Back to Journals » OncoTargets and Therapy » Volume 9
Ectopic adrenocorticotropic hormone syndrome caused by neuroendocrine tumors of the thymus: 30-year experience with 16 patients at a single institute in the People’s Republic of China
Authors Chen Y ![]() , Li S, Liu H, Qin Y, Li L, Huang C, Bi Y, Meng Y, He J, Zhou X, Ma D
, Li S, Liu H, Qin Y, Li L, Huang C, Bi Y, Meng Y, He J, Zhou X, Ma D
Received 15 November 2015
Accepted for publication 2 February 2016
Published 26 April 2016 Volume 2016:9 Pages 2193—2201
DOI https://doi.org/10.2147/OTT.S100585
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Min Li
Ye-ye Chen,1 Shan-qing Li,1,2 Hong-sheng Liu,1,2 Ying-zhi Qin,1 Li Li,1 Cheng Huang,1 Ya-lan Bi,3 Yun-xiao Meng,3 Jia He,1 Xiao-yun Zhou,1 Dong-jie Ma1
1Department of Thoracic Surgery, 2Key Laboratory of Endocrinology, 3Department of Pathology, Ministry of Health, Peking Union Medical College Hospital, Peking Union Medical College, Chinese Academy of Medical Science, Beijing, People’s Republic of China
Background and purpose: Thymic neuroendocrine carcinomas (TNECs) are extremely uncommon. Certain cases of TNECs can produce the adrenocorticotropic hormone (ACTH) and cause ectopic ACTH syndrome (EAS). The current literature on this topic consists mainly of case reports, and therapeutic guidelines are lacking. The aim of this study was to discuss the diagnosis, surgical management, and prognosis of EAS caused by TNECs to improve clinical experience with this rare disease.
Methods: From June 1984 to June 2014, at the Peking Union Medical College Hospital, the surgical interventions and follow-up outcomes of 16 consecutive patients (eight men and eight women) with EAS caused by TNECs were retrospectively analyzed.
Results: The median age was 32.5 years (range: 13–47 years), and the median disease duration was 8.5 months (range: 1–150 months). All patients presented with clinical and biochemical evidence indicating a diagnosis of Cushing’s syndrome. Contrast-enhanced thoracic computed tomography scans were critical to locating the ACTH-producing tumor and evaluating the feasibility of resection. All patients underwent surgery. One patient died of septicemia in the intensive care unit 2 weeks after surgery. No other morbidity or mortality occurred during the perioperative period. The median overall survival (OS) was 41 months (95% CI: 30.3–51.7 months), and the progression-free survival was 28 months (95% CI: 21.6–34.3 months). Both overall survival (P=0.002) and progression-free survival (P=0.030) improved significantly after complete resection.
Conclusion: TNEC is an extremely aggressive disease that should be considered when treating patients with Cushing’s syndrome due to ectopic ACTH secretion. In particular, all suspected patients should undergo contrast-enhanced thoracic computed tomography scans to facilitate early diagnosis. The current first-line treatment is surgical resection, and complete resection is a favorable prognostic factor. However, additional patients and a longer follow-up will be needed to determine the variables that are predictive of survival and to improve patient prognosis.
Keywords: ACTH syndrome, ectopic, neuroendocrine tumors, thymus gland, surgical procedures
Introduction
Ectopic adrenocorticotropic hormone (ACTH) production by nonpituitary tumors, referred to as the ectopic ACTH syndrome (EAS), accounts for ~10%–20% of Cushing’s syndrome (CS) cases.1,2 Thymic neuroendocrine carcinomas (TNECs) are very rare malignancies that comprise <5% of all thymic epithelial tumors2,3 but are known to be one of the most common causes of EAS (along with bronchial carcinoid tumors, small-cell lung cancer, and pancreatic carcinoid tumors), accounting for ~5%–42% of all EAS cases.4 Ectopic ACTH production, which leads to EAS, occurs with a frequency of ~10%–30% in all TNEC cases.5,6
However, EAS due to TNEC is an extremely rare condition. The current experience with TNEC associated with CS is mostly based on sporadic case reports and a few small case series. Therefore, the presentation, diagnostic process, therapeutic course, and prognosis are not well characterized. The aim of this study was to retrospectively analyze the clinical, endocrinological, and imagological features, the surgical management, and the prognosis of 16 EAS cases caused by TNEC. The patients were from the Peking Union Medical College Hospital (PUMCH). Here, we present our 30 years of experience with this rare disorder.
