Back to Journals » Neuropsychiatric Disease and Treatment » Volume 22
Echinocystic Acid Antagonizes Post-Stroke Depression in Mice by Suppressing the JNK/NF-κB Signaling Pathway
Authors Wang D ![]() , Li W, Zhang X, Zhu K
, Li W, Zhang X, Zhu K ![]() , Wang Y
, Wang Y ![]() , Cai Y, Chen H
, Cai Y, Chen H ![]() , Cai X
, Cai X ![]() , Sun J
, Sun J ![]()
Received 12 February 2026
Accepted for publication 21 May 2026
Published 5 June 2026 Volume 2026:22 596008
DOI https://doi.org/10.2147/NDT.S596008
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Dandan Wang,1,2,* Wei Li,1,2,* Xuan Zhang,1,2 Kaiqi Zhu,1,2 Yuzhen Wang,1,2 Yaozhuo Cai,1,2 Hao Chen,1,2 Xueli Cai,1,2 Jingping Sun1,2
1Department of Neurology, Lishui Central Hospital, The Fifth Affiliated Hospital of Wenzhou Medical University, Lishui, Zhejiang, People’s Republic of China; 2Lishui Clinical Research Center for Neurological Diseases, Lishui, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingping Sun, Department of Neurology, Lishui Central Hospital, The Fifth Affiliated Hospital of Wenzhou Medical University, No. 289, Kuocang Street, Lishui, Zhejiang, 323000, People’s Republic of China, Email [email protected] Xueli Cai, Department of Neurology, Lishui Central Hospital, The Fifth Affiliated Hospital of Wenzhou Medical University, No. 289, Kuocang Street, Lishui, Zhejiang, 323000, People’s Republic of China, Email [email protected]
Background: Post-stroke depression (PSD), a common neuropsychiatric complication after stroke, affects approximately one-third of stroke survivors and severely impairs recovery and quality of life. Echinocystic acid (EA), a pentacyclic triterpenoid from various medicinal herbs, exhibits anti-inflammatory and anti-depressant properties. However, whether EA exerts neuroprotective and antidepressant effects in PSD remains unknown.
Objective: This study aims to determine whether EA alleviates PSD and to explore its potential mechanisms involving the JNK/NF-κB pathway and inflammatory cytokines.
Methods: PSD was modeled using middle cerebral artery occlusion/reperfusion (MCAO/R) combined with chronic unpredictable mild stress (CUMS). Following MCAO/R surgery, animals received daily intraperitoneal injections of EA or EA combined with the JNK agonist anisomycin (AN) for 28 consecutive days. The expression of phosphorylated JNK (p-JNK) and phosphorylated NF-κB (p-NF-κB) was analyzed by Western blotting. The levels of inflammatory cytokines, including IL-1β, IL-6, and TNF-α, were measured using enzyme-linked immunosorbent assay (ELISA). Depressive-like behaviors and the therapeutic efficacy of EA were assessed through a series of behavioral tests. Furthermore, neuronal necrosis and Nissl body integrity were observed using hematoxylin-eosin (H&E) and Nissl staining, respectively.
Results: EA treatment significantly downregulated the expression of p-JNK, p-NF-κB, and inflammatory cytokines. H&E staining revealed that EA reduced neuronal necrosis in the hippocampal dentate gyrus. Consistently, Nissl staining demonstrated that EA increased the number of Nissl bodies in the same region. Furthermore, EA administration alleviated depressive-like behaviors in PSD mice. However, the administration of AN counteracted the suppressive effects of EA on the JNK/NF-κB signaling pathway and reversed the beneficial behavioral outcomes associated with EA treatment.
Conclusion: The results indicate that EA mitigates neuronal necrosis, alleviates depression-like behaviors, and alleviates PSD by suppressing JNK/NF-κB pathway activation and inflammatory cytokine production.
Keywords: echinocystic acid, JNK/NF-κB, anisomycin, inflammatory cytokines
Introduction
Post-stroke depression (PSD) is a complex mood disorder that develops following stroke, characterized by the depressive symptoms.1 PSD is one of the most common complications after stroke, affecting approximately one-third of stroke survivors. This condition adversely impacts both cognitive and physical functional recovery.2
The pathogenesis of PSD demonstrates remarkable complexity. Current evidence implicates multiple interrelated mechanisms including microglial activation, astrocytic response, NF-κB signaling pathway dysregulation, altered brain-derived neurotrophic factor (BDNF) expression, neurotransmitter imbalance, and oxidative stress in PSD development.3 Specifically, NF-κB activation promotes microglial polarization toward the pro-inflammatory M1 phenotype, leading to the overexpression of key inflammatory cytokines, including TNF-α, IL-1β, and IL-6. These cytokines subsequently contribute to PSD through multiple mechanisms, such as hypothalamic-pituitary-adrenal (HPA) axis dysfunction, disruption of neurotransmitter metabolism, reduction of BDNF levels, and induction of excitotoxicity and oxidative stress. Importantly, these pathological processes interact synergistically, leading to neuronal apoptosis and cellular necrosis in affected brain regions.4 Thus, the NF-κB-mediated inflammatory cascade represents a critical link between post-stroke immune responses and the development of depressive symptoms.
Besides, emerging evidence indicates that activation of the JNK signaling pathway plays a critical role in the pathogenesis of PSD. In PSD models, proBDNF/p75NTR signaling activates the RhoA-JNK pathway, promoting apoptosis-related protein expression and inhibiting synaptic regeneration.5 Additionally, ischemic stroke combined with chronic stress induces sustained JNK phosphorylation, leading to upregulation of c-Jun and AP-1, which impairs synaptic plasticity by reducing BDNF, PSD95, and synaptophysin, while also promoting neuroinflammation and neuronal apoptosis. Notably, inhibiting the JNK pathway alleviates these pathological changes and depressive-like behaviors.6 Therefore, JNK signaling represents a key mediator linking post-stroke neuroinflammatory and apoptotic cascades to PSD development. The dentate gyrus (DG) of the hippocampus has been identified as a key region involved in PSD. Direct evidence from a rat PSD model demonstrated that ischemia-induced neurodegeneration in the DG is causally linked to anhedonia, a core symptom of depression, and that antidepressant treatment prevents both DG neuronal loss and depressive-like behaviors.7 Furthermore, ketamine has been shown to exert lasting antidepressant effects in PSD mice by modulating NMDAR/CaMKII-mediated synaptic plasticity in the DG.8 Given that stroke-induced neuroinflammation critically regulates DG neurogenesis,9 we hypothesized that EA, a natural anti-inflammatory compound, may exert its therapeutic effects against PSD by protecting the DG from inflammation-induced damage through inhibition of the JNK/NF-κB pathway.
Taken together, we hypothesize that the development of PSD may be associated with activation of the JNK/NF-κB signaling pathways and inflammatory cytokines, along with cellular pathology in the hippocampal dentate gyrus.
Although drugs are currently available for the treatment of PSD in clinical practice, they are all associated with certain side effects. Studies indicate that patients with PSD face higher risks of mortality and disability compared to non-depressed stroke survivors.10 Meanwhile, drugs used for treating PSD generally have problems such as numerous side effects and slow onset of action, which result in poor patient compliance.11 Selective serotonin reuptake inhibitors (SSRIs) are the first-line clinical treatment for post-stroke anxiety and depression.12 Although SSRIs are used to treat PSD, they concurrently increase the risk of bleeding, which could potentially lead to higher mortality in patients taking these medications.13 Therefore, it is necessary to explore novel anti-PSD drugs that are safer, more effective, and have fewer side effects. However, the molecular mechanisms underlying PSD, particularly the relationship involving the JNK/NF-κB signaling pathway, inflammatory cytokines, and their link with cells in the hippocampal dentate gyrus, remain poorly understood. Moreover, whether echinocystic acid (EA), a natural pentacyclic triterpenoid with known anti-inflammatory and antidepressant properties, exerts neuroprotective effects against PSD through modulation of the JNK/NF-κB pathway has not yet been investigated.
