")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Early-onset Alzheimer’s disease patient with prion (PRNP) p.Val180Ile mutation
Authors Bagyinszky E, Kang MJ, Pyun J, Giau VV , An SSA , Kim S
Received 10 May 2019
Accepted for publication 18 June 2019
Published 16 July 2019 Volume 2019:15 Pages 2003—2013
DOI https://doi.org/10.2147/NDT.S215277
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Taro Kishi
Eva Bagyinszky1,*, Min Ju Kang2,*, Jungmin Pyun,3 Vo Van Giau,1 Seong Soo A An,1 SangYun Kim3
1Department of Bionano Technology, Gachon Medical Research Institute, Gachon University, Sungnamsi 461-701, Republic of Korea; 2Department of Neurology, Veterans Medical Research Institute, Veterans Health Service Medical Center, Seoul, Republic of Korea; 3Department of Neurology, Seoul National University College of Medicine and Neurocognitive Behavior Center, Seoul National University Bundang Hospital, Seongnam-si, Gyeonggi-do, 463-707, Republic of Korea
*These authors contributed equally to this work
Background: In this study, a known PRNP mutation, Val180Ile (c.G538A), was reported in a 58 years old female patient, clinically diagnosed with Alzheimer’s disease (AD).
Case report: The patient presented slowly progressive cognitive decline in memory and visuospatial domain. Neuroimaging showed hippocampal atrophy in MRI and mild amyloid positivity in PET scan. Even though her cerebrospinal fluid (CSF) was positive for 14–3-3 protein, no sign of Creutzfeldt-Jakob diseases symptoms was observed. In addition, reduced Aβ42 and elevated total-Tau and phospho-Tau in CSF also proved the AD diagnosis. The mutation may disturb the hydrophobic core of prion protein, and result in abnormal intramolecular interactions. Due to 23andMe, PRNP Val180Ile could not be categorized either as a mutation with complete penetrance, or as neutral variant, and could have a possible role in neurodegeneration. Pathological overlap was observed between prion diseases and other neurodegenerative diseases, including AD or frontotemporal dementia.
Conclusion: Whole exome sequencing and pathway analysis of patient revealed rare or possible risk variants in AD associated genes, such as SORL1 or ABCA7. Along with PRNP, AD risk genes may play a role in negative regulation of amyloid formation. Dysfunctions in these genes could possibly be associated in reduced neuroprotection and amyloid clearance.
Keywords: Alzheimer’s disease, prion, PRNP Val180Ile mutation, Creutzfelt -Jakob disease
Introduction
Human prion diseases are fatal and rare neurodegenerative diseases. The incidence of the disease is approximately one to two cases per million per year.1 Creutzfeldt-Jakob diseases (CJD) are the most prevalent subtype of human prion disease with rapidly progressive dementia, cerebellar ataxia, myoclonus, and behavioral changes.2 Although the majority of CJD cases may be sporadic, genetic prion disease could be caused by mutations in the prion protein gene (PRNP). Genetic CJD or familial CJD accounted for only 10–15% of all CJD cases.3 Prion diseases are characterized by an accumulation of an abnormally folded isoform (PrPSc) of the host-encoded cellular prion protein (PrPC). Up to now, about 50 different pathogenic mutations in PRNP have been addressed, which result in dominant hereditary prion diseases.4,5 Mutations showed diversity in clinical manifestations depending on each genotype.6 Prion diseases could also share similarities with additional neurodegenerative diseases, including Alzheimer’s disease (AD) or other types of dementia. AD and prion diseases were both verified as age-related diseases with possible genetic causative factors. In cases of genetic mutation, both of them could be inherited autosomal dominantly. In addition, both prion diseases and AD are based of aggregation of abnormally folding proteins, resulting in neurodegeneration. The abnormal protein assembly could be caused by aging, oxidative stress, abnormal inflammatory process, and reduced defensive mechanisms. The two main hallmarks of AD (senile plaques and neurofibrillary tangles) could also be presented in the brain of CJD patients.7,8
PRNP Val180Ile mutation was suggested as one of the causative mutations for familial CJD,4 and several cases were reported in South Korea.9 Herein, we reported the clinical phenotypes of a 58-year-old female patient with PRNP Val180Ile mutation, who developed progressive cognitive impairment. The disease progression was slow in her and did not present any typical neurologic features of CJD, such as myoclonus or parkinsonian features.
Materials and methods
Whole exome sequencing (WES) was performed on the proband patient by Novogene Inc (https://en.novogene.com; Beijing, China). The DNA was isolated from white blood cells, and 2 µg of genomic DNA was used for the sequencing process. After library preparation, the Illumina platform was used for WES. Whole annotation of data was received as an Excel file, and sequencing data was sent as a .bam file. The Integrative Genomics Viewer (IGV) tool was used to visualize the sequencing data. An extensive analysis of 100 possible causative or risk factor genes of different neurodegenerative diseases (such as Alzheimer’s disease, Parkinson’s disease, frontotemporal dementia, dementia with Lewy bodies, or prion diseases) was carried out on the sequencing data (Table S1).
