")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 11
Dysfunctional phagocytosis capacity, granulocyte recruitment and inflammatory factor secretion of Kupffer cells in diabetes mellitus reversed by Lidocaine
Authors Wang R , Sheng M, Shi F , Zhao Y , Zhao L, Wu J, Wu G, Song Q
Received 6 September 2018
Accepted for publication 17 October 2018
Published 26 November 2018 Volume 2018:11 Pages 827—834
DOI https://doi.org/10.2147/DMSO.S186695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Ruibin Wang,1 Minjia Sheng,2 Feng Shi,3 Yanjie Zhao,4 Lin Zhao,5 Jiangping Wu,4 Guangjiang Wu,6 Qingkun Song7–9
1Department of Emergency, Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, China; 2Department of Gynaecology and Obstetrics, China-Japan Union Hospital of Jilin University, Changchun, Jilin 130033, China; 3Department of Pathology, Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, China; 4Department of Medical Oncology, Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, China; 5Department of Medical Records and Statistics, Xuanwu Hospital, Capital Medical University, Beijing 100045, China; 6Department of Infection Control, Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, China; 7Department of Gynaecology and Obstetrics, Beijing Key Laboratory of Cancer Therapeutic Vaccine, Beijing 100038, China; 8Department of Evidence-based Medicine, Oncology School of Capital Medical University, Beijing 100038, China; 9Department of Science and Technology, Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, China
Purpose: Kupffer cells (KCs) present dysfunctional immunity capacity among the diabetes mellitus patients. This study aims to investigate whether Lidocaine could reverse dysfunctions of KCs, in terms of phagocytosis, granulocyte recruitment and inflammatory mediator secretion.
Methods: db/db and C57BL/6 mice were employed to establish diabetic and nondiabetic models. Upon intravenous injection of Lidocaine, KCs were isolated and cultured ex vivo. The functions of phagocytosis, recruiting granulocytes and inflammatory mediator secretion in KCs were compared between Lidocaine-treated and untreated (control) groups.
Results: Comparing with nondiabetic mice, KCs in diabetic mice presented reduced phagocytosis, activated granulocyte recruitment, increased expression of intercellular cell adhesion molecule-1 (ICAM-1) and activated levels of inflammatory mediators. With Lidocaine injection, phagocytic functions of KCs in diabetic mice were improved significantly; in contrast, recruitment of granulocytes, expression of ICAM-1 and secretion of inflammatory mediators were reduced markedly. However, Lidocaine intervention did not alter KC functions in phagocytosis, granulocyte recruitment, ICAM-1 expression or inflammatory mediator secretion among nondiabetic mice.
Conclusion: Lidocaine reversed diabetes-related dysfunctions of KCs in terms of phagocytosis, granulocyte recruitment, ICAM-1 expression or inflammatory mediator secretion.
Keywords: macrophages, diabetes, phagocytosis, granulocyte recruitment, inflammatory mediator, Lidocaine
Introduction
Diabetes mellitus (DM) is a chronic metabolic disease with lifetime hyperglycemia. According to 2015 International Diabetes Federation, 415 million people had been diagnosed with DM and 5 million people died of DM worldwide.1 In China, the number of DM patients accounts for 25% of global cases.2 In 2010, the prevalence of DM increased to 11.6% of the population in China.3 The age-standardized incidence of type 2 DM was 9.6 and 9.2 per 1,000 person-years in men and women, reported from the China Multicenter Collaborative Study of Cardiovascular Epidemiology and the China Cardiovascular Health Study.4 DM is a serious burden to public health, as well as economic and social development in China.5
Kupffer cells (KCs) are a group of mononuclear macrophages, engaging in removal of bacteria from the circulation and subsequent elimination of phagocytosed bacteria.6 KCs played an important role against bacterial infection from the gastrointestinal tract; however, in DM, the KCs were dysfunctional5 while the inflammatory factors were significantly activated.7,8 KCs activation was related with the altered level of tumor necrosis factor α (TNF-α), interferon (IFN), ILs and nitric oxide (NO).6,9 In our previous study, the number of KCs was comparable with non-DM patients; however, ICAM-1 level was significantly higher in DM patients. Lidocaine was reported to affect the inflammatory factors,10 but whether lidocaine can improve the KCs function of diabetic patients has not been reported.
This study aimed to investigate whether Lidocaine could recover functions of KCs in terms of phagocytosis, granulocyte recruitment and inflammatory factor secretion using diabetes murine models.
Methods
Ethical approval
All procedures performed in studies in were in accordance with the ethical standards of the ethical committee of Beijing Shijitan Hospital, Capital Medical University, and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The ethical committee of Beijing Shijitan Hospital, Capital Medical University approved this study.
Animal administration
Ten-week-old male C57BLKS/J db/m and C57BLKS/J db/db mice (Cavens Experimental Animal Co. Ltd.) were divided into nondiabetic (n=5, C57BLKS/J db/m mice), nondiabetic + lidocaine (n=5, C57BLKS/J db/db mice injected with lidocaine), diabetes (n=5, C57BLKS/J db/db mice) and diabetes + lidocaine (n=5, C57BLKS/J db/db mice injected with lidocaine) groups. Lidocaine (1 mg/kg) was injected intravenously once every 5 minutes for four times. After 1 hour, all mice were anesthetized and their livers, femur and tibia were aseptically harvested.
