Back to Journals » International Journal of Nanomedicine » Volume 13
Dysfunction of various organelles provokes multiple cell death after quantum dot exposure
Received 15 November 2017
Accepted for publication 12 January 2018
Published 7 May 2018 Volume 2018:13 Pages 2729—2742
DOI https://doi.org/10.2147/IJN.S157135
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Linlin Sun
Yan Wang,1,2 Meng Tang1,2
1Key Laboratory of Environmental Medicine Engineering of Ministry of Education, School of Public Health & Collaborative Innovation Center of Suzhou Nano Science and Technology, Southeast University, Nanjing, Jiangsu, People’s Republic of China; 2Jiangsu Key Laboratory for Biomaterials and Devices, Southeast University, Nanjing, Jiangsu, People’s Republic of China
Abstract: Quantum dots (QDs) are different from the materials with the micrometer scale. Owing to the superiority in fluorescence and optical stability, QDs act as possible diagnostic and therapeutic tools for application in biomedical field. However, potential threats of QDs to human health hamper their wide utilization in life sciences. It has been reported that oxidative stress and inflammation are involved in toxicity caused by QDs. Recently, accumulating research unveiled that disturbance of subcellular structures plays a magnificent role in cytotoxicity of QDs. Diverse organelles would collapse during QD treatment, including DNA damage, endoplasmic reticulum stress, mitochondrial dysfunction and lysosomal rupture. Different forms of cellular end points on the basis of recent research have been concluded. Apart from apoptosis and autophagy, a new form of cell death termed pyroptosis, which is finely orchestrated by inflammasome complex and gasdermin family with secretion of interleukin-1 beta and interleukin-18, was also summarized. Finally, several potential cellular signaling pathways were also listed. Activation of Toll-like receptor-4/myeloid differentiation primary response 88, nuclear factor kappa-light-chain-enhancer of activated B cells and NACHT, LRR and PYD domains-containing protein 3 inflammasome pathways by QD exposure is associated with regulation of cellular processes. With the development of QDs, toxicity evaluation is far behind its development, where specific mechanisms of toxic effects are not clearly defined. Further studies concerned with this promising area are urgently required.
Keywords: quantum dot (QD), autophagy, pyroptosis, inflammasome, endoplasmic reticulum (ER) stress, lysosomal rupture
Introduction
As a semiconductor nanomaterial, quantum dot (QD) consists of chemical elements from the chemical element group III–IV or the second group VI in the periodic table, whose size generally ranges from 1 to 10 nm. Different from bulk counterparts, QDs possess superiority in fluorescence and optical stability and, thus, act as a candidate for imaging and diagnosis.1 Compared with the conventional dyes, the utility model has unique photophysical and electrical properties with broad absorption, narrow emission spectra and resistance to photobleaching.2 Surface chemistry can help enhance the fluorescence efficiency and keep relatively low rate of quenching.3 A typical QD is made up of a core and a shell with different materials at large. Peptides, antibodies and nucleic acids can be used to modify primary QDs for special biomedical use. For example, a core of CdTe and a shell of ZnS constitute CdTe/ZnS QD, which can be further conjugated with polyethylene glycol (PEG) for better solubility. In addition, the emission wavelength of QDs can be easily controlled by sizes of the core. Figure 1 illustrates the typical basic structure of QDs with core/shell conjugate. Cadmium-containing QDs are the widely studied materials thus far in the field of biological imaging, such as CdSe/ZnS and CdTe/ZnS core/shell QDs. On account of a series of distinct size effects, quantum confinement effect, macroscopic quantum tunneling effect and specific surface effect, prolonged exposure of metal-containing QDs can stimulate the release of toxic metal ions to the surrounding biological fluids and, thus, exert adverse effect.4 Fortunately, surface modification is a commonly useful way to overcome this challenge. Besides diagnosis, QDs show great potential for application in tumor tracing, microbial detection and drug-targeted therapy.5,6 It has been reported that QDs can be used as probes for in vivo imaging. Therefore, labeled blood vessels and tumors can be clearly distinguished.2,7,8 Withal, application of QDs is capable of guiding medicines to specific foci for targeted drug therapy.6 Although much attention has been paid to the potential function of QDs, it is necessary to conduct safety evaluation of nanomaterials for medical application when the potential threats exist. Due to the special nanosize effect, physiochemical properties of QDs are different from those of micro-scaled particles. A large number of studies in vitro have shown that QDs are obviously toxic, which may be attributed to the metal ion and several undefined factors of nanoparticles themselves.9 In order to obtain comprehensive information for the safe application of QDs in medical field, it is particularly important to conduct in vitro and in vivo research in the absence of epidemiological data. A decade ago, researchers started to explore potential toxicity of QDs and a strip of mysteries have been disclosed.10,11 It has been demonstrated that QDs have the capacity to disrupt the cellular structure and function in a series of cell lines.12–14 In recent years, great importance has been attached to the concrete root of toxicity. However, the molecular basis of cytotoxicity, especially the dysfunction of organelles, has not been clearly defined. Moreover, there is no unambiguous procedure available for the safety evaluation of nanomaterials up to now. Therefore, a systematic summary is vital for the evaluation of underlying toxicity before the application of QDs to life sciences. At the same time, addition of various outer layers and modifications for biocompatibility are also of importance. In spite of the existence of several reviews regarding the toxicity of QDs, most of them have focused on their toxicity in vitro or in vivo,15–17 while potential mechanisms regarding the disruption of organelles as discussed above were not incorporated. Therefore, this review is intended to summarize diverse forms of cell death and pathways of cytotoxicity induced by QDs. Furthermore, the underlying mechanisms of disruption of subcellular structures have also been included.
 | Figure 1 The basic structure of QD is composed of a core and a shell. |
Forms of cell death caused by QDs
Currently, human-derived18 or rodent-derived immortalized cells19 and primary cells20,21 have been extensively used to investigate the adverse effects of nanomaterials. Cytotoxicity of QDs is generally characterized by diminished cell viability, elevation of apoptosis rate and dysfunction of immune responses. Indicators of oxidative stress are significantly changed in the form of increase in methane dicarboxylic aldehyde and reactive oxygen species (ROS) with impairment of the antioxidant system.22,23 Due to the lack of epidemiological data, experimental design for the safety evaluation of nanomaterials seems to be of importance. In vitro experiments possess plenty of advantages for the judgment of toxic effect of exogenous substances, including convenience and in-depth research, and extensive launch via diverse cultured cell models.24,25 Investigations show that QDs more or less interfere with cellular viability and proliferation, which may not exist at a low dose, but there is an overt transformation under high dose or prolonged duration.26 When the concentration and exposure time exceed the threshold, the detrimental effects of QDs emerge. However, it is hard to confirm the accurate threshold because it is difficult to guarantee the uniformity of various QDs from disparate preparation methods. Additionally, the threshold value of disturbance of organelles is also difficult to determine.
The extent of cell injury depends on the intensity of the stress. Ordinarily, low-level oxidative stress disrupts cellular redox homeostasis, and intermediate oxidative milieu causes inflammation. Also, high oxidative burst leads to cytotoxicity and even cell death.9 Based on a growing number of investigations concerning cellular outcomes in response to QDs, forms of cell death have been summed up, which could be categorized into four representative groups as follows.
Necrosis
Necrosis is a kind of cell death with pathologic changes, which can result from invasion by external chemicals. The features of necrotic cells are composed of increased membrane permeability, cell deformation and loss of membrane integrity without obvious morphologic changes in the nucleus in the early stage.27 In late necrosis, innate immune system is heavily activated by the leakage of cellular contents with inflammatory reaction. Afterward, the immune cells (monocytes, neutrophils and eosinophils) are recruited to the site of injury to help clear debris. Interestingly, recently, necrosis has been classified as accidental cell death (necrosis) and regulated necrotic cell death (necroptosis).28 The expression of receptor-interacting protein kinase 3 (RIP3/RIPK3) protein is a molecular switch for necrosis and apoptosis.29 Conventionally, necrosis is in contrast to programmed cell death and associated with passive cellular damage, which refers to malignant cell death with the occurrence of pyknosis and karyorrhexis.30 Necroptosis is a programmed form of necrosis which executes necrosis in a programmed fashion. The signaling pathway involves the stimulation of tumor necrosis factor receptor type 1-associated death domain protein, which signals to receptor-interacting protein kinase 1 (RIP1/RIPK1) in necroptosis. Recruitment of RIPK3 to RIPK1 forms the necrosome, which is the symbol of necrosis. Phosphorylation of mixed lineage kinase domain-like protein (MLKL) is mediated by RIPK3. Oligomerization of MLKL allows MLKL to insert into the cell membrane and permeabilize the membranes of organelles.31 Formation of holes in the cell membrane thus enhances permeability, which leads to lysis of cells and release of damage-associated molecular patterns (DAMPs). Several reports about ultrafine particulate matter-induced necrotic cell death have been published.32 Necrosis induced by QDs33,34 has also been found, while necroptosis has not been reported.