Methods
Data were analyzed retrospectively and permission to use and analyze this data was provided by The Scientific Research Department of Peking Union Medical College Hospital. Because this study was based on data extracted from registries from records preserved in the Medical Record Department with all data de-identified, this study was exempt from human subjects review, and members of the study population did not have to provide informed consent. In this study, 16 consecutive patients who underwent surgery and were pathologically diagnosed with TNEC-induced EAS at PUMCH between June 1984 and June 2014 were included. Patients diagnosed by biopsy were excluded. All case records were reviewed via follow-up assessments extending to June 2015. We analyzed demographic variables, endocrine laboratory results, imagological data, operative techniques, and the overall survival (OS). The particular laboratory data for CS, such as cortisol and ACTH levels, are described using the median values; Wilcoxon rank sum test was used for the analysis. The median OS and progression-free survival (PFS) were estimated using the Kaplan–Meier method. For OS and PFS, the time to death and the time to disease progression, respectively, were calculated as months after the date of thoracic surgery to the time of death. In the absence of death, survival was censored at the date of the last known follow-up. Univariate log-rank tests were used to assess variables such as age, sex, 2004 World Health Organization classification,7 Masaoka–Koga (MK) stage,8 and tumor diameter, which might potentially have influenced the survival time. Results with P-values <0.05 were considered significant. We performed the statistical analyses using the IBM SPSS Statistics Version 19.0 (IBM Corporation, Armonk, NY, USA).
Results
Clinical manifestations and endocrine laboratory data
From June 1984 to June 2014, 403 patients with ACTH-dependent CS were admitted to PUMCH. A total of 76 (18.8%) were diagnosed with EAS, and 16 cases (eight men, eight women) had a confirmed pathological diagnosis of a TNEC after surgery (21.0% among all EAS cases). The median age at the time of diagnosis was 32.5 years (range: 13–47 years), and the median disease duration was 8.5 months (range: 1–150 months) (Table 1). All 16 patients lacked a circadian cortisol rhythm and had different clinical manifestations of hypercortisolism (Table 2), such as hypertension (10/16), diabetes mellitus (11/16), hypokalemia (15/16), central obesity (11/16), a buffalo hump (12/16), a moon face (12/16), acne (11/16), and muscle weakness (9/16). Upon admission, the median serum cortisol concentration was 46.4 μg/dL (range: 23.8–87.0 μg/dL), and the median ACTH concentration was 197 pg/mL (range: 58.8–1,250.0 pg/mL). Only one patient exhibited morning cortisol suppression to <50% during a high-dose dexamethasone suppression test.
 | Table 1 Clinical, biochemical, and hormonal data of 16 patients with ACTH-producing TNEC |
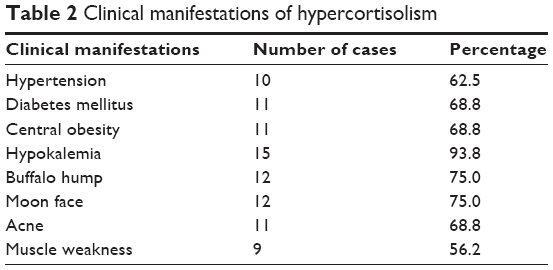 | Table 2 Clinical manifestations of hypercortisolism |
Imaging
Following the biochemical identification of ectopic ACTH secretion, imaging studies were performed to identify the source of the secretion. All the ACTH-producing thymic tumors were ultimately detected by thoracic computed tomography (CT), and the feasibility of resection was estimated prior to operation using the contrast-enhanced CT (Figure 1A and B). When the CS diagnosis was first established, a thoracic CT scan was performed on eleven of the 16 patients (68.8%), and thymic tumors were detected. Of the remaining five patients, two received chest X-rays, but no abnormalities were found. The remaining three patients did not receive any chest imagological examinations. Locating the thymic tumor in these five patients was delayed for 5 months, 30 months, 33 months, 44 months, or 146 months. Scintigraphy of the tumor’s somatostatin receptors was performed in nine patients, of which eight exhibited positive results (88.9%). The saddle area images from magnetic resonance imaging or CT scans were normal in 13 patients. Three patients underwent saddle area exploration before EAS was diagnosed. The pathological results were pituitary hyperplasia in two patients and microadenoma in one patient.
 | Figure 1 Thoracic CT scan with contrast. |
Surgical treatments and pathological examinations
Macroscopic radical tumor resection with pathologically confirmed negative resection margins (R0) was achieved in 13 patients (81.2%). In two patients, macroscopic residual (R2) resection was achieved. In another patient, we achieved a microscopic positive resection margin (R1) (Table 3). Eleven patients underwent operation via a median sternotomy, three via a right posterolateral thoracotomy, and two via video-assisted thoracoscopic surgery (VATS). Ten (62.5%) patients showed evidence of tumor invasion (Table 3). Superior vena cava or innominate vein resection with reconstruction was required in four patients. Unilateral phrenic nerve resection was performed in three patients. The mediastinal lymph nodes were resected if they appeared enlarged on the preoperative images or during surgery. Patient number 14 died of septicemia in the intensive care unit, 2 weeks after surgery. No other morbidity or mortality occurred during the perioperative period.
