")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Dual Role For A MEK Inhibitor As A Modulator Of Inflammation And Host Defense Mechanisms With Potential Therapeutic Application In COPD
Authors Kurian N , Cohen TS, Öberg L , De Zan E, Skogberg G, Vollmer S, Baturcam E , Svanberg P, Bonn B, Smith PD, Vaarala O, Cunoosamy DM
Received 9 April 2019
Accepted for publication 23 September 2019
Published 26 November 2019 Volume 2019:14 Pages 2611—2624
DOI https://doi.org/10.2147/COPD.S211619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Richard Russell
Nisha Kurian,1 Taylor S Cohen,2 Lisa Öberg,3 Erica De Zan,3 Gabriel Skogberg,4 Stefan Vollmer,3 Engin Baturcam,3 Petter Svanberg,5 Britta Bonn,5 Paul D Smith,6 Outi Vaarala,3 Danen M Cunoosamy3
1Respiratory Inflammation and Autoimmune (RIA) Precision Medicine Unit, Precision Medicine, Oncology R&D, AstraZeneca, Gothenburg, Sweden; 2Microbial Sciences, Medimmune, Gaithersburg, MD, USA; 3Translational Science and Experimental Medicine, Research and Early Development, RIA, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden; 4Bioscience, Research and Early Development, RIA, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden; 5Drug Metabolism and Pharmacokinetics, Research and Early Development, RIA, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden; 6Bioscience, Oncology R&D, AstraZeneca, Cambridge, UK
Correspondence: Nisha Kurian
Respiratory Inflammation and Autoimmune (RIA) Precision Medicine Unit, Precision Medicine, Oncology R&D, AstraZeneca, Gothenburg, Sweden
Tel +46 72 197 9662
Email [email protected]
Background: Unlike p38 mitogen-activated protein Kinases (MAPK) that has been extensively studied in the context of lung-associated pathologies in COPD, the role of the dual-specificity mitogen-activated protein kinase kinase (MEK1/2) or its downstream signaling molecule extracellular signal-regulated kinases 1/2 (ERK1/2) in COPD is poorly understood.
Objectives: The aim of this study was to address whether MEK1/2 pathway activation is linked to COPD and that targeting this pathway can improve lung inflammation through decreased immune-mediated inflammatory responses without compromising bacterial clearance.
Methods: Association of MEK1/2 pathway activation to COPD was investigated by immunohistochemistry using lung tissue biopsies from COPD and healthy individuals and through analysis of sputum gene expression data from COPD patients. The anti-inflammatory effect of MEK1/2 inhibition was assessed on cytokine release from lipopolysaccharide-stimulated alveolar macrophages. The effect of MEK1/2 inhibition on bacterial clearance was assessed using Staphylococcus aureus killing assays with RAW 264.7 macrophage cell line and human neutrophils.
Results: We report here MEK1/2 pathway activation demonstrated by increased pERK1/2 staining in bronchial epithelium and by the presence of MEK gene activation signature in sputum samples from COPD patients. Inhibition of MEK1/2 resulted in a superior anti-inflammatory effect in human alveolar macrophages in comparison to a p38 inhibitor. Furthermore, MEK1/2 inhibition led to an increase in bacterial killing in human neutrophils and RAW 264.7 cells that was not observed with the p38 inhibitor.
Conclusion: Our data demonstrate the activation of MEK1/2 pathway in COPD and highlight a dual function of MEK1/2 inhibition in improving host defense responses whilst also controlling inflammation.
Keywords: exacerbation, infection, alveolar macrophage, p38 MAPK, steroid, transcriptomics, sputum
Introduction
COPD is a multifactorial progressive disease characterized by an abnormal inflammatory response within the lung environment and destruction of the lung parenchyma resulting in “airflow limitation that is only partially reversible”.1 Despite the current standard of care including bronchodilators and anti-inflammatory therapies, COPD patients continue to experience disease exacerbations.2 Beyond the combination of therapies with bronchodilator and steroids, Roflumilast is currently the only “new” anti-inflammatory drug that has been approved by the regulatory agencies to reduce the incidence of exacerbations in adults with severe COPD.
COPD exacerbations are commonly associated with the presence of viral and/or bacterial pathogens that are thought to promote inflammation. The connection between pathogens and disease has recently been confirmed by a two-year longitudinal follow up study (AERIS) of well-characterized COPD patients. In this longitudinal study, it was observed that COPD exacerbation-related dysbiosis is a result of the outgrowth of pre-existing bacteria rather than colonization with new bacterial genera3 confirming the view that an interplay between host defense mechanisms and airway microbiome is driving disease in both stable state and in exacerbations. Experimental human infection models have been extensively utilized to study the role of pathogens in driving disease morbidity as reviewed in Gunawardana et al, 2014.4 These models have shown that following viral infection, an increase in secondary bacterial infections is observed with a concurrent decrease in antimicrobial peptide production, implicating again the importance of mucosal homeostasis as a regulator of infection-related exacerbations.5 Additional studies with experimental viral infections confirmed an increase in bacterial burden and an outgrowth of bacterial species already present in the lung.6
Inhaled and oral steroids have been a mainstay of care for treating inflammation in COPD, although the use of steroids has been linked to increased reports of pneumonia.7 Following an inflammatory insult, activation of pathogen pattern recognition receptors and/or damage-associated molecular patterns in immune cells or airway epithelial cells (AECs) ultimately converge to the nuclear factor kappa B (NF-κB) and MAPK signaling pathways. Amongst the modules of the MAPK cascade, p38 drew considerable interest as elevated levels of activated p38 were detected in alveolar macrophages from COPD patients.8–10 MAPK inhibitors particularly those targeting p38 MAPK pathway have been tested in COPD.9–12 Oral p38 inhibitors have shown marginal effects on lung function in early clinical development.11,12 However, targeting of this kinase pathway in the lung using an inhaled p38 inhibitor AZD7624 failed to show clinical efficacy by reducing exacerbations in patients.9
There is emerging indirect evidences linking the activation of another MAPK, the MEK pathway in COPD. Smoking is associated with increased expression of Epidermal Growth Factor Receptor (EGFR) in the bronchial epithelium13,14 and EGFR, through the MEK1/2-ERK1/2 pathway activation has been shown to contribute to the secretion of IL-8/CXCL8, a potent neutrophil chemoattractant and secretory mucin MUC5AC and its regulators.14 Chronic mucus hypersecretion and airway neutrophilia are key features of COPD. Furthermore, increased inactivation of glycogen synthase-3β (GSK-3 β) was reported in COPD patients, that is thought to play a key role in oxidative stress and subsequent steroid insensitivity.15 This inactivation was mechanistically linked to activation of the MEK/ERK pathway and the phosphatidylinositol 3-kinase/AKT pathways in epithelial and immune cells.15 In addition, Rhinovirus (RV), a respiratory virus that causes COPD exacerbations, induces the expression of IL-8/CXCL8 in a MEK-dependent manner.16 However, a more direct association between MEK pathway activation and COPD is yet to be established. This study was initiated to address the hypothesis that MEK1/2-ERK1/2 MAPK pathway activation is associated with COPD and that targeting this pathway will control inflammation without compromising host defense responses.
Materials And Methods
Ethical Approvals
Written informed consent was obtained from an exploratory study in patients undergoing lung transplantation or resection surgery in Sahlgrenska University Hospital, Gothenburg according to protocols approved by the local ethics committee in Gothenburg (Dnr: 657–12). The transplant donors were not from any vulnerable populations and all donors (or next of kin) provided written informed consent that was freely given. Use of healthy human blood samples was approved by the Medimmune Institutional Review Board. Informed consent was provided by all donors and the study was performed in accordance with the ethical principles in the Declaration of Helsinki.