Apoptosis
Apoptosis is a strictly regulated molecular process, which is characterized by chromosome fragmentation, membrane blebbing and shrinkage with the execution of caspase–cascade reaction.35 Generally, apoptosis is necessary for organism development, regulation of immune system and cellular refreshment. However, excessive apoptosis has implications in diverse diseases caused by stimuli of xenobiotics. When oxidative damage cannot be correctly repaired, apoptosis would appear. It has been found that CdTe QDs can induce upregulation of Fas receptor, which, in turn, leads to apoptosis of fibroblasts.26 Similar to cadmium-containing QDs, other types of QDs have similar effects on apoptosis.34,35
It was generally acknowledged that apoptosis usually takes place through mitochondrial or death receptor pathway with respective activation of caspase 9 and caspase 8. Initiation of extrinsic pathway by stimulation of QDs was attributed to release of tumor necrosis factor-α or contact of Fas ligand with Fas receptor.36 Activation of death receptor promotes cell death through phosphorylation of apoptotic factor c-Jun and cleavage of pro-caspase 8. With respect to intrinsic pathway, mitochondrial membrane potential (MMP) collapse through internalization of QDs and subsequent leakage of cytochrome c would give rise to cell apoptosis after activation of caspase 9. The convergence of intrinsic and extrinsic pathway is caspase 3 which is the downstream signal of caspase 9 and caspase 8 and meanwhile a key executor of apoptosis.37 Moreover, regulator proteins of B-cell lymphoma-2 (Bcl-2) family manipulate apoptosis by either induction or inhibition. Bcl-2 is one of the antiapoptotic proteins and is localized at the outer membrane of mitochondria, where it plays an important role in attenuating the actions of proapoptosis and promoting cellular survival. On the contrary, proapoptotic proteins including Bcl-2-associated X protein (Bax) and Bcl-2 antagonist/killer 1 promote mitochondrial membrane permeabilization, which, in turn, stimulates translocation of cytochrome c from the mitochondria into the cytoplasm, finally triggering cascade reaction.26 Apoptosis always accompanies the decline of Bcl-2/Bax ratio.38 Bcl-2 and its relative B-cell lymphoma-extra large (Bcl-xl) are negatively regulated by Bax. Recently, several researchers have found that in addition to these two classical pathways, endoplasmic reticulum (ER) stress also act as a sensitive early event for apoptosis. Dilatation of ER cisternae was observed in human umbilical vein endothelial cells after exposure to CdTe QDs, along with significantly elevated apoptosis rate and upregulation of ER stress markers.39 Withal, apoptosis induced by QDs can be significantly mitigated by inhibitors of ER stress, further suggesting that ER stress really participates in QD-induced apoptosis.39 The schematic representation of apoptotic pathways induced by QDs is exhibited in Figure 2.
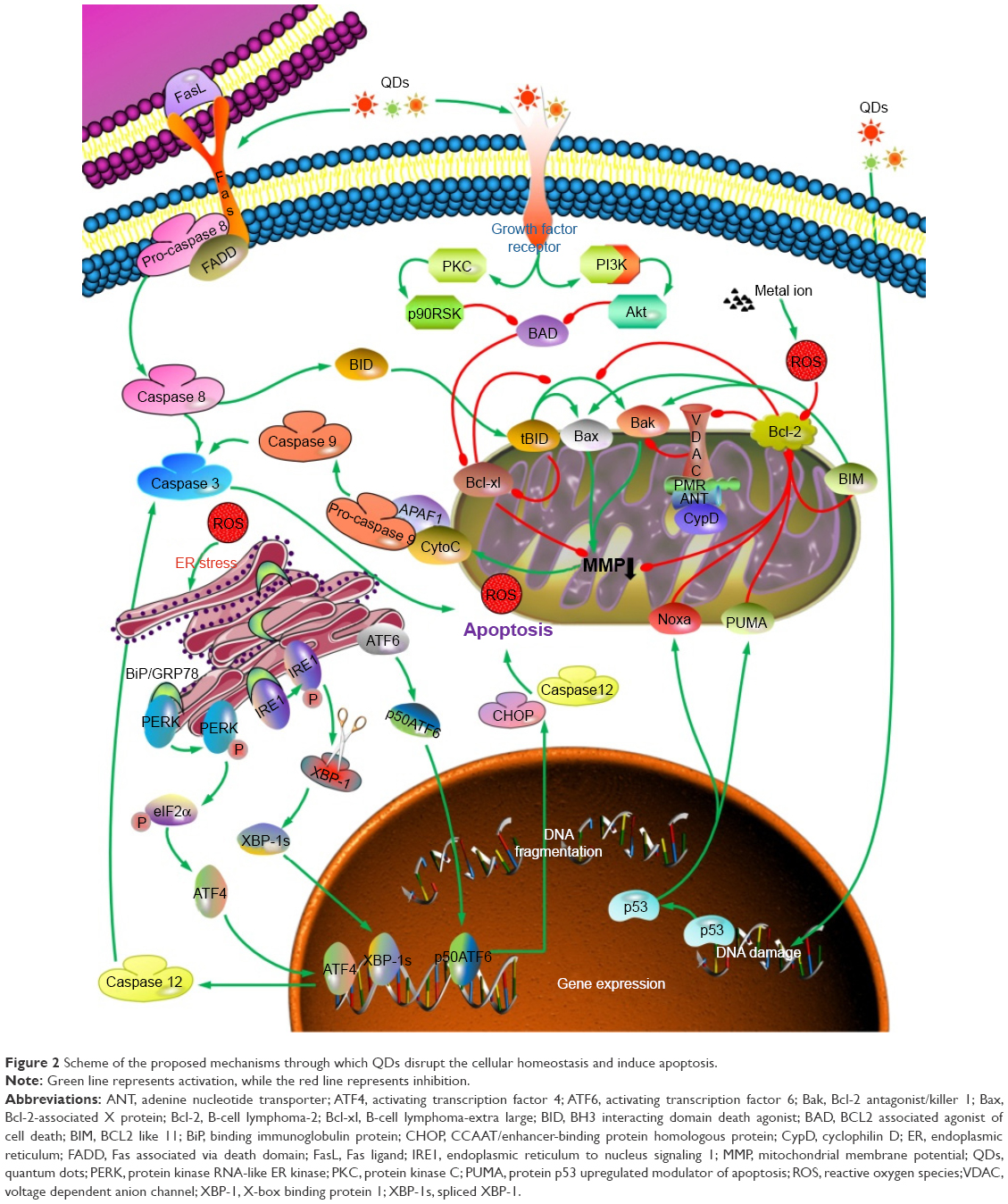 | Figure 2 Scheme of the proposed mechanisms through which QDs disrupt the cellular homeostasis and induce apoptosis. |
Autophagy
Autophagy is regarded as a prosurvival mechanism in response to cellular stress (starvation or injury), through which cells are engaged in clearance of pathogens and damaged organelles by means of autophagosome.40 In general, ubiquitin protein complexes and other cellular trashes can stimulate autophagy pathway. Initially, numerous autophagy-related proteins are activated and recruited to the phagophore structure. Next, double-membraned autophagic vacuoles, also called autophagosomes, are formed through sequestration of cytoplasmic contents. Meanwhile, lipidated microtubule-associated protein 1A/1B light chain 3B (MAP1LC3B/LC3-II) is assembled, and LC3-II complex is translocated from the cytosol to the membrane of autophagosome.41 Eventually, the autophagosome and its cargo undergo fusion with the lysosome for the synthesis of autolysosome. The total process is called autophagy flux. As reported, the process of autophagy is negatively regulated by phosphoinositide-3-kinase-I/protein kinase B/mammalian target of rapamycin pathway and positively regulated by Atgs.42,43 Under normal conditions, cellular damage is inhibited by moderate induction of autophagy through timely clearance of the waste. Nonetheless, excessive degradation of cellular components can promote a vicious loop. It is said that the protective effect of autophagy helps in cell survival in acute reaction, while long-term autophagy conversely gives rise to chronic damage.