EA, a pentacyclic triterpenoid, is a natural compound derived from various medicinal herbs including Codonopsis pilosula, Albizia julibrissin and Gleditsia sinensis fruit (Figure 1A).14–16 Studies have demonstrated that EA can be absorbed into the blood-brain barrier (BBB)17 and exhibits multiple beneficial pharmacological activities, including antidepressant, anti-inflammatory, and anti-apoptotic effects.16,18–20 Studies have suggested that neuroinflammation or the activation of immune cells in the brain may contribute to the pathogenesis and development of depression.21 Therefore, anti-inflammatory drugs could serve as potential antidepressants.22 He et al demonstrated that EA alleviates neuroinflammation in Parkinson’s disease mouse model, both in vivo and in vitro, by inhibiting the JNK signaling pathway and NF-κB activation.19 Previous studies from our group have demonstrated that EA alleviates cerebral ischemia-reperfusion injury in an ischemic stroke model by inhibiting the JNK signaling pathway, concurrently reducing the expression of pro-inflammatory cytokines, including IL-6 and IL-1β, following the injury.23 Complementing these findings, Joh et al reported that EA effectively inhibits the production of proinflammatory cytokines (TNF-α and IL-1β) as well as the activation of both JNK and NF-κB signaling pathways in an acute lung injury model.20 However, the underlying mechanism of EA in PSD mice has not been fully elucidated.
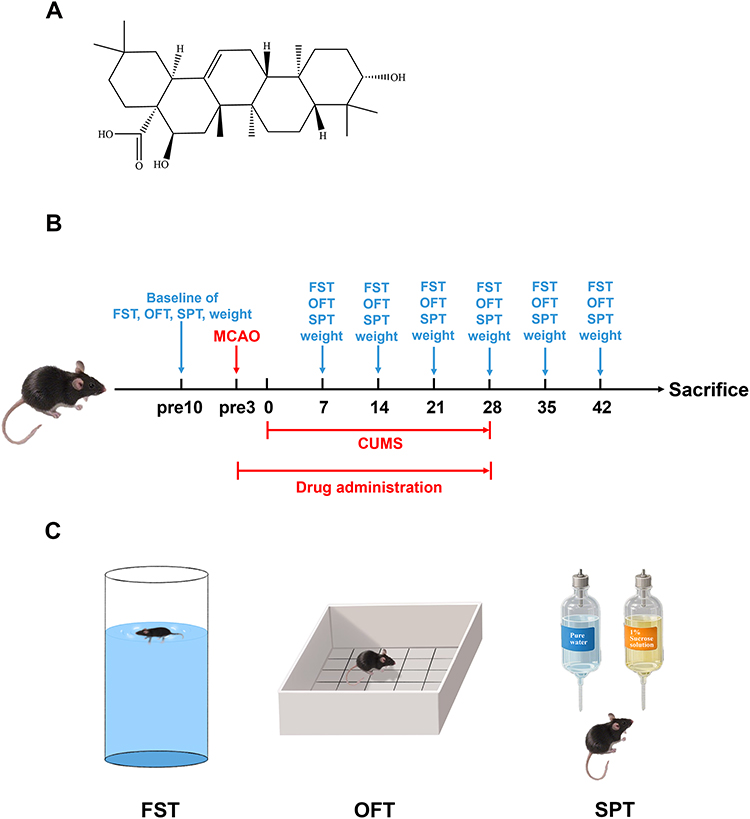 |
Figure 1 The experimental procedure of PSD and behavioral tests. (A) The structure of EA. (B) Schematic illustration on the PSD mouse model establishment and drug treatment. (C) The animals were subjected to the SPT, OFT and FST for each week. |
Collectively, our study elucidates that EA confers neuroprotective and antidepressant effects in PSD mice by inhibiting the activation of the JNK/NF-κB signaling pathway and suppressing the expression of inflammatory cytokines. These findings may offer novel therapeutic targets and provide new research perspectives for PSD treatment.
Materials and Methods
Experimental Animals
The specific pathogen-free (SPF) male C57BL/6 mice about 25–30g (9–10 weeks old) were purchased from PIZHOU Oriental & Xiaohe Technology (Production License No. SCXK (su) 2017-0003). All mice were randomly assigned to groups before modeling and maintained in an environment with appropriate humidity (55% ± 10%) and constant temperature (22°C ± 2°C). And the mice had free access to standard food and water, and were kept on a 12-hour light/12-hour dark cycle. All experimental procedures were conducted in accordance with the ethical guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Lishui University (Ethical Approval No. 2024YD0158).
Grouping and Drug Administration
The mice were randomly assigned to the following treatment groups (n = 10 per group). Study 1 aimed to investigate the therapeutic effects of EA on PSD mice: (i) vehicle-treated group (Sham group); (ii) EA-treated group (EA group); (iii) vehicle-treated PSD group (PSD group); (iv) EA-treated PSD group (PSD+EA group). Study 2 focused on elucidating the underlying mechanisms by which EA alleviates PSD: (i) vehicle-treated group (Sham group); (ii) vehicle-treated PSD group (PSD group); (iii) EA-treated PSD group (PSD+EA group); (iv) EA+AN (Anisomycin) -treated PSD group (PSD+EA+AN group). During the 28-day post-modeling period, PSD mice received EA (50 mg/kg, i.p.) (510-30-5, Nanjing Spring & Autumn Biological Engineering Co., Ltd, China) and AN (0.1 mg/kg, i.p.) (HY-18982, MCE, USA). The concentrations of EA and AN were determined based on previously described studies.23,24 The Sham group was treated with vehicle (2% DMSO in physiological saline) at 10 mL/kg body weight. To investigate the potential role of the JNK/NF-κB signaling pathway in PSD, mice were euthanized by cervical dislocation on day 42 following the approved animal protocol. Subsequently, the expression levels of p-JNK, p-NF-κB, IL-1β, IL-6, and TNF-α were measured.
MCAO/R Model
A mouse model of focal cerebral ischemia was established via left middle cerebral artery occlusion/reperfusion (MCAO/R) as previously described.25 Mice were anesthetized by intraperitoneal injection of tribromoethanol (30 μL/g) (BR4108423, Macklin, China) and fixed in a supine position. A small ventral midline incision was made on the neck skin to adequately expose the left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA). And a small incision was made approximately 2 mm proximal to the CCA bifurcation. A monofilament suture (A6-122250, Cinontech, China) was then inserted into the ICA and advanced 9–11 mm to achieve temporary MCA occlusion. After 45 minutes of focal cerebral ischemia, the filament was withdrawn to allow cerebral blood flow reperfusion. The Sham group animals underwent neck incision and ECA ligation only, without MCA occlusion. Core body temperature was maintained at 36.5–37.5°C throughout the procedure using a heating pad until animals recovered from anesthesia. Neurological deficits were assessed at 24 h and 48 h after reperfusion using the Longa score to confirm the successful establishment of MCAO.