In silico analysis was performed by online tools, including PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and SIFT (http://sift.jcvi.org/) software, PROVEAN, or ExPASY (https://www.expasy.org/) tools. Structures of normal and mutant prion proteins were constructed by the Raptor X (http://raptorx.uchicago.edu/) software and visualized by Discovery Studio 3.5 Visualizer.10 Genes, which were binding any mutations (either common or rare) were analyzed by ClueGO v2.0.5. This tool visualized biological networks of gene interactions, which would group the genes into a network with statistical evaluations, based on the annotations in the Gene Ontology.11
Case report
A 58-year-old right-handed woman (II-2) with 12 years of education presented progressive memory difficulties in the past year. She reported that the symptoms occurred insidiously, with disorganization and inconsistency in the performance of her duties at work. She regularly misplaced items, showed signs of short-term memory loss, and could not find the way home. Difficulties were also observed in managing her daily activities. No additional family members were reported with similar neurodegenerative disease (Figure 1). Her mother died in her 80s of heart disease (I-1), her father was neurologically normal until his death in his 90s (I-2). One of her brothers (II-4) had died of an accident (in his 40s) and did not present any known neurological abnormality before his death. She had three siblings, one sister (67 years, II-1) and two brothers (62 years and 60 years, II-3 and II-5, respectively), who were healthy and cognitively normal. Negative family history could not be proven, since all living family members refused the genetic test.
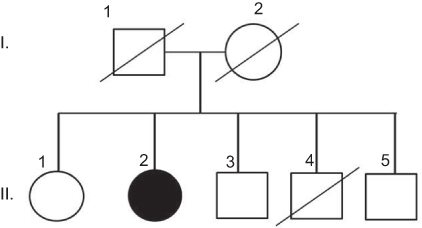 |
Figure 1 Family history of patient. She did not have any family history of disease. Notes: Parents I (I-1 and I-2) and 3 siblings (II-1, II-3, II-5) of patient (II-2) were unaffected. The 3rd sibling (II-4) died in his 40s. |
Upon cognitive testing, the patient was scored 15/30 on the Mini-Mental State Examination (MMSE) test (impairment in memory, language, visuospatial, and frontal domain) Her initial investigation, electrolytes, renal function, thyroid function, and complete blood count were within normal ranges. The patient’s MRI revealed global atrophy with medial temporal lobe and hippocampal atrophy predominance (Figures 2A–C). Aggravated diffuse brain atrophy and small vessel ischemic lesion were observed in follow-up MRI, but no specific abnormality was detected in diffusion-weighted image (DWI, Figures 2A–C). There was no DWI, which implied possible CJD. The Axial 18F-fluorbetaben positron emission tomography (FDG-PET) of the patient shows mild amyloid positivity, and increased diffusion uptake over the bilateral cortices, and increased cerebral cortex (Figure 2D. Electroencephalography (EEG) was also normal without periodic synchronous discharge. The patient was initially diagnosed as probable Alzheimer’s disease by the National Institute on Aging–Alzheimer’s Association (NIA–AA) criteria.12 She was prescribed with an anti-acetylcholinesterase inhibitor, donepezil. One year later, her cognitive deficits were more noticeable. Her husband reported that the patient stopped her daily activities outside her house such as exercise and social activity, and she became unable to perform her activities of daily living. Her MMSE score was reduced to 13/30. Two years later, the MMSE score was dropped to 10 out of 30, and global clinical dementia rating was 2 and sum of box score was 11. Her visuospatial function impairment became more noticeable. She was unable to notice the food on the table, and also developed prosopagnosia. Cerebrospinal fluid (CSF) analysis, including cell count, and venereal disease research laboratory (VDRL) test, protein, and glucose, was normal. However, the CSF 14–3-3 protein was positive. Enzyme-linked immunosorbent assays (ELISA) analyses were performed on Ab42, total tau, and phospho-tau in CSF. Reduced levels of Ab42 (326.8 pg/mL) with elevated total tau (732.9 pg/mL) and phospho-Tau (90.6 pg/µL) protein concentrations were observed. For the accurate disease diagnosis, extensive genetic screening was performed on the patient.