Intriguingly, despite lack of corroborant evidence, a few hypotheses have been put forward that autophagy has an association with apoptosis.44 Also, the interdependent relationship between these two items is clarified. First, autophagy usually occurs ahead of apoptosis. The second one is that degradation of detrimental components by autophagy helps mitigate ensuing cascade reaction, through which apoptosis can be inhibited. Finally, autophagy can be transformed into apoptosis with which autophagy promotes cell death together.45 Recent studies have demonstrated that Beclin-1, a protein involved in autophagy and apoptosis, can increase the activity of caspase 9 to increase the rate of apoptosis. However, the contradiction is that cleavage of Beclin-1 from Beclin-1/Bcl-2 complex liberates Bcl-2, which consequently inactivates apoptosis and autophagy.46 Bcl-2 is considered as an important antiapoptotic protein. Notably, Bcl-2 not only plays a role in antiapoptosis but also is implicated in anti-autophagy and was thus classified as an oncogene.47 It also reveals that apoptosis could be regulated by autophagy through the selective degradation of Fas-associated phosphatase 1 and tumor protein p53 upregulated modulator of apoptosis (PUMA). It is supposed that knockdown of PUMA can rescue the cell viability as PUMA facilitates cellular apoptosis. However, cell viability is not improved, but significantly decreased after PUMA is knocked down. Actually, besides apoptosis-related pathway, PUMA is also involved in autophagy, which plays a prosurvival role. Under this condition, PUMA accounts for a larger quotient in autophagy than apoptosis. Hence, once PUMA is eliminated, the protective function of PUMA is overwhelmingly attenuated as a cause of cell damage.48 In general, both autophagy and apoptosis exist. With regard to the end point of cells, it depends on which effect is greater than the other. The relationship between autophagy and apoptosis is still confusing. Whether autophagy is a friend or a foe under different conditions needs to be determined in the future.
Mitophagy, a subset of self-protection mechanism, refers to the process of damaged mitochondria being targeted for degradation, so as to maintain storage of healthy mitochondria.49 Restraint of mitophagy brings about aggregation of dysfunctional mitochondria. Recent research has revealed that environmental factor-induced cellular aging and mitochondriopathies have been linked to inhibition of mitophagy, which would exert huge influence on cellular stability.50 Caspase 1-mediated cell death can be partially recovered through mitophagy. On the contrary, inhibition of mitophagy may further deteriorate cellular function, confirming the positive role of mitophagy.51 Of note, the excessive mitophagy is also detrimental. Some nanomaterials such as nano-silver52 and nano-rare earth metal oxide53 have a destructive impact on autophagy influx on account of disruption of autophagic–lysosomal axis. The capability of QDs to induce autophagy has been verified in a series of cell lines from multiple systems.19
Autophagy is one form of cell death. Inhibition of autophagy can alleviate cellular lesion, which further confirms the role of autophagy in cellular impairment.54,55 Whether the stress will culminate into fatal point is dependent on the inherent properties of nanomaterials and the strength of accumulative upstream events, such as increased ROS production and superfluous cytoplasmic concentration of Ca2+. The cellular end point, survival or death, is synthetically determined by multiple factors, such as cell type, intensity and duration of stress and physiochemical characteristics of xenobiotics. Nevertheless, the methods for detection of QD-induced autophagy are always confined to the observation of autophagosome by an electron microscope and the detection of classical protein expression via Western blot, such as LC3 and nucleoporin p62 (SQSTM1/p62). The molecular basis of autophagy toward protective function is not well defined. Importantly, the key to modulate autophagy toward protective function rather than the opposite is a meaningful topic which deserves in-depth exploration.
Pyroptosis
In the past decades, a series of research studies focused on apoptosis and then great attention was paid to autophagy. However, the question remaining is that nanomaterial-induced toxicity is also accompanied by inflammation, which could not be elicited by apoptosis itself. With research going on, the mysteries that were initially complicated are gradually being unveiled. In this decade, a new form of cell death termed pyroptosis has been widely investigated, which is finely orchestrated by inflammasome complex and gasdermin family with the secretion of interleukin (IL)-1β and IL-18.56 Lately, it has been found that pyroptosis is not only dependent on caspase family but also on gasdermin family through formation of membrane pore. Moreover, caspase family is not limited to apoptosis and is associated with pyroptosis, as gasdermin D (GSDMD) can be cleaved by caspase 1. Translocation of gasdermin-N domain, which is formed through cleavage of GSDMD by caspase 1 or cleavage of gasdermin E by caspase 3, from the cytoplasm to the membrane is the cause for formation of pore at the membrane with the release of lactate dehydrogenase and secretion of IL-1β.57 Actually, these findings show that pyroptosis is directly related to gasdermin family instead of caspase. What is remarkable is that pyroptosis simultaneously shares the features with necrosis and apoptosis, which means that it possesses the characteristics of necrosis (rupture of cell membrane, liberation of intracellular substances) and apoptosis (DNA fragmentation and nuclear condensation).58 Innate immunity could be activated by a plethora of stimuli including exogenous and endogenous factors called pathogen-associated molecular patterns (PAMPs) and DAMPs.59 Stimulation of the immune system initially starts from the interaction of pattern-recognition receptors (PRRs) with PAMPs or DAMPs. PRRs are composed of Toll-like receptors (TLRs), retinoic acid-inducible gene I-like receptors, nucleotide oligomerization domain-like receptors (NLRs) and C-type lectin receptors. The inflammasome is a multiprotein oligomer consisting of caspase 1, apoptosis-associated speck-like protein containing a CARD (PYCARD), NLRs and sometimes caspase 5, including NLR family pyrin domain containing 3 (NLRP3), absent in melanoma 2 (AIM2) and so forth. Resting in physical state, they are usually composed of NLRs, adaptor protein apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) and the effector pro-caspase 1 which connects with ASC via caspase activation and recruitment domain (CARD) and a cytosolic sensor. Pyroptosis through the noncanonical pathways of caspase 4/5/11 has also been studied.60 All pathways toward the end point of pyroptosis should undergo the process of cleavage of gasdermin family, for example, cleavage of GSDMD by caspase 1.56
Conventionally, secretion of IL-1β should undergo two steps via pyroptosis pathway. The first signal is implicated in activation of TLRs and stimulation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)/activator protein 1 transcription factor triggered by PAMPs or DAMPs following the synthesis of pro-IL-1β. The second signal is provided by the assembly of inflammasome complex with cleavage of pro-caspase 1 and pro-IL-1β to their active forms. Notably, there is just production of IL-1β after these two steps. Furthermore, secretion of IL-1β needs another step. With the help of caspase, gasdermin-N is separated from gasdermin protein and translocated to the membrane, which results in pore formation and ensuing secretion of IL-1β from the cytosol to the supernatant. Among the homologous inflammasomes, NLRP3 is intensively studied. Redundant production of IL-1β subsequent to the assembly of inflammasomes conversely stimulates PRRs, further promoting the activation of inflammasomes and the expression of inflammatory genes such as tumor necrosis factor-α and inducible nitric oxide synthase. Inflammasome-mediated inflammation originally served as a protective measure to clear the antigens, which seems to be a benign operation to the body. It also does harm to organisms once beyond control. Research have revealed that NLRP3 inflammasome participates in immune response and plays a pivotal role in nanoparticle-caused inflammation. Activation of NLRP3 by environmental irritants such as silica,58 asbestos58 and alum61 has been authenticated.62 Furthermore, structure-varied nanoparticles such as monosodium urate,63 nano-SiO2,58 black carbon64 and carbon nanotubes65 are able to actuate NLRP3 inflammasome. NLRP3 activation induced by QDs was first reported in hepatic cells and the liver of mice.18 It suggested that ROS,65 lysosomal rupture,66 mitochondrial damage18 and K+ efflux67 are interrelated cellular events for NLRP3 activation. As the damaged mitochondria can be normally eliminated by mitophagy, it is reasonable to hypothesize that moderate mitophagy assists with the maintenance of steady state, whereas inhibition of mitophagy might bring about adverse effect. However, the role of multiple signals in the activation of NLRP3 inflammasome is not yet clear. Accumulating research have discussed various cellular end points after incubation with QDs; still there are potential molecular mechanisms under investigation though.