Neurological Deficit Score
Assessment of neurological deficits was performed according to the Longa scoring system.26 Mice scoring 2–3 points were considered successful models and included in subsequent experiments. Scores 1–2 indicated mild neurological injury, while 3–4 represented severe injury. Mice were pre-trained before surgery and evaluated at the designated timepoints. The Longa scale was defined as follows: score 0: no deficit; score 1: failure to fully extend the contralateral forelimb; score 2: circling to the contralateral side when pulled by tail; score 3: falling to the contralateral side at rest; score 4: no spontaneous walking with depressed consciousness. Furthermore, the modified Neurological Severity Score (mNSS) was evaluated at 24 h and 48 h after MCAO to complement the Longa score (Supplementary Figure 1).27
Establishment of PSD Model
Following MCAO/R modeling, mice were subjected to chronic unpredictable mild stress (CUMS) to establish PSD models according to the previous study.28 PSD animals were individually housed and exposed daily to one of the following stressors: 24-h cage tilting, 5-min forced swim, damp bedding (200 mL water per 100 g bedding), 24-h food deprivation, 24-h water deprivation, 5-min shaking, 1-min tail clamping, and 24-h light/dark cycle inversion. This protocol was maintained for 4 weeks with no consecutive repetition of the same stressor (Figure 1B). The Sham and EA groups were maintained under standard conditions.
Forced Swimming Test (FST)
Immobility time served as an indicator of depressive-like behavior in stroke-injured animals (Figure 1C). Mice were placed individually in a transparent cylindrical tank (height: 60 cm and diameter: 18 cm) filled with water to a depth of 20 cm (temperature: 23 ± 2 °C). After a 1-min acclimation period, the test session was recorded by a video camera, and total immobility time (defined as passive floating without struggling) was measured during the 5-min trial using a stopwatch.
Open Field Test (OFT)
This test assesses the exploratory adaptability to novel environments, locomotor activity, and interest levels in mice (Figure 1C). A white open-field box (80 cm length × 80 cm width × 40 cm height) with the floor divided into 25 equal-area squares was used as the apparatus. Under quiet conditions, each mouse was placed in the chamber, and its behavior was recorded for 5 min after a 1-min acclimation period. Horizontal activity was scored as the number of squares crossed (four paws entering a new square), while vertical activity was counted as rearing events (both forelimbs lifted off the ground). The experiment box was cleaned with 75% ethanol between trials to eliminate residual odors.
Sucrose Preference Test (SPT)
Reduced sucrose consumption indicated depressive-like behavior (Figure 1C). Mice were placed in a quiet room and allowed free access to two bottles of 1% sucrose (S8271, Solarbio, China) solution on day 1. The next day, they were given both 1% sucrose solution and plain water simultaneously. Following 24-hour food/water deprivation on day 3, the sucrose and water bottles were provided on day 4 with their positions alternated every 2 hours. Bottle weights were measured before and after testing, and sucrose preference was calculated as: (sucrose consumption) / (total liquid consumption) × 100%.
Body Weight
To assess the effect of EA on the bodyweight, the body weight of mice was measured at baseline and weekly thereafter for a total of 42 days post-modeling.
Western Blot (WB)
On day 42 after PSD induction, the mice were euthanized, and the hippocampal and cerebral cortical tissues were collected. Protein concentrations in the supernatants were determined using a BCA protein assay kit (P0010, Beyotime, China). Protein samples were separated by electrophoresis and transferred onto polyvinylidene fluoride (PVDF) membranes (IPVH00010, Millipore, USA). The membranes were blocked with 5% non-fat milk or 5% BSA in TBST for 1 h at room temperature, followed by overnight incubation at 4°C with the following primary antibodies: anti-GAPDH (1:1000; 5174S, CST, USA), anti-phospho-SAPK/JNK (p-JNK, 1:1000; 9255S, CST, USA), anti-SAPK/JNK (JNK, 1:1000; 9252S, CST, USA), and anti-phospho-NF-κB p65 (p-NF-κB; 1:1000; 3033S, CST, USA), anti-NF-κB p65 (NF-κB, 1:1000; 8242S, CST, USA). After washing, the membranes were incubated with horseradish peroxidase (HRP)-conjugated horse anti-mouse (1:10,000; 7076S, CST, USA) or goat anti-rabbit secondary antibodies (1:10,000; 7074S, CST, USA) for 1 h at room temperature. The protein bands were visualized using the Invitrogen iBright FL1500 Imaging System (Thermo Fisher Scientific) and quantified with ImageJ analysis software.
Enzyme-Linked Immunosorbent Assay (ELISA)
Ischemic brain tissues of mice were collected, and the levels of IL-1β, IL-6 and TNF-α in the brain homogenates were measured using commercial ELISA kits according to the manufacturer’s instructions (MM-0040M1, MM-0163M1and MM-0132M1, Meimian, China). Cytokine concentrations were expressed as pg/mg protein.
Hematoxylin-Eosin (H&E) Staining
Brain samples were fixed in 4% paraformaldehyde for 24 hours, washed with phosphate-buffered saline (PBS), and embedded in paraffin. The embedded tissues were then sectioned into approximately 3-μm-thick slices and stained with hematoxylin and eosin. Following staining, histopathological and morphological changes were observed under a light microscope.
Nissl Staining
Nissl staining was employed to detect Nissl bodies within the neuronal cytoplasm and dendrites. Paraffin-embedded mouse hippocampal tissues were sectioned into 3-μm-thick slices. Following deparaffinization and rehydration, the sections were stained with a Nissl staining solution at 56 °C for 1 hour. Hippocampal regions were imaged under a light microscope, with three random fields selected per specimen. Nissl-positive cells were quantified using ImageJ software (National Institutes of Health, USA).
Statistical Analysis
All data were analyzed using Graphpad prism 9.5 and presented as means ± standard deviation (SD). The normality of the data was assessed prior to analysis. One or two-way analysis of variance (ANOVA) followed by a Tukey post hoc test was then used for statistical analysis. In all cases, p < 0.05 was considered statistically significant.
Results
EA Suppresses JNK/NF-κB Signaling Pathway Activation in PSD Mice
Western blot analysis confirmed that the expression levels of p-JNK and p-NF-κB were significantly increased in PSD model mice, respectively, compared with the Sham and EA groups. In contrast, the PSD+EA group exhibited a marked reduction in p-JNK and p-NF-κB expression relative to the PSD group (p < 0.05) (Figure 2A and B).
 |
Figure 2 EA suppressed the upregulation of p-JNK and p-NF-κB in the ischemic brain tissue and decreased immobility time in the FST in PSD mice. (A) Representative Western blot analysis of p-JNK in hippocampus (n = 3). (B) Representative Western blot analysis of p-NF-κB in hippocampus (n = 3). (C) Immobility time in FST (n = 6). Values are presented as mean ± SD. *p < 0.05, **p < 0.01. |
EA Alleviated Depression-Like Behaviors Induced by PSD Mice
EA treatment significantly improved depression-like behaviors and physiological indicators in PSD mice. In the FST, the immobility time of the Sham, EA, and PSD+EA groups was significantly shorter than that of the PSD group from day 7 to day 42 (p < 0.05) (Figure 2C). In the OFT, the PSD+EA group exhibited significantly increased horizontal activity at days 7, 28, and 42, and higher vertical activity at days 7 and 28 compared to the PSD group (p < 0.05) (Tables 1 and 2). Body weight was also significantly higher in the PSD+EA group from day 7 to day 42 compared to the PSD group (p < 0.05) (Figure 3C). Similarly, in the SPT, EA administration restored sucrose preference at day 7 to day 42 compared to the PSD group (p < 0.05) (Figure 3E).