KCs isolation and culture and detection
The liver tissue was smashed to the size of 1 mm3. 0.1% type IV collagenase was added for digestion at 37°C for 40 minutes. The sample was blown gently with a pipette for about 100 times. The cell suspension was collected by a 100 µm cell strainer, and the filtrate was centrifuged and re-suspended in the medium of DMEM supplement with 10% FBS and 1% Penicillin-Streptomycin (PS).
Number of KCs detection
The number of KCs was detected by immunohistochemistry. With formalin fixation, paraffin-embedded samples were collected from all mice. A serial of 4 µm thick slides were used to determine the number of KCs of each specimen. CD68 monoclonal antibody (sc-20060; Santa Cruz Biotechnology Inc., Dallas, TX, USA) was purchased from Beijing Zhong Shan Golden Bridge Biotechnology Co. Ltd. Sections were baked at 60°C in a dehydration oven for 60 minutes, dewaxed and rehydrated with xylene and graded alcohol washes. Antigen retrieval and deparaffinization were processed with the EnVision FLEX Target Retrieval Solutions, cooled to room temperature in Tris Buffered Saline Tween wash buffer for 5 minutes and then incubated with the primary antibodies. CD68 detection was visualized with diaminobenzidine. Slides were counterstained with hematoxylin. CD68 was expressed in the cytoplasm of KCs, and the dark brown granules in cytoplasm were taken as CD68-positive reaction cells. The average number of KCs was counted in five randomly selected high-power fields (HPF) under a microscope (×400) for each specimen.
Electron microscopy to detect cellular ultrastructure
The liver tissue was transferred to an Eppendorf (Hamburg, Germany) tube containing 2.5% glutaraldehyde solution, fixed for ~1 hour and processed a 5-minute wash for three times by 1 M phosphate buffer. With fixation in 1% osmic acid for 30 minutes, the sample was processed a 5-minute wash twice by 0.1 M phosphate buffer. 3%–4% agar was used to pre-embed the sample and after solidification, the agar was cut into small pieces of about 1 mm3. 50%, 70% and 90% ethanol solutions, as well as 90% acetone and 10% acetone were applied for dehydration. Propylene oxide transition, epoxy resin impregnation, embedding, polymerization, ultrathin microtome section, uranyl acetate and lead citrate double staining, and drying were performed orderly. The sample was observed under FE I Tecnai G 212 transmission electron microscope at magnification of ×5,000.
Phagocytosis assay
KCs (1×108) and polystyrene beads were seeded on coverslips. After incubation for 60 minutes, the coverslips were rinsed three times with PBS, fixed in 4% paraformaldehyde for 30 minutes and stained with Giemsa. KCs phagocytosing polystyrene beads under a microscope were observed. On each coverslip, five fields were selected randomly and ten KCs randomly selected in each field of view. The mean number of polystyrene beads phagocytosed by KCs represented phagocytosis function.
Granulocyte recruitment
The recruitment capacity was measured by Transwell assay. The femur and tibia were flushed with culture medium in a 1 mL sterile syringe to obtain bone marrow, and single neutrophil cell suspension was prepared at the concentration of 2×108–1×109/mL. A suitable amount of separation liquid was added to the centrifuge tube, and the cell suspension was spread above the level of separation liquid, and centrifuged for 20–30 minutes. After centrifugation, a clear stratification was obtained. The granulocytes were in the middle layer of separation liquid. Cells were cultured in medium (DMEM + 10% FBS + 1% PS), in humidified atmosphere of 5% CO2 at 37°C.
Cell migration assays were operated with a modified Boyden chamber (Costar-Corning, New York, NY, USA). Five hundred microliter KC suspensions with a density of 2×105 cells/mL were added in the lower chamber. When cells growing to 80% confluence, the supernatant was discarded, and 500 µL complete medium (DMEM + 10% FBS + 1% PS) was added. Two hundred microliter neutrophil suspension with cells at a 2×105/mL density were added to the upper chamber and incubated at 37°C, 5% CO2 and saturated humidity for 2 hours. Liquid in the upper chamber was discarded, and the cells were washed twice with PBS. The chamber was inverted and air dried. Five hundred microliter 0.1% crystal violet dye solution was added in the lower chamber to submerge the chamber in the dye solution. After incubated for 30 minutes at 37°C, the crystal violet staining solution was discarded. The cells were washed twice with PBS and captured using photographed under a microscope (Olympus IX51) equipped with an Olympus color 3 digital camera (Olympus). Migration was assessed by counting the number of stained cells from five random HPF at ×200 magnification.
ICAM-1 detection
ICAM-1 expression in KCs was detected by Western blot. Cells were collected and lysed on ice with RIPA lysis buffer containing protease inhibitors. The extracted proteins were quantified with bicinchoninic acid quantification assay. Total cellular proteins were subjected to SDS-PAGE gel and transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat milk for 2 hours and then incubated with respective primary antibody overnight at 4°C. Following washing for three times with TBS-T for 10 minutes, the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibody for 1.5 hour at room temperature. The bands were captured with SuperSignal West Pico substrate (Thermo Fisher Scientific, Rockford, IL, USA). β-Actin expression was used as the internal reference, and the expression of ICAM-1 was estimated by the absorbance (OD) ratio to β-actin, semiquantitatively.