Disturbance of subcellular structures
It is apparent that whichever system has been discernibly influenced by that QDs.20,68,69 Further research found that various organelles would collapse during QD treatment. Lipid peroxidation of membrane and liberation of substrates would occur when the buffering system is extremely collapsed. Leakage of several classes of proteases from the ruptured organelles is responsible for the observed cell death, for example, Ca2+ from the ER,34 mitochondrial DNA from the mitochondria70 or cathepsin from the disrupted lysosome,71 which were authenticated by several studies with different types of cells. However, contributions of certain physicochemical property to the impairment of organelles are not completely explained.
DNA damage
Entry and egression of substances are rigorously regulated by the nucleus in order to maintain a steady milieu. Several studies revealed QDs can enter into the nucleus, whereas, on the contrary, others found that QDs are just located around the perinuclear area. Dissolution of the metal ion from QDs has the ability to influence the nucleus and cause genotoxicity, including oxidative modifications and DNA strand breaks.26 No obvious cytotoxicity was observed in the early stage of DNA damage, indicating that DNA damage could act as a sensitive marker for evaluation of QD-induced toxicity. Of note, cell cycle is arrested in G2/M period, and cell proliferation is obviously inhibited when DNA is damaged, suggesting that a protective mechanism has been activated to maintain the genome stability.72 PEG-coated silianized CdSe/ZnS QDs were incubated with IMR-90 cells up to 48 h, and the result implied that cell cycle was significantly arrested to the M phase.11 The single cell gel electrophoresis assay (also known as comet assay) is a sensitive technique for detection of DNA damage at the level of a single eukaryotic cell. Due to the existence of cadmium ions and ROS, appearance of obvious nuclear shrinkage and chromatin condensation would lead to increased nuclear deformation. CdTe QDs can enhance intracellular ROS and result in increased formation of γ-H2Ax foci in human umbilical vein endothelial cells. γ-H2Ax foci are a common marker for DNA double-strand breaks (DSBs), which can be inhibited by pretreatment with N-acetyl-L-cysteine, suggesting that oxidative stress is an instigator of DNA damage.73 Treatment of A549 cell line with CdSe QDs induces nuclear condensation and DNA fragmentation after 24 h, which are associated with intracellular ROS level.14 However, another literature reported that DSBs have been minimally triggered by PEG-coating QDs as no significant phosphorylation of the histone protein γ-H2Ax has been found.74 The opposite consequence can be explained by disparate QD types, different doses and cell lines.
It has been reported that negatively charged QDs are relatively noncytotoxic compared with positive QDs for the reason that positively charged QDs can be easily internalized. Even in the absence of cytotoxicity, a significant proportion of DNA strand breaks was found in the group of negatively charged QDs without compensation of DNA repair, suggesting that occurrence of genotoxicity was earlier than cytotoxicity and may be regarded as a sensitive indicator.75 CdSe QDs were respectively functionalized with mercaptopropionic acid (MPA) and cysteamine (CYST). The charge of MPA-CdSe QDs is negative and that of CYST-CdSe QDs is positive. Both types have similar size. Interestingly, cytotoxicity was only produced in CYST-CdSe QDs, while a high number of DSBs was displayed in the two groups. This experimental result also corresponds with the hypothesis that negatively charged QDs are less hazardous than positive modification. Moreover, increased expression of p53-binding protein 1 in the nuclei and decrease of p53 from the cytoplasmic extracts further confirmed that QDs can induce DNA damage. As a result, DNA could be seriously damaged by MPA-CdSe QDs in the absence of cytotoxicity. These data show that cytotoxic analysis must be supplemented with genotoxic potential assays to fully understand the cellular responses when recommendations would be made for the safety evaluation of QDs.
ER stress
The ER is an essential organelle in most types of eukaryotic cells for detoxification. It is not only a site for modification, assembly and transfer of proteins but also it is a reservoir of intracellular Ca2+. Besides, it is closely related to the concentration of blood glucose, synthesis of steroid hormone and lipid metabolism.76 Folding and assembly of cellular proteins could hardly proceed when ER has suffered from impairment. ER stress is supposed to tackle excessive unfolded proteins, which usually occurs under the conditions of hypoxia, oxidative stress, abnormal glycosylation and disturbance of calcium homeostasis in order to restore the stability of ER.77 Timely repair is essential to counteract this crisis; otherwise, cells would get impaired and even go to death. Concretely, further tissue damage could be induced when the unfolded proteins persistently accumulate.
Unfolded protein reaction (UPR) is well regulated by three signals, namely, inositol-requiring enzyme-1α (IRE-1α), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6). Under physical conditions, IRE-1α and PERK, respectively, bind to the ER resident chaperone-binding immunoglobulin protein (BiP/GRP78) to form an inactive complex.78 Huge aggregation of unfolded proteins facilitates separation of Bip/GRP78 from the complex, which strikes the activation of canonical signal transduction pathway of UPR, so as to degrade misfolded proteins.76 Three pathways concerning UPR could occur as follows: 1) activation of IRE-1α plays a stimulative role for endonuclease which cleaves X-box binding protein 1 (XBP-1) into spliced XBP-1 (XBP-1s) in the cytoplasm. Then, the spliced isoform XBP-1s enters the nucleus to regulate transcription of downstream genes such as Bip/GRP78 and other cytoprotective genes, accelerating the ubiquitination of misfolded proteins through ER-assisted degradation. 2) Signal transduction of PERK stimulates the phosphorylation of the α subunit of eukaryotic initiation factor 2α, followed by activation of activating transcription factor 4 (ATF4) to restore ER homeostasis, which can be ascribed to the blockage of translation. 3) Separation of ATF6 from the ATF6/Bip/GRP78 complex and translocation of ATF6 to the Golgi will help its activation by site-1 and site-2 proteases, and afterward, transfer of cleaved p50ATF6 to the nucleus promotes the expression of genes which are in charge of cellular self-healing.79
Researchers nowadays believe that ER plays an important role in the regulation of apoptosis. Theoretically, ER predominates in the regulation of intracellular Ca2+.80 In addition, it is also involved in the synthesis and modification of receptors at the cell membrane and organelle membrane. For example, caspase 12 is a protein from the caspase family, which is located at the surface of ER-cytoplasm, which cleaves its substrates at the C-terminal aspartic acid residues. Generally, caspase 12 could be activated by upstream signals such as three classical UPR signaling pathways (PERK, ATF6 and IRE-1α) under the state of sustained accumulation of unfolded proteins and disturbance of Ca2+ homeostasis.81 Additionally, it cooperates well with inflammatory caspases in the secretion of inflammatory cytokines. Hence, caspase 12 plays a vital role in ER signaling pathways through which external chemicals lead to cellular apoptosis or pyroptosis.76 Tang et al confirmed that QDs could lead to increased intracellular Ca2+ concentration and further demonstrated that this Ca2+ is released from the ER.82 Elevated expression of marker protein BiP/GRP78 and glucose-regulated protein 94 occurs in vascular endothelial cells after CdTe QD treatment, suggesting that ER stress can be triggered by CdTe QDs in the endothelial cells.39 Moreover, induction of apoptosis by QDs can be effectively inhibited after use of ER stress inhibitor salubrinal and caspase inhibitors Z-VAD-FMK and Ac-DEVD-CHO, which further confirmed that apoptosis can be ascribed to ER stress.39 Actually, normal cells possess strong resistance to adverse impacts for maintenance of stability. If the damage could not be restored in a timely manner, apoptosis would be induced through PERK-eukaryotic initiation factor 2α-ATF4, phosphorylation of c-JUN NH2-terminal kinase and caspase 4.39 Although apoptosis-related signaling pathways of ER stress have been uncovered, the extent of ER stress involved in the damage by QDs is still obscure. Withal, correlation between ER stress and other cellular organelles remains unclear and the underlying mechanisms of cellular damage also demand efforts in the future.