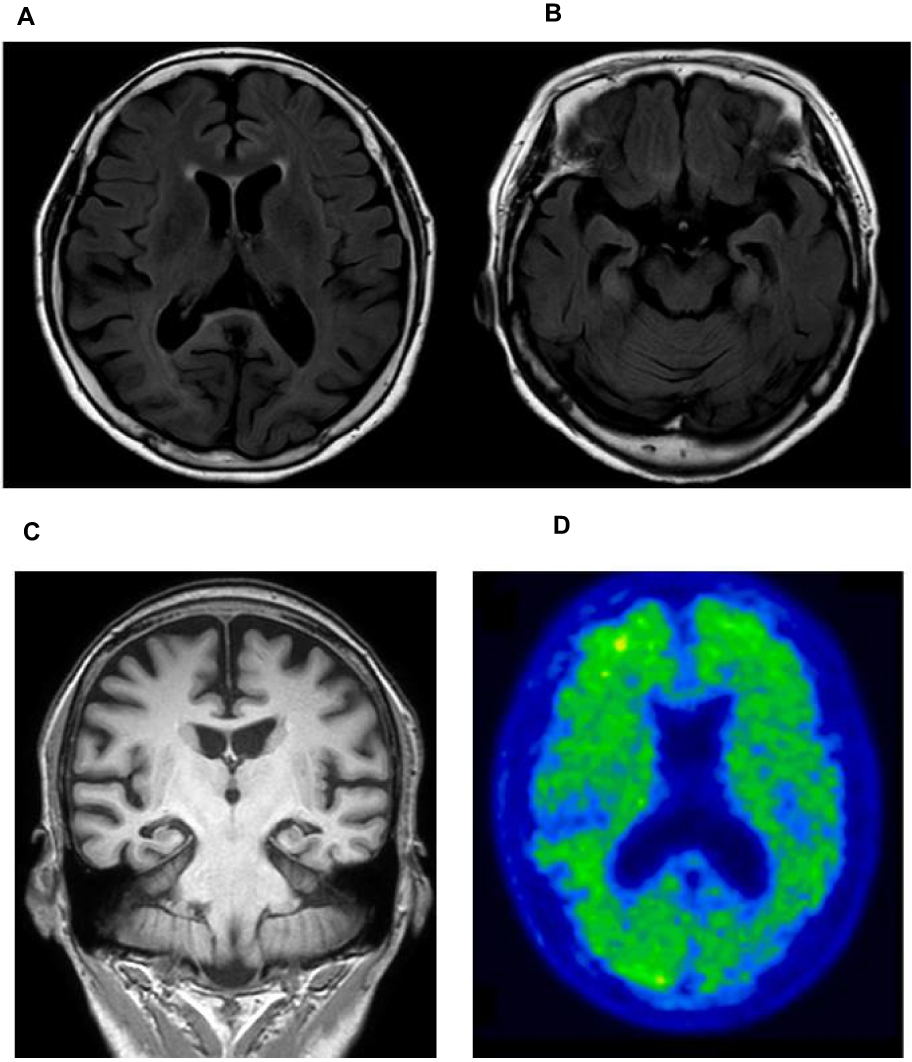 |
Figure 2 (A) Axial FLAIR image of the patient shows moderate atrophy in the bilateral cortices and (B) moderate atrophy in the hippocampus. (C) Coronal MRI image of patient. (D) Axial 18F-fluorbetaben positron emission tomography of the patient shows increased diffusion uptake over the bilateral cortices. |
Written informed consent was obtained from the patient for publication of this case report. This report was approved by the Institutional Review Board of Seoul National University Bundang Hospital (B-1612/376–701).
Results
Whole exome analysis did not reveal any mutations in APP, PSEN1, or PSEN2. A silent mutation (c.120G>A, p.Gly40, Figure 3A) and a known possible pathogenic mutation in PRNP at codon 180 (c.538G>A; p.Val180Ile Figure 3B) was detected. In addition, several common and rare mutations were found in additional risk genes (Table S1).
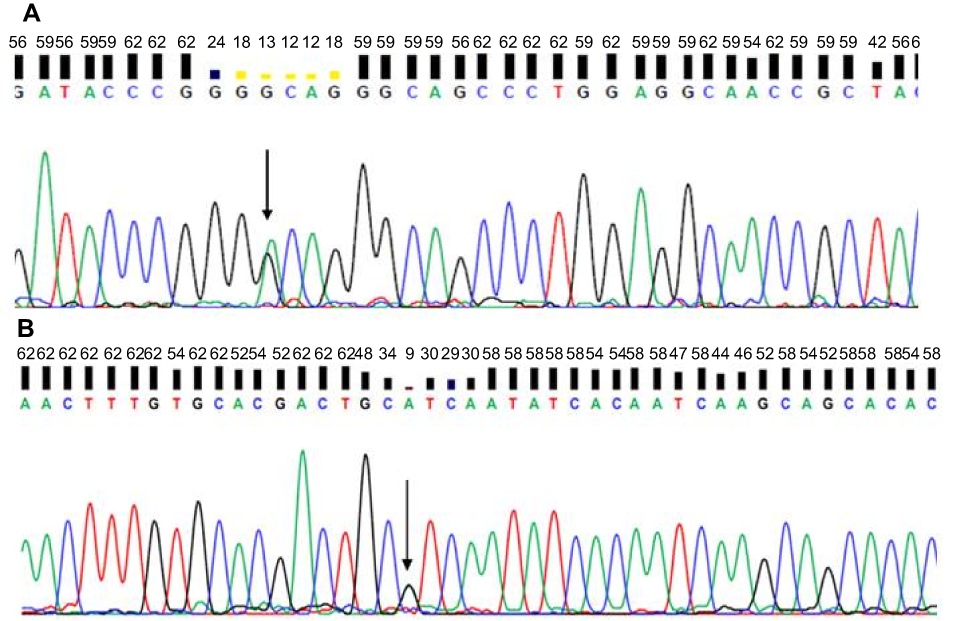 |
Figure 3 Sequencing data of patient with PRNP. (A) G40 and (B) V180I.Note: Arrows indicate mutation. |
Frequencies of PRNP Val180Ile were 0.00019 and 0.000049, in 1000Genomes and EXAC, respectively. ExAC frequency in East Asia was 0.0003, while in South-East Asia it was 0.0002. Both PolyPhen2 and SIFT revealed the mutation as possibly/probably damaging. PolyPhen2 HumDiv and HumVar scores were 0.937 and 0.887, respectively. SIFT prediction was also damaging, with the score of 0.014, while PROVEAN revealed this mutation as neutral with a score of −0.11. The ExPASY (Figure 4) tool did not reveal significant changes due to Val180Ile. A low increase was found in Kyte and Dootile hydrophobicity scores (V180: 1; I180: 1.033). No significant reduction was seen in bulkiness scores (V180: 17.04, I180: 17.021). In polarity scores (Grantham), low reduction could be seen due to mutation (V180: 7.922; I180: 7.844).