Detection of TNF-α, IL-6 and IFN-γ
Quantitation of TNF-α, IL-6 and IFN-γ was conducted with ELISA kits. Supernatant was obtained by centrifugation of cell culture medium at 1,000 g for 20 minutes. Equal amounts of cell culture medium were added in ELISA kits and reacted for 20–25 minutes at room temperature. The OD of TNF-α, IL-6 and IFN-γ was measured with a microplate reader at a wavelength of 450 nm to calculate the sample concentration.
Detection of NO
Equal amounts of cell culture medium and Griess reagent were mixed and reacted for 20–25 minutes at room temperature. The OD of NO was read at 546–550 nm wavelength to calculate the concentration.
Statistical analysis
GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA) was used for data analyses. All data were expressed as the median and IQR. Kruskal–Wallis test was used to estimate the difference between groups. Pair-wise comparisons were conducted by Wilcoxon tests with Bonferroni adjustments. All tests were two-sided, and a P<0.05 was considered statistically significant.
Results
Number of KCs in DM
The median number of KCs was 6/HPF (IQR =1), comparable between diabetic and nondiabetic mice (Figure 1A). However, KCs in diabetic mice had less mitochondria and endoplasmic reticulum (ER) than nondiabetic mice (Figure 1B, C).
 | Figure 1 Quantification of Kupffer cells, mitochondria and endoplasmic reticulum in Kupffer cells in nondiabetic vs diabetic mice. Notes: (A) Number of Kupffer cells (median ± IQR); (B) mitochondria (red arrow) and endoplasmic reticulum (green arrow) in Kupffer cells in nondiabetic mice; (C) mitochondria (red arrow) and endoplasmic reticulum (green arrow) in Kupffer cells in diabetic mice. Magnification ×5000. Abbreviation: HPF, high-power fields. |
Phagocytosis capacity
The phagocytic capacity was reduced in KCs from diabetic mice, in contrast to nondiabetic mice (P<0.05, Figure 2A, B). The number of beads phagocytosed by KCs was 10 (IQR =4) and 59 (IQR =5) in diabetic and nondiabetic mice (P<0.05, Figure 2C). KCs from nondiabetic mice had a similar phagocytic ability, the number of beads phagocytosed by KCs from nondiabetic and nondiabetic plus Lidocaine mice being comparable (Figure 2C, D). Phagocytic ability was improved significantly in diabetic mice receiving Lidocaine administration (Figure 2E), with a median phagocytosis of 49 (IQR =5) (Figure 2C). The phagocytic capacity of KCs from diabetic mice treated with Lidocaine was still lower than nondiabetic mice (Figure 2C).
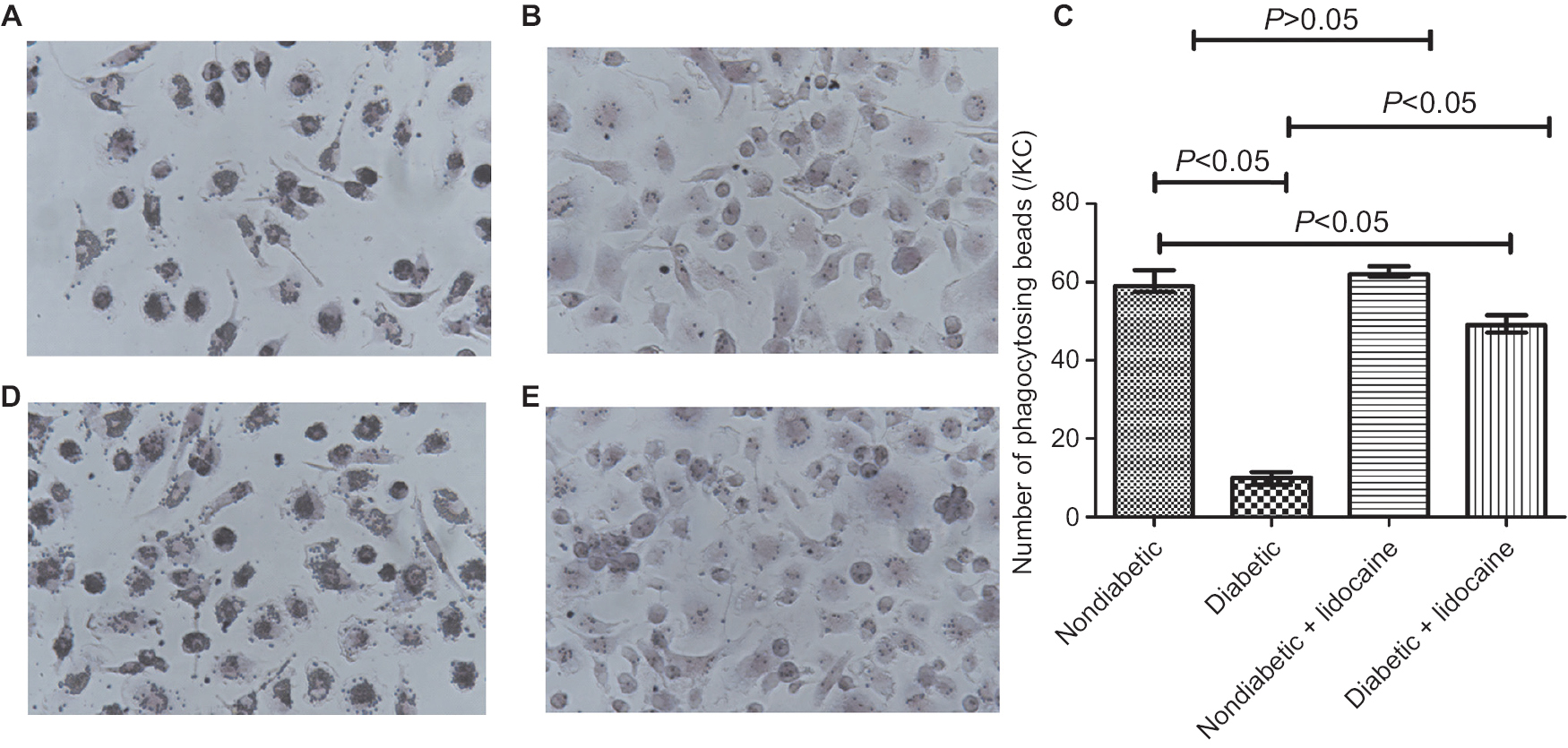 | Figure 2 Phagocytosis of KCs in nondiabetic and diabetic mice. Notes: (A) Baseline phagocytosis in nondiabetic mice; (B) baseline phagocytosis in diabetic mice; (C) comparison of phagocytosis of KCs among different groups (median ± IQR, n=5); (D) phagocytosis in nondiabetic mice treated with Lidocaine; (E) phagocytosis in diabetic mice treated with Lidocaine. Magnification ×200. Abbreviation: KC, Kupffer cell. |
