Back to Journals » Journal of Inflammation Research » Volume 19
Dysregulated Polarized Secretion of Small Extracellular Vesicles in the Retinal Pigment Epithelium: Mechanisms and Pathological Roles in Age-Related Macular Degeneration
Authors Guo H, Yan R ![]() , Han X, Yu W
, Han X, Yu W
Received 29 March 2026
Accepted for publication 14 May 2026
Published 27 May 2026 Volume 2026:19 612984
DOI https://doi.org/10.2147/JIR.S612984
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Haoxin Guo,1– 3 Ruiyi Yan,1– 3 Xiaoxu Han,1– 3 Weihong Yu1– 3
1Department of Ophthalmology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100730, People’s Republic of China; 2Beijing Key Laboratory of Fundus Diseases Intelligent Diagnosis & Drug/Device Development and Translation, Beijing, 100730, People’s Republic of China; 3Key Laboratory of Ocular Fundus Diseases, Chinese Academy of Medical Sciences, Beijing, 100730, People’s Republic of China
Correspondence: Weihong Yu, Email [email protected]
Abstract: The pathogenesis of age-related macular degeneration (AMD) is intrinsically driven by retinal pigment epithelium (RPE) dysfunction. Under physiological conditions, the strictly polarized secretion of small extracellular vesicles (sEVs) by the RPE dictates outer retinal homeostasis. In response to oxidative and hypoxic stress, this secretory architecture is profoundly disrupted, transforming sEVs into mediators of drusen formation, inflammation, and neovascularization. This review systematically delineates the molecular machinery governing RPE-sEV trafficking, unveiling the distinct protein and miRNA cargo profiles segregated between the apical and basolateral domains. We highlight the unique secretory features of RPE and elucidate how AMD stressors disrupt this polarity via cytoskeletal collapse, secretory autophagy, and Rab GTPase dysregulation. Consequently, this altered sEV secretion abolishes apical neurotrophic support while deteriorating the basolateral microenvironment. Crucially, this establishes a vicious pathological loop where microenvironmental deterioration and sEV dysregulation are mutually causative. Recognizing dysregulated sEV polarity as a contributing factor to AMD, we propose that repairing RPE intracellular trafficking offers a fundamental strategy to restore secretory homeostasis and impede disease progression.
Keywords: small extracellular vesicles, age-related macular degeneration, retinal pigment epithelium, vesicular trafficking, polarization
Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss among the aging population worldwide.1 AMD is categorized into drusen-associated dry AMD and choroidal neovascularization (CNV)-driven neovascular AMD. Therapeutic options for AMD remain restricted: anti-VEGF injections for neovascular AMD only suppress downstream angiogenesis without rescuing cellular degeneration, while effective therapies for dry AMD are largely unavailable.2,3 These therapeutic limitations underscore the need to target early pathogenic events.
The pathogenesis of AMD is intrinsically related to the dysfunction of the retinal pigment epithelium (RPE).4 As a highly polarized epithelial monolayer, the RPE features distinct apical and basolateral membrane domains partitioned by tight junctions. At the apical domain, microvilli interdigitate with photoreceptor outer segments (POS) to drive both the visual cycle and rhythmic POS phagocytosis. At the opposite side, the basolateral surface anchors firmly to Bruch’s membrane via complex plasma membrane infoldings, facilitating nutrient uptake and the clearance of intracellular metabolic end-products into the choroidal circulation.5 Such exquisite structural asymmetry serves as the anatomical prerequisite for RPE functionality. It not only maintains the integrity of the outer blood-retinal barrier but also enables the establishment of distinct molecular microenvironments at opposing domains through precisely targeted secretion.4 Crucially, the breakdown of RPE polarity homeostasis acts as a shared pathological basis for both dry and neovascular AMD, driven by a complex interplay of oxidative stress, chronic inflammation, complement activation, and lipid dysregulation.2,6,7
Small extracellular vesicles (sEVs), historically referred to as “exosomes,” are 30–150 nm membranous vesicles released upon the fusion of multivesicular bodies (MVBs) with the plasma membrane.8 sEVs can selectively package proteins, lipids, and nucleic acids to facilitate information transfer and material exchange between donor and recipient cells.9 Rather than a stochastic diffusion process, sEV release in highly polarized epithelia such as the RPE is a strictly directional event orchestrated by the cytoskeleton and Rab GTPases.10 This polarized secretion establishes distinct, region-specific signaling microenvironments at the apical neuroretinal and basolateral choroidal compartments.11 Emerging evidence suggests that sEV secretion in the RPE exhibits distinctive regulatory features compared to typical epithelia, and that the dysregulation of this polarized secretion plays a pivotal role in AMD development.11–13 In this context, this review aims to systematically elucidate the mechanisms underlying the dysregulated polarized secretion of RPE-sEVs and their pathological roles in AMD, thereby providing a conceptual framework to identify novel therapeutic targets aimed at restoring secretory homeostasis for impeding disease progression.
Molecular Regulatory Mechanisms of Polarized sEV Secretion in RPE
MVB Heterogeneity and Differential Cargo Sorting
The foundation for polarized sEV secretion is established early during MVB biogenesis. This process is driven by the coordinated interplay between the endosomal sorting complex required for transport (ESCRT)-dependent machinery and ESCRT-independent pathways, such as ceramide- and tetraspanin-enriched microdomains.14 Concomitant with MVB maturation, the invagination of the limiting endosomal membrane generates intraluminal vesicles (ILVs), a process coupled to the selective package of bioactive molecules.15 Crucially, the specific cargo repertoire packaged within these ILVs acts as the primary determinant of the MVB trajectory, dictating whether the late endosome is committed to lysosomal degradation or routed to the plasma membrane for exocytosis.15–17
Upstream of MVB biogenesis, the initial sorting of protein cargoes at the trans-Golgi network (TGN) predetermines their ultimate extracellular fate.18 A defining feature of the RPE is the intrinsic absence of the adaptor protein (AP)-1B, a complex that classically segregates cargoes in typical epithelia by recognizing basolateral motifs.19,20 This absence abrogates canonical basolateral retention, redirecting these cargoes to the apical membrane.21 This enables the apical release of sEVs enriched with basolateral signature proteins, allowing them to exert specialized functions within the neuroretinal microenvironment. Furthermore, Golgi-mediated post-translational modifications function as molecular tags, partitioning specific cargoes into distinct secretory ILV subpopulations.22,23
Cytoskeleton-Guided Directional Transport Networks
The intracellular trafficking of mature MVBs is governed by a highly orchestrated cytoskeletal network comprising microtubules and actin filaments.24 RPE cells exhibit a robust cytoskeletal polarity: the microtubule-organizing center (MTOC) is anchored at the apical subcortex, establishing a longitudinal array where microtubule minus-ends face apically and plus-ends extend basolaterally.25 This polarity dictates the long-range trajectory of MVBs via specific motor proteins: dyneins mediate minus-end-directed transit toward the apical pole, whereas kinesins drive plus-end-directed transport toward the basolateral membrane.25,26
Upon arriving at the subcortical region, MVBs transition from microtubule tracks to the cortical actin network.27 This handover relies on unconventional myosins, which mediate the short-range vesicular displacement along actin filaments required to reach the plasma membrane.16 At the apical domain, the effector protein Myrip recruits the actin motor Myosin VIIa, orchestrating the transfer of MVBs from microtubules and tethering them to parallel F-actin bundles within the microvilli.28,29 Although this tethering machinery was classically characterized in the context of melanosome transport—where MYO7A mutations precipitate Usher syndrome type 1B30 —MVBs share extensive biogenetic and transport machineries with melanosomes.31,32 Consequently, the Rab27a-Myrip-Myosin VIIa axis is hypothesized to function as the central molecular nexus governing apical MVB exocytosis. Ultimately, this bipartite system of long-range microtubule navigation coupled with short-range actin tethering forms the structural scaffold for polarized sEV release.