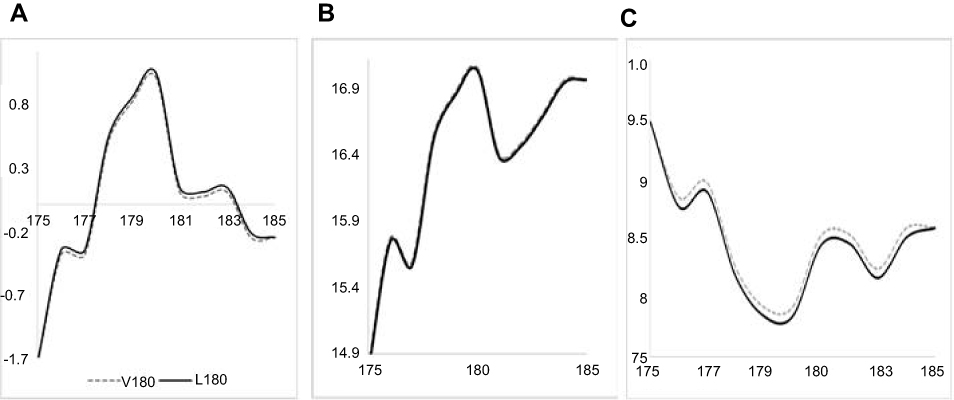 |
Figure 4 ExPASY prediction for PRNP V180I mutation. (A) Kyte Dootile hydrophobicity scores. (B) Bulkiness scores. (C) Polarity scores. |
The in silico 3D model predicted that mutation could disturb the protein structure and interactions. A 3D model was performed on normal and mutant prion protein. Our in silico analysis did not predict any significant helix torsion due to Val180Ile, but interactions between α2 and α3 helices changed significantly. Val180 was predicted to be on hydrophobic contact with Cys214 on the α3 helix. Ile180 enhanced the contact of α2–α3 helices, since it could form a hydrogen bond with Glu211 and additional hydrophobic contact with Val210. Orientation of Ile180 was also changed, which could result in reduced distance and stronger contact between α2–α3 helices (Figure 5).
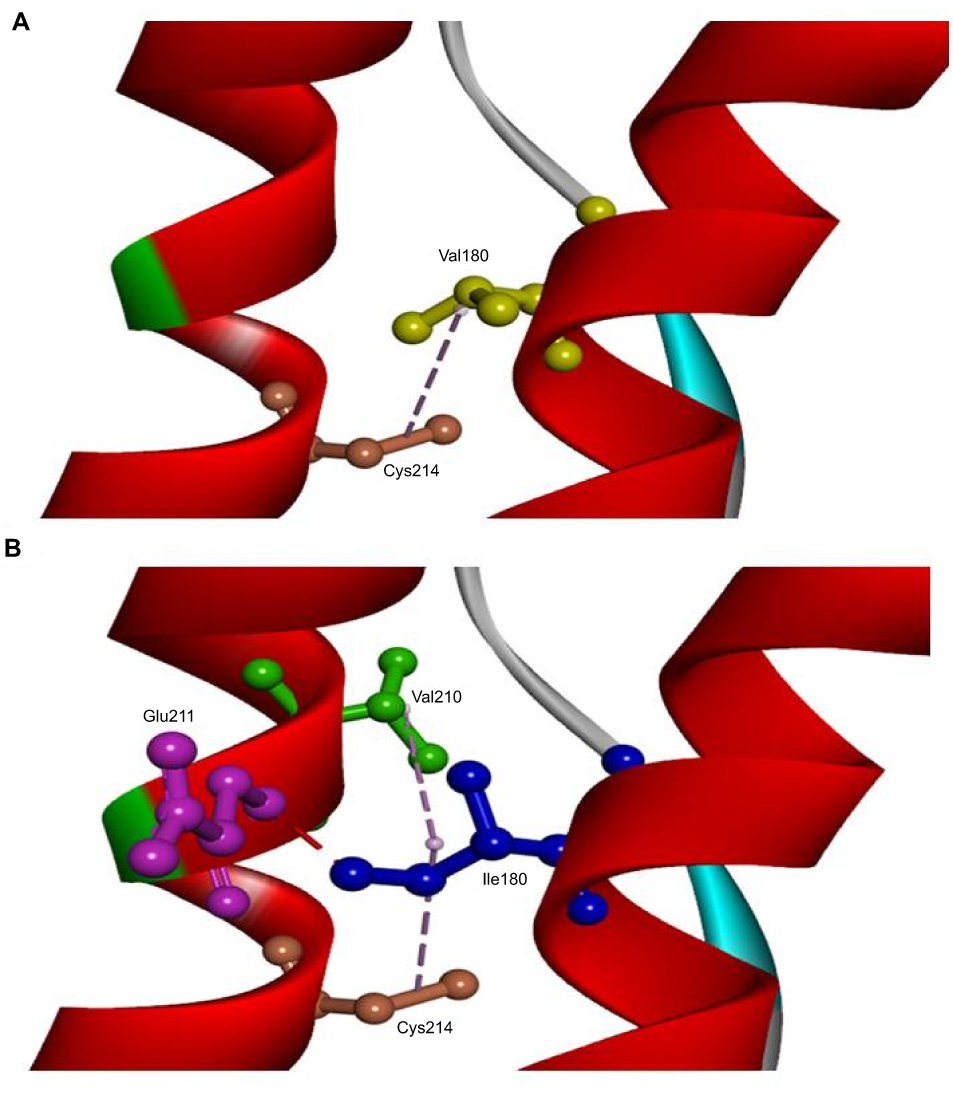 |
Figure 5 3D model of normal PRNP and PRNP with V180I mutation. (A) Val180 (yellow) was predicted to be on hydrophobic contact with Cys214 (orange) on α3 helix. (B) Ile180 (blue) enhanced the contact of α2-α3 helices, since it could form a hydrogen bond with Glu211 (purple) and additional hydrophobic contact with Val210 (green). The orientation of Ile180 was also changed, which could result in a reduced distance between α2–α3 helices. |