Granulocyte recruitment
Compared to nondiabetic mice, KCs recruited more granulocytes in diabetic mice (Figure 3A, B). The median of granulocytes recruited by KCs in nondiabetic and diabetic mice was 15/HPF (IQR =7) and 96/HPF (IQR =19), respectively (P<0.05, Figure 3C). Lidocaine administration did not change the number of granulocytes in nondiabetic mice (Figure 3C, D). However, diabetic mice received Lidocaine had a significant reduction of granulocytes with a median of 33/HPF (IQR =11) (Figure 3C, E). The median number of granulocytes recruited by KCs in diabetic mice treated with Lidocaine was still higher than nondiabetic mice (Figure 3C, E).
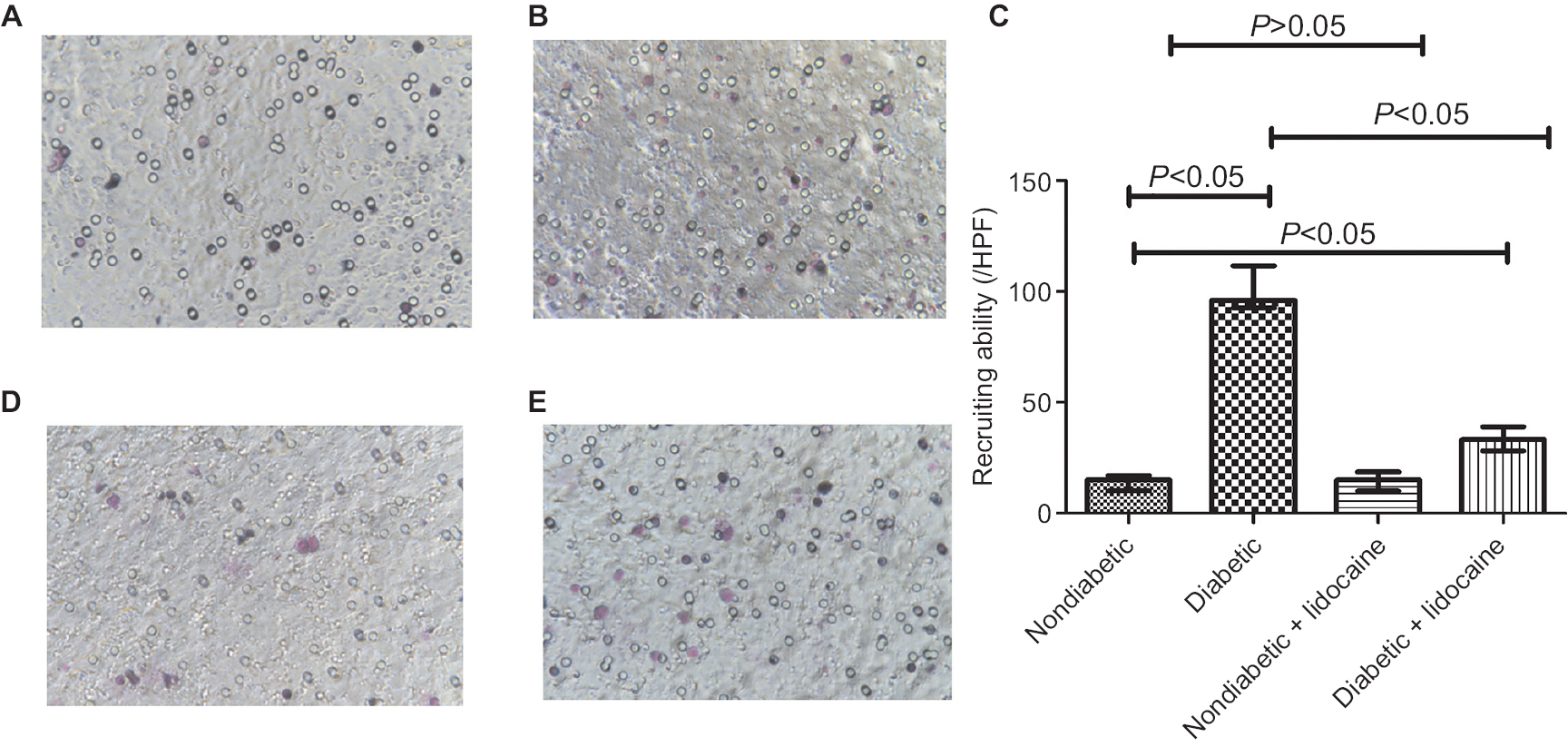 | Figure 3 Granulocyte recruitment by Kupffer cells in nondiabetic and diabetic mice. Notes: (A) Baseline granulocyte recruitment in nondiabetic mice; (B) baseline granulocyte recruitment in diabetic mice; (C) comparison of granulocyte recruitment by Kupffer cells among different groups (median ± IQR, n=5); (D) granulocyte recruitment in nondiabetic mice treated with Lidocaine; (E) granulocyte recruitment in diabetic mice treated with Lidocaine. Magnification ×200. Abbreviation: HPF, high-power fields. |
ICAM-1 production
The level of ICAM-1 was 2.41 (IQR =0.03) and 1.33 (IQR =0.02) in diabetic and nondiabetic mice, respectively (P<0.05, Figure 4A, B). The ICAM-1 level was reduced to 1.51 (IQR =0.03) upon Lidocaine intervention in diabetic mice (P<0.05, Figure 4B), which was not affected in nondiabetic mice (P>0.05, Figure 4A, B).