Human Tissue And Blood Cells Isolation
Human alveolar macrophages were derived from COPD Gold 4 lung transplant tissue or from lung resection surgery by flushing tissue with sterile Ca2+ Mg2+-free PBS (Life Technologies cat # 14200166) using a 19-gauge needle (BD cat# 305187). For functional assays, cells were seeded (200,000 cells per well) in a 96-well tissue-culture grade flat bottom plate (Costar cat # 3596). Following plating, non-adherent cells were removed by copious washing with serum-free RPMI 1640 (Life Technologies cat # 32404014) following an hour’s rest at 37°C. After the final wash, the cells were incubated overnight in XVivo10 media (Lonza cat # 04-743Q) supplemented with 4mM L-glutamine (Life Technologies cat #25030081) and 1% penicillin-streptomycin (Sigma cat# 59202C).
Neutrophils were isolated from the blood of healthy volunteers by density gradient separation following a routine protocol. Briefly, blood was overlaid on a Histopaque 1077 (Sigma cat #1077) gradient and centrifuged for 30 mins at 500g. Neutrophils were collected from beneath the Histopaque layer and red blood cells were lysed with ACK lysing buffer (Gibco cat# A1049201). Neutrophils were kept on ice until use.
Immunohistochemistry, Image Acquisition And Analysis
Paraffin-embedded formalin-fixed lung tissue was cut into 4 µm sections (ultra plus glasses, ThermoFisher Scientific cat # 10417002). The sections were placed over night in 37ᵒC. The next day, slides were deparaffinized and rehydrated by the following washes: 3 times 5 min in xylene, two times 10 mins in 100% ethanol, two times 10 mins in 95% ethanol and finally two times 5 mins in dH2O. Slides were stained on an intellipath flx autostainer system (Biocare Medical) using mach3 anti rabbit pERK1/2 detection kit (Biocare Medical cat # M3R531) with clone 20G11 (Cell Signaling Technology cat # 4376) as primary antibodies at a concentration of 1 µg/mL. Nasal polyp sections were used as positive control and rabbit IgG isotype control (Dako cat # X0903) was performed at matching concentration. Slides were mounted using pertex (Histolab Products AB).
Images were captured on a Scanscope (Aperio, Leica Biosystems) at 20x magnification and imported into the Biopix image analysis software (Biopix AB). In all sections for accurate image quantification purposes, the airway epithelial layer was encircled and subsequently analysed for positive DAB (3,3ʹ-diaminobenzidine) nuclei signal using a set threshold based on negative signal determined in isotype control as cut off for a positive pERK1/2 signal.
MEK Activity Gene Sets And Gene Set Variation Analysis
The primary MEK activity gene set utilized consisted of 58 genes identified in a panel of cancer cell lines.17 Furthermore, two additional MEK activity gene sets were defined using a) Ingenuity Pathway Analysis (IPA)18 as genes found down-stream of MEK1/2 and where the expression or the transcription was increased by MEK1/2, all findings from human and b) Pathway Studio (PS)19 as genes activated by MEK1/2.
The activity of MEK was assessed in a publicly available gene expression data set from sputum samples from the ECLIPSE clinical study20,21 which spans moderate to severe COPD. Gene expression data in the ECLIPSE study were generated from all cells obtained following sputum induction. CEL files were downloaded from Gene Expression Omnibus (GSE22148) and analyzed using ArrayStudio version 10 (OmicSoft, Cary, NC). The data were processed with Robust Multiarray Average (RMA)22 using a cfd-file from BrainArray23 version 21 (HGU133Plus2_Hs_ENSG.cdf). The effect of three confounding factors (age, gender, and batch) was removed with the Remove Batch Effect function. All 58 genes from the MEK activity gene set from Dry et al17 mapped to the ECLIPSE sputum dataset variables and the corresponding numbers for the MEK activity IPA and PS sets were 58 and 141 genes, respectively.
Gene set variation analysis (GSVA)24 was used to quantify the activation state of gene sets across COPD severity groups in sputum samples. GSVA-scores were generated using the normalized enrichment score option (R, version 3.3.3) on the expression data where the effect of three confounding factors (age, gender, and batch) had been removed. The GSVA-scores were compared across severity groups using Kruskal–Wallis test (R, version 3.3.3).
Hierarchical clustering was used to group COPD sputum samples (ECLIPSE) based on the MEK activation gene set expression. The effect of three confounding factors (age, gender, and batch) was removed from the gene expression data prior to clustering, correlation was used as the distance metrics, and for visualization purposes, data were normalized using robust center scale (the row median is subtracted from each individual value and then scaled by dividing by the row median absolute deviation) for heat map generation. Two (TYR, HMCN1) of the 58 genes in the MEK gene set had a substantially much lower expression (not shown) in these sputum samples indicative of not being expressed and were therefore excluded from clustering.
MEK Pathway Signaling
Media from overnight rested alveolar macrophage cells were removed and fresh media were added to the cells in the presence or absence of MEK (PD0325901), p38 inhibitor (AZ12904231) and steroid (Fluticasone Propionate) for 1h at 37°C. DMSO (0.1%)-treated cells were used as a vehicle control in all experiments. The MEK inhibitor utilized in the study is a highly selective and potent kinase inhibitor.25 The p38 inhibitor is an p38α selective in-house compound with less activity against p38β (data not shown). Fluticasone Propionate utilized in this study is a commercially marketed steroid licensed for use in COPD. After 1h, cells were stimulated with lipopolysaccharide (LPS) from E coli (serotype 026: B6, Sigma-Aldrich cat # L8274) at a final concentration of 100ng/mL per well and cells were incubated for 18h at 37°C for cytokine analysis, 30mins or other stipulated time points at 37°C for ERK or p38 phosphorylation assays. Unstimulated cells were additionally included in all assays, as control. For cytokine analysis, the supernatants were collected and cytokine levels analysed by multiplex immunoassay from MSD™ (Meso Scale Discovery). For calculating percentage inhibition of compounds on cytokine release the following formula was used ((maximal LPS response – LPS response in the presence of inhibitors)/(maximal LPS response – unstimulated response))*100.
For quantification of ERK or p38 phosphorylation, SureFire® ERK (pThr202/Tyr204, Perkin Elmer cat # TGRES10K) or SureFire® p38 (pThr180/Tyr182, Perkin Elmer cat #TGRE38S10K) was used as per the manufacturer’s instructions. For data analysis, phosphorylation was calculated relative to total ERK levels (SureFire® ERK1/2, Perkin Elmer cat # TGRETS10K) or total p38 levels (SureFire® p38 kit, Perkin Elmer cat # TGRET38S10K) in each well to normalize for any well to well variation in the assays. Fold increase over basal unstimulated cells was then calculated and the effect of compounds related as a percentage inhibition of LPS-mediated maximum ERK phosphorylation.
All concentrations and time points analysed were carried out in duplicates in each individual donor, with a total number of donors tested in each experiment specified in figures. To obtain concentration-responses, curve fitting was carried out using non-linear regression, four-parameter equation with variable slope (GraphPad Prism 8). No constraints were placed on curve fitting. Curve fitted donor data are presented as mean± S.E.M.
Cellular Viability Assay
Cellular viability was assessed using WST-1 reagent (Roche cat # 5015944001) as per the manufacturer’s instructions. The absorbance was read at 450nm and at 650nm (reference wavelength). The data used for analysis were the corrected absorbance (abs 450nm-abs 650nm). Background absorption values were negligible in comparison to test conditions. Treatment with LPS resulted in an apparent increase in absorbance in comparison to untreated cells. Therefore, cell viability on compound treatment was calculated as a percentage of LPS-only control cells.