Rab GTPase Conversion and SNARE-Mediated Fusion
The terminal subcortical tethering and exocytosis of MVBs are governed by Rab GTPase molecular switches and the SNARE fusion machinery. Following their maturation from Rab5-positive early endosomes, MVBs undergo a critical Rab identity switch—transitioning from a Rab7-dominant state to a Rab27a-positive state, concomitant with the recruitment of Arl8b. This conversion endows them with the secretory competence required to target specific membrane domains.33,34 Within this cascade, Rab27 paralogs exhibit a functional and spatial dichotomy: perinuclear Rab27b propels MVB transport toward the microtubule-actin interface, whereas peripherally localized Rab27a coordinates precise vesicular tethering and plasma membrane fusion.35 Studies in Madin-Darby Canine Kidney (MDCK) cells have revealed that polarized sEV secretion is governed by distinct Rab subsets, with Rab27a/b and Rab37 primarily directing apical release, and Rab39 regulating basolateral exocytosis.36 This paradigm implies that RPE may deploy similarly compartmentalized Rab isoforms to dictate the polarized sEV routing, but this extrapolation remains hypothetical and requires further validation. Furthermore, the apical recycling endosome resident Rab11a acts as a central regulatory node in the RPE apical secretory pathway, operating in concert with the kinesin motor KIF16B to drive directed vesicular delivery and tethering at the apical plasmalemma.21
In the terminal fusion phase, the mutual recognition and zippering of vesicular v-SNAREs and target-membrane t-SNAREs into a stable four-helix bundle catalyzes the fusion of the MVB limiting membrane with the plasma membrane. This dynamic rearrangement dilates a fusion pore, extruding ILVs into the extracellular space as mature sEVs.37 Notably, the RPE exhibits a highly atypical SNARE profile compared to canonical epithelia. As illuminated by Low et al,13 the RPE lacks the apical t-SNARE Syntaxin 3 and instead expresses Syntaxin 1A and 1B, isoforms classically restricted to neuronal and neuroendocrine lineages. Furthermore, the polarity of Syntaxin 2 is inverted in the RPE, shifting from the apical surface to co-assemble with Syntaxin 4 at the lateral membrane domain, immediately subjacent to the tight junctions. This highly specialized SNARE topology implies that RPE cells harbor a dedicated secretory locus within this lateral sub-tight-junction region, explicitly tailored to mediate the basolateral exocytosis of sEVs.
Collectively, the polarized secretion of RPE-sEVs requires the precise integration of selective cargo sorting, cytoskeletal navigation, and Rab/SNARE-mediated fusion to sustain the physiological homeostasis of the outer retina. Figure 1 depicts the molecular regulatory mechanisms of polarized sEV secretion in RPE.
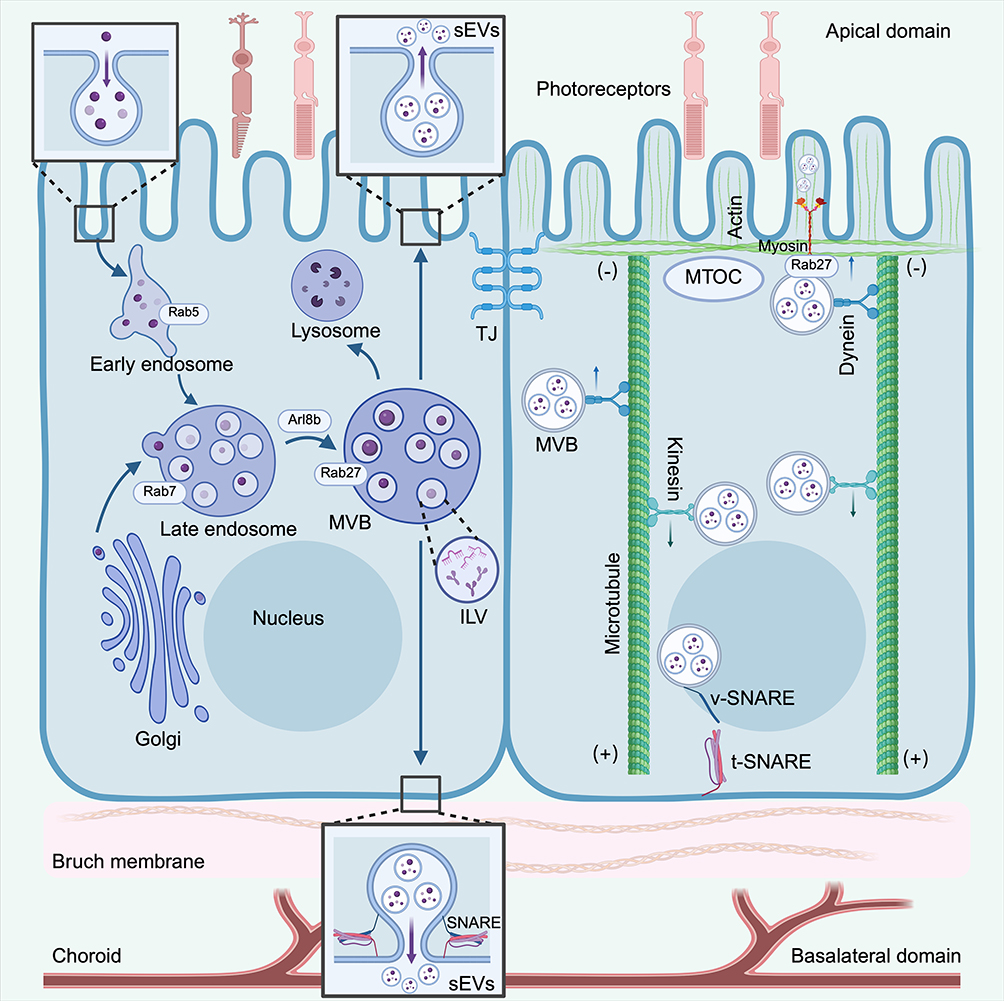 |
Figure 1 Molecular mechanisms of polarized sEV secretion in RPE. The left panel illustrates the biogenesis and maturation of MVBs. Cargo from the Golgi network transitions through Rab5-positive early endosomes to Rab7-positive late endosomes, eventually forming Rab27/Arl8b-positive secretory MVBs packed with ILVs. These MVBs are either targeted for lysosomal degradation or routed to the plasma membrane. The right panel depicts the bipartite cytoskeleton-guided transport network. With the MTOC anchored apically, dynein motors drive MVB transit toward the apical domain (minus-end), whereas kinesin motors mediate basolateral transport (plus-end). Terminal short-range transport and membrane fusion rely on myosin-actin interactions and specific v-SNARE/t-SNARE complex assembly, ultimately facilitating the polarized release of sEVs into either the subretinal space (facing photoreceptors) or the basolateral microenvironment (facing Bruch’s membrane and choroid). (Created with BioRender.com). Abbreviation: TJ, tight junction. |
Polarized Cargo Signatures of RPE-sEVs and Their Physiological Implications
sEVs secreted from the apical and basolateral domains of RPE cells exhibit strictly compartmentalized molecular signatures. By leveraging Transwell culture systems to recapitulate the native RPE monolayer, researchers can effectively isolate these domain-specific sEV populations. Proteomic analysis by Klingeborn et al38 of primary porcine RPE revealed that among 631 identified sEV proteins, over half exhibited strictly unidirectional release: 299 were exclusively partitioned to the apical domain, whereas 94 were restricted to the basolateral compartment. This stringent spatial segregation extends to the miRNA repertoire, underscoring the intrinsic capacity of the RPE for spatially resolved, bidirectional signaling. Table 1 summarizes the representative protein and miRNA cargoes differentially segregated into apical and basolateral sEVs.