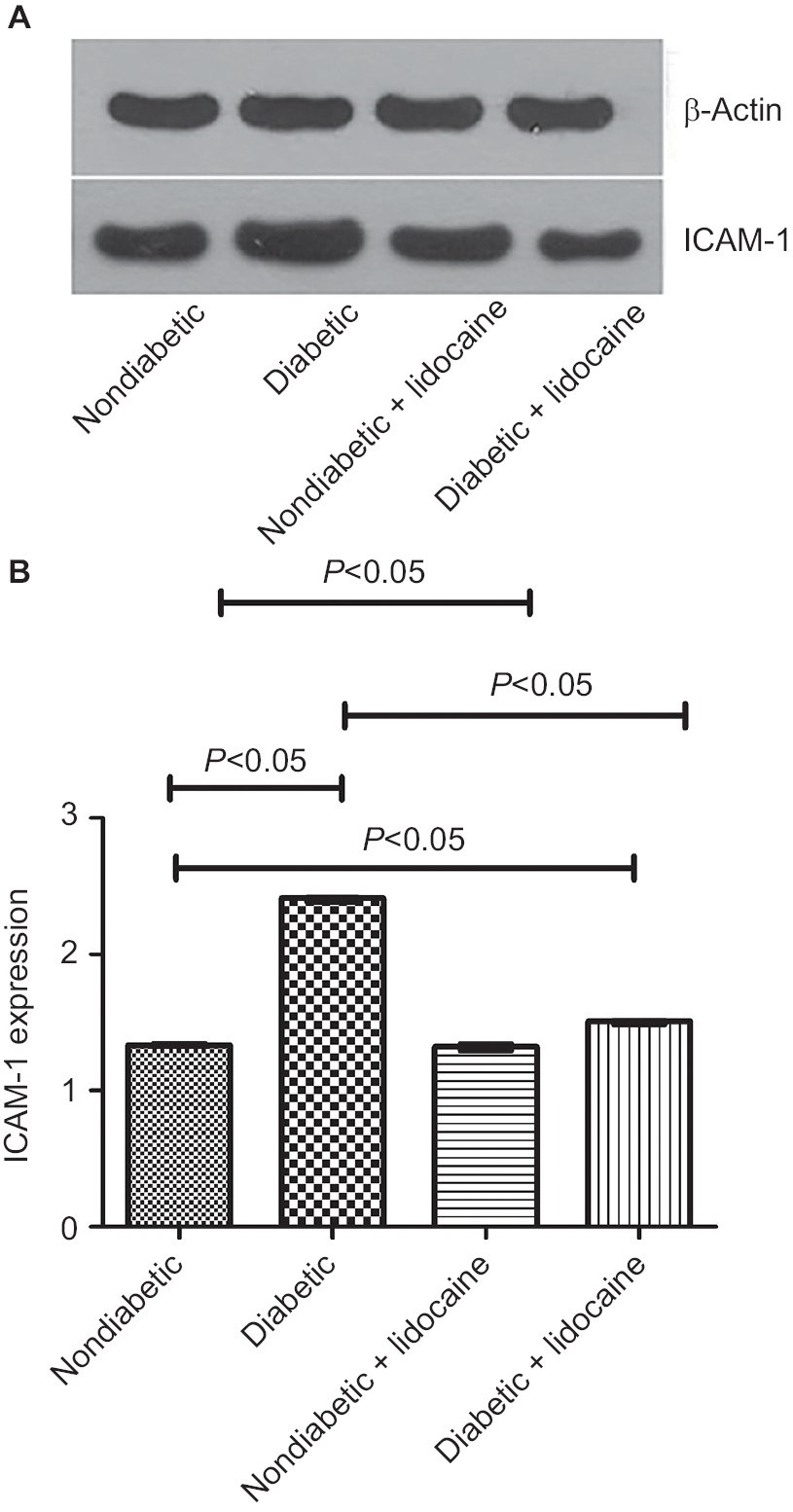 | Figure 4 ICAM-1 expression on the surface of Kupffer cells in nondiabetic and diabetic mice. Notes: (A) Detection of ICAM-1 using Western blot; (B) comparison of ICAM-1 expression levels among different groups (median ± IQR, n=5). |
NO production
The level of NO was 1.32 mmol/L (IQR =0.22) in nondiabetic mice, significantly lower than that of diabetic mice (4.97 mmol/L, IQR =0.10) (Figure 5A). In response to Lidocaine administration, NO was reduced significantly to 3.37 mmol/L (IQR =0.54) in diabetic mice, which was still higher than nondiabetic mice (P<0.05, Figure 5A). The level of NO did not change significantly in nondiabetic mice with Lidocaine intervention (Figure 5A).
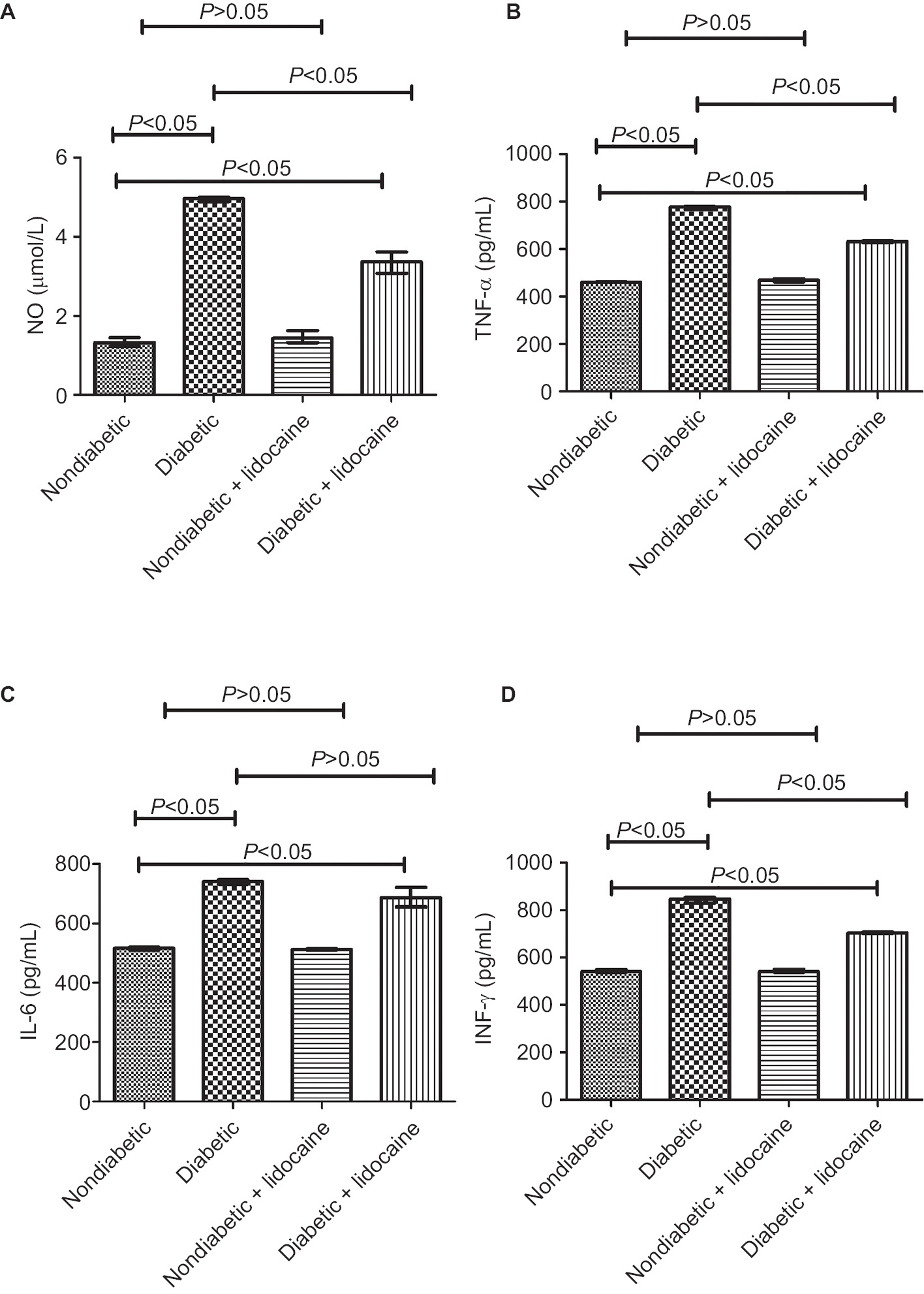 | Figure 5 Secretion of inflammatory mediators by Kupffer cells (median ± IQR, n=5). Notes: (A) Nitric oxide secretion; (B) tumor necrosis factor alpha secretion; (C) interleukin-6 secretion; (D) interferon-gamma secretion. |
TNF-α production
TNF-α level was significantly higher (776.91 pg/mL, IQR =14.52) in diabetic mice compared with nondiabetic mice (461.61 pg/mL, IQR =3.35) (P<0.05, Figure 5B). In response to Lidocaine intervention, TNF-α level was reduced to 631.44 pg/mL (IQR =7.70) in diabetic mice (P<0.05), still higher than nondiabetic mice (Figure 5B). Lidocaine intervention did not affect TNF-α level in nondiabetic mice (Figure 5B).