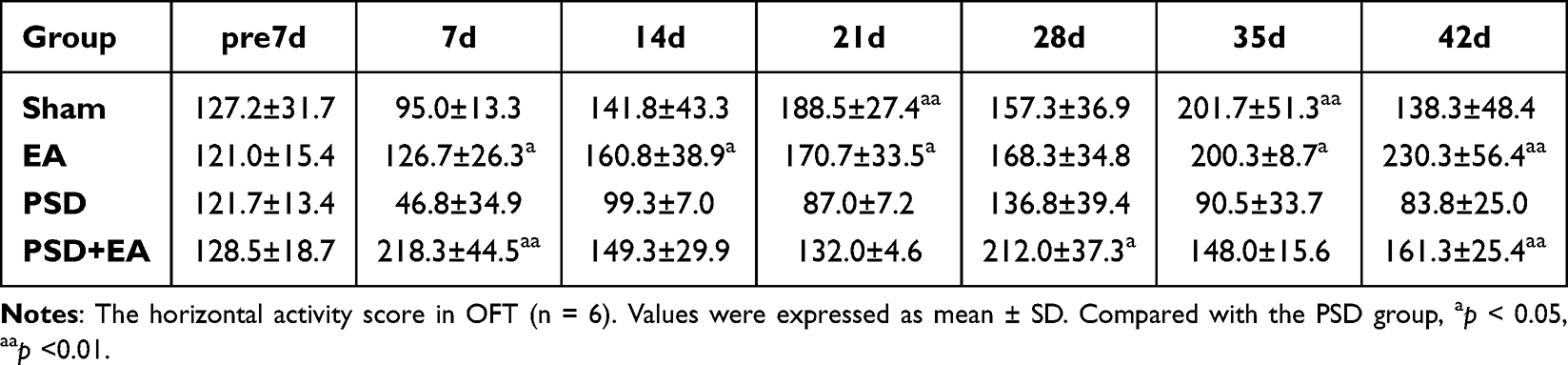 |
Table 1 Horizontal Activity Score of Study 1 |
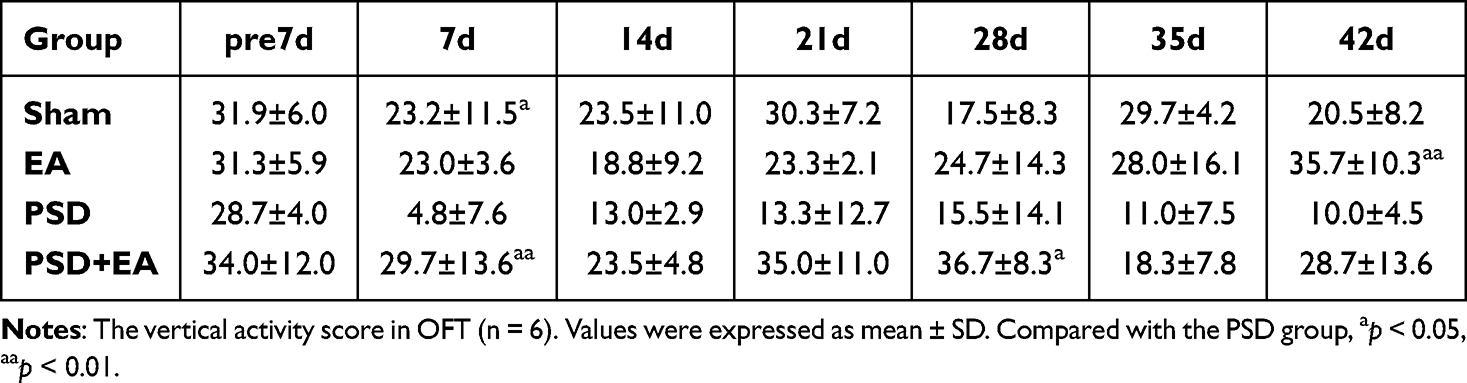 |
Table 2 Vertical Activity Score of Study 1 |
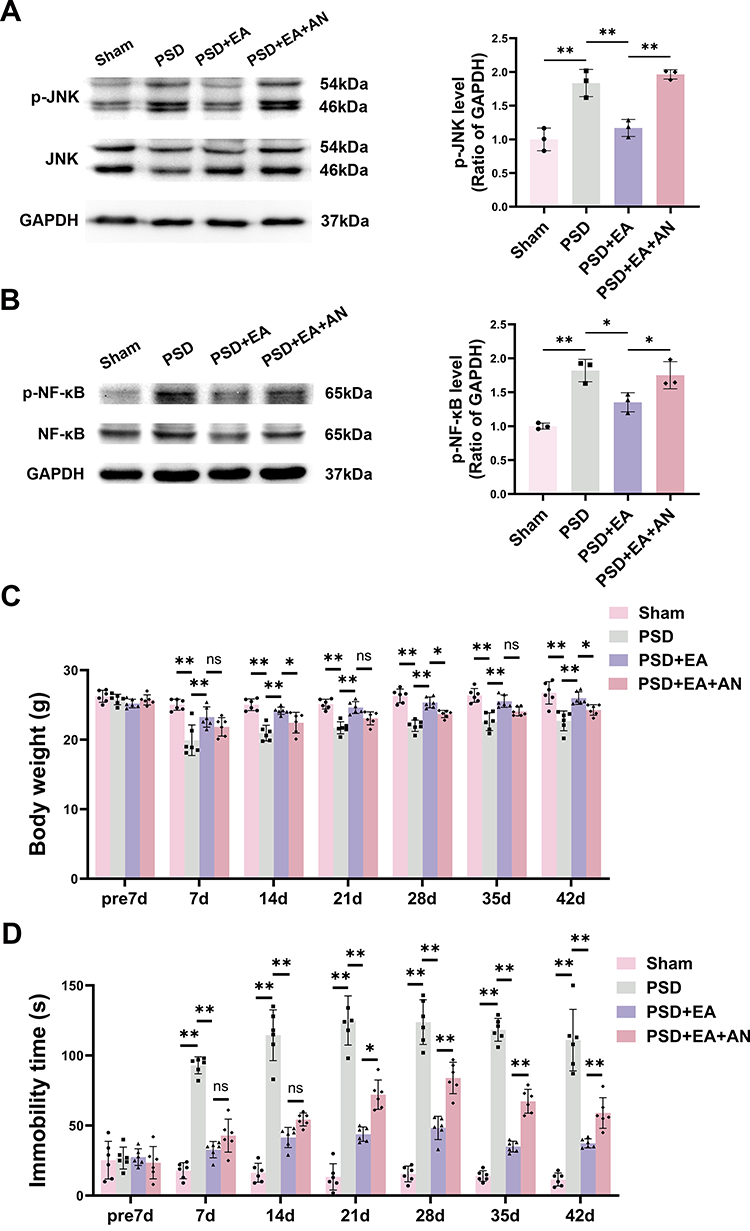 |
Figure 3 AN attenuated the therapeutic effects of EA via activation of the JNK pathway. (A) Representative Western blot analysis of p-JNK in hippocampus (n = 3). (B) Representative Western blot analysis of p-NF-κB in hippocampus (n = 3). (C) Body weight changes in mice at different time points (n = 6). (D) Immobility time in FST (n = 6). (E) Sucrose preference (%) in SPT (n = 6). Values are presented as mean ± SD. *p < 0.05, **p < 0.01, ns indicates not significant. |
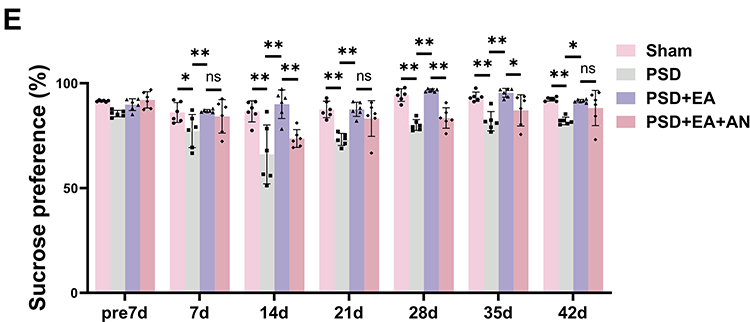 |
Figure 3 Continued. |
JNK Activator AN Reverses EA-Induced JNK/NF-κB Signaling Pathway Suppression
Western blot analysis demonstrated that AN (0.1 mg/kg, i.p.) significantly increased the expression levels of both p-JNK and p-NF-κB in the PSD+EA+AN group compared to the PSD+EA group (p < 0.05) (Figure 3A and B), indicating that AN administration reversed the EA-induced downregulation of these phosphorylated signaling molecules.