Bacterial Killing Assays
Commercially purchased RAW 264.7 cells (ATCC® cat# TIB-71™) were cultured in RPMI 1640 with 10% fetal bovine serum (Life Technologies cat # 16000044) and penicillin and streptomycin (100U/mL). Cells were plated (200,000 cells per well) in a 96-well tissue-culture U-bottom plate (Costar cat # 3799) and allowed to adhere overnight in culture media. Neutrophils were plated and used immediately after isolation (100,000 cells per well). Cells were washed with antibiotic-free media and pretreated with inhibitors for 2 hrs prior to incubation with Staphylococcus aureus (MOI-1). SF8300 frozen stock cultures were thawed and diluted to the appropriate inoculum in sterile PBS, pH7.2 (Invitrogen cat # 14040117), and 1 hr following bacterial inoculation, the entire contents of the well (cells and media) were removed and bacterial CFU enumerated by serial dilution.
Statistics
Pharmacological data were analyzed using Prism 8 (GraphPad Prism). Comparison of more than one group was done with ANOVA followed by Dunnett's multiple comparison test. A p-value of less than 0.05 was considered statistically significant.
Results
MEK Pathway Activation In COPD Lung
To investigate the possible enhanced activation of MEK-pERK1/2 pathways in severe COPD lung tissue, we performed immunohistochemistry in lung tissue sections from patients with end-stage COPD (GOLD stage 4) undergoing lung transplantation and from healthy donors. Demographic information of subjects utilized in the study are detailed in Table 1. The staining revealed that p-ERK1/2 nuclear expression was higher in the airway epithelium in COPD sections as compared to the controls (p= 0.029; Mann–Whitney test) (Figure 1A–C). Also, it was noted that p-ERK1/2 expressions were extensive in areas of tissue remodeling close to airways in COPD sections (Figure 1D). Interestingly, the COPD group also shows ubiquitous staining for p-ERK1/2 in alveolar macrophages in comparison to healthy controls (Figure 1E and F).
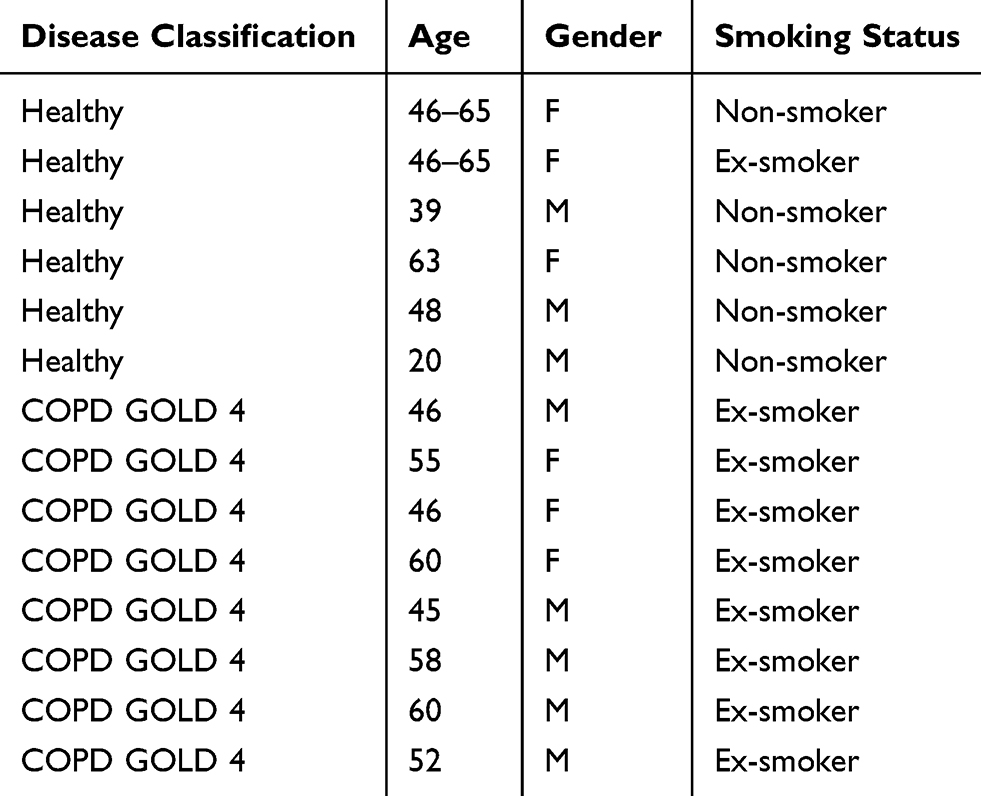 |
Table 1 Demographics Of Subjects Utilized In Histological Analysis Of MEK Pathway Activation |
 |
Figure 1 Expression of the activated MEK pathway evidenced through ERK 1/2 phosphorylation in COPD. (A–B) Increased activation of the pathway was observed in lung tissue from patients with end-stage COPD (GOLD 4) in comparison to healthy lung tissue. (C) Quantification of staining pattern in bronchial epithelia showed a significant increase (*p 0.0293 Mann–Whitney test) in pERK1/2 in severe COPD. (D) pERK1/2 staining was also observed in areas of remodeling in GOLD4 (black arrows) and (F) in alveolar macrophages in GOLD4 but not in (E) healthy lung tissue. Sections shown are representative. |
Transcriptomic Analysis Of The MEK Pathway Activation In Sputum From COPD Patients
To further explore the activation of the MEK pathway in COPD patients across varying disease severity, we utilized a MEK1/2 activity gene signature17 and used Gene Set Variation analysis (GSVA) to calculate activity signature scores in a publicly available sputum transcriptional dataset. This relatively large COPD transcriptional cohort from the ECLIPSE study was conceived as a valuable sentinel for investigation of MEK-activity in COPD.21The MEK activity GSVA-score was found to be significantly higher in severe compared to moderate COPD (p=0.0016; Kruskal–Wallis test) (Figure 2A).