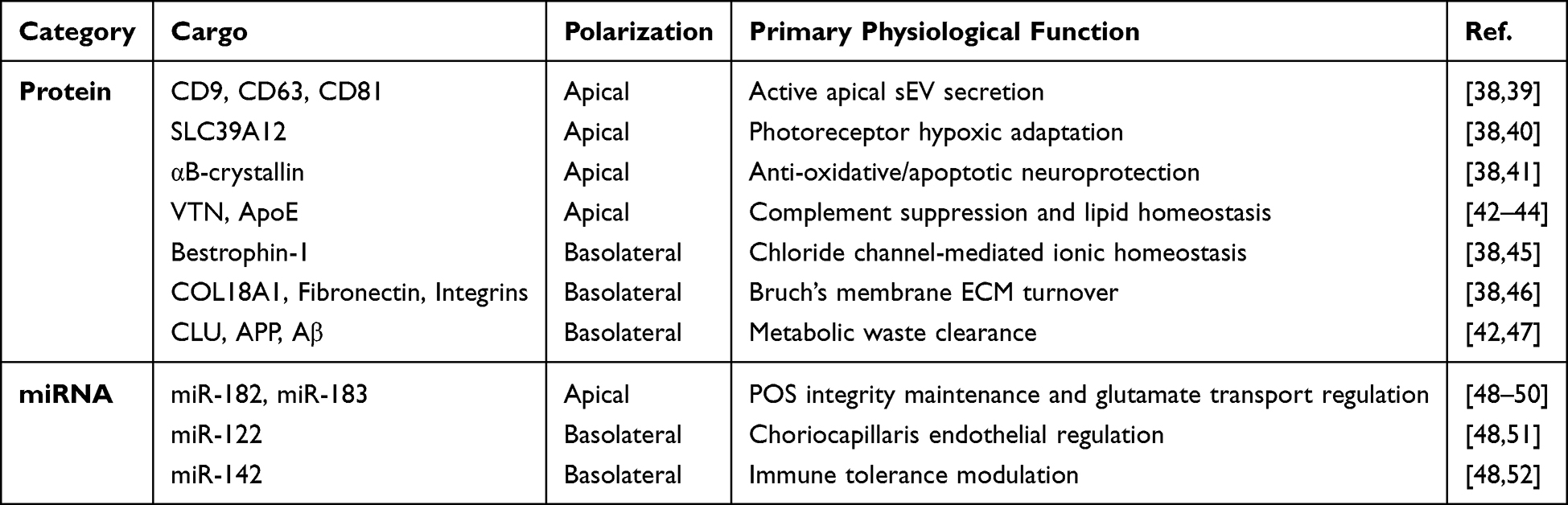 |
Table 1 Representative Protein and miRNA Cargoes in Apical and Basolateral sEVs |
The apical sEV profile is exquisitely tailored to confer neurotrophic and structural support to the overlying photoreceptors. Specifically, apical sEVs are enriched in classical sEV markers (eg, CD9, CD63, and CD81), suggesting a more active sEV secretion process at the apical surface.38,39 Furthermore, they selectively package the zinc transporter SLC39A12, which mediates photoreceptor adaptation to chronic hypoxia,40 and αB-crystallin, a heat shock protein predominantly released apically to shield photoreceptors against oxidative insult.41 Additionally, human iPSC-derived RPE models exhibit preferential apical secretion of vitronectin (VTN) and apolipoprotein E (ApoE), which function to suppress subretinal complement overactivation and maintain lipid homeostasis.42–44 Synergizing with this protein landscape, apical sEVs harbor a highly polarized miRNA payload, notably enriched in miR-182 and miR-183.48 These miRNAs are essential for the maintenance and differentiation of POS49 and putatively modulate photoreceptor glutamate transporters (eg, Slc1a1) to sustain optimal visual signal transduction.50
At the opposing pole, basolaterally secreted sEVs are dedicated to the structural and homeostatic maintenance of the underlying Bruch’s membrane and the choroid. Basolateral sEVs are specifically enriched in the chloride channel Bestrophin-1 and extracellular matrix (ECM) constituents (eg, COL18A1, Fibronectin, Integrins), underscoring their active involvement in BrM remodeling and regional ionic balance.38,45,46 Notably, while proteins such as clusterin (CLU), amyloid precursor protein (APP), and amyloid-beta (Aβ) are core constituents of drusen, they exhibit a homeostatic basolateral secretion preference. This physiological extrusion acts as an essential clearance mechanism to mediate BrM turnover and prevent intracellular metabolic toxicity.42,47 Paralleling this protein asymmetry, basolateral sEVs selectively package specific miRNAs, notably miR-122 and miR-142.48 Given that miR-122 regulates choriocapillaris endothelial cell function51 and miR-142 modulates angiogenesis and microglial activation,52 their directional basolateral delivery indicates that the RPE tonically modulates choroidal vascular homeostasis and immune tolerance.
Mechanisms of Dysregulated sEV Secretion Under AMD Stress
Cytoskeletal Collapse
Elevated reactive oxygen species (ROS) can dismantle the microtubule network, causing its fragmentation and disorganization by impairing the nucleating capacity of the MTOC and reducing tubulin acetylation levels.53–55 Specifically, 4-hydroxynonenal (4-HNE), a highly electrophilic byproduct of lipid peroxidation, targets tubulin by forming irreversible covalent adducts via Michael addition. This pathological modification triggers aberrant tubulin cross-linking, potently paralyzing the polymerization of functional microtubules.56,57
Concurrently, oxidative stress ruptures the homeostatic rheostat of the Rho GTPase family. The activities of Cdc42 and Rac1—key orchestrators of epithelial polarity and cortical actin assembly—are suppressed, whereas the RhoA/ROCK pathway, which drives cellular contractility, is aberrantly hyperactivated. This signaling inversion precipitates the breakdown of the characteristic circumferential actin belt in RPE cells, driving the reorganization of the F-actin network into thick, transcellular stress fibers.58–60 Ultimately, the collapse of these microtubule and microfilament architectures deprives sEVs of the structural tracks required for polarized trafficking, resulting in either their intracellular retention or non-directional, stochastic release.