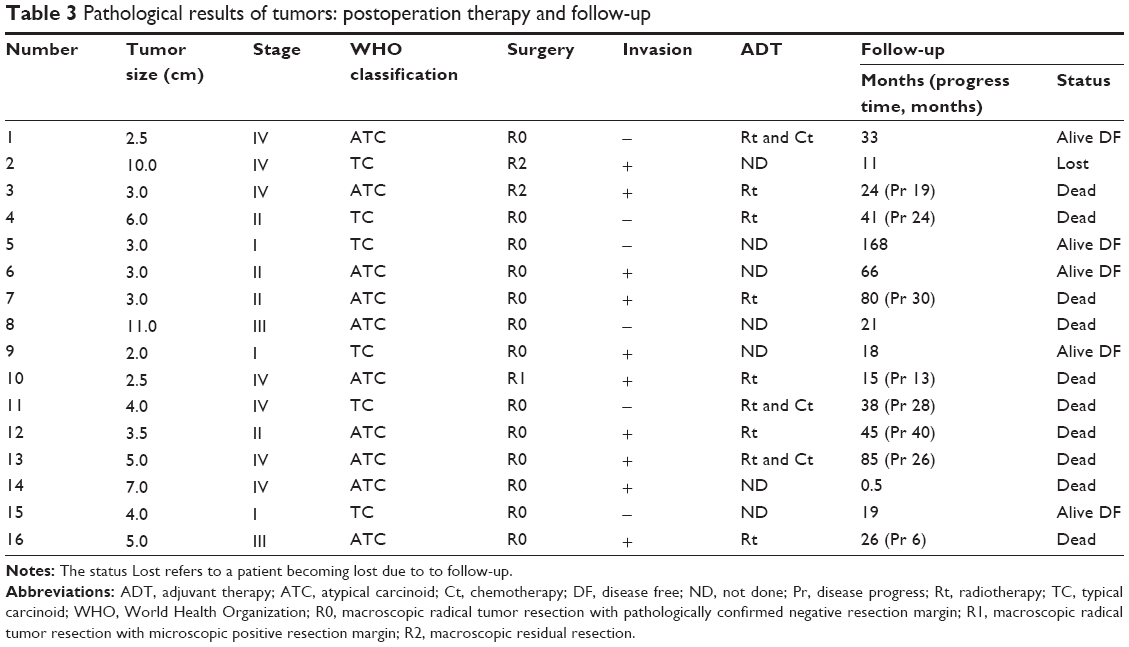 | Table 3 Pathological results of tumors: postoperation therapy and follow-up |
The serum cortisol and plasma ACTH concentrations significantly decreased after surgery (median cortisol decreased from 46.4 μg/dL to 8.7 μg/dL, P=0.001; median ACTH decreased from 197.0 pg/mL to 52.2 pg/mL, P=0.001; nonparametric tests). The maximum tumor diameter was 2.0–11.0 cm, and the median diameter was 3.8 cm. The MK stages were I, II, III, and IV in three, four, two, and seven patients, respectively (Table 3). According to the histopathological examinations, six tumors were typical carcinoid tumors, and ten tumors were atypical carcinoid tumors. Hematoxylin and eosin staining (Figure 2A) revealed the typical neuroendocrine tumor manifestation of irregularly shaped nests of tumor cells with predominantly round to oval bland nuclei. Positive immunohistochemical staining for chromogranin (Figure 2B), synaptophysin (Figure 2C), and CD56 (Figure 2D) supported the diagnosis of neuroendocrine tumors. Nine of ten (90%) tumors stained positive for ACTH (Figure 2E). No small-cell or large-cell neuroendocrine carcinomas were observed in this series.
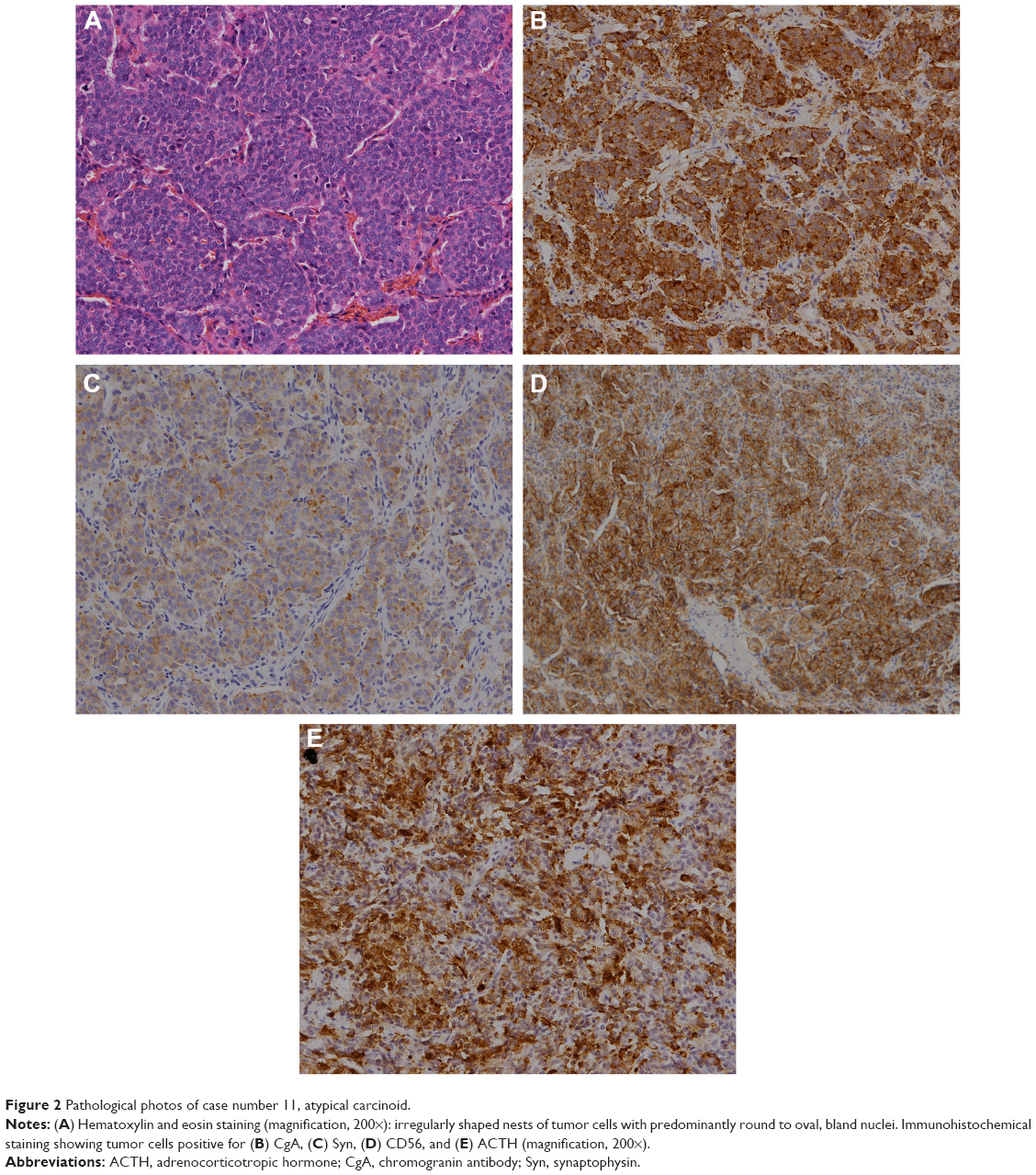 | Figure 2 Pathological photos of case number 11, atypical carcinoid. |
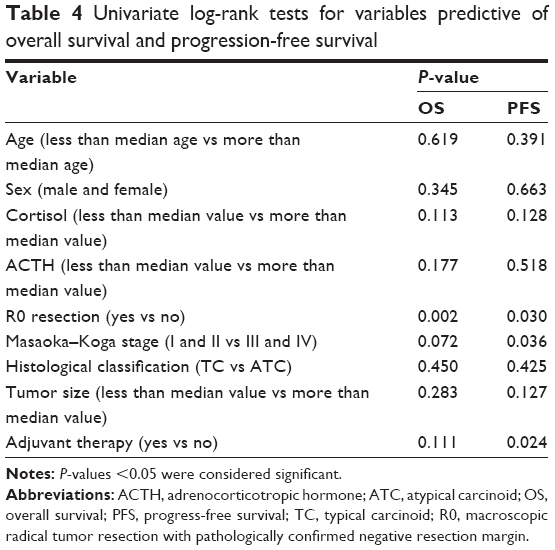
Patients were followed up mainly at the outpatient clinic of PUMCH. Patients numbers 2 and 8 did not routinely participate in the clinical follow-ups and received no adjuvant therapies. Patient number 2 was lost during follow-up 11 months after surgery, and patient number 8 died from disease progression 21 months after surgery. Four patients received no adjuvant therapy and had no sign of relapse at their last follow-ups at ~18 months, 19 months, 66 months, and 168 months. Six patients received adjuvant mediastinal radiation, and the remaining three patients received both radiation and chemotherapy (etoposide and cisplatin). In the nine patients who received adjuvant therapy, only one was alive without disease at the last follow-up, and the other eight had died. Of these eight patients, one patient died of complications due to chemotherapy 15 months after surgery, and the other seven died due to disease progression caused by metastatic recurrence. The cumulative survival of all patients, calculated using the Kaplan–Meier method, is shown in Figure 3. The median OS was 41 months (95% CI: 30.3–51.7 months), and the PFS was 28 months (95% CI: 21.6–34.3 months). Table 4 shows the association between certain variables and OS and PFS using univariate log-rank tests. Only the R0 resection was significantly associated with a better prognosis for both OS (P=0.002) and PFS (P=0.030).