Mitochondrial dysfunction
Mitochondrion is the main site of energy production in cells, which functions in oxidative phosphorylation, electron transfer and catabolism. As well, mitochondria are the cellular resources of adenosine triphosphate and ROS. Abnormalities in mitochondrial structure usually occur once the superoxide levels are above the intrinsic ability of clearance. Due to the different changes occurring in mitochondrial morphology between apoptosis and necrosis, it is an important approach to distinguish apoptosis from necrosis. In the early stage of apoptosis, the mitochondrial structure remains intact but is swollen in necrosis. Mitochondria are commonly involved in important events of apoptosis, including mitochondrial membrane permeability, cytochrome c release and expression changes of apoptosis-related proteins (Bcl-2, Bax).68,83 Cytoplasm condensation, large-scaled DNA fragmentation and formation of apoptotic bodies along with cell membrane invagination are the typical morphologic features of apoptosis. Our group found that CdTe/CdS QDs induced AML12 cell apoptosis through alteration in permeability of mitochondria, disappearance of mitochondrial membrane integrity, increased cytoplasmic Ca2+ concentration and release of apoptotic factors.22 Additionally, CdSe QDs can cause a series of biochemical changes through a mitochondria-dependent pathway in SH-SY5Y cells, consisting of activation of c-Jun N-terminal kinase, loss of MMP and increase of ROS.84 Adverse far-reaching effects on cell stability and tissue integrity are caused, unless the damaged mitochondria are promptly cleared through mitophagy.51 Under intense and persistent stress, release of ROS from NADPH oxidase and electron transfer chain overwhelmingly collapses the buffering system and severely abates the expectancy of cells.85 Similar to other organelles, mitochondrion is a potential target for prevention of damage from xenobiotics.83 However, the complicated mechanism in mitochondrial dysfunction remains obscure. Concrete details of how mitochondria manipulate cellular events remain to be explored.
Owing to the complex courses of cellular activity, it is difficult to judge which events take place initially. The sequence of Ca2+ accumulation and ROS release is still under debate. An experiment concerning hepatotoxicity of QDs in vitro showed that mitochondrial ROS (mtROS) can be partially alleviated by Ca2+ chelator, but Ca2+ mobilization was not abridged when mtROS inhibitor had been pretreated, which draws a conclusion, at least in this case, that QDs partially cause mtROS via upstream Ca2+ mobilization in L02 cells.18 Nevertheless, since the gaps between in vitro and in vivo could not be neglected, further studies should be carried out in vivo for more clarification. Although both disruption of calcium homeostasis and mtROS production are closely related to mitochondrial dysfunction, conflicts still exist concerning whether Ca2+ influx is a cause or a downstream signal of mtROS generation. As the cause–effect relationship is abstruse, the exact sequence of diverse events is mysterious so far, implying the necessity of further unequivocal studies on this area. It has also been reported lately that mitochondria could finely tune the innate immune response with recruitment of inflammasome complex and generation of inflammatory cytokines.86 Mitochondrial outer membrane permeabilization usually culminates in apoptosis via extrinsic programmed cell death pathways, which depends on the relative activity of pro- or antiapoptotic proteins in Bcl-2 family, whereas mitochondrial permeability transition (MPT) commits cells to death through intrinsic pathway under the control of permeability transition pore at the mitochondrial inner membrane. Ca2+ from the ER or cytoplasm to the mitochondria strengthens the process of oxidative phosphorylation with sensitization of dehydrogenases in the matrix, accelerating the course of MPT.87 Moreover, promotion of mtROS via MPT was due to the derangement of electron transport chain. Meanwhile, elevated mtROS reversely oxidizes some receptors at the membrane surface to promote mitochondrial Ca2+ uptake and touch off MPT. Hence, a vicious loop among Ca2+, MPT and mtROS will eventually contribute to irreversible mitochondrial lesion.88 It is worth noting that mitochondrial toxicity induced by QDs is closely related to specific physicochemical properties. Compared with MPA CdTe QDs, bovine serum albumin CdTe QDs induced severe mitochondrial swelling and had a greater influence on membrane fluidity, which may be attributed to the stronger lipophilicity of bovine serum albumin.89 Intriguingly, the interaction between QDs and membrane proteins leads to the increase in mitochondrial membrane fluidity and the binding affinity of the QDs with pore-forming proteins is closely pertinent to size.90 As aforementioned, ligands91 and size90 are two factors having capability to influence mitochondrial toxicity.
In short, on the one hand, internalization of QDs contributes to morphologic changes of mitochondria with reduction of the MMP and subsequent release of cytochrome c, which strikes protease cascade reaction and eventually leads to cell damage. On the other hand, mitochondrial dysfunction has a connection with mtROS production and mitochondrial DNA release, abetting oligomerization of ASC and assembly of inflammasome complex, which activates the proteolytic activity of caspase 1 and facilitates the secretion of inflammatory cytokines related to a new form of cell death termed pyroptosis.
Lysosomal rupture
In the setting of photocatalytic oxidation, swelling and breakage of lysosomal membrane can be observed. Lysosomes generally participate in waste disposal. Recent studies disclosed that lysosome also functions in the late stage of autophagy and ensures the integrity of autophagy flux, which incorporates the steps of initiation, elongation, maturation and degradation.40 The stage of maturation refers to the formation of autolysosome through the fusion of autophagosome with lysosome, which is beneficial for acceleration of waste degradation, so as to maintain equilibrium of the organism. If lysosome damage was extremely evoked, a cascade reaction would be easily initiated through the leakage of acid materials. Meanwhile, disruption of autophagic–lysosomal axis accelerates the assembly of NLRP3 inflammasome, subsequently triggering cleavage of gasdermin, which eventually results in cellular pyroptosis.71 Activation of NLRP3 inflammasome during lysosomal damage can be alleviated through either inhibitor of cathepsin or inhibition of phagosomal acidification, demonstrating that leakage from lysosomal compartments into cytosol could trigger NLRP3 inflammasome activation. Moreover, cathepsin plays an indispensable role in this process.62 It has been reported that nano-silver52 and nano-rare earth metal oxide53 have the capability to stimulate inflammasome activation due to disruption of lysosomal homeostasis. In addition, lysosomal damage induced by nanomaterials is closely related to the characteristics of nanoparticles. It has been postulated that the long aspect ratio nanomaterials, such as nanorods, nanotubes and nanowires, tend to activate inflammasome for the reason that needle-shaped nanoparticles have the capacity to pierce lysosomal membrane, which consequently causes liberation of contents from the lysosome.92 Although a battery of nanomaterials are able to provide signals for inflammasome assembly through lysosomal enzymes, research regarding lysosomal rupture caused by QDs has not been reported thus far. Interestingly, a study conducted by Lima et al illustrated that extensive NLRP3 signaling ignited by pyroptosis agonist was prior to lysosome rupture, which is contradictory to NLRP3 activation as a result of lysosomal disruption. Meanwhile, they claimed that the lysosomotropic drug Leu-Leu-OMe with the ability of disturbance of lysosome failed to cause pyroptosis with minimal cleavage of caspase 1.93 The relationship between lysosomal rupture and NLRP3 activation remains to be clarified.