Secretory Autophagy
Lysosomal overload is a hallmark of RPE senescence and AMD pathogenesis.61 The relentless accumulation of lipofuscin, stemming from incomplete POS digestion, impairs lysosomal proton pump function, elevates luminal pH, and curtails hydrolase activity.62 This degradation deficit not only stalls normal autophagic flux but also induces RPE cells to initiate a compensatory secretory autophagy pathway.63 This process redirects the vesicular trajectory, forcing the extracellular release of metabolic waste via sEVs—thereby acting as an inciting factor for drusen biogenesis and the inflammatory priming of the retinal microenvironment.64,65
Mechanistically, Ghosh et al66 demonstrated that the dysregulation of the AKT2/SIRT5/TFEB signaling axis serves as the upstream driver that simultaneously impedes lysosomal biogenesis and activates secretory autophagy. This rerouting dictates a precise molecular reprogramming: the cargo receptor TRIM16 selectively sorts pathological cargo into secretory autophagosomes, while the autophagosomal membrane recruits the secretory SNARE SEC22B instead of the lysosome-fusion protein syntaxin 17 (STX17), thereby sealing a secretory fate.64 Furthermore, autophagosomes can fuse with MVBs to form amphisomes. This event marks a pathogenic convergence of the autophagic and sEV biogenesis pathways. Ultimately, orchestrated by key molecules including Rab8a, Rab27a, and GRASP55, these amphisomes bypass the Golgi apparatus to fuse directly with the RPE plasma membrane, co-releasing cytosolic pathological aggregates alongside sEVs.64,65
Although this cargo extrusion mechanism temporarily alleviates intracellular stress within the RPE under conditions of lysosomal suppression,67 the basolateral discharge of sEVs enriched with Aβ, lipofuscin, and damaged mitochondrial components—accompanied by the aberrant modification of complement C365 —contributes directly to drusen formation. In addition, this pathway mediates the unconventional secretion of leaderless inflammatory cytokines and angiogenesis-related factors,64 thereby establishing the pathological foundation for chronic outer retinal inflammation and choroidal CNV.
Rab GTPase Aberration
Oxidative stress and hypoxic microenvironments can directly derail the core molecular switches governing vesicular routing. Rab GTPases are acutely susceptible to oxidative perturbation. Hoppe et al68 demonstrated that oxidized lipoproteins impair normal phagosome maturation, postulating that this defect arises from the disrupted membrane recruitment and activation of Rab5. This dysfunction is particularly pronounced during POS phagocytosis. Keeling et al69 observed that oxidative stress triggers the premature derailment of POS-containing phagosomes from the classical Rab5/Rab7 degradation pathway, inducing pathological vesicular swelling. This oxidative derailment dismantles the homeostatic control of Rab GTPases, rendering the highly metabolic RPE exquisitely vulnerable to vesicular trafficking failure.
The modulatory impact of hypoxia on Rab GTPases, which have been extensively delineated in tumor biology, provides a compelling mechanistic framework for AMD pathogenesis. In the tumor microenvironment, hypoxia-inducible factors fundamentally reprogram the expression and spatial distribution of Rab proteins.70 Analogous to the coordinated upregulation of Rab27a and downregulation of Rab7 in ovarian cancer cells,71 alongside the perinuclear clustering of Rab5-positive vesicles in prostate cancer cells,72 the RPE likely activates a similar molecular program under hypoxic stress to hyperactivate sEV secretion. Nevertheless, translating these mechanisms from tumor microenvironments to retinal physiology is inherently speculative and necessitates targeted confirmation. While this vesicular extrusion initially serves as an adaptive cellular triage to purge metabolic waste during oxygen deprivation, it becomes maladaptive within the specialized retinal niche. The hypoxia-driven deluge of RPE-sEVs carrying pro-angiogenic payloads, notably VEGF, acts as a molecular catalyst for CNV initiation.
Taken together, AMD-related stress dismantles the RPE intracellular trafficking machinery via cytoskeletal collapse, Rab GTPase derailment, and secretory autophagy. This collective breakdown abolishes apical-basolateral polarity, exacerbating cellular stress and driving a vicious cycle. Figure 2 illustrates the mechanisms of dysregulated sEV secretion under AMD-associated stress.
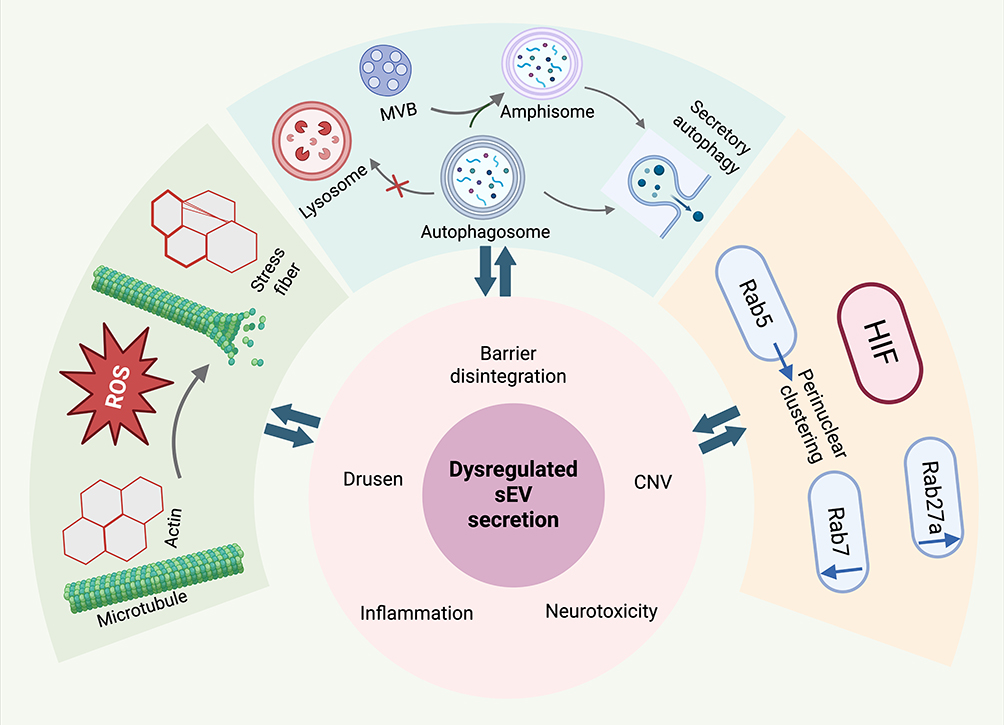 |
Figure 2 Mechanisms of dysregulated sEV secretion under AMD-associated stress. The outer panels depict the tripartite collapse of the RPE intracellular trafficking machinery in response to cellular stressors. (Left) ROS destabilizes microtubule networks and remodels actin cytoskeletons into stress fibers. (Top) Impaired lysosomal degradation drives secretory autophagy, leading to the exocytosis of either autophagosomes or amphisomes formed via MVB fusion. (Right) HIF induces abnormal Rab GTPase signaling, characterized by the perinuclear clustering of Rab5, alongside the concurrent downregulation of Rab7 and upregulation of Rab27a. As shown in the central core, this collective breakdown leads to dysregulated sEV secretion, directly contributing to hallmark AMD pathologies including barrier disintegration, CNV, neurotoxicity, inflammation, and drusen formation. The bidirectional arrows highlight a vicious feedback loop where these microenvironmental deteriorations further amplify the initial RPE cellular stress. (Created with BioRender.com). |
Dysregulated sEV Secretion Induces Outer Retinal Pathology
Drusen Formation
The pathological deposition of drusen along Bruch’s membrane represents the clinical hallmark of dry AMD. Existing evidence indicates that drusen are not merely passive extracellular precipitates, but rather actively extruded metabolic byproducts, a process triggered by the disruption of intracellular proteostasis within the RPE.64 Wang et al73 revealed that in the eyes of aged mice and human AMD donors, drusen is not only enriched with the sEV markers CD63 and CD81, but also exhibits colocalization with autophagic markers, complement components, and Aβ. This discovery validates that sEVs extruded via secretory autophagy directly constitute drusen. Paralleling these in vivo findings, utilizing a polarized iPSC-RPE cell model, Flores-Bellver et al42 demonstrated that exposure to cigarette smoke extract (CSE) significantly augmented the basolateral secretion of RPE-sEVs. These sEVs are highly enriched with CLU, Aβ, vinculin (VIN), VTN, and ApoE—all of which are hallmark protein constituents of drusen.