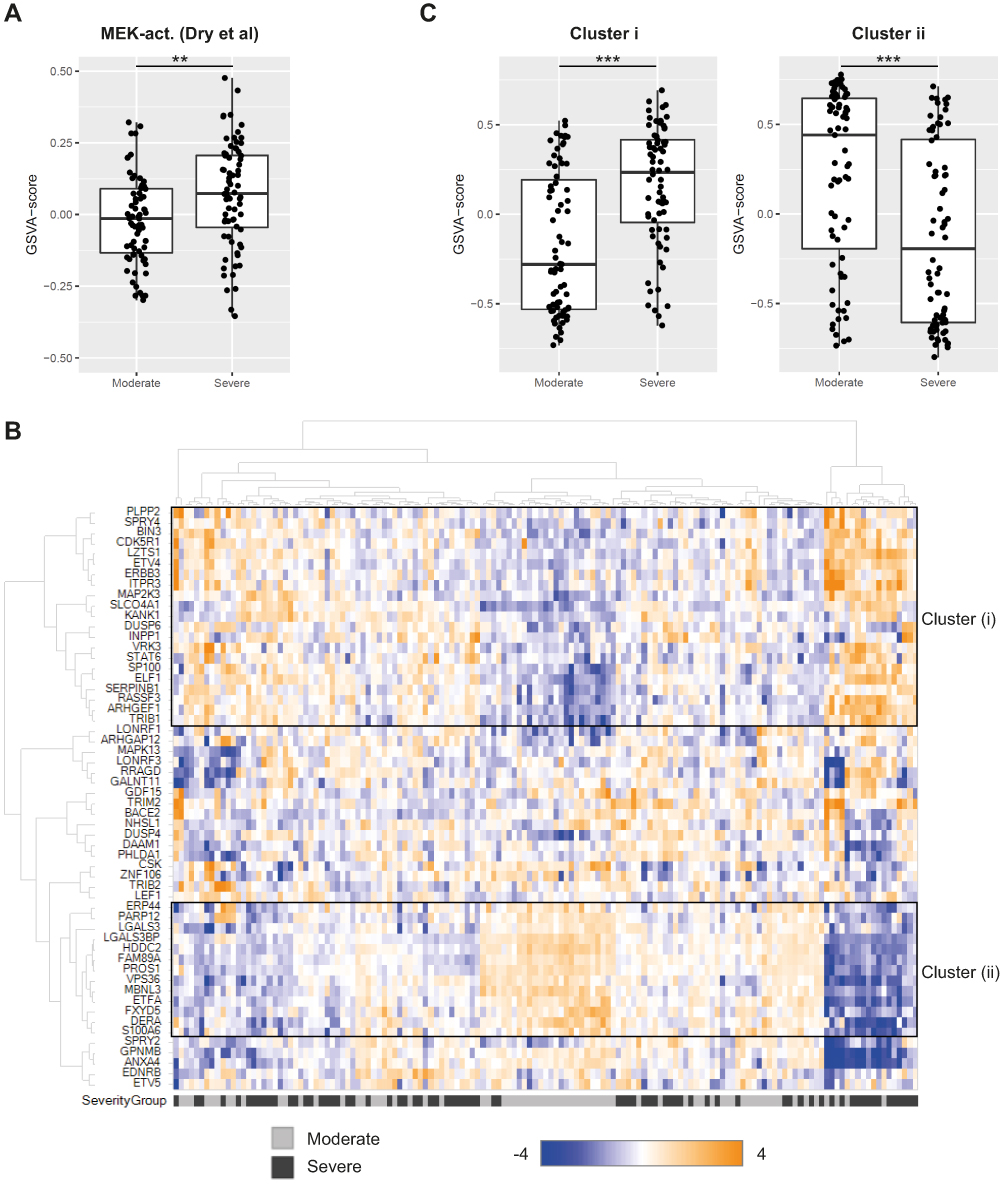 |
Figure 2 Transcriptomic profiling of MEK activation. (A) MEK activity in sputum from moderate and severe COPD (GSE22148) assessed as a GSVA-score using the MEK activity gene signature from Dry et al (2010).17 **p<0.01 (Kruskal–Wallis test). (B) Hierarchical clustering of genes in the MEK activity gene set from Dry et al (2010)17 in COPD sputum samples (GSE22148). Expression of each gene was scaled across the samples with expression going from low (blue) to high (orange). Samples are color-coded at the bottom to indicate disease severity, moderate COPD in grey and severe COPD in black. Two sub-clusters are highlighted with black boxes and denoted (i) and (ii), respectively. (C) GSVA-scores for sub-cluster (i) and (ii) gene sets from the hierarchical clustering in (B) compared across disease severity (moderate, severe) in the sputum transcriptomics dataset GSE22148. ***p<0.001 (Kruskal–Wallis test). |
As an approach to validate our observations further, two additional MEK activity gene sets were defined using Ingenuity Pathway Analysis (IPA) and Pathway Studio (PS), respectively, and these alternative MEK activity gene sets also resulted in significantly higher GSVA-scores in severe COPD (p=1.2e-2 (IPA) and p=7e-4 (PS), Kruskal–Wallis test) (Supplementary Figure 1A, B). The three independently defined MEK activity gene sets were quite distinct with only a small overlap (Supplementary Figure 1C) but each of them indicated increased MEK activity in more severe COPD.
To investigate the heterogeneity in COPD of the experimentally more tightly defined gene set of MEK activity,17 we performed unsupervised clustering of MEK activity signature genes in the ECLIPSE sputum samples. Hierarchical clustering revealed two clusters, cluster (i) and cluster (ii), with reciprocal expression pattern across the sputum samples (Figure 2B). Again, we used GSVA to summarize the activity of gene sets and found that cluster (i) genes were increased with COPD severity (p=2.0e-6, Kruskal–Wallis test) while genes in cluster (ii) decreased with COPD severity (p=4.0e-5, Kruskal–Wallis test) (Figure 2C). To characterize these clusters further, IPA was used to predict upstream regulators as well as enrichment of functional terms and MEK was found as the top upstream regulator of a cluster (i) which was also enriched for terms related to the respiratory system as well as immune response. Cluster (ii) had TRIM24 as the top upstream regulator with non-respiratory organ systems terms found to be enriched for this cluster (Table 2).
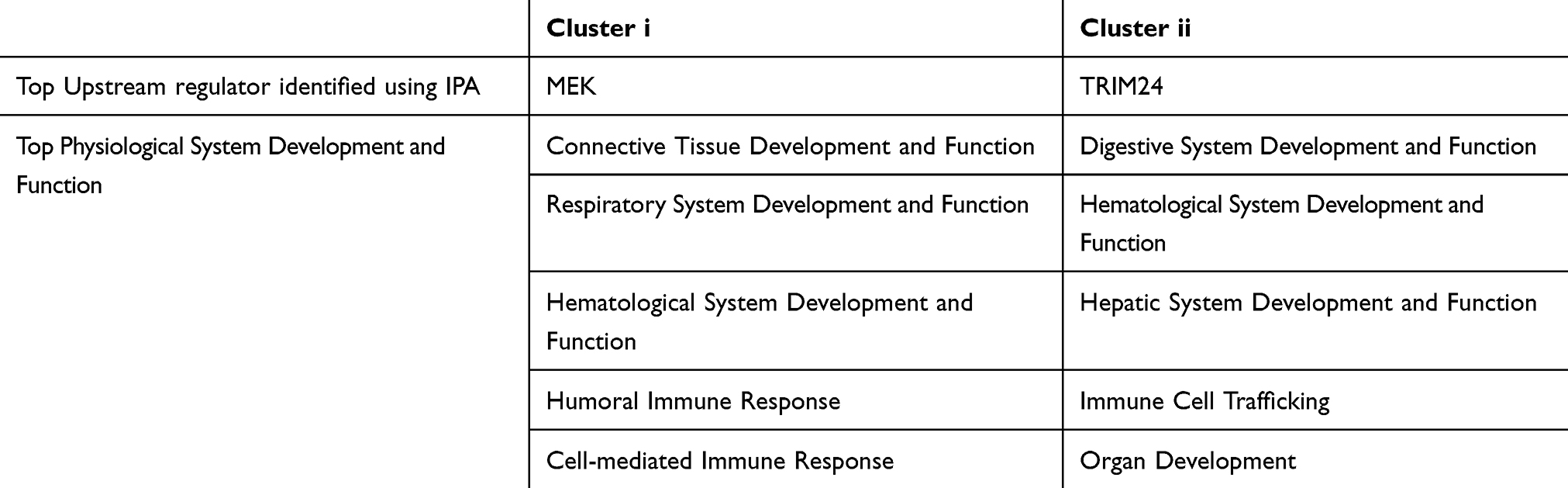 |
Table 2 Pathway Analysis Of MEK Activity Signature Genes. Predicted Top Upstream Regulator And Enriched Physiological Functional Terms For Genes In Cluster (i) And Cluster (ii), Respectively, Using IPA |
MEK Inhibition Shows A Superior Anti-Inflammatory Effect On TLR4 Signaling Mediated Activation Of Human Alveolar Macrophages
Data generated by our study have demonstrated activation of the MEK pathway in COPD both at protein and transcriptional levels in severe COPD patients. To address whether the MEK pathway is activated following an inflammatory insult, we exposed human alveolar macrophages that are key innate immune cell population contributing to inflammation in the lung of COPD patients26–28 to LPS endotoxin challenge. Stimulation of human alveolar macrophages with LPS resulted in a robust phosphorylation of ERK1/2 in all donors tested with significant peak phosphorylation responses 30 mins post-stimulation. This response continued to decline up to a period of 90 mins post-stimulation (Figure 3A; p= 0.01; p<0.0001 One-way Anova with Dunnett multiple comparison test). LPS treatment similarly resulted in a significant phosphorylation of p38 peaking at 30 mins in all donors tested mirroring the pERK kinetic response (Supplementary Figure 2; p=0.01; p=0.003; p<0.0001 One-way Anova with Dunnett multiple comparison test). No basal phosphorylation of either p38 or ERK1/2 could be quantified in unstimulated samples utilizing this assay technique.