JNK Activator AN Reverses EA’s Antidepressant Effects in PSD Mice
Compared to the PSD group, the PSD+EA group showed significantly higher body weight and sucrose consumption from day 7 to day 42. Conversely, the PSD+EA+AN group exhibited significantly lower body weight at days 14, 28, 42 and reduced sucrose preference at days 14, 28 and 35 compared to the PSD+EA group, indicating that AN attenuated the antidepressant effect of EA (p < 0.05) (Figure 3C and E).
In the FST, EA treatment significantly reduced immobility time from day 7 to day 42 compared to the PSD group (p < 0.05). However, PSD+EA+AN group counteracted this effect, increasing immobility time at days 21, 28, 35, and 42 compared to the PSD+EA group (p < 0.05) (Figure 3D).
In the OFT, EA ameliorated PSD-induced reductions in horizontal and vertical activity. Notably, AN administration attenuated these beneficial effects, leading to significantly decreased horizontal activity at days 21 and 28, and reduced vertical activity at the same time points compared to the PSD+EA group (p < 0.05) (Tables 3 and 4).
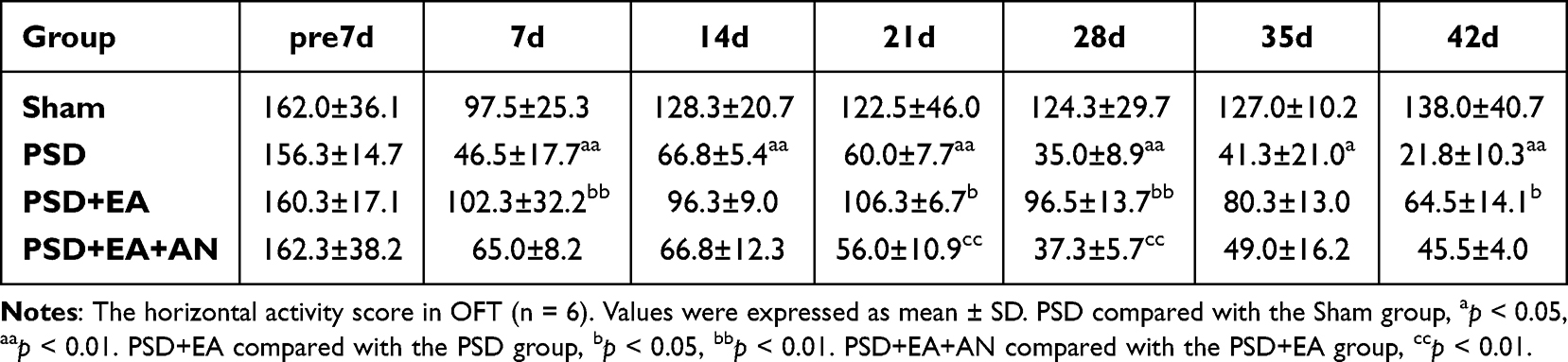 |
Table 3 Horizontal Activity Score of Study 2 |
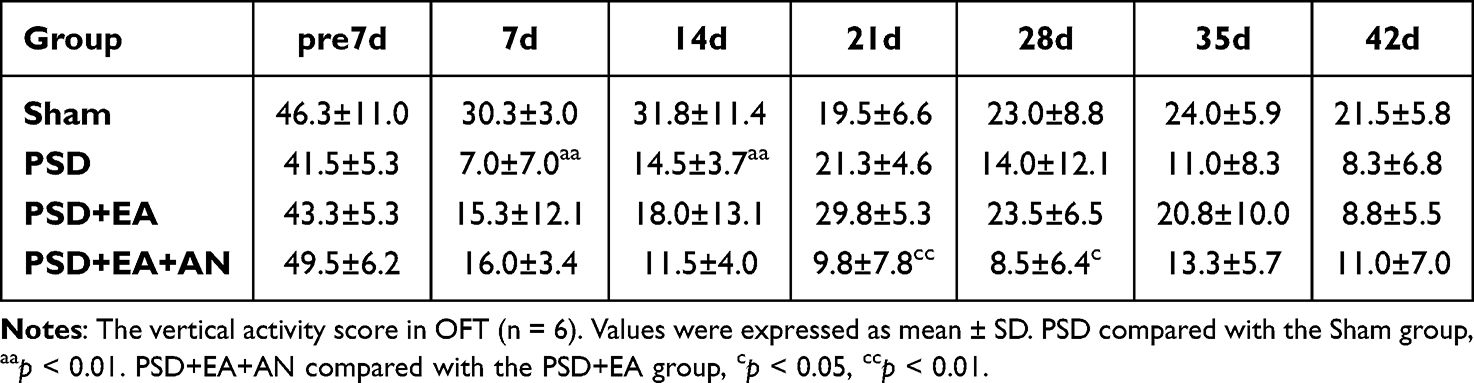 |
Table 4 Vertical Activity Score of Study 2 |
EA Reduces the Expression of Inflammatory Cytokines Induced by PSD Mice
ELISA analysis revealed that the levels of IL-1β, IL-6, and TNF-α in the homogenates of the ischemic hemisphere were distinctly increased in the PSD group compared to the Sham group (p < 0.01). Notably, PSD+EA group markedly reduced these inflammatory cytokines relative to untreated PSD group (p < 0.01), whereas PSD+EA+AN group reversed EA’s anti-inflammatory effects (p < 0.05) (Figure 4A–C).