Neurotoxicity and Inflammatory Propagation
Under AMD-associated stress conditions, RPE-sEVs act as pathogenic messengers that drive neuroretinal degeneration and the pro-inflammatory remodeling of the outer retinal microenvironment. Zhang Z. et al74 revealed that under oxidative stress, RPE-sEVs exhibit a profound depletion of neuroprotective miRNAs (eg, miR-125b-5p, miR-128-3p) alongside an aberrant upregulation of pro-apoptotic miRNAs such as miR-7-5p. These differentially expressed miRNAs predominantly target cell survival pathways, including MAPK and PI3K-Akt. Subretinal injection of these stress-derived sEVs in vivo directly provokes outer nuclear layer apoptosis, exacerbates ROS production, and triggers a cascading release of pro-inflammatory cytokines, including IL-1β and IL-6. Corroborating the role of sEVs in inflammatory propagation, Zhang W. et al75 demonstrated that photo-oxidative stress activates the NLRP3 inflammasome, prompting the sEV-mediated secretion of active caspase-1, IL-1β, and IL-18, thereby sustaining the chronic inflammatory milieu of the outer retina.
Barrier Disintegration and CNV Initiation
The hallmark of neovascular AMD is the formation of CNV. During this pathogenesis, RPE-sEVs mediate the disintegration of the RPE barrier and trigger the proliferation and migration of choroidal endothelial cells. Atienzar-Aroca et al76 and Maisto et al77 demonstrated that oxidative stress induces RPE cells to secrete sEVs enriched in angiogenic factors, including VEGFA and VEGFR, establishing the molecular foundation for CNV initiation. Furthermore, these sEVs orchestrate a pathological bidirectional crosstalk between the RPE and macrophages. Otsuki et al78 revealed that stress-derived RPE-sEVs exhibit a pro-inflammatory molecular signature, instructing macrophages to upregulate tumor necrosis factor-α (TNF-α). This upregulation, in turn, reciprocally stimulates the RPE to release monocyte chemotactic protein-1 (MCP-1). The subsequently recruited and activated macrophages release matrix metalloproteinases (MMPs) to proteolytically degrade Bruch’s membrane, thereby eliminating structural constraints to facilitate neovascular invasion. Klingeborn et al79 proposed that oxidative stress prompts the RPE to basolaterally shed desmosomes and hemidesmosomes via sEVs. This extracellular expulsion of junctional proteins directly compromises RPE barrier integrity and serves as a prelude to epithelial-mesenchymal transition (EMT), rendering the subretinal space permissive to CNV ingrowth.
Table 2 shows the key cargo alterations of RPE-sEVs under AMD-associated stress and their primary pathogenic roles in AMD.
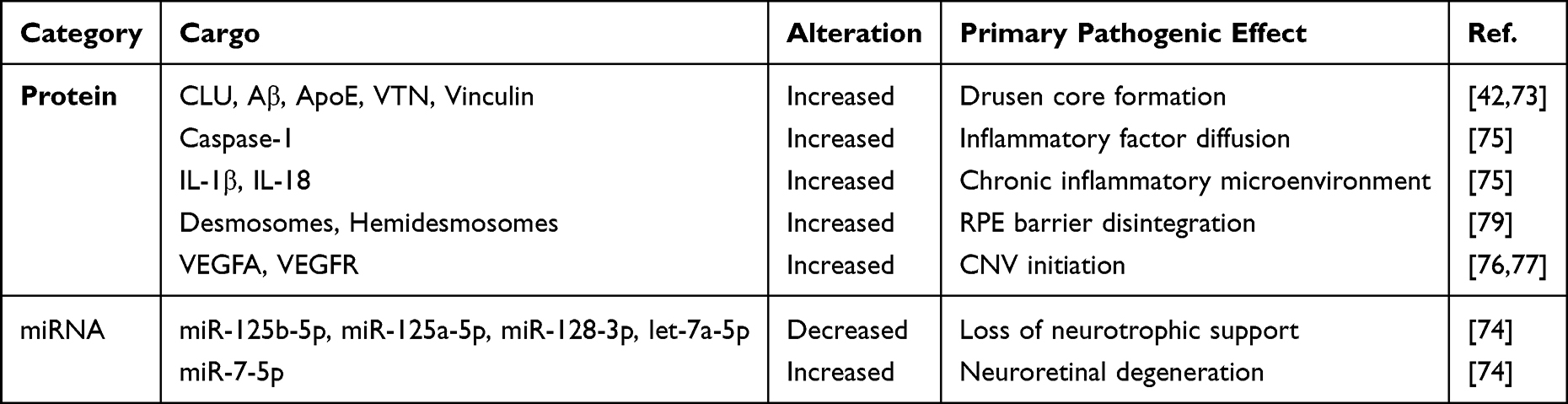 |
Table 2 Stress-Induced Cargo Alterations in RPE-sEVs and Their Pathogenic Roles in AMD |
Conclusion and Perspectives
In conclusion, the dysregulated polarized secretion of RPE-sEVs is a critical contributing factor to both dry and neovascular AMD, rather than a mere bystander phenomenon. As synthesized above, the stress-induced collapse of intracellular trafficking machineries abolishes secretory polarity, perpetuating a vicious feedback loop of microenvironmental deterioration.
To advance the understanding of the mechanisms of RPE-sEV secretory dysfunction, future research must pivot away from paradigms extrapolated from canonical epithelia and tumor models to construct an RPE-specific polarized regulatory atlas. Currently, the isoform-specific functional partitioning among Rab GTPases dictating apical versus basolateral sEV routing remains elusive. For instance, whether the RPE-specific melanosome transport apparatus (Rab27a-Myrip-Myosin VIIa axis) genuinely orchestrates apical sEV tethering requires rigorous validation. Furthermore, the RPE exhibits a unique distribution of SNARE proteins, such as the polarized inversion of Syntaxin 2. This distinct molecular architecture likely underpins the specific sEV secretory profile that distinguishes the RPE from other epithelial tissues. Systematically deconstructing the RPE-specific molecular circuitries governing sEV polarity will establish the theoretical cornerstone required to precisely intercept AMD progression.