ClueGo (Figure 6) mapping was also performed on the patient, and revealed a complex network of genes. Pathway analysis suggested that PRNP could interact with several genes. PRNP could directly play a role negative regulation of amyloid beta formation along with several AD risk genes (such as ABCA7, SORL1, and BACE1), regulation of calcium mediated signaling with different disease risk genes such as AD (CR1), and PD (BST1, SYT11, PRK2B). Gene interactions revealed additional possible pathways, in which PRNP could contribute, such as lysosome organization, cell response to hydrogen peroxide, or regulation of mitochondrion autophagy.
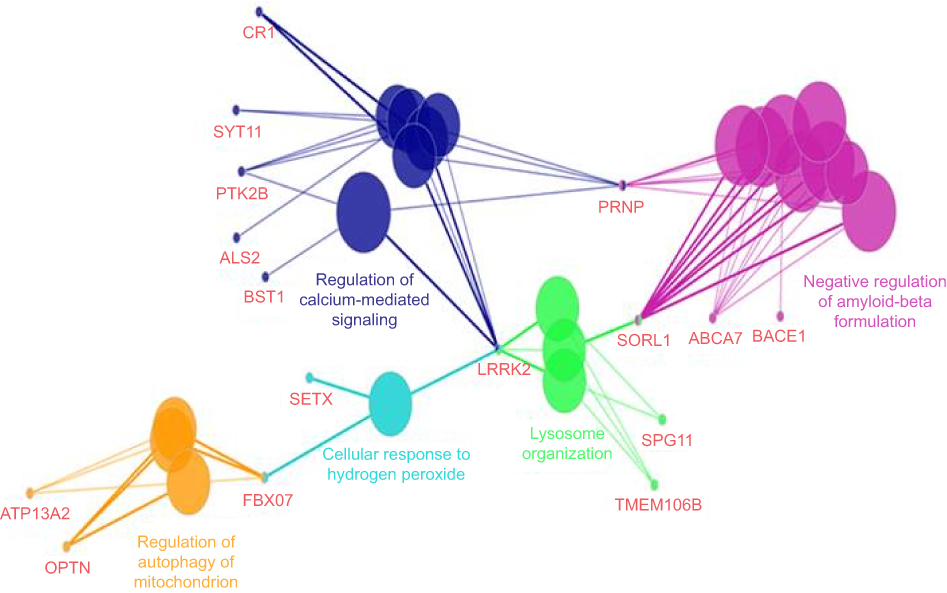 |
Figure 6 ClueGo mapping of mutations, found in the proband patient. |
Discussion
PRNP Val180Ile was commonly observed in East-Asian (Korean, Chinese, and Japanese) patients, but it was also found in a few European CJD cases.4,12 Val180Ile mutation could be causative mutations for CJD, and its occurrence was rare. There are two previously reported cases of sporadic CJD (sCJD) with PRNP Val180Ile mutation in South Korea.13,14 The clinical and laboratory characteristics of CJD patients with the Val180Ile mutation have been described by Jin et al.12 The features could include 1) late age of onset, 2) slow disease progression, 3) unique clinical symptoms such as frequent cognitive dysfunction, but no visual or cerebellar symptoms, 4) reduced rate of brain-specific proteins such as neuron-specific enolase (NSE) and 14–3-3 protein in CSF, and 5) a lack of periodic synchronous discharge in electroencephalogram (EEG) throughout the course of disease. The proband patient in this study did not match any of these features of sCJD, associated with PRNP Val180Ile.