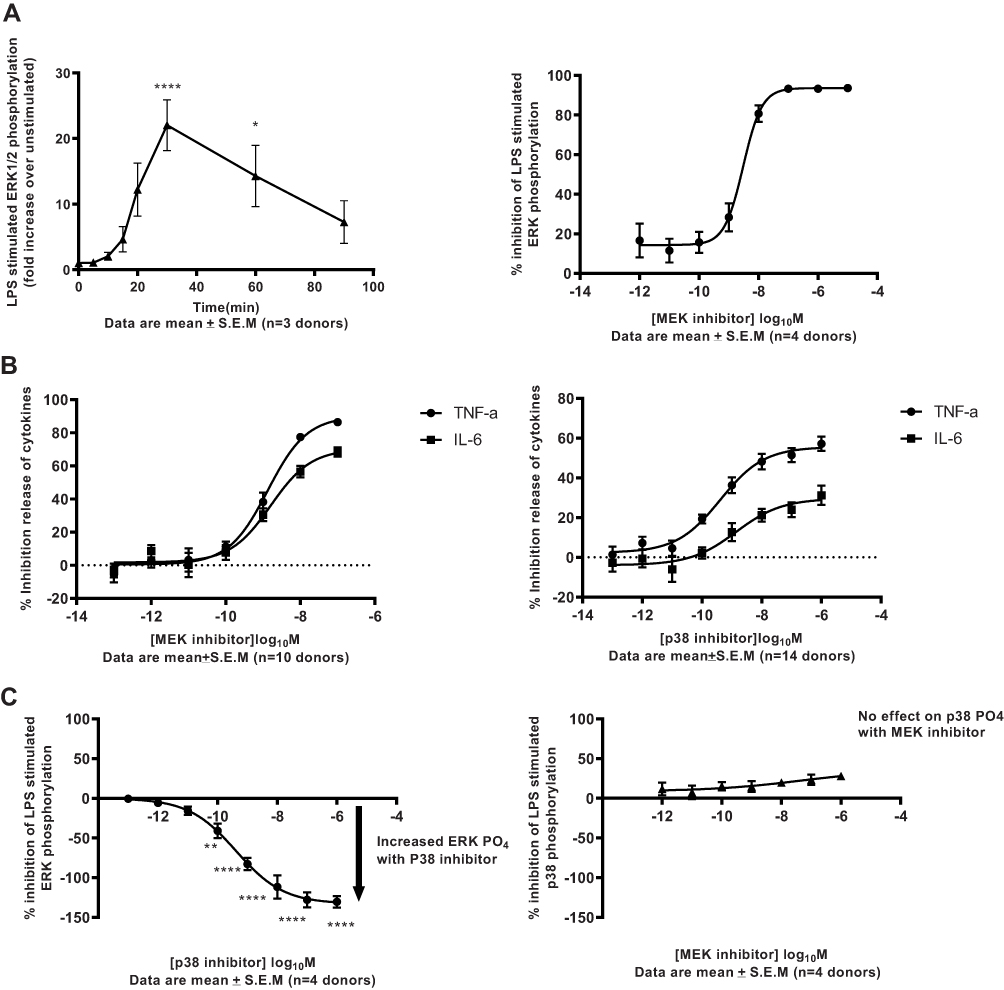 |
Figure 3 Functional characterisation of MEK pathway activation in human alveolar macrophages. (A) Robust time-dependent phosphorylation of ERK1/2 was observed in human alveolar macrophages stimulated with 100ng/mL LPS (*p 0.01, ****p<0.0001 One-way Anova with Dunnett multiple comparison test). Peak phosphorylation of ERK 1/2 measured at 30min-post LPS challenge was potently inhibited by the MEK inhibitor. Data are mean ± S.E.M of 3/4 donors. (B) Potent inhibition of inflammatory cytokine release on LPS challenge in human alveolar macrophages treated with MEK or p38 kinase inhibitors. Data are mean ± S.E.M of 10/14 donors. (C) Activation of the ERK1/2 pathway through phosphorylation is observed in a concentration-dependent manner in human alveolar macrophages on treatment with p38 kinase inhibitors at 90mins post-LPS challenge (**p 0.007, ****p<0.0001 One-way Anova with Dunnett multiple comparison test). No significant activation of the p38 pathway measured through phosphorylation is observed in human alveolar macrophages on treatment with MEK kinase inhibitors at 90mins post-LPS challenge. Data are mean ± S.E.M of 4 donors. |
Treatment of cells with the MEK inhibitor resulted in a concentration-dependent inhibition of peak ERK1/2 phosphorylation with an IC50 value of 3.05nM (Figure 3A) confirming the ability of the inhibitors to block pathway activation in alveolar macrophages following inflammatory insult with endotoxin. In addition to the effect of MEK inhibition on ERK1/2 phosphorylation, a potent inhibition of LPS-induced TNF-α and IL6 (IC50 of 1.4 and 1.6nM, respectively; Figure 3B) responses were observed in alveolar macrophages. For curve-fitting, concentrations of MEK inhibitor where cellular viability was impacted by more than 40% (10 and 1μM concentrations) were excluded from the analysis (Supplementary Figure 3). Interestingly, although p38 inhibition resulted in an inhibition of TNF-α and IL6 with an IC50 of 0.4 and 1.4nM, respectively, a decreased maximal inhibition of TNF-α and IL6 release (59% and 37%) by the p38 inhibitor in comparison to the MEK inhibitor (90% and 70%) was observed (Figure 3B). No apparent effect of p38 inhibitor was observed on cellular viability, therefore, no data points were excluded from the curve fitting (Supplementary Figure 3). Additionally, the effect of steroid Fluticasone Propionate was determined in this assay system. A potent inhibition of LPS-induced TNF-α and IL6 (IC50 of 0.8 and 1.5nM, respectively; Supplementary Figure 4) responses was observed in alveolar macrophages with a maximal inhibition of 90% and 85%, respectively, for TNF-α and IL6 release. There was no apparent dose-dependent effect of the steroid observed on cellular viability and therefore no data points were excluded from the curve fitting (Supplementary Figure 4).
P38 But Not MEK Inhibition Results In MAPK Pathway Shunting In Human Alveolar Macrophages
To understand the differences in the observed maximal inhibition between MEK and p38 inhibitors, alveolar macrophages were treated with MEK or p38 compounds and kinase pathway activation was investigated at 90 mins post-LPS-challenge. This time point was chosen for testing pathway re-activation as our data indicated that by 90 min, peak phosphorylation declined close to baseline (Figure 3A, Supplementary Figure 2). In cells treated with a p38 inhibitor, a significant concentration-dependent increase of ERK1/2 phosphorylation was observed in comparison to untreated cells (Figure 3C; p=0.007; p<0.0001 One-way Anova with Dunnett multiple comparison test). In contrast, in cells treated with a MEK inhibitor, there was no significant increase in p38 phosphorylation in treated or untreated cells (Figure 3C). These data indicate that ERK pathway shunting occurs in alveolar macrophages upon p38 inhibition and could be one of the drivers to the decreased maximal responses of cytokine inhibition observed in comparison to MEK inhibitors.
MEK Inhibition Results In Enhanced Bacterial Killing In Murine Macrophages And Human Neutrophils
To confirm that MEK inhibition was reducing inflammation without compromising host defense, we initially tested the ability of the murine macrophage cell line RAW 264.7 to kill Staphylococcus aureus one of the major opportunistic human airway pathogen in the presence of Fluticasone Propionate, p38 inhibitor or MEK inhibitor. Control macrophages treated with DMSO were able to kill approximately 20% of the bacterial inocula over the hour incubation period. Neither the steroid nor the p38 inhibitor increased bacterial killing above this baseline level. MEK inhibition, however, significantly increased bacterial killing in a dose-dependent manner at concentrations that also induced a potent anti-inflammatory effect in alveolar macrophages (Figure 4A; p=0.025; p=0.01 One-way Anova with Dunnett multiple comparison test). MEK pathway activation on the bacterial challenge was confirmed by Western blot. Furthermore, pathway activation was inhibited by 1μM concentration of the MEK inhibitor (Figure 4B) at all time points tested.