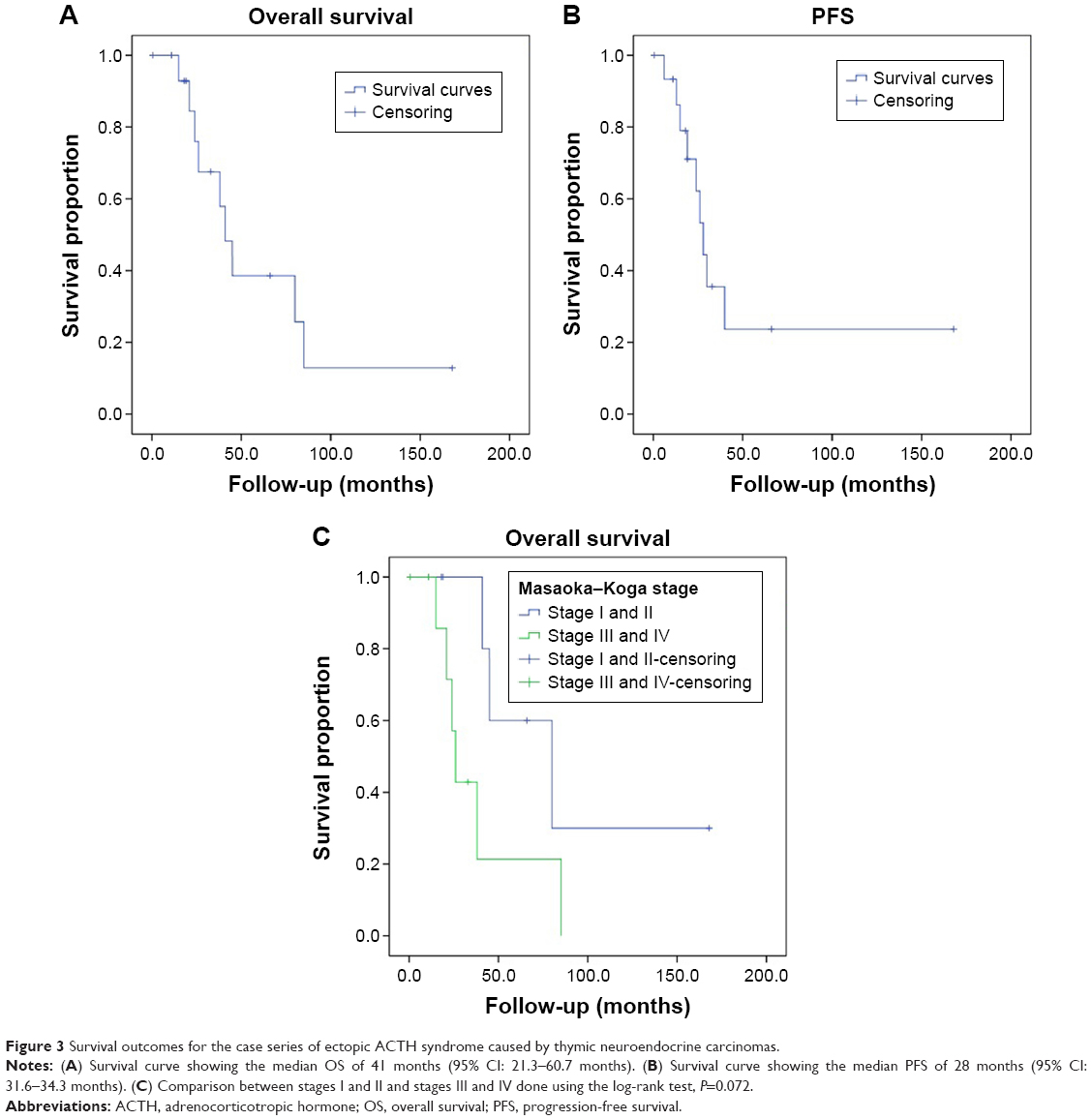 | Figure 3 Survival outcomes for the case series of ectopic ACTH syndrome caused by thymic neuroendocrine carcinomas. |
 | Table 4 Univariate log-rank tests for variables predictive of overall survival and progression-free survival |
Comment
TNEC-induced EAS is an extremely rare clinical condition. To the best of our knowledge, the 16 patients whom we have described here represent the largest case series reported from a single center. When compared with the data in the meta-analysis conducted by Neary et al,9 we obtained similar outcomes, such as a median age of 32.5 years (compared to 34 years), a sex ratio (male:female) of 1:1 (1.5:1), and a median length of survival of 41 months (35 months).
The clinical signs of EAS are easily confused with those of CS, in which ACTH is produced by a pituitary adenoma, making EAS diagnosis very challenging for clinicians. The diagnosis should be established using biochemical confirmation of the hypercortisolemic state and by locating the ACTH source.2,3,10,11 All our patients had elevated serum cortisol and ACTH levels, which suggested a diagnosis of ACTH-dependent CS. Serum cortisol levels remained elevated in 15 patients (93.8%) after a high-dose dexamethasone suppression test, which suggested EAS. The rate of cortisol elevation was similar to those reported in the literature (90%–91%).12–14
For EAS cases, the key point for diagnosis is locating the ectopic ACTH-producing tumor. According to previous reports, tumors originating in the chest cavity are most commonly associated with EAS and account for nearly two-thirds of all ectopic ACTH sources.2,3,12,14,15 Thus, chest imaging is extremely important. In our series, all thymic tumors were eventually identified with thoracic CT scans. However, the identification of thymic tumors was delayed in five cases because the initial medical institutions did not consider EAS when the CS diagnosis was first established. Of these five patients, three had no chest imaging done at all, and the other two patients received chest X-rays during their regular admission examinations. However, the X-ray findings were unremarkable, likely due to overlapping features of the spine or other organs in the mediastinum and small-diameter early stage tumors (Figure 1A). As a result, bilateral adrenalectomy was performed in two patients to control serum cortisol levels, and saddle area exploration was performed in three patients with suspected pituitary microadenoma. In these five patients, the tumor identification was delayed by 5–146 months. In addition to facilitating tumor location, CT with contrast for mediastinal masses could help surgeons to assess the invasion of tumors and estimate the feasibility of complete resection prior to surgery (Figure 1B). Therefore, we strongly suggest that thoracic CT scans with contrast (rather than chest X-rays) be performed as part of the routine examination for all patients with CS to increase the likelihood of an early diagnosis of and early therapy for EAS and to simultaneously avoid unnecessary adrenalectomy or hypophysectomy. Scintigraphy of the tumor’s somatostatin receptors may be beneficial in locating the tumors. In eight of nine patients (88.9%) who underwent the scintigraphy examination in the present study, the tumor location was consistent with the CT scan. However, we could not observe a clear tumor margin or vascular involvement, and so could not assess the feasibility of complete resection. In addition, the sensitivity of this examination ranges from 60% to 85%,16–18 so this examination is not advantageous for determining diagnosis and treatment.