IL-6 production
The baseline level of IL-6 was 516.32 pg/mL (IQR =8.58) and 741.84 pg/mL (IQR =15.21) in nondiabetic and diabetic mice, respectively (P<0.05, Figure 5C). In diabetic mice, in response to Lidocaine administration, IL-6 level was reduced to 687.66 pg/mL (IQR =64.54), still higher than nondiabetic mice (Figure 5C). Lidocaine intervention did not change IL-6 level in nondiabetic mice (Figure 5C).
INF-γ production
The level of INF-γ was 541.60 pg/mL (IQR =11.66) in nondiabetic mice, significantly lower than that of diabetic mice (845.33 pg/mL, IQR =25.01, Figure 5D). Upon Lidocaine treatment, INF-γ level was reduced to 704.46 pg/mL (IQR =6.86) in diabetic mice (Figure 5D). Lidocaine intervention did not influence INF-γ level significantly in nondiabetic mice (Figure 5D).
Discussion
Worldwide, 284 million people have been affected by DM and have an increasing prevalence.11 DM patients are vulnerable to infections due to dysfunctional immunity, neuropathy and poor circulation.12 Hyperglycemia contributes to dysfunctional immunity and bacterial growth.5 35.3% of DM patients have an increased risk for pyogenic liver abscess, compared with normal people.13 KCs are the first line of defense for pathogens entering the liver and involved in elimination of resisting bacteria and viruses invading into liver sinus.6,14,15 The number of KCs was similar between diabetic and nondiabetic mice. The amounts of mitochondria, lysosome and ER in KCs in the DM group were significantly lower than the control group. ER connects inflammatory and stress signals with metabolism of cells, engaged in the development of type 2 DM.16 Reduction in mitochondria and ER may imply diminished phagocytosis of KCs.17 The functions rather than the number of KCs are significantly affected in DM. The KCs of diabetic mice were dysfunctional, with markedly reduced phagocytic ability than nondiabetic mice. This was consistent with previous reports that the function of diabetic macrophages was depressed.5,18,19 Hyperglycemia may lead to dysfunctional macrophages through anoxic stress.18–20 Therefore, Lidocaine may improve phagocytosis of KCs by inhibiting inflammatory response in diabetic mice. Lidocaine may restore phagocytosis of KCs in DM through suppressing the release of inflammatory factors.
KCs involve in direct clearance and recruit neutrophils to eliminate invading bacteria.21,22 Pathological exacerbation of endotoxin, TNF-α, TGF, IFN, ILs, oxygen-free radicals, NO and other inflammatory mediators contribute to the activation of KCs.6,9
The inflammatory mediators upregulate ICAM-1 expression in KCs.23 Higher expression of ICAM-1 on the surface of KCs was observed only in DM. KCs were significantly activated among DM patients. DM patients always have chronic inflammation24,25 under an aberrant immunity microenvironment.26,27 Our study identified increased secretion of NO, IL-6, INF-γ and TNF-α, as well as enhanced expression of ICAM-1 in KCs of diabetic mice compared with nondiabetic mice. Therefore, KCs of diabetic mice were activated and recruited more granulocytes. We found that Lidocaine could inhibit the release of inflammatory mediators, downregulate expression of ICAM-1 on the surface of KCs and reduce granulocyte recruitment in diabetic mice.
Lidocaine is widely used as a local anesthetic and antiarrhythmic agent. Recent studies have indicated that Lidocaine exerts a protective effect on vasculature system,28 inhibits the expression of inflammatory factors10 and presents an anti-tumor effect.29 These previous findings were similar as our study. Amide-linked local anesthetics have well-established anti-inflammatory effects, attenuate inflammatory Src signaling and disrupt the PI3K–Akt–NO pathway, thus blocking Src-dependent neutrophil adhesion and endothelial hyperpermeability.28 Transcription factor NF-κB is indicated to be involved in the development of DM and chronic inflammation.30,31 Lidocaine could regulate NF-κB signaling pathway and inhibit cellular inflammatory response.10 Lidocaine could protect endothelial cell function by inhibiting inflammatory mediators.28
Major limitations of this study include a relatively small sample size and lack of functional validation for hypothesized molecular mechanisms. ICAM-1 expression was only detected at the translation level but not the transcription.