The clinical course was distinctively slower than other CJD cases, which were indicated by the absence of the marked drop of MMSE over 4 years (15 to 10 in 4 years). Additionally, the patient was still alive at the time of 48-month follow-up. Cardinal symptoms of CJD such as myoclonic jerk or parkinsonism were not observed. Moreover, the typical MRI abnormalities for sCJD patients with Val180Ile, such as diffuse cortical high-intensity DWI signal, were not revealed in the proband. The florbetaben PET image showed mild diffuse amyloid deposit on bilateral cortices. CSF biomarker of total Ab42, total Tau and phospho-Tau analyses also correlated with AD, which also proved that disease diagnosis.15 The overall clinical characteristics, laboratory and imaging data corresponded with early onset AD. It remained uncertain whether the PRNP mutation was incidentally found in the early onset AD patient or the sCJD with the Val180Ile patient would have atypical clinical course. Furthermore, it remained questionable whether the Val180Ile mutation could always be responsible for the expression of CJD. Nevertheless, several previous reports support the hypothesis that PRNP mutation is not always associated with a prion disease. According to the genotype analysis of PRNP in a South Korean population, including prion disease patients group (n=22), suspected prion diseases patient group (n=163) and Korea Association Resource (KARE) data group, three cases of Val180Ile mutations were reported, and two cases from the KARE data group did not display characteristics or symptoms of the neurodegenerative disorder.16 Prion diseases may share some features such as dementia phenotype and pathological characteristics with AD. A series of studies suggests the hypothesis that the mutations of PRNP also account for other neurodegenerative diseases, including AD. For example, patients with PRNP Ser17Gly,17 Tyr145Ter,18 Gln160Ter,19 Tyr163Ter,20 or Ile215Val21 were diagnosed with AD or AD-like phenotypes. PRNP Val180Ile has low frequency in the ExAC database (0.00004942, http://exac.broadinstitute.org/). In the 23andMe project, Val180Ile was suggested as a mutation with contradictory pathogenic nature. Even though the pathogenicity of mutation should not be ignored, it was suggested to have low penetrance.22 Val180Ile was reported in a patient with CJD and Alzheimer’s type pathology. In the brain of the patient, several senile plaques appeared with a moderate number of neurofibrillary tangles. The degree of spongiosis was lower than the typical CJD cases. AD and CJD pathology may coexist in a few patients.23 The rlationship between AD and PRNP has been investigated, and prion disease may share some overlap with AD. Both of them are neurodegenerative diseases, based on the abnormal protein conformation and accumulation. In addition, they could have familial and sporadic forms.17
PRNP Val180Ile could be involved in a late onset form of CJD or other neurodegenerative diseases. Alzheimer’s like phenotypes were also found in several cases of mutation. AD with PRNP V180I mutation occurred in patients in their 70s, and disease duration was long, since patients survived for 4–8 years after onset. Imaging data presented hippocampal atropy, and EEG did not present periodic sharp wave complexes.24 It may be possible that additional genetic factors or genetic interactions could act as disease-modifiers, and result in earlier disease onset. This proband patient presented similar phenotypes to previous cases, but disease onset was much younger in her. Pathway analysis (Figure 6) with the additional variants in different risk genes revealed a complex network of disease-causing and risk genes, found in the patients (Table S1). Interacting with SORL1 and ABCA7, PRNP could play a role in negative regulation of Aβ formation. In the patient, a rare mutation was found in ABCA7 (Ala583Asp) and a possible AD risk mutation was found in SORL1 (Ala528Thr). Both ABCA7 and SORL1 could impact the amyloid metabolism, and their role in the early onset form of AD has been investigated.25,26 Interaction between SORL1, and amyloid peptides were proven, since sortilin may play a role in the defense against amyloid aggregation by promoting α-secretase mechanisms. It could also be involved in amyloid trafficking and clearance.27,28 SORL1 could also protect against prion infection, since the VPS10 domain of sortilin receptor could play a role trafficking prions into lysosomes. However, the abnormal prion proteins could promote the disruption of sortilin in lysosomes, reducing the sortilin levels and lysosomal degradation.29,30 Prion induced SORL1 dysfunctions may affect the amyloid production and aggregation. No direct interaction was found between prions and ABCA7; however, they may be related through amyloid-associated mechanisms, such as amyloid clearance and microglial mechanisms. Microglia may be protective against amyloid aggregations. ABCA7 was expressed in microglia, resulting in elevated amyloid clearance and phagocytosis.31 Microglia could also play a possible role in the defense against prion diseases by the clearance of PrPSc.32,33 The ability of interaction between Aβ and prion proteins has been proven, and its interaction may be involved in neurodegeneration. Amyloid peptide aggregates may inhibit prion propagation. Normal prion protein may have neuroprotective effects against oxidative stress and amyloid deposition, by inhibiting the β-secretase activity. Normal prion protein could be down-regulated in sporadic AD.34It may be possible that PRNP Val180Ile could interact with additional AD risk genes, such as with ABCA7 and/or SORL1, which also carried a rare/verified risk mutation. This interaction could result in reduced neuroprotection, amyloid clearance and enhanced amyloid deposition in the patient, which they may also contribute to the earlier disease onset.
Limitations of this study were that we could not perform a brain biopsy on the patient. In addition, even though other affected family members were not affected, a de novo case of mutation could not be verified, since all living family members refused the genetic test. Although PRNP Val180Ile mutation has been reported to cause CJD, findings in the presented patient suggest that Val180Ile mutation may not be causative for AD. However, the role of Val180Ile in disease should not be ignored, and it could be a possible risk factor for AD. PRNP Val180Ile mutation may reduce the neuroprotective effects in AD patients, and, along with other AD risk genes, it may result in increased Aβ42 production. To confirm the functional role of Val180Ile mutation and its pathogenicity, further investigation may be required.