Signaling pathway
Signal pathway is the process involved in transmission of signals from upstream event to downstream, which ultimately results in a cellular response. Regulation of inflammatory responses, induction of apoptosis and occurrence of excessive autophagy are inseparable from mediation of signaling transduction. Notably, complicated networks are composed of diverse signaling pathways which are complementary aspects of each other. Elucidation of cellular signaling pathways can not only help to disclose the mechanisms of toxicity but also provide clues for exploration of measures against potential damage caused by engineering nanoparticles. One possible connection between nanoparticle exposure and inflammatory responses could be NF-κB, which is the key regulator of the inflammatory cascade. In addition, stimulation of QDs always regulates immunoreaction and inflammatory reaction through NF-κB/inflammasome pathway.94 Production of pro-inflammatory cytokines involves the cascade activation from TLR-2/MyD88 to NF-κB pathway in J774A.1 macrophages after treatment with adenosine 5′-monophosphate–conjugated CdSe/CdS/ZnS QDs. The same goes for MPA-capped CdSe/CdS/ZnS QDs, which is even more serious.95 Moreover, QD705-COOH and QD705-PEG have the ability to enhance the secretion of interferon-β in RAW264.7 cells via TLR-4/TIR-domain-containing adapter-inducing interferon-β-dependent signaling pathway,96 which can also increase the release of chemoattractant protein 1 through TLR-4/MyD88/NF-κB pathway.97 Inflammatory response, apoptosis and autophagy were mediated by the signaling pathway of P38 mitogen-activated protein kinases/NF-κB in activated THP-1 macrophages after exposure of graphene quantum dots (GQDs).98 In the meantime, the antioxidant system is triggered for inherent clearance once xenobiotics are recognized by the organisms. Nuclear factor (erythroid-derived 2)-like 2 (Nrf2)/antioxidant response element (ARE)/heme oxygenase-1 pathway exerts its important action on detoxification for maintenance of redox balance. For instance, experiments conducted by our group have illustrated that activation of Nrf2/ARE signaling pathway confers protection against apoptosis in AML12 cells induced by CdTe QDs.22 Withal, pathways of ER stress can be activated when unfolded proteins are difficult to be degraded.76 Signaling transduction of cytotoxicity induced by QDs is sketched in Figure 3.
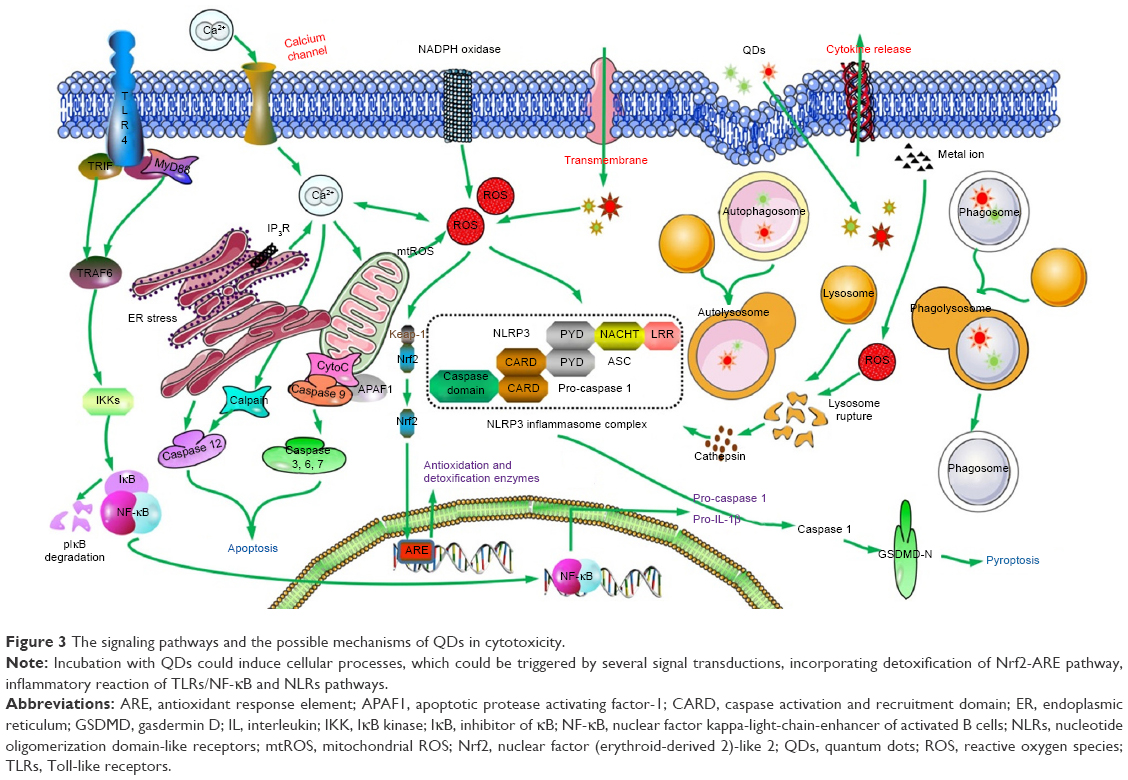 | Figure 3 The signaling pathways and the possible mechanisms of QDs in cytotoxicity. |
Perspectives
Morphologic alteration, metabolic disorder as well as diverse forms of cell death are obvious evidence for the existence of toxicity in QDs. The presence of toxicity has hindered application of QDs in life sciences, even though QDs are abundant with unique properties. Therefore, it is crucial to elucidate the mechanisms involved in QD-induced toxicity, which have not been completely clarified. It has been reported that oxidative stress, inflammation and disturbance of organelles are related to toxicity caused by QDs. Cellular end points, including necrosis, apoptosis, autophagy and pyroptosis, are induced by QD exposure. As each type is a unique nanocrystal in the light of size, shape, components and surface chemistry, it is a great challenge to comprehensively understand the toxicity mechanism of different types of QDs. Multiple target organs should be adopted for safety evaluation of various kinds of QDs. The evaluation from a single cell level to the whole animal model should also be emphasized. Likewise, distinct exposure routes should be considered. Moreover, it is necessary to assess the risk of QDs through both acute and chronic exposure. Of note, decrease in toxicity should be the prerequisite for utility of QDs in diagnosis and therapy. Due to the potential social benefits and medical assistance of QDs, studies about this promising area should be further strengthened. We hope that approval of compatibly designed QDs will make a contribution to the therapy of certain diseases in the near future.
Acknowledgments
This work was supported by the National Important Project on Scientific Research of China (No 2011CB933404), National Natural Science Foundation of China (No 81172697, 81302461, 81473003, 81502783, 81573186, 31671034 and 81673218) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJZZ16_0033).
Disclosure
The authors report no conflicts of interest in this work.