Methodologically, future investigations should transition from phenomenological descriptions of sEV cargo alterations to high-resolution mechanistic decoding. A critical imperative is to elucidate exactly how pathological stressors remodel the endolysosomal sorting machinery to force the aberrant rerouting of sEVs. This necessitates transcending the inherent limitations of bulk vesicle population analyses. By leveraging emerging single-vesicle analysis technologies,80 researchers can untangle the heterogeneous vesicular subpopulations within the RPE. Identifying endogenous sorting motifs and their cognate chaperone networks that dictate cargo entry into distinct secretory pathways will clarify the fundamental differences between physiological and stress-induced secretion, ultimately unraveling the intricate interplay between physiological maintenance and pathological aggravation.
From a translational perspective, current therapeutic strategies predominantly focus on the regenerative potential and delivery vehicle utility of exogenous sEVs. For instance, using engineered or stem cell-derived sEVs to deliver therapeutic agents has demonstrated robust efficacy in preclinical models.81,82 In contrast, the mechanisms elucidated here propose a shift toward endogenous functional rescue. In this regard, subthreshold retinal laser has been clinically established as a physical modality to normalize general RPE function by utilizing sublethal thermal stress to trigger adaptive hormesis.83,84 Investigating how such established physical treatments modulate RPE microvesicle physiology may represent an essential research frontier. Ultimately, the strategy of rectifying apical-basal secretory flux at its intracellular origin offers a theoretical framework to halt microenvironmental deterioration, potentially advancing AMD therapy from symptomatic management to structural and functional restoration.
Data Sharing Statement
Data sharing is not applicable to this article as no new data was created or analyzed in this study.
Author Contributions
Haoxin Guo: Conceptualization, Investigation, Formal analysis, Writing – original draft. Ruiyi Yan: Data curation, Investigation, Writing – review & editing. Xiaoxu Han: Investigation, Writing – review & editing. Weihong Yu: Conceptualization, Supervision, Writing – review & editing. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Funded by National High Level Hospital Clinical Research Funding (2025-PUMCH-C-027).
Disclosure
Dr. Haoxin Guo, Dr. Ruiyi Yan, and Dr. Weihong Yu received support from the National High Level Hospital Clinical Research Funding during the conduct of this study.
Dr. Ruiyi Yan’s research was funded by National High Level Hospital Clinical Research Funding (grant number 2025-PUMCH-C-027).
All funding was received for research support only and had no influence on the study design, data collection, analysis, interpretation of results, manuscript preparation, or the decision to submit for publication.
The authors report no other conflicts of interest in this study.
References
1. Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–12. doi:10.1016/s2214-109x(13)70145-1
2. Fleckenstein M, Keenan TDL, Guymer RH, et al. Age-related macular degeneration. Nat Rev Dis Primers. 2021;7(1):31. doi:10.1038/s41572-021-00265-2
3. Maguire MG, Martin DF, Ying GS, et al. Five-year outcomes with anti-vascular endothelial growth factor treatment of neovascular age-related macular degeneration: the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2016;123(8):1751–1761. doi:10.1016/j.ophtha.2016.03.045
4. Lakkaraju A, Umapathy A, Tan LX, et al. The cell biology of the retinal pigment epithelium. Prog Retin Eye Res. 2020:100846. doi:10.1016/j.preteyeres.2020.100846
5. Caceres PS, Rodriguez-Boulan E. Retinal pigment epithelium polarity in health and blinding diseases. Curr Opin Cell Biol. 2020;62:37–45. doi:10.1016/j.ceb.2019.08.001
6. Tang S, Yang J, Xiao B, et al. Aberrant lipid metabolism and complement activation in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2024;65(12):20. doi:10.1167/iovs.65.12.20
7. Guymer RH, Campbell TG. Age-related macular degeneration. Lancet. 2023;401(10386):1459–1472. doi:10.1016/s0140-6736(22)02609-5
8. Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. doi:10.1080/20013078.2018.1535750
9. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478). doi:10.1126/science.aau6977
10. Klingeborn M, Stamer WD, Bowes Rickman C. Polarized exosome release from the retinal pigmented epithelium. Adv Exp Med Biol. 2018;1074:539–544. doi:10.1007/978-3-319-75402-4_65
11. Klingeborn M, Dismuke WM, Bowes Rickman C, Stamer WD. Roles of exosomes in the normal and diseased eye. Prog Retin Eye Res. 2017;59:158–177. doi:10.1016/j.preteyeres.2017.04.004
12. Tarau IS, Berlin A, Curcio CA, Ach T. The cytoskeleton of the retinal pigment epithelium: from normal aging to age-related macular degeneration. Int J Mol Sci. 2019;20(14). doi:10.3390/ijms20143578
13. Low SH, Marmorstein LY, Miura M, et al. Retinal pigment epithelial cells exhibit unique expression and localization of plasma membrane syntaxins which may contribute to their trafficking phenotype. J Cell Sci. 2002;115(Pt 23):4545–4553. doi:10.1242/jcs.00116
14. Hanson PI, Cashikar A. Multivesicular body morphogenesis. Annu Rev Cell Dev Biol. 2012;28:337–362. doi:10.1146/annurev-cellbio-092910-154152
15. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–228. doi:10.1038/nrm.2017.125
16. Arya SB, Collie SP, Parent CA. The ins-and-outs of exosome biogenesis, secretion, and internalization. Trends Cell Biol. 2024;34(2):90–108. doi:10.1016/j.tcb.2023.06.006
17. Mathieu M, Martin-Jaular L, Lavieu G, Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21(1):9–17. doi:10.1038/s41556-018-0250-9
18. Loh YP, Xiao L, Park JJ. Trafficking of hormones and trophic factors to secretory and extracellular vesicles: a historical perspective and new hypothesis. Extracell Vesicles Circ Nucl Acids. 2023;4(4):568–587. doi:10.20517/evcna.2023.34
19. Diaz F, Gravotta D, Deora A, et al. Clathrin adaptor AP1B controls adenovirus infectivity of epithelial cells. Proc Natl Acad Sci U S A. 2009;106(27):11143–11148. doi:10.1073/pnas.0811227106
20. Fölsch H, Ohno H, Bonifacino JS, Mellman I. A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell. 1999;99(2):189–198. doi:10.1016/s0092-8674(00)81650-5
21. Perez Bay AE, Schreiner R, Mazzoni F, et al. The kinesin KIF16B mediates apical transcytosis of transferrin receptor in AP-1B-deficient epithelia. EMBO j. 2013;32(15):2125–2139. doi:10.1038/emboj.2013.130