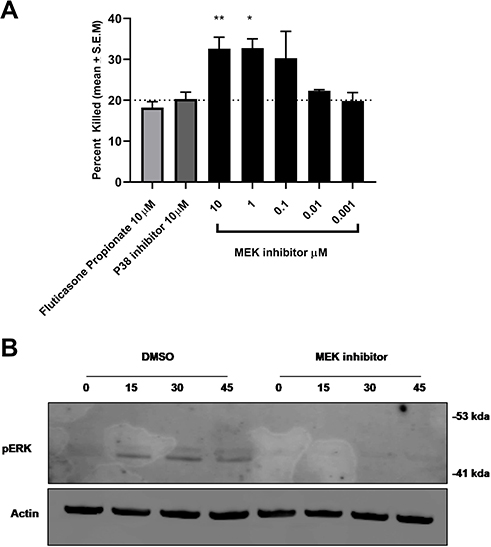 |
Figure 4 MEK inhibition enhances bacterial killing in RAW264.7 cells. (A) MEK inhibition results in enhanced S.aureus killing in RAW264.7 cells (*p 0.01, **p 0.002 One-way Anova with Dunnett multiple comparison test). This was not observed with p38 inhibitor or steroid Fluticasone Propionate. (B) Time-dependent activation of the MEK-pERK 1/2 pathway on S. aureus exposure in RAW264.7 cells was confirmed by Western blot analysis. Activation of the cascade was inhibited by treatment with the MEK inhibitor. Data are mean + S.E.M of 3 different experiments. |
In the airways, neutrophils also play a major role against bacterial pathogens; therefore, it was important to confirm that MEK inhibition does not adversely influence the killing function of these cells. For testing this principle, human neutrophils were purified from healthy donors and incubated with S. aureus in the presence of the same inhibitors. Once again, only MEK inhibition increased bacterial killing above DMSO control (Figure 5A; p=0.01 One-way Anova with Dunnett multiple comparison test). Inhibition of the MEK pathway was assessed by Western blot for changes in phosphorylation of ERK1/2 (Figure 5B) confirming that the observed effect of enhanced bacterial killing is driven through inhibition of the MEK pathway.
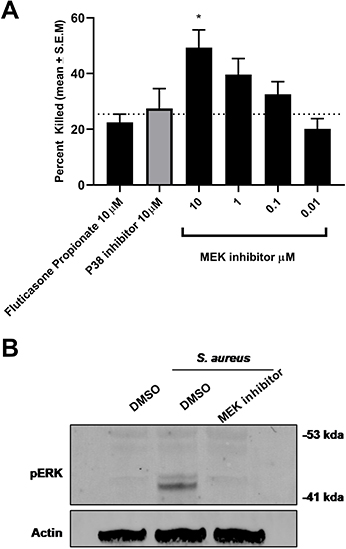 |
Figure 5 MEK inhibition enhances bacterial killing in human neutrophils. (A) MEK inhibition resulted in an increase in S.aureus killing in human neutrophils in a concentration-dependent manner (*p 0.01 One-way Anova with Dunnett multiple comparison test). No effect of p38 inhibitor or steroid was observed in bacterial killing in neutrophils at the concentrations tested. (B) MEK-pERK 1/2 pathway on S. aureus exposure in neutrophils was confirmed by Western blot analysis. Activation of the cascade was inhibited by treatment with the MEK inhibitor. Data are mean + S.E.M of 4 donors. |
Discussion
COPD is a chronic inflammatory disease characterized by the incidence of exacerbations triggered by respiratory pathogens3 On the current standard of care treatment, COPD patients continue to experience exacerbations driving disease morbidity. Thus, there is a need for new therapeutics that can dampen inflammation while also boosting host defense. We report here enhanced activation of MEK pathway in severe COPD patients and reveal a novel dual function of MEK inhibition in modulating inflammation without compromising phagocytosis-mediated pathogen-clearance. The MEK signaling pathway is recognized as being important in cancer, as such, several small molecule inhibitors of MEK have demonstrated clinical efficacy in certain cancers29 providing an opportunity for potential repurposing of these assets in COPD.
Our study is the first to demonstrate an enhanced MEK activation gene signature in COPD based on GSVA and unsupervised clustering of the sputum transcriptomic datasets available from the ECLIPSE cohort consisting only of ex-smoker COPD patients. Induced sputum samples consist of a mixture of inflammatory cells such as neutrophils and macrophages sampled from the airways of patients and are therefore highly relevant to investigate from the context of this study. Interrogation of the MEK signature sub-clusters using Ingenuity Pathway Analysis demonstrated that MEK was the top predicted upstream regulator of genes in cluster (i) while cluster (ii) has TRIM24 as the top upstream regulator. To date, no reports exist on a direct link between TRIM24 and COPD, but up-regulation of TRIM24 expression has been implicated in several human cancers, including lung cancer30 and in some instances shown to be MEK inhibitor sensitive. Thus, it is not unexpected that TRIM24 was identified as a key regulator of MEK signature genes in cluster (ii). While the use of this signature highlights a potential implication for genes regulated by the MEK pathway being active in subsets of patients with severe disease, this needs to be confirmed utilizing a gene signature that is generated in human primary cells relevant to COPD.
We also observed a prominent p-ERK1/2 protein nuclear expression in the airway epithelium and a ubiquitous expression in alveolar macrophages in COPD lung tissue, indicating that activation of MEK1/2-ERK1/2 signaling pathway could play an important role in COPD disease pathology. To our knowledge, this is the first report demonstrating an activated MEK1/2 pathway at the protein level in COPD. Although we report a significant upregulation at the protein level of the MEK pathway in GOLD 4 lung sections obtained from COPD transplant patients with end-stage disease, we have not examined pathway activation in mild or moderate COPD lung sections; therefore, it is unclear whether pathway activation could be a driver of disease progression and this important aspect warrants further investigation. Expression of other MAPK, particularly p38, has been reported in alveolar macrophages and bronchial epithelia of COPD patients8–10 implicating the importance of other MAPK pathways in driving inflammation. Enhanced p-ERK1/2 and p-p38 expression was also reported in airway epithelium in biopsy specimens from asthmatics compared to non-asthmatic subjects, indicating that the activation of these signaling pathways may have a broader implication in airway inflammatory diseases.31 Indeed, in an allergic asthma ovalbumin mouse model, a widely used MEK1/2 inhibitor U0126 demonstrated a beneficial effect on airway inflammatory responses and airway hyperresponsiveness.32 Furthermore, U0126 also provided protection against stretch-induced epithelial barrier dysfunction in healthy and septic models.33,34 These data provide further evidence for a role for these kinase pathways in airway diseases.