References
Yang L, Wang Z, Wang J, et al. Doxorubicin conjugated functionalizable carbon dots for nucleus targeted delivery and enhanced therapeutic efficacy. Nanoscale. 2016;8(12):6801–6809. | ||
Zhao MX, Zhu BJ, Yao WJ, Chen DF, Wang C. The delivery of doxorubicin of multifunctional beta-cyclodextrin-modified CdSe/ZnS quantum dots for bioactivity and nano-probing. Chem Biol Drug Des. 2018;91(1):285–293. | ||
Su Y, He Y, Lu H, et al. The cytotoxicity of cadmium based, aqueous phase–synthesized, quantum dots and its modulation by surface coating. Biomaterials. 2009;30(1):19–25. | ||
Wu T, He K, Ang S, et al. Impairments of spatial learning and memory following intrahippocampal injection in rats of 3-mercaptopropionic acid-modified CdTe quantum dots and molecular mechanisms. Int J Nanomed. 2016;11:2737–2755. | ||
Matea CT, Mocan T, Tabaran F, et al. Quantum dots in imaging, drug delivery and sensor applications. Int J Nanomed. 2017;12:5421–5431. | ||
Pohanka M. Quantum dots in the therapy: current trends and perspectives. Mini Rev Med Chem. 2017;17(8):650–656. | ||
Wu C, Shi L, Li Q, et al. Probing the dynamic effect of cys-CdTe quantum dots toward cancer cells in vitro. Chem Res Toxicol. 2010;23(1):82–88. | ||
Jain MP, Choi AO, Neibert KD, Maysinger D. Probing and preventing quantum dot-induced cytotoxicity with multimodal alpha-lipoic acid in multiple dimensions of the peripheral nervous system. Nanomedicine (London, England). 2009;4(3):277–290. | ||
Li KG, Chen JT, Bai SS, et al. Intracellular oxidative stress and cadmium ions release induce cytotoxicity of unmodified cadmium sulfide quantum dots. Toxicol In Vitro. 2009;23(6):1007–1013. | ||
Lovric J, Bazzi HS, Cuie Y, Fortin GR, Winnik FM, Maysinger D. Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots. J Mol Med (Berl). 2005;83(5):377–385. | ||
Zhang T, Stilwell JL, Gerion D, et al. Cellular effect of high doses of silica-coated quantum dot profiled with high throughput gene expression analysis and high content cellomics measurements. Nano Lett. 2006;6(4):800–808. | ||
Lee EY, Bae HC, Lee H, et al. Intracellular ROS levels determine the apoptotic potential of keratinocyte by Quantum Dot via blockade of AKT Phosphorylation. Exp Dermatol. 2017;26(11):1046–1052. | ||
Paesano L, Perotti A, Buschini A, et al. Data on HepG2 cells changes following exposure to cadmium sulphide quantum dots (CdS QDs). Data Brief. 2017;11:72–97. | ||
Jigyasu AK, Siddiqui S, Lohani M, Khan IA, Arshad M. Chemically synthesized CdSe quantum dots inhibit growth of human lung carcinoma cells via ROS generation. EXCLI J. 2016;15:54–63. | ||
Wu T, Zhang T, Chen Y, Tang M. Research advances on potential neurotoxicity of quantum dots. J Appl Toxicol. 2016;36(3):345–351. | ||
Chen N, He Y, Su Y, et al. The cytotoxicity of cadmium-based quantum dots. Biomaterials. 2012;33(5):1238–1244. | ||
Wu T, Tang M. Toxicity of quantum dots on respiratory system. Inhal Toxicol. 2014;26(2):128–139. | ||
Lu Y, Xu S, Chen H, et al. CdSe/ZnS quantum dots induce hepatocyte pyroptosis and liver inflammation via NLRP3 inflammasome activation. Biomaterials. 2016;90:27–39. | ||
Ji X, Xu B, Yao M, et al. Graphene oxide quantum dots disrupt autophagic flux by inhibiting lysosome activity in GC-2 and TM4 cell lines. Toxicology. 2016;374:10–17. | ||
Wu TS, He KY, Zhan QL, et al. Partial protection of N-acetylcysteine against MPA-capped CdTe quantum dot-induced neurotoxicity in rat primary cultured hippocampal neurons. Toxicol Res. 2015;4(6):1613–1622. | ||
Sun H, Cui E, Liu R. Molecular mechanism of copper-zinc superoxide dismutase activity change exposed to N-acetyl-L-cysteine-capped CdTe quantum dots-induced oxidative damage in mouse primary hepatocytes and nephrocytes. Environ Sci Pollut Res Int. 2015;22(22):18267–18277. | ||
Zhang T, Hu Y, Tang M, et al. Liver toxicity of cadmium telluride quantum dots (CdTe QDs) due to oxidative stress in vitro and in vivo. Int J Mol Sci. 2015;16(10):23279–23299. | ||
Stan MS, Memet I, Sima C, et al. Si/SiO2 quantum dots cause cytotoxicity in lung cells through redox homeostasis imbalance. Chem Biol Interact. 2014;220:102–115. | ||
Dunpall R, Nejo AA, Pullabhotla VS, Opoku AR, Revaprasadu N, Shonhai A. An in vitro assessment of the interaction of cadmium selenide quantum dots with DNA, iron, and blood platelets. IUBMB Life. 2012;64(12):995–1002. | ||
Smith WE, Brownell J, White CC, et al. In vitro toxicity assessment of amphiphillic polymer-coated CdSe/ZnS quantum dots in two human liver cell models. ACS Nano. 2012;6(11):9475–9484. | ||
Zhang T, Wang Y, Kong L, Xue Y, Tang M. Threshold dose of three types of quantum dots (QDs) induces oxidative stress triggers DNA damage and apoptosis in mouse fibroblast L929 cells. Int J Environ Res Public Health. 2015;12(10):13435–13454. | ||
Henriquez M, Armisen R, Stutzin A, Quest AF. Cell death by necrosis, a regulated way to go. Curr Mol Med. 2008;8(3):187–206. | ||
Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151–164. | ||
Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. | ||
Feoktistova M, Leverkus M. Programmed necrosis and necroptosis signalling. FEBS J. 2015;282(1):19–31. | ||
Zhou W, Yuan J. SnapShot: necroptosis. Cell. 2014;158(2):464–464.e461. | ||
Peixoto MS, de Oliveira Galvao MF, Batistuzzo de Medeiros SR. Cell death pathways of particulate matter toxicity. Chemosphere. 2017;188:32–48. | ||
Lai L, Jin JC, Xu ZQ, Mei P, Jiang FL, Liu Y. Necrotic cell death induced by the protein-mediated intercellular uptake of CdTe quantum dots. Chemosphere. 2015;135:240–249. | ||
Wang H, Liu Z, Gou Y, et al. Apoptosis and necrosis induced by novel realgar quantum dots in human endometrial cancer cells via endoplasmic reticulum stress signaling pathway. Int J Nanomed. 2015;10:5505–5512. | ||
Amna T, Van Ba H, Vaseem M, et al. Apoptosis induced by copper oxide quantum dots in cultured C2C12 cells via caspase 3 and caspase 7: a study on cytotoxicity assessment. Appl Microbiol Biotechnol. 2013;97(12):5545–5553. | ||
Nguyen KC, Willmore WG, Tayabali AF. Cadmium telluride quantum dots cause oxidative stress leading to extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2 cells. Toxicology. 2013;306:114–123. | ||
Yan M, Zhang Y, Xu K, Fu T, Qin H, Zheng X. An in vitro study of vascular endothelial toxicity of CdTe quantum dots. Toxicology. 2011;282(3):94–103. | ||
Zhan Q, Tang M. Research advances on apoptosis caused by quantum dots. Biol Trace Elem Res. 2014;161(1):3–12. | ||
Yan M, Zhang Y, Qin H, et al. Cytotoxicity of CdTe quantum dots in human umbilical vein endothelial cells: the involvement of cellular uptake and induction of pro-apoptotic endoplasmic reticulum stress. Int J Nanomed. 2016;11:529–542. | ||
Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9:20. | ||
Hasanain M, Bhattacharjee A, Pandey P, et al. α-Solanine induces ROS-mediated autophagy through activation of endoplasmic reticulum stress and inhibition of Akt/mTOR pathway. Cell Death Dis. 2015;6:e1860. | ||
Guo C, Yang M, Jing L, et al. Amorphous silica nanoparticles trigger vascular endothelial cell injury through apoptosis and autophagy via reactive oxygen species-mediated MAPK/Bcl-2 and PI3K/Akt/mTOR signaling. Int J Nanomed. 2016;11:5257–5276. | ||
Xia J, Guo S, Fang T, et al. Dihydromyricetin induces autophagy in HepG2 cells involved in inhibition of mTOR and regulating its upstream pathways. Food Chem Toxicol. 2014;66:7–13. | ||
Kasprowska-Liskiewicz D. The cell on the edge of life and death: crosstalk between autophagy and apoptosis. Postepy Hig Med Dosw (Online). 2017;71(0):825–841. | ||
Kemp MG. Crosstalk between apoptosis and autophagy: environmental genotoxins, infection, and innate immunity. J Cell Death. 2017;9:1179670716685085. eCollection 2017. | ||
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37(3):299–310. | ||
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. | ||
Zhang Y, Shen K, Bai Y, et al. Mir143-BBC3 cascade reduces microglial survival via interplay between apoptosis and autophagy: implications for methamphetamine-mediated neurotoxicity. Autophagy. 2016;12(9):1538–1559. | ||