The optimal treatment of EAS involves resection or complete destruction of the ACTH-secreting neoplasm. TNECs are very aggressive, and patients have a poor long-term prognosis overall. TNECs are also resistant to standard chemotherapy regimens and radiation.4,9,19,20 Thus, it is widely agreed that radical surgical excision as a first-line treatment during the early stage offers the best chance of prolonged survival and may be the only curative therapy.9,20–22 However, curative resection of the tumor producing ectopic ACTH is possible in only a small proportion of patients (12%–13% in certain studies) due to extremely aggressive metastatic disease or the inability to locate the tumor at the time of EAS diagnosis.2,3,12,23 These factors again remind us of the importance of being more aware of and vigilant about this rare situation. Surgery should include aggressive lymph node dissection to achieve a radical resection to ACTH source to allow remission with little chance of recurrence and to permit more accurate clinical staging.9,20–22,24,25 In accordance with the aggressive nature of this tumor, ten of 16 cases (62.5%) in our series were invasive and required extended resection of the pericardium, pleura, lung, or even the phrenic nerve and great vessels, such as the innominate vein and superior vena cava, to achieve radical resection (Figure 1B). However, the percentage of R0 resection in our study was only 81.2%.
The midline sternotomy approach is recommended because this enables excision of the entire thymus, the perithymic fat, and the mediastinal lymph nodes and allows for concomitant pulmonary or great vessel reconstruction. Minimally invasive approaches, including VATS and robotic-assisted thoracoscopic surgery, for early stage thymomas are becoming more common26,27 in recent years. Certain institutions have also reported performing minimally invasive resection of thymic carcinomas, including TNECs, to achieve R0 resection.4,28,29 Whether minimally invasive methods are appropriate for thymic carcinomas is still controversial, so these methods should be used only for highly selected cases, with the clinical consideration mainly based on imagological examinations in the early stage and a lack of invasion (Figure 1A). In our series, VATS was widely used for thoracic diseases. We performed VATS only twice in this series, and those patients were stage I, as confirmed by pathological analysis in both cases. However, neither patient had a sufficiently long follow-up period to allow assessment of disease recurrence. Operations in patients with CS can be very troublesome due to the risk of complications during the perioperative period; these risks are caused by perioperative hypercortisolism. For certain cases of early stage, noninvasive, small tumors, minimally invasive surgery could be an optional operating method to achieve R0 resection and to reduce the risk of complications. However, whether minimally invasive surgery is better than an open procedure is still unknown. More cases with long-term follow-ups will be needed to obtain an accurate assessment.
In our series, the median OS was 41 months and the median PFS was 28 months. The prognostic factors for survival in TNEC patients, such as the Ki67 index, macroscopically radical primary surgery, tumor size, MK stage, and the 2004 World Health Organization classification, are still under debate.4,6,21,22,25 The only validated prognostic factors for survival among patients with TNECs are the stage upon presentation and the completeness of resection of the thymic tumor.26 We analyzed certain variables, such as the age, sex, the MK stage, the histological classification, the R0 resection, the tumor size, and adjuvant therapy, that might predict survival (Table 4). However, the results of a univariate analysis were disappointing: only R0 resection was significant for both OS (P=0.002) and PFS (P=0.030). Adjuvant therapy might be beneficial for PFS (Table 4; P=0.024) but not for OS (Table 4; P=0.111). The validity of chemotherapy and radiotherapy for TNEC is still unclear, and most studies have reported a negative effect on long-term survival.4,22,25,30–32 Certain researchers have noted that adjuvant therapy might reduce the risk of local relapse but has little effect on the long-term survival of patients with TNECs.21,22 Moreover, whether the MK stage plays a role in OS is still very controversial.5,20,25 By using a log-rank test to compare the survival curves between stages I and II and stages III and IV, we could observe an obvious difference between the two curves (Figure 3C; P=0.072) for OS, and the difference in PFS was significant (P=0.036). The MK stage and adjuvant therapy might be prognostic indicators for survival, but the rarity of this disease resulted in only 16 patients being enrolled in our series, and so we could not draw a definitive conclusion. Further work is thus needed to better understand the characteristics of this rare condition and to improve systemic therapies.