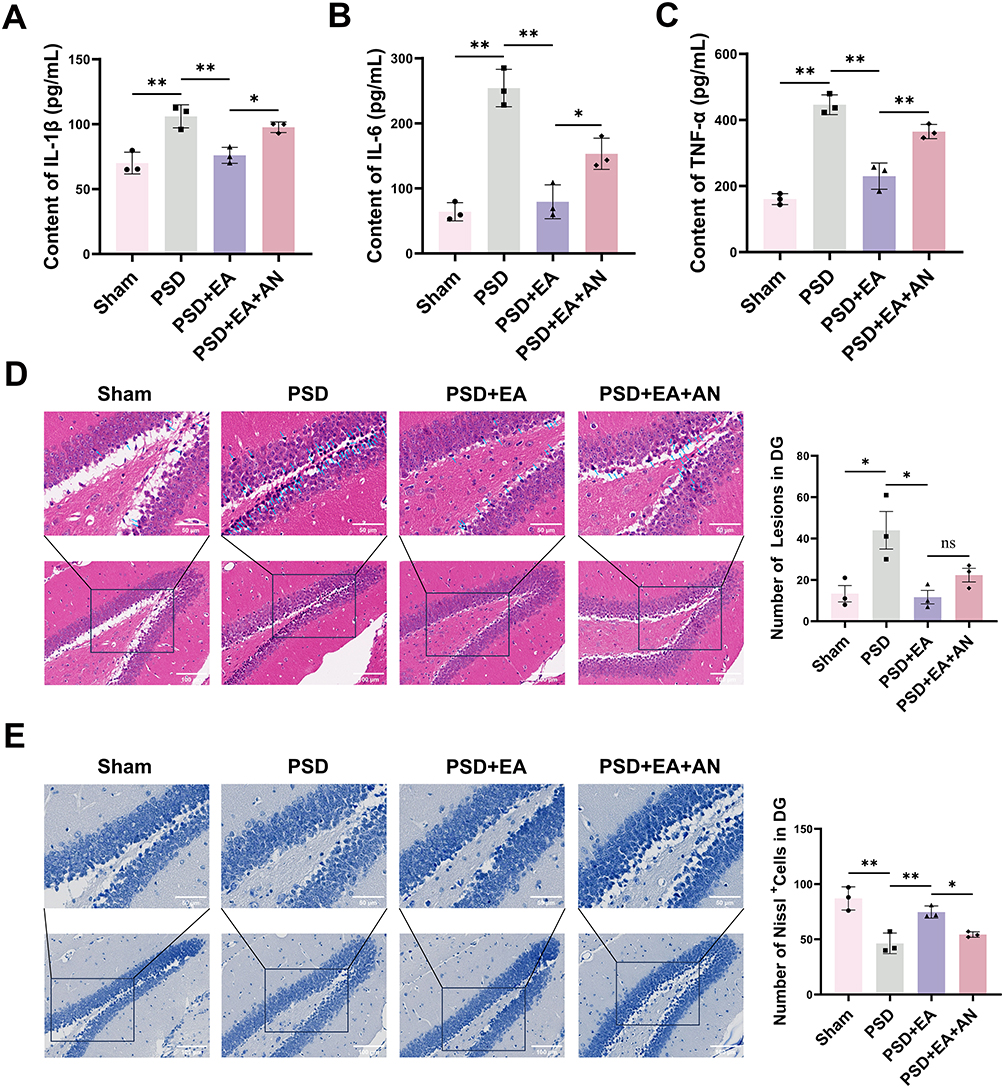 |
Figure 4 The expression of inflammatory factors and neuronal survival in the hippocampal dentate gyrus. (A–C) Levels of IL1β, IL-6 and TNF-α in the hippocampal dentate gyrus of mice measured by ELISA (n = 3). (D) H&E staining demonstrated neuronal degeneration and necrosis (n = 3). Magnification 200 × and 400 ×, Scale bar = 50 μm and 100 μm. (E) Nissl staining and quantitative analysis (n = 3). Magnification 200 × and 400 ×, scale bar = 50 μm and 100 μm. Measurement data were expressed as mean ± SD. *p < 0.05, **p < 0.01, ns indicates not significant. |
EA Reduces Neuronal Necrosis in the Dentate Gyrus of Hippocampus
H&E staining revealed a significant increase in granulosa cells with nuclear pyknosis in the dentate gyrus (DG) of the PSD group compared to the Sham group (p < 0.05). This increase was significantly reduced by EA treatment (p < 0.05). However, the PSD+EA+AN group showed a non-significant upward trend in the number of pyknotic neurons compared to the PSD+EA group (Figure 4D). Nissl staining demonstrated a significant reduction of Nissl bodies in the DG region of PSD mice compared to Sham controls (p < 0.01). This reduction was effectively reversed by EA administration (p < 0.01). However, co-treatment with AN significantly diminished the protective effect of EA, leading to a notable decrease in Nissl bodies compared to the PSD+EA group (p < 0.05) (Figure 4E). Besides, to evaluate the effects of EA on post-stroke physical disability, we assessed mNSS scores in PSD and PSD+EA mice. As shown in Supplementary Figure 1, EA treatment significantly reduced mNSS scores at 24 h and 48 h post-MCAO compared to the PSD group (p < 0.05), indicating early recovery of neurological function.
Discussion
PSD is the most prevalent neuropsychological disorder among stroke survivors, which is primarily characterized by persistent low mood and diminished interest.29 PSD significantly impedes rehabilitation and overall quality of life, with evidence linking it to increased post-stroke mortality.30 Consequently, the development of novel therapeutic strategies for PSD has become an urgent priority.
JNK, a member of the mitogen-activated protein kinase (MAPK) family, plays a pivotal role in cellular stress response, apoptosis, and inflammation regulation.31 Studies have demonstrated that hyperactivation of the JNK signaling pathway is closely associated with the pathogenesis and progression of depression. It contributes to the pathological processes of PSD by modulating the expression of downstream pro-inflammatory cytokines, apoptotic proteins, and neurotrophic factors.3 After cerebral ischemia-reperfusion injury, the inflammatory response rapidly activates microglia, which in turn produce cytokines that contribute to secondary inflammation-related brain damage.32 Among these, the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α can activate the JNK signaling pathway. In turn, activated JNK signaling further promotes the expression of inflammatory cytokines via NF-κB activation (Figure 5).33 NF-κB, a ubiquitously expressed transcriptional regulator involved in immune responses and inflammatory reactions, typically exists in the cytoplasm of most cells as an NF-κB1 dimeric protein composed of p65/p50 subunits.34
 |
Figure 5 EA attenuates PSD progression by suppressing the JNK/NF-κB signaling pathway. In this model, EA inhibits the expression of p-JNK and p-NF-κB, resulting in reduced levels of the inflammatory cytokines IL-1β, IL-6, and TNF-α, which impedes the development of PSD. “-” indicates an inhibitory effect. |
Traditional Chinese Medicine is gaining increased attention for its desirable attributes, including a high response rate, rapid onset, persistent antidepressant effects, and a low incidence of adverse effects.35,36 As mentioned previously, EA, which is widely found in various common Chinese medicinal herbs, has been proven to exert robust neuroprotective effects in various neurological diseases. Li et al found that EA alleviates hypoxic-ischemic brain injury in neonatal mice by activating the PI3K/Akt/Nrf2 signaling pathway.37 Yu et al found that EA alleviates cerebral ischemia/reperfusion injury by inhibiting the JNK signaling pathway.23 Another study showed that EA contributed to the reduction in the reserpine-induced pain/depression dyad in mice.18 Moreover, Joh et al found that EA ameliorates pulmonary inflammation in mice and alveolar macrophages by inhibiting the NF-κB and MAPK pathways.20 Based on the aforementioned findings, we investigated the therapeutic potential of EA on PSD using a well-established mouse model combining MCAO/R with CUMS. For the first time, we explored its potential efficacy and underlying mechanisms against PSD.
It is well established that the JNK/NF-κB pathway in hippocampal neurons is a critical mediator of stress- and ischemia-induced inflammatory responses and cell death. For instance, Khan et al demonstrated that chronic cerebral ischemia induces oxidative stress-mediated JNK phosphorylation, which subsequently regulates NF-κB signaling and neuroinflammation in the hippocampus; inhibition of JNK significantly reduced neuronal loss.38 Similarly, Lv et al showed that NF-κB activation directly mediates JNK pathway signaling in hippocampal neurons, leading to increased expression of TNF-α, IL-6, and iNOS, as well as neuronal apoptosis.39 Furthermore, Kang et al reported that the ASK1/JNK signaling cascade in the hippocampus is associated with inflammatory cytokine elevation and neuronal apoptosis under chronic ischemic conditions.40 Collectively, these studies establish that the JNK/NF-κB pathway in hippocampal neurons is a key mediator of inflammation and apoptosis under ischemic and stress conditions. Consistent with this mechanistic framework, our findings demonstrate that EA treatment significantly reduced the expression of p-JNK and p-NF-κB in the hippocampus compared to the PSD group. Moreover, we further validated the anti-inflammatory properties of EA and found that it decreased the expression of IL-1β, IL-6, and TNF-α. Notably, these therapeutic effects were reversed by the AN. In conclusion, these results indicate that EA exerts its neuroprotective and antidepressant effects by inhibiting the JNK signaling pathway and reducing the production of inflammatory cytokines, which likely contributes to its therapeutic effects against PSD.