Zhu J, Wang KZ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy. 2013;9(11):1663–1676. | ||
Zhang HT, Mi L, Wang T, et al. PINK1/Parkin-mediated mitophagy play a protective role in manganese induced apoptosis in SH-SY5Y cells. Toxicol In Vitro. 2016;34:212–219. | ||
Kim MJ, Yoon JH, Ryu JH. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016;49(10):529–535. | ||
Mishra AR, Zheng J, Tang X, Goering PL. Silver nanoparticle-induced autophagic-lysosomal disruption and NLRP3-inflammasome activation in HepG2 cells is size-dependent. Toxicol Sci. 2016;150(2):473–487. | ||
Li R, Ji Z, Qin H, et al. Interference in autophagosome fusion by rare earth nanoparticles disrupts autophagic flux and regulation of an interleukin-1beta producing inflammasome. ACS Nano. 2014;8(10):10280–10292. | ||
Li X, Chen N, Su Y, et al. Autophagy-sensitized cytotoxicity of quantum dots in PC12 cells. Adv Healthc Mater. 2014;3(3):354–359. | ||
Fan J, Sun Y, Wang S, et al. Inhibition of autophagy overcomes the nanotoxicity elicited by cadmium-based quantum dots. Biomaterials. 2016;78:102–114. | ||
Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. | ||
Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. | ||
Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. | ||
Bauer RN, Diaz-Sanchez D, Jaspers I. Effects of air pollutants on innate immunity: the role of Toll-like receptors and nucleotide-binding oligomerization domain-like receptors. J Allergy Clin Immunol. 2012;129(1):14–24; quiz 25–6. | ||
Ding J, Shao F. SnapShot: the noncanonical inflammasome. Cell. 2017;168(3):544–544.e541. | ||
Mustafa Rizvi SH, Parveen A, Verma AK, Ahmad I, Arshad M, Mahdi AA. Aluminium induced endoplasmic reticulum stress mediated cell death in SH-SY5Y neuroblastoma cell line is independent of p53. PLoS One. 2014;9(5):e98409. | ||
Jessop F, Hamilton RF Jr, Rhoderick JF, Fletcher P, Holian A. Phagolysosome acidification is required for silica and engineered nanoparticle-induced lysosome membrane permeabilization and resultant NLRP3 inflammasome activity. Toxicol Appl Pharmacol. 2017;318:58–68. | ||
Giamarellos-Bourboulis EJ, Mouktaroudi M, Bodar E, et al. Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 beta by mononuclear cells through a caspase 1-mediated process. Ann Rheum Dis. 2009;68(2):273–278. | ||
Long CM, Nascarella MA, Valberg PA. Carbon black vs. black carbon and other airborne materials containing elemental carbon: physical and chemical distinctions. Environ Pollut. 2013;181:271–286. | ||
Sun B, Wang X, Ji Z, et al. NADPH oxidase-dependent NLRP3 inflammasome activation and its important role in lung fibrosis by multiwalled carbon nanotubes. Small. 2015;11(17):2087–2097. | ||
Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and inflammasomes. Mol Immunol. 2017;86:10–15. | ||
Rivers-Auty J, Brough D. Potassium efflux fires the canon: potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol. 2015;45(10):2758–2761. | ||
Nguyen KC, Rippstein P, Tayabali AF, Willmore WG. Mitochondrial toxicity of cadmium telluride quantum dot nanoparticles in mammalian hepatocytes. Toxicol Sci. 2015;146(1):31–42. | ||
Wang X, Tian J, Yong KT, et al. Immunotoxicity assessment of CdSe/ZnS quantum dots in macrophages, lymphocytes and BALB/c mice. J Nanobiotechnol. 2016;14:10. | ||
Paesano L, Perotti A, Buschini A, et al. Markers for toxicity to HepG2 exposed to cadmium sulphide quantum dots; damage to mitochondria. Toxicology. 2016;374:18–28. | ||
Orlowski GM, Sharma S, Colbert JD, et al. Frontline science: multiple cathepsins promote inflammasome-independent, particle-induced cell death during NLRP3-dependent IL-1beta activation. J Leukoc Biol. 2017;102(1):7–17. | ||
Li Y-B, Zhang H-X, Guo C-X, et al. Cytotoxicity and DNA damage effect of TGA-capped CdTe quantum dots. Chem Res Chin Univ. 2012;28(2):276–281. | ||
Wang L, Zhang J, Zheng Y, Yang J, Zhang Q, Zhu X. Bioeffects of CdTe quantum dots on human umbilical vein endothelial cells. J Nanosci Nanotechnol. 2010;10(12):8591–8596. | ||
Ju L, Zhang G, Zhang C, et al. Quantum dot-related genotoxicity perturbation can be attenuated by PEG encapsulation. Mutati Res. 2013;753(1):54–64. | ||
Nagy A, Hollingsworth JA, Hu B, et al. Functionalization-dependent induction of cellular survival pathways by CdSe quantum dots in primary normal human bronchial epithelial cells. ACS Nano. 2013;7(10):8397–8411. | ||
Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Inte J Mol Sci. 2016;17(3):327. | ||
Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283(14):2640–2652. | ||
Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta. 2014;1843(10):2150–2163. | ||
Chen R, Huo L, Shi X, et al. Endoplasmic reticulum stress induced by zinc oxide nanoparticles is an earlier biomarker for nanotoxicological evaluation. ACS Nano. 2014;8(3):2562–2574. | ||
Kuum M, Veksler V, Kaasik A. Potassium fluxes across the endoplasmic reticulum and their role in endoplasmic reticulum calcium homeostasis. Cell Calcium. 2015;58(1):79–85. | ||
Voccoli V, Mazzoni F, Garcia-Gil M, Colombaioni L. Serum-withdrawal-dependent apoptosis of hippocampal neuroblasts involves Ca++ release by endoplasmic reticulum and caspase-12 activation. Brain Res. 2007;1147:1–11. | ||
Tang M, Xing T, Zeng J, et al. Unmodified CdSe quantum dots induce elevation of cytoplasmic calcium levels and impairment of functional properties of sodium channels in rat primary cultured hippocampal neurons. Environ Health Perspect. 2008;116(7):915–922. | ||
Li J, Zhang Y, Xiao Q, et al. Mitochondria as target of quantum dots toxicity. J Hazard Mater. 2011;194:440–444. | ||
Choi AO, Cho SJ, Desbarats J, Lovric J, Maysinger D. Quantum dot-induced cell death involves Fas upregulation and lipid peroxidation in human neuroblastoma cells. J Nanobiotechnol. 2007;5:1. | ||
Wang Y, Xiong L, Tang M. Toxicity of inhaled particulate matter on the central nervous system: neuroinflammation, neuropsychological effects and neurodegenerative disease. J Appl Toxicol. 2017;37(6):644–667. | ||
Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015;21(3):193–201. | ||
Armstrong JS. The role of the mitochondrial permeability transition in cell death. Mitochondrion. 2006;6(5):225–234. | ||
Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014;35(6):253–261. | ||
Lai L, Jin JC, Xu ZQ, Ge YS, Jiang FL, Liu Y. Spectroscopic and microscopic studies on the mechanism of mitochondrial toxicity induced by CdTe QDs modified with different ligands. J Membr Biol. 2015;248(4):727–740. | ||
Lai L, Li YP, Mei P, Chen W, Jiang FL, Liu Y. Size effects on the interaction of QDs with the mitochondrial membrane in vitro. J Membr Biol. 2016;249(6):757–767. | ||
Xiang X, Wu C, Zhang BR, et al. The relationship between the length of surface ligand and effects of CdTe quantum dots on the physiological functions of isolated mitochondria. Chemosphere. 2017;184:1108–1116. | ||
Sun B, Wang X, Ji Z, Li R, Xia T. NLRP3 inflammasome activation induced by engineered nanomaterials. Small. 2013;9(9–10):1595–1607. | ||
Lima H Jr, Jacobson LS, Goldberg MF, et al. Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle. 2013;12(12):1868–1878. | ||
Romoser AA, Chen PL, Berg JM, et al. Quantum dots trigger immunomodulation of the NFkappaB pathway in human skin cells. Mol Immunol. 2011;48(12–13):1349–1359. | ||
Dai T, Li N, Liu L, Liu Q, Zhang Y. AMP-conjugated quantum dots: low immunotoxicity both in vitro and in vivo. Nanoscale Res Lett. 2015;10(1):434. | ||
Ho CC, Luo YH, Chuang TH, Lin P. Quantum dots induced interferon beta expression via TRIF-dependent signaling pathways by promoting endocytosis of TLR4. Toxicology. 2016;344–346:61–70. | ||
Ho CC, Luo YH, Chuang TH, Yang CS, Ling YC, Lin P. Quantum dots induced monocyte chemotactic protein-1 expression via MyD88-dependent Toll-like receptor signaling pathways in macrophages. Toxicology. 2013;308:1–9. | ||
Qin Y, Zhou ZW, Pan ST, et al. Graphene quantum dots induce apoptosis, autophagy, and inflammatory response via p38 mitogen-activated protein kinase and nuclear factor-kappaB mediated signaling pathways in activated THP-1 macrophages. Toxicology. 2015;327:62–76. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.