In this study, expression of pERK1/2 was noted in alveolar macrophages in GOLD 4 COPD lung sections. Alveolar macrophages are implicated as key drivers of inflammation in COPD,35 their numbers are reported to be increased in COPD patients26,27,36 and linked to disease severity.28 We demonstrate that MEK inhibitor is a potent down-regulator of LPS-stimulated inflammatory response in alveolar macrophages. This is in line with previous reports of activation of the pathway in human, rodent macrophages and other immune cells.37–40 Recent work in rodent macrophages and human peripheral blood mononuclear cells have similarly reported a beneficial effect of MEK inhibition on suppressing inflammation utilizing other MEK inhibitors.37,38,41
Activation of the MAPK pathways, particularly p38, is a key driver of LPS-stimulated cytokine release.42,43 When we compared the effect of the inhibition of p38, steroid and MEK, we found a superior anti-inflammatory effect of MEK and steroid inhibition on human alveolar macrophages in comparison to the tested p38 inhibitor. In our experimental setup, cells were sensitive to steroid inhibition for these cytokines. This observation is consistent with other reports in lung macrophages from either healthy or COPD patients to the effect of steroids.44 However, the authors44 highlight that the sensitivity of steroid inhibition to the neutrophil chemoattractant IL-8/CXCL8 was lower in some patients. We have not measured IL-8/CXCL8 in our studies and in future studies it would be important to address if MEK inhibition could indeed overcome this loss of sensitivity to IL-8/CXCL8 inhibition observed to steroids. The level of inhibition of inflammatory cytokines observed due to p38 inhibition in this study was similar to other reports9,45,46 suggesting that there might be a ceiling effect for inflammatory cytokine inhibition by p38 inhibitors in this cell type. MEK inhibition therefore may be an alternative strategy to overcome this ceiling effect observed with p38 inhibitors.
An explanation for the ceiling effect could be due to the “shunting” effect when p38 inhibitors are utilized in this assay system at the level of MAPK phosphorylation. This cross-talk or shunting between the ERK and p38 pathways have been previously reported in osteosarcoma cells,47 PC12 cells,48 bronchial epithelial cells49 and in corneal epithelial cells.50 In corneal epithelial cells and osteosarcoma cells, it was reported that inhibition of either one of the MAPK pathways could result in the activation of the other. However, in our studies, we observed a mono-directional effect on ERK activation upon p38 inhibition at the 90-min time-point analysed, consistent with the reports in bronchial epithelial cells and PC-12 cells. Activation of the ERK pathway is critical to the regulation of transcription factors such as AP-1, C/EBPB and thereby inflammatory gene transcription in LPS-stimulated human alveolar macrophages.51 Therefore, shunting to the MEK-ERK pathway in the context of p38 inhibition could result in altered transcriptional regulation of the inflammatory cytokines.
Interestingly, we observed a beneficial effect of MEK inhibition on bacterial clearance in both RAW 264.7 macrophage cell line and in primary human neutrophils in-vitro. However, these findings have to be confirmed in patient-derived cells, since clearance defects have been reported in alveolar macrophages from COPD patients in several studies in COPD and reviewed by Jubrail et al.52 Defective clearance could be one of the driver contributing to increased bacterial colonization. Bacterial colonization in COPD is increasingly being recognized as a driver of disease morbidity with reports of higher bacterial load resulting in greater inflammatory load and increased risk of acute exacerbation in COPD patients.53,54 The ability to spare or even enhance host-defense responses while dampening inflammation is an attractive therapeutic strategy and is our key finding.
Furthermore, our observations of enhanced bacterial clearance are in line with reports of the beneficial effect of MEK inhibitors in murine sepsis and in endotoxin shock37,55 and more recently in an experimental Pseudomonas aeruginosa model.56 The effect on bacterial clearance was consistent regardless of the inhibitor utilized (U0126, Trametinib or PD0325901) suggesting that this may be a “class-effect” of MEK inhibition. Attenuation of inflammation, measured by either impact on inflammatory cell recruitment in lungs or on cytokine release was reported in these in-vivo studies with both prophylactic or therapeutic treatment with the MEK inhibitor, consistent with our in-vitro observations. Although there are some suggestions for MEK inhibitors in influencing macrophage polarization56,57 the exact mechanism by which these inhibitors spare or enhance bacterial clearance is not fully elucidated.
The beneficial effect of MEK inhibitors on host defense may not be limited to bacterial infections as several reports suggest a direct or indirect antiviral effect on different respiratory viruses, including influenza58,59 and rhinovirus.60 The completion of the life cycle of influenza is dependent on the MEK pathway as it facilitates the nuclear export of the viral ribonucleoprotein.61 We recently identified MEK as a key regulator of interferon response in airway epithelial cells and inhibition of MEK led to enhanced IFN response, which translated into a reduction of rhinovirus replication.60 Since these viruses are commonly associated with COPD exacerbations, the inhibition of MEK1/2 pathway may represent a new strategy for treating virus-induced exacerbations in COPD.
Despite some limitations discussed, our study has a unique strength in that we have utilized target validation in human tissue and cells providing a translational model to further interrogate the importance of this pathway in disease.
Conclusion
In conclusion, our study provides the first direct translational in-vitro evidence for the presence of an activated MEK-ERK pathway in COPD and that utilization of a MEK inhibitor may have a dual-role in the treatment of COPD by controlling inflammation while sparing pathogen-clearance mechanisms.
Acknowledgments
We thank the Transplant Institute and Department of Cardiothoracic Surgery in the Sahlgrenska University Hospital for excellent collaboration on tissue acquisition led by Dr Göran Dellgren. A special gratitude to Linda Thimour-Bergström for her great contribution to the smooth logistics which made this work possible. We thank the patients that took part in the study without which this work would not have been possible.
Author Contributions
Conceptualization and study design: NK, TSC, PDS, BB, OV, DMC. Data acquisition and interpretation: NK, TSC, LÖ, EDZ, GS, PS, EB, SV, DMC. Drafting and review of manuscript: NK, TSC, GS, LÖ, EDZ, EB, PS, SV, PDS, BB, OV, DMC. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
All authors (NK, LÖ, EDZ, GS, SV, EB, PS, BB, PDS, OV, and DMC) were full-time employees of the commercial company AstraZeneca and its subsidiary Medimmune (TSC). The Authors report no other conflicts of interest in this work.
References
1. Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. doi:10.1056/NEJMra0804752
2. Barnes PJ, Burney PGJ, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076. doi:10.1038/nrdp.2015.76
3. Wilkinson TMA, Aris E, Bourne S, et al. A prospective, observational cohort study of the seasonal dynamics of airway pathogens in the aetiology of exacerbations in COPD. Thorax. 2017;72(10):919–927. doi:10.1136/thoraxjnl-2016-209023
4. Gunawardana N, Finney L, Johnston SL, Mallia P. Experimental rhinovirus infection in COPD: implications for antiviral therapies. Antiviral Res. 2014;102:95–105. doi:10.1016/j.antiviral.2013.12.006
5. Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(11):1117–1124. doi:10.1164/rccm.201205-0806OC
6. Molyneaux PL, Mallia P, Cox MJ, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(10):1224–1231. doi:10.1164/rccm.201302-0341OC
7. Kew KM, Seniukovich A. Inhaled steroids and risk of pneumonia for chronic obstructive pulmonary disease. Cochrane Database Sys Rev. 2014;(3). doi:10.1002/14651858.CD010115.pub2
8. Renda T, Baraldo S, Pelaia G, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31(1):62–69. doi:10.1183/09031936.00036707
9. Patel NR, Cunoosamy DM, Fagerås M, et al. The development of AZD7624 for prevention of exacerbations in COPD: a randomized controlled trial. Int J Chron Obstruct Pulmon Dis. 2018;13:1009–1019. doi:10.2147/COPD.S150576
10. Gaffey K, Reynolds S, Plumb J, Kaur M, Singh D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42(1):28–41. doi:10.1183/09031936.00170711
11. MacNee W, Allan RJ, Jones I, De Salvo MC, Tan LF. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: a randomised clinical trial. Thorax. 2013;68(8):738–745. doi:10.1136/thoraxjnl-2012-202744
12. Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(1):63–72. doi:10.1016/S2213-2600(13)70200-5
13. O’Donnell RA, Richter A, Ward J, et al. Expression of ErbB receptors and mucins in the airways of long term current smokers. Thorax. 2004;59(12):1032. doi:10.1136/thx.2004.028043
14. Hussain SS, George S, Singh S, et al. A small molecule BH3-mimetic suppresses cigarette smoke-induced mucous expression in airway epithelial cells. Sci Rep. 2018;8(1):13796. doi:10.1038/s41598-018-32114-w
15. Ngkelo A, Hoffmann RF, Durham AL, et al. Glycogen synthase kinase-3β modulation of glucocorticoid responsiveness in COPD. Am J Physiol Lung Cell Mol Physiol. 2015;309(10):L1112–L1123. doi:10.1152/ajplung.00077.2015
16. Liu K, Gualano RC, Hibbs ML, Anderson GP, Bozinovski S. Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J Biol Chem. 2008;283(15):9977–9985. doi:10.1074/jbc.M710257200
17. Dry JR, Pavey S, Pratilas CA, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res. 2010;70(6):2264–2273. doi:10.1158/0008-5472.CAN-09-1577