To evaluate antidepressant effects, we conducted body weight monitoring, FST, SPT and OFT. The body weight changes reflect the overall status of mice under stress conditions. The FST remains one of the most frequently employed behavioral paradigms in depression research, especially for the purpose of screening potential antidepressants,41 while the SPT provides a sensitive measure of depressive-like states through anhedonia assessment.42 The OFT, as a well-established behavioral assay in rodents, enables simultaneous evaluation of locomotor activity and anxiety-like behaviors.43 Under CUMS exposure, the PSD group exhibited notably less weight gain compared to the Sham group, and EA treatment reversed this weight reduction in PSD mice, consistent with most conventional antidepressants. The PSD group demonstrated significantly decreased sucrose preference (%) (indicating anhedonia), distinctly increased immobility time in FST (suggesting despair-like behavior), and significantly reduced horizontal and vertical scores in OFT (reflecting anxiety-like behavior). Following EA intervention, all measured parameters—including immobility time in the FST, sucrose preference (%), and horizontal/vertical scores in the OFT—were significantly improved. These results collectively demonstrate that EA rescued PSD-induced depressive-like behaviors, including anhedonia, behavioral despair, and anxiety-like behavior. However, AN administration reversed these therapeutic effects of EA.
Meanwhile, the results demonstrated that EA treatment significantly reduced nuclear pyknosis in granulosa cells and increased the number of Nissl bodies in the hippocampal DG region, suggesting a neuroprotective effect against ischemic brain injury.
However, several limitations of this study should be acknowledged. First, we did not directly measure neurotransmitter levels. Whether EA restores monoamine balance beyond its anti-inflammatory effects remains to be determined. Second, our histological quantification focused only on the DG. The potential neuroprotective effects of EA in CA1, CA2, and CA3 cannot be excluded and require future investigation. Third, mNSS was assessed only at 24 h and 48 h. The long-term effects of EA on motor function warrant further study with extended observation.
Conclusion
EA attenuates PSD in a mouse model by inhibiting the JNK/NF-κB pathway and inflammatory cytokines. This effect is associated with reduced neuronal necrosis and also with the amelioration of depressive-like behaviors. These research findings provide preliminary evidence for further exploration of the therapeutic potential of EA in treating PSD.
Abbreviations
PSD, post-stroke depression; BDNF, brain-derived neurotrophic factor; SSRIs, selective serotonin reuptake inhibitors; EA, echinocystic acid; BBB, blood-brain barrier; AN, anisomycin; CUMS, chronic unpredictable mild stress; FST, forced swimming test; OFT, open field test; SPT, sucrose preference test; PVDF, polyvinylidene fluoride; PBS, phosphate-buffered saline; ANOVA, analysis of variance; DG, dentate gyrus; MAPK, mitogen-activated protein kinase.
Data Sharing Statement
The data that support the findings of this study are available from both corresponding authors.
Author Contributions
Dandan Wang: Writing – review & editing, Writing – original draft, Validation, Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Wei Li: Writing – review & editing, Writing – original draft, Validation, Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Xuan Zhang: Writing – review & editing, Investigation. Kaiqi Zhu: Writing – review & editing, Investigation. Yuzhen Wang: Writing – review & editing, Formal analysis. Yaozhuo Cai: Writing – review & editing, Visualization. Hao Chen: Writing – review & editing, Formal analysis. Xueli Cai: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Project administration, Funding acquisition, Methodology, Data curation, Conceptualization. Jingping Sun: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Conceptualization.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Zhejiang Provincial Traditional Chinese Medicine Health Service Research Project (Grant No. 2023ZF062) and the Zhejiang Medical Association Project (Grant No. 2022ZYC-Z41).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferrari F, Villa RF. The neurobiology of depression: an integrated overview from biological theories to clinical evidence. Mol Neurobiol. 2017;54(7):4847–14. doi:10.1007/s12035-016-0032-y
2. Mitchell AJ, Sheth B, Gill J, et al. Prevalence and predictors of post-stroke mood disorders: a meta-analysis and meta-regression of depression, anxiety and adjustment disorder. Gen Hosp Psychiatry. 2017;47:48–60. doi:10.1016/j.genhosppsych.2017.04.001
3. Feng X, Ma X, Li J, et al. Inflammatory pathogenesis of post-stroke depression. Aging Dis. 2024;16(1):209–238. doi:10.14336/AD.2024.0203
4. Zhang Y, Yang Y, Li H, Feng Q, Ge W, Xu X. Investigating the potential mechanisms and therapeutic targets of inflammatory cytokines in post-stroke depression. Mol Neurobiol. 2024;61(1):132–147. doi:10.1007/s12035-023-03563-w
5. Yang B, Wang L, Nie Y, Wei W, Xiong W. proBDNF expression induces apoptosis and inhibits synaptic regeneration by regulating the RhoA-JNK pathway in an in vitro post-stroke depression model. Transl Psychiatry. 2021;11(1):578. doi:10.1038/s41398-021-01667-2.
6. Lai C, He W, Yang H, Lai J, Huang S. Electroacupuncture improved depressive behaviors and synaptic plasticity of post-stroke depressed mice via inhibiting the JNK signaling pathway. Neurological Res. 2026;48(1):12–27. doi:10.1080/01616412.2025.2520017
7. Kasahara J, Uchida H, Tezuka K, Oka N. Postischemic anhedonia associated with neurodegenerative changes in the hippocampal dentate gyrus of rats. Neural Plast. 2016;2016:1–20. doi:10.1155/2016/5054275
8. Abdoulaye IA, Wu SS, Chibaatar E, et al. Ketamine induces lasting antidepressant effects by modulating the NMDAR/CaMKII-mediated synaptic plasticity of the hippocampal dentate gyrus in depressive stroke model. Neural Plast. 2021;2021(1):6635084. doi:10.1155/2021/6635084
9. Dos Santos IRC, Dias MNC, Gomes-Leal W. Microglial activation and adult neurogenesis after brain stroke. Neural Regen Res. 2021;16(3):456–459. doi:10.4103/1673-5374.291383
10. Liu L, Marshall IJ, Li X, et al. Long-term outcomes of depression up to 10-years after stroke in the South London Stroke Register: a population-based study. Lancet Reg Health Eur. 2025;54:101324. doi:10.1016/j.lanepe.2025.101324
11. Castilla-Guerra L, Fernandez Moreno MDC, Esparrago-Llorca G, Colmenero-Camacho MA. Pharmacological management of post-stroke depression. Expert Rev Neurother. 2020;20(2):157–166. doi:10.1080/14737175.2020.1707666
