Burrage LC, Miller MJ, et al. Lysinuric protein intolerance presenting with multiple fractures. Mol Genet Metab Rep. 2014;1(1):176–183. doi:10.1016/j.ymgmr.2014.03.004
17. Cimbalistienè L, Lehnert W, Huoponen K, Kučinskas V. First reported case of lysinuric protein intolerance (LPI) in Lithuania, confirmed biochemically and by DNA analysis. J Appl Genet. 2007;48(3):277–280. doi:10.1007/bf03195224
18. Ravenscroft G, Pannell S, O’Grady G, et al. Variants in ACTG2 underlie a substantial number of Australasian patients with primary chronic intestinal pseudo-obstruction. Neurogastroenterol Motil. 2018;30(9):e13371. doi:10.1111/nmo.13371
19. Wangler MF, Beaudet AL. ACTG2-Related Disorders. University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/pubmed/26072522.
20. Sticova E, Jirsa M, Pawłowska J. New Insights in Genetic Cholestasis: from Molecular Mechanisms to Clinical Implications. Canadian Journal of Gastroenterology and Hepatology. 2018;2018:1–12. doi:10.1155/2018/2313675
21. Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R. Brain-derived neurotrophic factor: A bridge between inflammation and neuroplasticity. Front Cell Neurosci. 2014;8(DEC). doi:10.3389/fncel.2014.00430
22. Nicastro E, Di Giorgio A, Marchetti D, et al. Diagnostic Yield of an Algorithm for Neonatal and Infantile Cholestasis Integrating Next-Generation Sequencing. J Pediatr. 2019;211(54):54–62.e4. doi:10.1016/j.jpeds.2019.04.016
23. Nicastro E, D’Antiga L. Next generation sequencing in pediatric hepatology and liver transplantation. Liver Transplant. 2018;24(2):282–293. doi:10.1002/lt.24964
24. Huang HY, Zhou H, Wang H, Chen YX, Fang F. Novel mutations in the 3β-hydroxy-Δ5-C27-steroid dehydrogenase gene (HSD3B7) in a patient with neonatal cholestasis. Chin Med J. 2016;129(1):98–100. doi:10.4103/0366-6999.172603
25. Cheng JB, Jacquemin E, Gerhardt M, et al. Molecular genetics of 3β-hydroxy-Δ5-C27-steroid oxidoreductase deficiency in 16 patients with loss of bile acid synthesis and liver disease. J Clin Endocrinol Metab. 2003;88(4):1833–1841. doi:10.1210/jc.2002-021580
26. Yuste-Checa P, Vega AI, Martín-Higueras C, et al. DPAGT1-CDG: functional analysis of disease-causing pathogenic mutations and role of endoplasmic reticulum stress. Lewin AS, ed. PLoS One. 2017;12(6):e0179456. doi:10.1371/journal.pone.0179456
27. Carrera IA, Matthijs G, Perez B, Cerdá CP. DPAGT1-CDG: report of a patient with fetal hypokinesia phenotype. Am J Med Genet Part A. 2012;158A(8):2027–2030. doi:10.1002/ajmg.a.35472
28. Imtiaz F, Al-Mostafa A, Al-Hassnan ZN. Further delineation of the phenotype of congenital disorder of glycosylation DPAGT1-CDG (CDG-Ij) identified by homozygosity mapping. JIMD Rep. 2012;2:107–111. doi:10.1007/8904_2011_57
29. Kumar P, Radhakrishnan J, Chowdhary MA, Giampietro PF. Prevalence and patterns of presentation of genetic disorders in a pediatric emergency department. Mayo Clin Proc. 2001;76(8):777–783. doi:10.1016/S0025-6196(11)63220-5
30. Stark Z, Tan TY, Chong B, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18(11):1090–1096. doi:10.1038/gim.2016.1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.