22. Giovannone AJ, Reales E, Bhattaram P, Fraile-Ramos A, Weimbs T. Monoubiquitination of syntaxin 3 leads to retrieval from the basolateral plasma membrane and facilitates cargo recruitment to exosomes. Mol Biol Cell. 2017;28(21):2843–2853. doi:10.1091/mbc.E17-07-0461
23. MacDonald C, Payne JA, Aboian M, Smith W, Katzmann DJ, Piper RC. A family of tetraspans organizes cargo for sorting into multivesicular bodies. Dev Cell. 2015;33(3):328–342. doi:10.1016/j.devcel.2015.03.007
24. Xu M, Ji J, Jin D, et al. The biogenesis and secretion of exosomes and multivesicular bodies (MVBs): intercellular shuttles and implications in human diseases. Genes Dis. 2023;10(5):1894–1907. doi:10.1016/j.gendis.2022.03.021
25. Hazim RA, Williams DS. Microtubule motor transport of organelles in a specialized epithelium: the RPE. Front Cell Dev Biol. 2022;10:852468. doi:10.3389/fcell.2022.852468
26. Robinson BP, Bass NR, Bhakt P, Spiliotis ET. Septin-coated microtubules promote maturation of multivesicular bodies by inhibiting their motility. J Cell Biol. 2024;223(8). doi:10.1083/jcb.202308049
27. Li P, Bademosi AT, Luo J, Meunier FA. Actin remodeling in regulated exocytosis: toward a mesoscopic view. Trends Cell Biol. 2018;28(9):685–697. doi:10.1016/j.tcb.2018.04.004
28. Gibbs D, Diemer T, Khanobdee K, Hu J, Bok D, Williams DS. Function of MYO7A in the human RPE and the validity of shaker1 mice as a model for Usher syndrome 1B. Invest Ophthalmol Vis Sci. 2010;51(2):1130–1135. doi:10.1167/iovs.09-4032
29. Schwander M, Lopes V, Sczaniecka A, et al. A novel allele of myosin VIIa reveals a critical function for the C-terminal FERM domain for melanosome transport in retinal pigment epithelial cells. J Neurosci. 2009;29(50):15810–15818. doi:10.1523/jneurosci.4876-09.2009
30. Leong YC, Di Foggia V, Pramod H, Bitner-Glindzicz M, Patel A, Sowden JC. Molecular pathology of Usher 1B patient-derived retinal organoids at single cell resolution. Stem Cell Rep. 2022;17(11):2421–2437. doi:10.1016/j.stemcr.2022.09.006
31. Burgoyne T, Jolly R, Martin-Martin B, et al. Expression of OA1 limits the fusion of a subset of MVBs with lysosomes - a mechanism potentially involved in the initial biogenesis of melanosomes. J Cell Sci. 2013;126(Pt 22):5143–5152. doi:10.1242/jcs.128561
32. Raposo G, Marks MS. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8(10):786–797. doi:10.1038/nrm2258
33. Jongsma ML, Bakker J, Cabukusta B, et al. SKIP-HOPS recruits TBC1D15 for a Rab7-to-Arl8b identity switch to control late endosome transport. EMBO j. 2020;39(6):e102301. doi:10.15252/embj.2019102301
34. Verweij FJ, Bebelman MP, George AE, et al. ER membrane contact sites support endosomal small GTPase conversion for exosome secretion. J Cell Biol. 2022;221(12). doi:10.1083/jcb.202112032
35. Ostrowski M, Carmo NB, Krumeich S, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(1):
36. Matsui T, Sakamaki Y, Nakashima S, Fukuda M. Rab39 and its effector UACA regulate basolateral exosome release from polarized epithelial cells. Cell Rep. 2022;39(9):110875. doi:10.1016/j.celrep.2022.110875
37. Baker RW, Hughson FM. Chaperoning SNARE assembly and disassembly. Nat Rev Mol Cell Biol. 2016;17(8):465–479. doi:10.1038/nrm.2016.65
38. Klingeborn M, Dismuke WM, Skiba NP, Kelly U, Stamer WD, Bowes Rickman C. Directional exosome proteomes reflect polarity-specific functions in retinal pigmented epithelium monolayers. Sci Rep. 2017;7(1):4901. doi:10.1038/s41598-017-05102-9
39. Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–E977. doi:10.1073/pnas.1521230113
40. Zhao L, Oliver E, Maratou K, et al. The zinc transporter ZIP12 regulates the pulmonary vascular response to chronic hypoxia. Nature. 2015;524(7565):356–360. doi:10.1038/nature14620
41. Sreekumar PG, Kannan R, Kitamura M, et al. αB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS One. 2010;5(10):e12578. doi:10.1371/journal.pone.0012578
42. Flores-Bellver M, Mighty J, Aparicio-Domingo S, et al. Extracellular vesicles released by human retinal pigment epithelium mediate increased polarised secretion of drusen proteins in response to AMD stressors. J Extracell Vesicles. 2021;10(13):e12165. doi:10.1002/jev2.12165
43. Curcio CA, Johnson M, Huang JD, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010;51(3):451–467. doi:10.1194/jlr.R002238
44. Anderson DH, Radeke MJ, Gallo NB, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29(2):95–112. doi:10.1016/j.preteyeres.2009.11.003
45. Marmorstein AD, Kinnick TR, Stanton JB, Johnson AA, Lynch RM, Marmorstein LY. Bestrophin-1 influences transepithelial electrical properties and Ca2+ signaling in human retinal pigment epithelium. Mol Vis. 2015;21:347–359.
46. Marneros AG, Keene DR, Hansen U, et al. Collagen XVIII/endostatin is essential for vision and retinal pigment epithelial function. EMBO J. 2004;23(1):89–99. doi:10.1038/sj.emboj.7600014
47. Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99(18):11830–11835. doi:10.1073/pnas.192203399
48. Hernandez BJ, Strain M, Suarez MF, et al. Small extracellular vesicle-associated MiRNAs in polarized retinal pigmented epithelium. Invest Ophthalmol Vis Sci. 2024;65(13):57. doi:10.1167/iovs.65.13.57
49. Busskamp V, Krol J, Nelidova D, et al. miRNAs 182 and 183 are necessary to maintain adult cone photoreceptor outer segments and visual function. Neuron. 2014;83(3):586–600. doi:10.1016/j.neuron.2014.06.020
50. Krol J, Busskamp V, Markiewicz I, et al. Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell. 2010;141(4):618–631. doi:10.1016/j.cell.2010.03.039
51. Oltra M, Vidal-Gil L, Maisto R, et al. miR302a and 122 are deregulated in small extracellular vesicles from ARPE-19 cells cultured with H(2)O(2). Sci Rep. 2019;9(1):17954. doi:10.1038/s41598-019-54373-x
52. Roblain Q, Louis T, Yip C, et al. Intravitreal injection of anti-miRs against miR-142-3p reduces angiogenesis and microglia activation in a mouse model of laser-induced choroidal neovascularization. Aging. 2021;13(9):12359–12377. doi:10.18632/aging.203035
53. Vishwakarma A, Chihki L, Todkar K, Ouellet M, Germain M. Mitochondrial dysfunction alters early endosome trafficking via microtubule reorganization. Life Sci Alliance. 2026;9(1). doi:10.26508/lsa.202403020
54. Eshun-Wilson L, Zhang R, Portran D, et al. Effects of α-tubulin acetylation on microtubule structure and stability. Proc Natl Acad Sci U S A. 2019;116(21):10366–10371. doi:10.1073/pnas.1900441116
55. Yan K, Cui K, Nie J, et al. Mogroside V protects porcine oocytes from lipopolysaccharide-induced meiotic defects. Front Cell Dev Biol. 2021;9:639691. doi:10.3389/fcell.2021.639691