12. Dold M, Kasper S. Dose-response relationship of selective serotonin reuptake inhibitors. Acta Psychiatr Scand. 2020;142(6):427–429. doi:10.1111/acps.13262
13. Zhang Y, Liu X, Wu J, et al. Possible association between concomitant use of SSRIs with NSAIDs and an increased risk of adverse events among people with depressive disorders: data mining of FDA adverse event reporting system. Pharmaceuticals. 2025;18(7):1062. doi:10.3390/ph18071062
14. Park HJ, Kwon H, Lee S, et al. Echinocystic acid facilitates neurite outgrowth in neuroblastoma Neuro2a cells and enhances spatial memory in aged mice. Biol Pharm Bull. 2017;40(10):1724–1729. doi:10.1248/bpb.b17-00324
15. Jæger D, Ndi CP, Crocoll C, et al. Isolation and structural characterization of echinocystic acid triterpenoid saponins from the australian medicinal and food plant acacia ligulata. J Nat Prod. 2017;80(10):2692–2698. doi:10.1021/acs.jnatprod.7b00437
16. Lai P, Liu Y. Echinocystic acid, isolated from gleditsia sinensis fruit, protects endothelial progenitor cells from damage caused by oxLDL via the akt/eNOS pathway. Life Sci. 2014;114(2):62–69. doi:10.1016/j.lfs.2014.07.026
17. Jung IH, Jang SE, Joh EH, Chung J, Han MJ, Kim DH. Lancemaside a isolated from codonopsis lanceolata and its metabolite echinocystic acid ameliorate scopolamine-induced memory and learning deficits in mice. Phytomedicine. 2012;20(1):84–88. doi:10.1016/j.phymed.2012.09.005
18. Li S, Han J, Wang D, et al. Echinocystic acid reduces reserpine-induced pain/depression dyad in mice. Metab Brain Dis. 2016;31(2):455–463. doi:10.1007/s11011-015-9786-6
19. He D, Hu G, Zhou A, et al. Echinocystic acid inhibits inflammation and exerts neuroprotective effects in MPTP-induced Parkinson’s disease model mice. Front Pharmacol. 2022;12:787771. doi:10.3389/fphar.2021.787771
20. Joh EH, Gu W, Kim DH. Echinocystic acid ameliorates lung inflammation in mice and alveolar macrophages by inhibiting the binding of LPS to TLR4 in NF-κB and MAPK pathways. Biochem Pharmacol. 2012;84(3):331–340. doi:10.1016/j.bcp.2012.04.020
21. Kuhn M, Sühs KW, Akmatov MK, et al. Mass-spectrometric profiling of cerebrospinal fluid reveals metabolite biomarkers for CNS involvement in varicella zoster virus reactivation. J Neuroinflammation. 2018;15(1):20. doi:10.1186/s12974-017-1041-0
22. Guo B, Zhao C, Zhang C, et al. Elucidation of the anti-inflammatory mechanism of er miao san by integrative approach of network pharmacology and experimental verification. Pharmacol Res. 2022;175:106000. doi:10.1016/j.phrs.2021.106000
23. Yu H, Li W, Cao X, et al. Echinocystic acid, a natural plant extract, alleviates cerebral ischemia/reperfusion injury via inhibiting the JNK signaling pathway. Eur J Pharmacol. 2019;861:172610. doi:10.1016/j.ejphar.2019.172610
24. Zhao TC, Zhang L, Liu JT, Guo TL. Disruption of Nox2 and TNFRp55/p75 eliminates cardioprotection induced by anisomycin. Am J Physiol Heart Circ Physiol. 2012;303(10):H1263–H1272. doi:10.1152/ajpheart.00306.2012
25. Chen Y, Wang L, Zhang L, et al. Inhibition of connexin 43 hemichannels alleviates cerebral ischemia/reperfusion injury via the TLR4 signaling pathway. Front Cell Neurosci. 2018;12:372. doi:10.3389/fncel.2018.00372
26. Ou Z, Li P, Wu L, et al. Albiflorin alleviates neuroinflammation of rats after MCAO via PGK1/Nrf2/HO-1 signaling pathway. Int Immunopharmacol. 2024;137:112439. doi:10.1016/j.intimp.2024.112439
27. Ge Y, Yang J, Chen J, et al. Absence in
28. Qian L, Huang S, Liu X, et al. Morroniside improves the symptoms of post-stroke depression in mice through the BDNF signaling pathway mediated by MiR-409-3p. Phytomedicine. 2023;123:155224. doi:10.1016/j.phymed.2023.155224
29. Zhou H, Wei YJ, Xie GY. Research progress on post-stroke depression. Exp Neurol. 2024;373:114660. doi:10.1016/j.expneurol.2023.114660
30. Villa RF, Ferrari F, Moretti A. Post-stroke depression: mechanisms and pharmacological treatment. Pharmacol Ther. 2018;184:131–144. doi:10.1016/j.pharmthera.2017.11.005
31. De Los Reyes Corrales T, Losada-Pérez M, Casas-Tintó S. JNK pathway in CNS pathologies. IJMS. 2021;22(8):3883. doi:10.3390/ijms22083883
32. Xu L, Li Y, Fu Q, Ma S. Perillaldehyde attenuates cerebral ischemia–reperfusion injury-triggered overexpression of inflammatory cytokines via modulating akt/JNK pathway in the rat brain cortex. Biochem Biophys Res Commun. 2014;454(1):65–70. doi:10.1016/j.bbrc.2014.10.025
33. Wen Z, Zhang Y, Luo M, et al. Microcystin-LR drives hepatic meta-inflammation and insulin resistance by hijacking the PP2A-JNK signaling axis. Int Immunopharmacol. 2026;168:115832. doi:10.1016/j.intimp.2025.115832
34. Alipourgivi F, Lu T. Celebrating the 40-year milestone: NF-ĸB in oncoimmunity. Cancer Lett. 2026;636:218087. doi:10.1016/j.canlet.2025.218087
35. Ma R, Ma J, Zhang H. Management of depression utilizing Traditional Chinese Medicine. Front Pharmacol. 2026;17:1763214. doi:10.3389/fphar.2026.1763214
36. Liu J, Fang Y, Yang L, Qin X, Du G, Gao X. A qualitative, and quantitative determination and pharmacokinetic study of four polyacetylenes from radix bupleuri by UPLC-PDA–MS. J Pharm Biomed Anal. 2015;111:257–265. doi:10.1016/j.jpba.2015.04.002
37. Li Y, Chen L, Zheng D, et al. Echinocystic acid alleviated hypoxic-ischemic brain damage in neonatal mice by activating the PI3K/akt/Nrf2 signaling pathway. Front Pharmacol. 2023;14:1103265. doi:10.3389/fphar.2023.1103265
38. Khan MS, Khan A, Ahmad S, et al. Inhibition of JNK alleviates chronic hypoperfusion-related ischemia induces oxidative stress and brain degeneration via Nrf2/HO-1 and NF- κ B signaling. Oxid Med Cell Longev. 2020;2020:1–18. doi:10.1155/2020/5291852
39. Liu F, Liu TW, Kang J. The role of NF-κB-mediated JNK pathway in cognitive impairment in a rat model of sleep apnea. J Thorac Dis. 2018;10(12):6921–6931. doi:10.21037/jtd.2018.12.05
40. Kang K, Chen SH, Wang DP, Chen F. Inhibition of endoplasmic reticulum stress improves chronic ischemic hippocampal damage associated with suppression of IRE1α/TRAF2/ASK1/JNK-dependent apoptosis. Inflammation. 2024;47(4):1479–1490. doi:10.1007/s10753-024-01989-5
41. Hammo A, Wisser S, Cichon J. Single-dose psilocybin rapidly and sustainably relieves allodynia and anxiodepressive-like behaviors in mouse models of chronic pain. Nat Neurosci. 2025;28(11):2285–2295. doi:10.1038/s41593-025-02068-0
42. Liu MY, Yin CY, Zhu LJ, et al. Sucrose preference test for measurement of stress-induced anhedonia in mice. Nat Protoc. 2018;13(7):1686–1698. doi:10.1038/s41596-018-0011-z
43. Ferreira J. Factors contributing to the open-field test variability. Lab Anim. 2025;54(6):135. doi:10.1038/s41684-025-01570-z
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.