Acknowledgments
This case report was supported by a National Research Foundation of Korea (NRF) Grants awarded by the Korean government (Ministry of Education, Science and Technology, Nos. 2017R1A2B4012636 and 2017R1C1B5017807). The authors would like to express their gratitude to the proband patient and her family members for their time and support.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi:10.1073/pnas.95.23.13363
2. Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006;66:286–287. doi:10.1212/01.wnl.0000196440.00297.67
3. Heinemann U, Krasnianski A, Meissner B, Grasbon-Frodl EM, Kretzschmar HA, Zerr I. Novel PRNP mutation in a patient with a slow progressive dementia syndrome. Med Sci Monit. 2008;14:CS41–CS43.
4. Kovács GG
5. Schmitz M, Dittmar K, Llorens F, et al. Hereditary human prion diseases: an update. MolNeurobiol. 2017;54:4138–4149. doi:10.1007/s12035-016-9918-y
6. Bagyinszky E, Giau VV, Youn YC, An SSA, Kim S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr Dis Treat. 2018;14:2067–2085. doi:10.2147/NDT.S165445
7. Castellani RJ, Perry G, Smith MA. Prion disease and Alzheimer’s disease: pathogenic overlap. Acta Neurobiol Exp (Wars). 2004;64:11–17.
8. Tousseyn T, Bajsarowicz K, Sánchez H, et al. Prion disease induces alzheimer disease-like neuropathologic changes. JNeuropatholExpNeurol. 2015;74:873–888. doi:10.1097/NEN.0000000000000228
9. Lee SM, Chung M, Hyeon JW, et al. Genomic characteristics of genetic creutzfeldt-jakob disease patients with V180I mutation and associations with other neurodegenerative disorders. Plos One. 2016;11:e0157540. doi:10.1371/journal.pone.0157540
10. Kallberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi:10.1038/nprot.2012.085
11. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi:10.1093/bioinformatics/btp101
12. Jin K, Shiga Y, Shibuya S, et al. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology. 2004;62:502–505. doi:10.1212/01.wnl.0000106954.54011.80
13. Kim J, Lee D-S, Park KW. Familial creutzfeldt-jakob disease with V180I mutation presented with broca’s aphasia. J Korean Neurol Assoc. 2018;36:345–349. doi:10.17340/jkna.2018.4.15
14. Yang TI, Jung DS, Ahn BY, et al. Familial Creutzfeldt-Jakob disease with V180I mutation. J Korean Med Sci. 2010;25:1097–1100. doi:10.3346/jkms.2010.25.7.1097
15. Paterson RW, Slattery CF, Poole T, et al. Cerebrospinal fluid in the differential diagnosis of alzheimer’s disease: clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res Ther. 2018;10(1):32. doi:10.1186/s13195-018-0361-3
16. Moe Lee S, Ju Y R, Choi BY, et al. Genotype patterns and characteristics of PRNP in the Korean population. Prion. 2012;6:375–382. doi:10.4161/pri.20195
17. Zhang W, Jiao B, Xiao T, et al. Mutational analysis of PRNP in Alzheimer’s disease and frontotemporal dementia in China. Sci Rep. 2016;6:38435. doi:10.1038/srep38435
18. Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in gerstmann-sträussler syndrome with mutant PrP plaques. Biochem Biophys Res Commun. 1993;192:525–531. doi:10.1006/bbrc.1993.1447
19. Jayadev S, Nochlin D, Poorkaj P, et al. Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol. 2011;69(4):712–720. doi:10.1002/ana.22264
20. Mead S, Gandhi S, Beck J, et al. A novel prion disease associated with diarrhea and autonomic neuropathy. N Engl J Med. 2013;369:1904–1914. doi:10.1056/NEJMoa1214747
21. Muñoz-Nieto M, Ramonet N, López-Gastón JI, et al. A novel mutation I215V in the PRNP gene associated with creutzfeldt-jakob and Alzheimer’s diseases in three patients with divergent clinical phenotypes. J Neurol. 2013;260:77–84. doi:10.1007/s00415-012-6683-3
22. Minikel EV, Vallabh SM, Lek M, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra9. doi:10.1126/scitranslmed.aaf0746
23. Yoshida H, Terada S, Ishizu H, et al. An autopsy case of creutzfeldt-jakob disease with a V180I mutation of the PrP gene and alzheimer-type pathology. Neuropathology. 2010;30:159–164. doi:10.1111/j.1440-1789.2009.01048.x
24. Qina T, Sanjo N, Hizume M, et al. Clinical features of genetic creutzfeldt-jakob disease with V180I mutation in the prion protein gene. BMJ Open. 2014;4:e004968. doi:10.1136/bmjopen-2014-004968
25. Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and alzheimer disease. Ann Neurol. 2015;77(2):215–227. doi:10.1002/ana.24305
26. Aikawa T, Holm ML, Kanekiyo T. ABCA7 and pathogenic pathways of alzheimer’s disease. Brain Sci. 2018;8:
27. Cuccaro ML, Carney RM, Zhang Y, et al. SORL1 mutations in early- and late-onset alzheimer disease. Neurol Genet. 2016;2(6):e116. doi:10.1212/NXG.0000000000000116
28. Gustafsen C, Glerup S, Pallesen LT, et al. Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. J Neurosci. 2013;33(1):64–71. doi:10.1523/JNEUROSCI.2371-12.2013
29. Sakaguchi S, Uchiyama K. Novel amplification mechanism of prions through disrupting sortilin-mediated trafficking. Prion. 2017;11(6):398–404. doi:10.1080/19336896.2017.1391435
30. Uchiyama K, Tomita M, Yano M, et al. Prions amplify through degradation of the VPS10P sorting receptor sortilin. PLoS Pathog. 2017;13(6):e1006470. doi:10.1371/journal.ppat.1006470
31. Fu Y, Hsiao JH, Paxinos G, Halliday GM, Kim WS. ABCA7 mediates phagocytic clearance of amyloid-β in the brain. J Alzheimers Dis. 2016;54(2):569–584. doi:10.3233/JAD-160456
32. Carroll JA, Race B, Williams K, Striebel J, Chesebro B. Microglia are critical in host defense against prion disease. J Virol. 2018;92(15):
33. Carroll JA
34. Griffiths HH, Whitehouse IJ, Hooper NM. Regulation of amyloid-β production by the prion protein. Prion. 2012;6:217–222. doi:10.4161/pri.18988
Supplementary materials
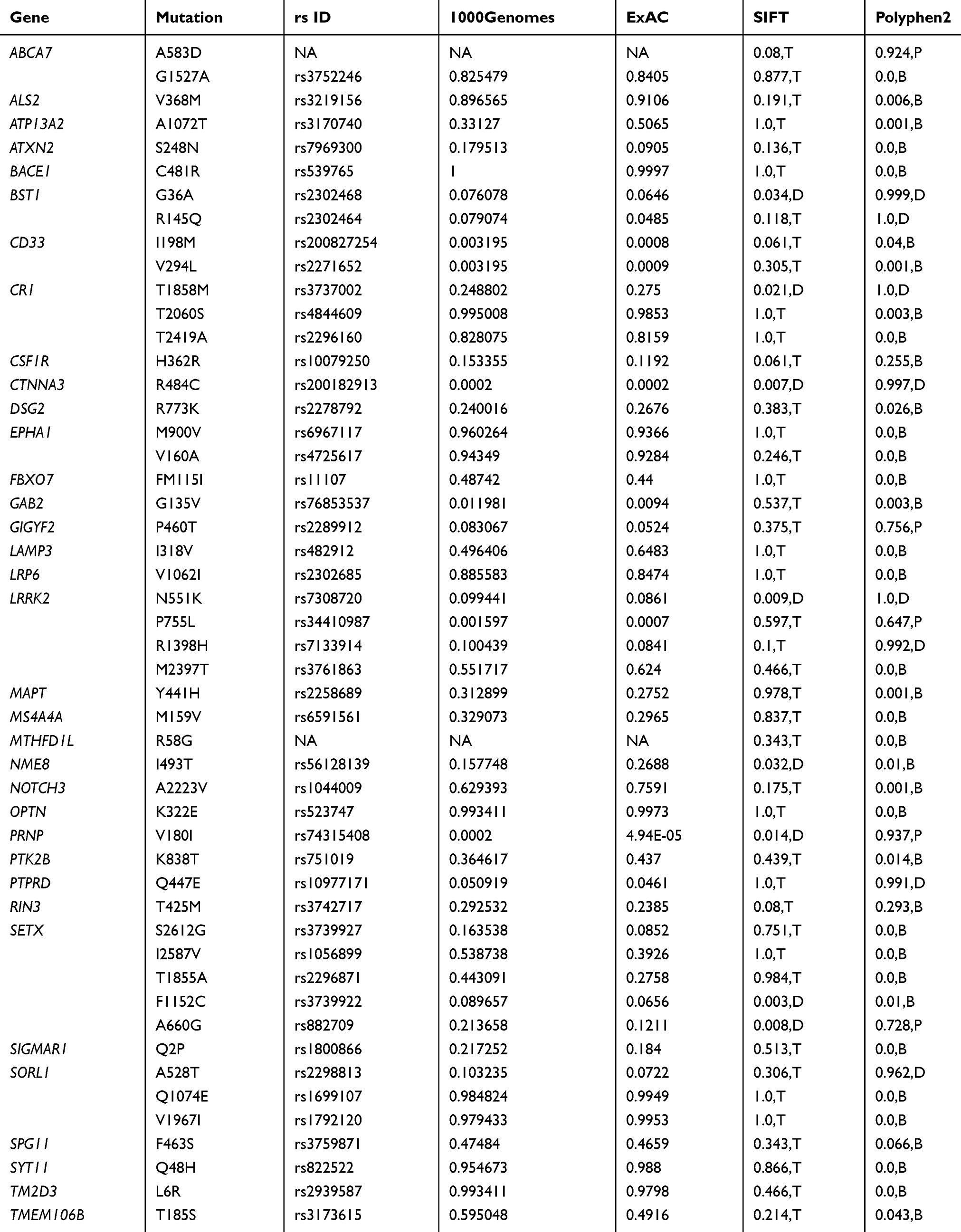 |
Table S1 Mutations, found in the patient by 100 gene panel analysis |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.