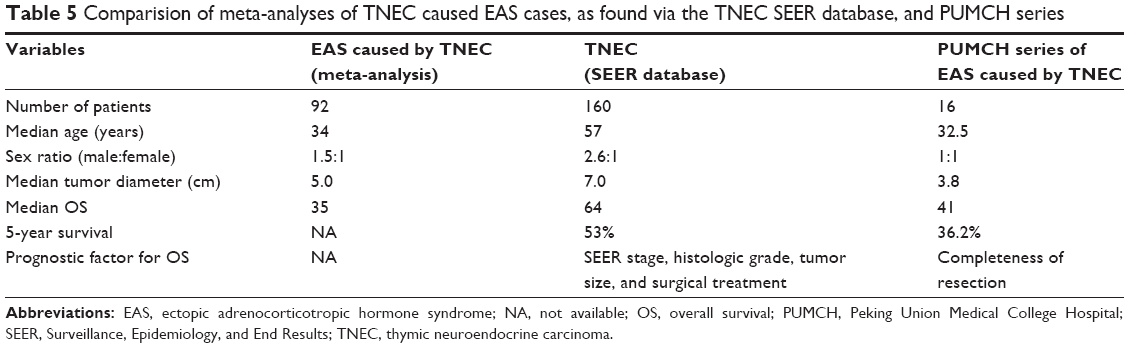
In addition, we compared our results with the results of the only published meta-analysis of TNEC-induced ACTH, which included 92 cases.9 We also compared our results with the Surveillance, Epidemiology, and End Results (SEER) database analysis of 160 TNEC patients by Gaur et al,20 which is the largest multicenter series with nearly complete surgical data (Table 5). We determined intuitively that the median age was much lower and that male predominance was not clearly associated with a smaller tumor size in ACTH-producing TNECs compared with the TNECs more generally; perhaps TNECs that secrete ACTH have different characteristics. The median OS in our series was 41 months, which is similar to the 35-month OS in the case of the meta-analysis but much less than the 64-month OS reported in the SEER database of TNECs. In the SEER database, the median OS decreased with advanced stage: it was 110 months for localized (remaining in situ or confined to the organ) cases, 59 months for regional (locally invasive or metastasized to the lymph nodes) cases, and 35 months for distant (distant metastasis) cases. The majority of our patients were stage III or IV (56.2%), which, likely due to the aggressive nature of TNECs and the occult characteristics of EAS, might explain the poor outcome. In particular, ACTH secretion or hypercortisolism might affect the OS of patients with TNECs.
 | Table 5 Comparision of meta-analyses of TNEC caused EAS cases, as found via the TNEC SEER database, and PUMCH series |
Conclusion
In summary, ACTH-producing TNECs are an extremely rare disease that should be considered in CS due to ectopic ACTH secretion. Obtaining a confirmed diagnosis and initiating therapy early are still very challenging for clinicians. Specifically, all suspected patients should undergo thoracic CT scans with contrast to achieve an early diagnosis and to estimate the feasibility of complete resection. The current first-line treatment is surgical resection, and a minimally invasive technique is recommended only in highly selected cases in the early stage. Resection completeness is an indicator of prognosis, and the MK stage may also affect prognosis. However, there are certain limitations to this study. This was a retrospective study, and bias could have resulted from the data collection. In addition, because of the rarity of the disease, no reliable statistical analysis could be performed. Additional cases at multiple centers with longer follow-ups will thus be needed to determine which variables predict survival and the ideal multidisciplinary treatment for EAS caused by TNECs.
Disclosure
The authors report no conflicts of interest in this work.
References
Wajchenberg BL, Mendonca BB, Liberman B, et al. Ectopic adrenocorticotropic hormone syndrome. Endocr Rev. 1994;15(6):752–787. | ||
Isidori AM, Lenzi A. Ectopic ACTH syndrome. Arq Bras Endocrinol Metabol. 2007;51(8):1217–1225. | ||
Alexandraki KI, Grossman AB. The ectopic ACTH syndrome. Rev Endocr Metab Disord. 2010;11(2):117–126. | ||
Filosso PL, Yao X, Ahmad U, et al; European Society of Thoracic Surgeons Thymic Group Steering Committee. Outcome of primary neuroendocrine tumors of the thymus: a joint analysis of the International Thymic Malignancy Interest Group and the European Society of Thoracic Surgeons databases. J Thorac Cardiovasc Surg. 2015;149(1):103.e2–109.e2. | ||
Crona J, Bjrklund P, Welin S, Kozlovacki G, Oberg K, Granberg D. Treatment, prognostic markers and survival in thymic neuroendocrine tumours. A study from a single tertiary referral centre. Lung Cancer. 2013;79(3):289–293. | ||
Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–3072. | ||
Travis W, Brambilla E, Müller-Hermelink H, Harris C, editors. World Health Organization Classification of Tumours, Pathology and Genetics: Tumours of the Lung, Pleura, Thymus and Heart (Who Classification of Tumours). Lyon: IARC Press; 2004. | ||
Detterbeck FC, Nicholson AG, Kondo K, Van Schil P, Moran C. The Masaoka–Koga stage classification for thymic malignancies: clarification and definition of terms. J Thorac Oncol. 2011;6(7 Suppl 3):S1710–S1716. | ||