In DM, KCs actively recruit granulates, markedly release inflammation mediators and dysfunctionally phagocytose. Lidocaine was capable of recovering phagocytosis and inhibiting the release of inflammatory mediators.
Acknowledgments
This study was supported by Beijing Shijitan Hospital (RW. 2016-Q18), Ministry of Railway (QS. grant number J2017Z604) and Beijing Municipal Administration of Hospitals (QS. grant number PX2018029). The supporting organizations had no role in study design, data collection, analysis and interpretation.
Author contributions
Study design: QS, MS and RW. Data acquisition: FS, YZ, LZ and GW. Data analysis: QS, LZ and JW. Manuscript writing and modification: QS, FS, RW, YZ, LZ, JW, MS and GW. Submission approval: QS, FS, RW, YZ, LZ, JW, MS and GW. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The author reports no conflicts of interest in this work.
References
Diabetes: a global emergency. IDF Diabetes Atlas Seventh Edition. 2015:11–13. Available from: http://www.diabetesatlas.org/resources/previous-editions.html. Accessed November 19, 2018. | ||
Zhou B, Lu Y, Hajifathalian K; NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet. 2016;387(10027):1513–1530. | ||
Xu Y, Wang L, He J, et al. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310(9):948–958. | ||
Xue H, Wang C, Li Y, et al. Incidence of type 2 diabetes and number of events attributable to abdominal obesity in China: a cohort study. J Diabetes. 2016;8(2):190–198. | ||
Joshi N, Caputo GM, Weitekamp MR, Karchmer AW. Infections in patients with diabetes mellitus. N Engl J Med. 1999;341(25):1906–1912. | ||
Wohlleber D, Knolle PA. The role of liver sinusoidal cells in local hepatic immune surveillance. Clin Transl Immunology. 2016;5(12):e117. | ||
Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8(12):923–934. | ||
Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185. | ||
Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26(10):1175–1186. | ||
Yuan T, Li Z, Li X, Yu G, Wang N, Yang X. Lidocaine attenuates lipopolysaccharide-induced inflammatory responses in microglia. J Surg Res. 2014;192(1):150–162. | ||
Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87(1):4–14. | ||
Dryden M, Baguneid M, Eckmann C, et al. Pathophysiology and burden of infection in patients with diabetes mellitus and peripheral vascular disease: focus on skin and soft-tissue infections. Clin Microbiol Infect. 2015;21(Suppl 2):S27–S32. | ||
Chen YC, Lin CH, Chang SN, Shi ZY. Epidemiology and clinical outcome of pyogenic liver abscess: an analysis from the National Health Insurance Research Database of Taiwan, 2000-2011. J Microbiol Immunol Infect. 2016;49(5):646–653. | ||
Doorduijn DJ, Rooijakkers SH, van Schaik W, Bardoel BW. Complement resistance mechanisms of Klebsiella pneumoniae. Immunobiology. 2016;221(10):1102–1109. | ||
Balmer ML, Slack E, de Gottardi A, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. 2014;6(237):237ra266. | ||
Özcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. | ||
Lohmann-Matthes ML, Steinmüller C, Franke-Ullmann G. Pulmonary macrophages. Eur Respir J. 1994;7(9):1678–1689. | ||
Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010;2010:453892–11. | ||
Tabet F, Lambert G, Cuesta Torres LF, et al. Lipid-free apolipoprotein A-I and discoidal reconstituted high-density lipoproteins differentially inhibit glucose-induced oxidative stress in human macrophages. Arterioscler Thromb Vasc Biol. 2011;31(5):1192–1200. | ||
Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes. 2008;57(11):3090–3098. | ||
Gregory SH, Wing EJ. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J Leukoc Biol. 2002;72(2):239–248. | ||
Gregory SH, Cousens LP, van Rooijen N, Döpp EA, Carlos TM, Wing EJ. Complementary adhesion molecules promote neutrophil-Kupffer cell interaction and the elimination of bacteria taken up by the liver. J Immunol. 2002;168(1):308–315. | ||
Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J Immunol. 1986;137(1):245–254. | ||
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119. | ||
Galassetti P. Inflammation and oxidative stress in obesity, metabolic syndrome, and diabetes. Exp Diabetes Res. 2012;2012:943706–2. | ||
Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014;105(2):141–150. | ||
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. | ||
Piegeler T, Votta-Velis EG, Bakhshi FR, et al. Endothelial barrier protection by local anesthetics: ropivacaine and lidocaine block tumor necrosis factor-α-induced endothelial cell Src activation. Anesthesiology. 2014;120(6):1414–1428. | ||
Piegeler T, Votta-Velis EG, Liu G, et al. Antimetastatic potential of amide-linked local anesthetics: inhibition of lung adenocarcinoma cell migration and inflammatory Src signaling independent of sodium channel blockade. Anesthesiology. 2012;117(3):548–559. | ||
Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13(1):11–22. | ||
Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25(10):1939–1948. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.