56. Stewart BJ, Doorn JA, Petersen DR. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem Res Toxicol. 2007;20(8):1111–1119. doi:10.1021/tx700106v
57. Wang X, Yang Y, Moore DR, Nimmo SL, Lightfoot SA, Huycke MM. 4-hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by Enterococcus faecalis-infected macrophages. Gastroenterology. 2012;142(3):543–551.e7. doi:10.1053/j.gastro.2011.11.020
58. Ruiz-Loredo AY, López E, López-Colomé AM. Thrombin promotes actin stress fiber formation in RPE through Rho/ROCK-mediated MLC phosphorylation. J Cell Physiol. 2011;226(2):414–423. doi:10.1002/jcp.22347
59. Huang ZX, Mao XM, Wu RF, et al. RhoA/ROCK pathway mediates the effect of oestrogen on regulating epithelial-mesenchymal transition and proliferation in endometriosis. J Cell Mol Med. 2020;24(18):10693–10704. doi:10.1111/jcmm.15689
60. He Y, Northey JJ, Pelletier A, et al. The Cdc42/Rac1 regulator CdGAP is a novel E-cadherin transcriptional co-repressor with Zeb2 in breast cancer. Oncogene. 2017;36(24):3490–3503. doi:10.1038/onc.2016.492
61. Falcão AS, Pedro ML, Tenreiro S, Seabra MC. Targeting lysosomal dysfunction and oxidative stress in age-related macular degeneration. Antioxidants. 2025;14(5). doi:10.3390/antiox14050596
62. Bergmann M, Schütt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB j. 2004;18(3):562–564. doi:10.1096/fj.03-0289fje
63. Kaarniranta K, Blasiak J, Liton P, Boulton M, Klionsky DJ, Sinha D. Autophagy in age-related macular degeneration. Autophagy. 2023;19(2):388–400. doi:10.1080/15548627.2022.2069437
64. Hyttinen JMT, Niittykoski M, Kaarniranta K. The role of secretory autophagy and exosomes in the accumulation of drusen during the development of age-related macular degeneration (AMD). Ageing Res Rev. 2025;110:102796. doi:10.1016/j.arr.2025.102796
65. Blasiak J, Kaarniranta K. Secretory autophagy: a turn key for understanding AMD pathology and developing new therapeutic targets? Expert Opin Ther Targets. 2022;26(10):883–895. doi:10.1080/14728222.2022.2157260
66. Ghosh S, Sharma R, Bammidi S, et al. The AKT2/SIRT5/TFEB pathway as a potential therapeutic target in non-neovascular AMD. Nat Commun. 2024;15(1):6150. doi:10.1038/s41467-024-50500-z
67. Solvik TA, Nguyen TA, Tony Lin YH, et al. Secretory autophagy maintains proteostasis upon lysosome inhibition. J Cell Biol. 2022;221(6). doi:10.1083/jcb.202110151
68. Hoppe G, O’Neil J, Hoff HF, Sears J. Products of lipid peroxidation induce missorting of the principal lysosomal protease in retinal pigment epithelium. Biochim Biophys Acta. 2004;1689(1):33–41. doi:10.1016/j.bbadis.2004.01.004
69. Keeling E, Chatelet DS, Johnston DA, et al. Oxidative stress and dysfunctional intracellular traffic linked to an unhealthy diet results in impaired cargo transport in the Retinal Pigment Epithelium (RPE). Mol Nutr Food Res. 2019;63(15):e1800951. doi:10.1002/mnfr.201800951
70. Kumar A, Deep G. Hypoxia in tumor microenvironment regulates exosome biogenesis: molecular mechanisms and translational opportunities. Cancer Lett. 2020;479:23–30. doi:10.1016/j.canlet.2020.03.017
71. Dorayappan KDP, Wanner R, Wallbillich JJ, et al. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: a novel mechanism linking STAT3/Rab proteins. Oncogene. 2018;37(28):3806–3821. doi:10.1038/s41388-018-0189-0
72. Panigrahi GK, Praharaj PP, Peak TC, et al. Hypoxia-induced exosome secretion promotes survival of African-American and Caucasian prostate cancer cells. Sci Rep. 2018;8(1):3853. doi:10.1038/s41598-018-22068-4
73. Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One. 2009;4(1):e4160. doi:10.1371/journal.pone.0004160
74. Zhang Z, Gu Q, Chen L, et al. Selective microRNA expression of exosomes from retinal pigment epithelial cells by oxidative stress. Vision Res. 2024;220:108388. doi:10.1016/j.visres.2024.108388
75. Zhang W, Ma Y, Zhang Y, Yang J, He G, Chen S. Photo-oxidative blue-light stimulation in retinal pigment epithelium cells promotes exosome secretion and increases the activity of the NLRP3 inflammasome. Curr Eye Res. 2019;44(1):67–75. doi:10.1080/02713683.2018.1518458
76. Atienzar-Aroca S, Flores-Bellver M, Serrano-Heras G, et al. Oxidative stress in retinal pigment epithelium cells increases exosome secretion and promotes angiogenesis in endothelial cells. J Cell Mol Med. 2016;20(8):1457–1466. doi:10.1111/jcmm.12834
77. Maisto R, Oltra M, Vidal-Gil L, et al. ARPE-19-derived VEGF-containing exosomes promote neovascularization in HUVEC: the role of the melanocortin receptor 5. Cell Cycle. 2019;18(4):413–424. doi:10.1080/15384101.2019.1568745
78. Otsuki Y, Ito E, Mukai A, et al. CD63(+) extracellular vesicles from retinal pigment epithelial cells participate in crosstalk with macrophages in the innate inflammatory axis. Exp Eye Res. 2021;205:108496. doi:10.1016/j.exer.2021.108496
79. Klingeborn M, Arora V, Chung C, et al. Desmosome and hemidesmosome release via exosomes from retinal pigmented epithelium - a precursor to epithelial-mesenchymal transition in early AMD? Curr Eye Res. 2025;50(12):1279–1288. doi:10.1080/02713683.2025.2469235
80. Rajendran RL, Gangadaran P, Ghosh S, Nagarajan AK, Batabyal R, Ahn BC. Unlocking the secrets of single extracellular vesicles by cutting-edge technologies. Pathol Res Pract. 2025;269:155878. doi:10.1016/j.prp.2025.155878
81. Tang Y, Kang Y, Zhang X, Cheng C. Mesenchymal stem cell exosomes as nanotherapeutics for dry age-related macular degeneration. J Control Release. 2023;357:356–370. doi:10.1016/j.jconrel.2023.04.003
82. Guan J, Meng F, Wang C, Zhang B, Chen J, Han J. Recent advances in engineered exosome-based therapies for ocular vascular disease. J Nanobiotechnology. 2025;23(1):526. doi:10.1186/s12951-025-03589-3
83. Chang DB, Luttrull JK. Comparison of subthreshold 577 and 810 nm micropulse laser effects on heat-shock protein activation kinetics: implications for treatment efficacy and safety. Transl Vis Sci Technol. 2020;9(5):23. doi:10.1167/tvst.9.5.23
84. Inagaki K, Shuo T, Katakura K, Ebihara N, Murakami A, Ohkoshi K. Sublethal photothermal stimulation with a micropulse laser induces heat shock protein expression in ARPE-19 cells. J Ophthalmol. 2015;2015:729792. doi:10.1155/2015/729792
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Expression Characteristics of Soluble sCD13 in Wet Age-Related Macular Degeneration and Its Diagnostic Value and Correlation Study
Zhang B, Tian F, Hu X, Hu Y, Li W
International Journal of General Medicine 2026, 19:553165
Published Date: 28 January 2026