Neary NM, Lopez-Chavez A, Abel BS, et al. Neuroendocrine ACTH-producing tumor of the thymus – experience with 12 patients over 25 years. J Clin Endocrinol Metab. 2012;97(7):2223–2230. | ||
Isidori AM, Kaltsas GA, Mohammed S, et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 2003;88(11):5299–5306. | ||
Newell-Price J, Trainer P, Besser M, Grossman A. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19(5):647–672. | ||
Isidori AM, Kaltsas GA, Pozza C, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab. 2006;91(2):371–377. | ||
Ejaz S, Vassilopoulou-Sellin R, Busaidy NL, et al. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion: the University of Texas MD Anderson Cancer Center experience. Cancer. 2011;117(19):4381–4389. | ||
Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab. 2005;90(8):4955–4962. | ||
Doi M, Sugiyama T, Izumiyama H, Yoshimoto T, Hirata Y. Clinical features and management of ectopic ACTH syndrome at a single institute in Japan. Endocr J. 2010;57(12):1061–1069. | ||
Tsagarakis S, Christoforaki M, Giannopoulou H, et al. A reappraisal of the utility of somatostatin receptor scintigraphy in patients with ectopic adrenocorticotropin Cushing’s syndrome. J Clin Endocrinol Metab. 2003;88(10):4754–4758. | ||
Pacak K, Ilias I, Chen CC, Carrasquillo JA, Whatley M, Nieman LK. The role of [(18)F]fluorodeoxyglucose positron emission tomography and [(111)In]-diethylenetriaminepentaacetate-D-Phe-pentetreotide scintigraphy in the localization of ectopic adrenocorticotropin-secreting tumors causing Cushing’s syndrome. J Clin Endocrinol Metab. 2004;89(5):2214–2221. | ||
Tabarin A, Valli N, Chanson P, et al. Usefulness of somatostatin receptor scintigraphy in patients with occult ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab. 1999;84(4):1193–1202. | ||
Oberg K, Hellman P, Ferolla P, Papotti M. Neuroendocrine bronchial and thymic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii120–vii123. | ||
Gaur P, Leary C, Yao JC. Thymic neuroendocrine tumors: a SEER database analysis of 160 patients. Ann Surg. 2010;251(6):1117–1121. | ||
Ruffini E, Oliaro A, Novero D, Campisi P, Filosso PL. Neuroendocrine tumors of the thymus. Thorac Surg Clin. 2011;21(1):13–23. | ||
Cardillo G, Treggiari S, Paul MA, et al. Primary neuroendocrine tumours of the thymus: a clinicopathologic and prognostic study in 19 patients. Eur J Cardiothorac Surg. 2010;37(4):814–818. | ||
Aniszewski JP, Young WF Jr, Thompson GB, Grant CS, van Heerden JA. Gushing syndrome due to ectopic adrenocorticotropic hormone secretion. World J Surg. 2001;25(7):934–940. | ||
Pass HI, Doppman JL, Nieman L, et al. Management of the ectopic ACTH syndrome due to thoracic carcinoids. Ann Thorac Surg. 1990;50(1):52–57. | ||
Cardillo G, Rea F, Lucchi M, et al. Primary neuroendocrine tumors of the thymus: a multicenter experience of 35 patients. Ann Thorac Surg. 2012;94(1):241–245; discussion: 245–246. | ||
Ruffini E, Venuta F. Management of thymic tumors: a European perspective. J Thorac Dis. 2014;6(Suppl 2):S228–S237. | ||
Toker A, Sonett J, Zielinski M, Rea F, Tomulescu V, Detterbeck FC. Standard terms, definitions, and policies for minimally invasive resection of thymoma. J Thorac Oncol. 2011;6(7 Suppl 3):S1739–S1742. | ||
Okereke IC, Kesler KA, Freeman RK, et al. Thymic carcinoma: outcomes after surgical resection. Ann Thorac Surg. 2012;93(5):1668–1672; discussion: 1672–1663. | ||
Mao T, Gu ZT, Fang WT, Chen WH. [Comparison of surgical approaches for thymic disorders: feasibility of VATS thymectomy and comparison with small incision and median sternotomy]. Zhonghua Wai Ke Za Zhi. 2013;51(8):737–740. | ||
de Montpréville VT, Macchiarini P, Dulmet E. Thymic neuroendocrine carcinoma (carcinoid): a clinicopathologic study of fourteen cases. J Thorac Cardiovasc Surg. 1996;111(1):134–141. | ||
Moran CA, Suster S. Neuroendocrine carcinomas (carcinoid tumor) of the thymus. A clinicopathologic analysis of 80 cases. Am J Clin Pathol. 2000;114(1):100–110. | ||
Gal AA, Kornstein MJ, Cohen C, Duarte IG, Miller JI, Mansour KA. Neuroendocrine tumors of the thymus: a clinicopathological and prognostic study. Ann Thorac Surg. 2001;72(4):1179–1182. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.