18. Qiagen. Available from: https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/.
19. Studio P. Available from: https://www.elsevier.com/solutions/pathway-studio-biological-research.
20. Vestbo J, Anderson W, Coxson HO, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur Respir J. 2008;31(4):869–873. doi:10.1183/09031936.00111707
21. Singh D, Fox SM, Tal-Singer R, et al. Induced sputum genes associated with spirometric and radiological disease severity in COPD ex-smokers. Thorax. 2011;66(6):489–495. doi:10.1136/thx.2010.153767
22. Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi:10.1093/biostatistics/4.2.249
23. Dai M, Wang P, Boyd AD, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33(20):e175–e175. doi:10.1093/nar/gni179
24. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14:7. doi:10.1186/1471-2105-14-7
25. Barrett SD, Bridges AJ, Dudley DT, et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett. 2008;18(24):6501–6504. doi:10.1016/j.bmcl.2008.10.054
26. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
27. Stefano AD, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158(4):1277–1285. doi:10.1164/ajrccm.158.4.9802078
28. Kaku Y, Imaoka H, Morimatsu Y, et al. Overexpression of CD163, CD204 and CD206 on alveolar macrophages in the lungs of patients with severe chronic obstructive pulmonary disease. PLoS One. 2014;9(1):e87400. doi:10.1371/journal.pone.0087400
29. Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577. doi:10.1038/nrc4000
30. Liu X, Huang Y, Yang D, et al. Overexpression of TRIM24 is associated with the onset and progress of human hepatocellular carcinoma. PLoS One. 2014;9(1):e85462. doi:10.1371/journal.pone.0085462
31. Liu W, Liang Q, Balzar S, Wenzel S, Gorska M, Alam R. Cell-specific activation profile of extracellular signal-regulated kinase 1/2, Jun N-terminal kinase, and p38 mitogen-activated protein kinases in asthmatic airways. J Allergy Clin Immunol. 2008;121(4):893–902.e892. doi:10.1016/j.jaci.2008.02.004
32. Duan W, Chan JHP, Wong CH, Leung BP, Wong WSF. Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J Immunol. 2004;172(11):7053–7059. doi:10.4049/jimmunol.172.11.7053
33. Cohen TS, Gray Lawrence G, Khasgiwala A, Margulies SS. MAPk activation modulates permeability of isolated rat alveolar epithelial cell monolayers following cyclic stretch. PLoS One. 2010;5(4):e10385. doi:10.1371/journal.pone.0010385
34. Cohen TS, Gray Lawrence G, Margulies SS. Cultured alveolar epithelial cells from septic rats mimic in vivo septic lung. PLoS One. 2010;5(6):e11322. doi:10.1371/journal.pone.0011322
35. Barnes PJ. Alveolar macrophages as orchestrators of COPD. J Chron Obstruct Pulmon Dis. 2004;1(1):59–70. doi:10.1081/COPD-120028701
36. Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1995;152(5):1666–1672. doi:10.1164/ajrccm.152.5.7582312
37. Shi-Lin D, Yuan X, Zhan S, Luo-Jia T, Chao-Yang T. Trametinib, a novel MEK kinase inhibitor, suppresses lipopolysaccharide-induced tumor necrosis factor (TNF)-α production and endotoxin shock. Biochem Biophys Res Commun. 2015;458(3):667–673. doi:10.1016/j.bbrc.2015.01.160
38. Li P, Wu Y, Li M, Qiu X, Bai X, Zhao X. AS-703026 inhibits LPS-induced TNFα production through MEK/ERK dependent and independent mechanisms. PLoS One. 2015;10(9):e0137107. doi:10.1371/journal.pone.0137107
39. Monick MM, Powers LS, Barrett CW, et al. Constitutive ERK MAP kinase activity regulates macrophage ATP production and mitochondrial integrity. J Immunol. 2008;180(11):7485–7496. doi:10.4049/jimmunol.180.11.7485
40. Chanteux H, Guisset AC, Pilette C, Sibille Y. LPS induces IL-10 production by human alveolar macrophages via MAPKinases- and Sp1-dependent mechanisms. Respir Res. 2007;8(1):71. doi:10.1186/1465-9921-8-71
41. Xu Y, Ito T, Fushimi S, et al. Spred-2 deficiency exacerbates lipopolysaccharide-induced acute lung inflammation in mice. PLoS One. 2014;9(10):e108914. doi:10.1371/journal.pone.0108914
42. Bode JG, Ehlting C, Häussinger D. The macrophage response towards LPS and its control through the p38MAPK–STAT3 axis. Cell Signal. 2012;24(6):1185–1194. doi:10.1016/j.cellsig.2012.01.018
43. Hoogerwerf JJ, AFd V, Cvt V, et al. Priming of alveolar macrophages upon instillation of lipopolysaccharide in the human lung. Am J Respir Cell Mol Biol. 2010;42(3):349–356. doi:10.1165/rcmb.2008-0362OC
44. Higham A, Booth G, Lea S, Southworth T, Plumb J, Singh DJRR. The effects of corticosteroids on COPD lung macrophages: a pooled analysis. Respir Res. 2015;16(1):98. doi:10.1186/s12931-015-0260-0
45. Armstrong J, Harbron C, Lea S, et al. Synergistic effects of p38 mitogen-activated protein kinase inhibition with a corticosteroid in alveolar macrophages from patients with chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2011;338(3):732–740. doi:10.1124/jpet.111.180737
46. Ratcliffe MJ, Dougall IG. Comparison of the anti-inflammatory effects of cilomilast, budesonide and a p38 mitogen activated protein kinase inhibitor in COPD lung tissue macrophages. BMC Pharmacol Toxicol. 2012;13:15. doi:10.1186/2050-6511-13-15
47. Shimo T, Matsumura S, Ibaragi S, et al. Specific inhibitor of MEK-mediated cross-talk between ERK and p38 MAPK during differentiation of human osteosarcoma cells. J Cell Commun Signal. 2007;1(2):103–111. doi:10.1007/s12079-007-0010-2
48. New L, Li Y, Ge B, et al. SB203580 promotes EGF-stimulated early morphological differentiation in PC12 cell through activating ERK pathway. J Cell Biochem. 2001;83(4):585–596.
49. Bowers EC, McCullough SD, Morgan DS, Dailey LA, Diaz-Sanchez D. ERK1/2 and p38 regulate inter-individual variability in ozone-mediated IL-8 gene expression in primary human bronchial epithelial cells. Sci Rep. 2018;8:9398. doi:10.1038/s41598-018-27662-0
50. Sharma G-D, He J, Bazan HEP. p38 and ERK1/2 Coordinate Cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between map kinase cascades. J Biol Chem. 2003;278(24):21989–21997. doi:10.1074/jbc.M302650200
51. Carter AB, Monick MM, Hunninghake GW. Both Erk and p38 Kinases are necessary for cytokine gene transcription. Am J Respir Cell Mol Biol. 1999;20(4):751–758. doi:10.1165/ajrcmb.20.4.3420
52. Jubrail J, Kurian N, Niedergang F. Macrophage phagocytosis cracking the defect code in COPD. Biomed J. 2017;40(6):305–312. doi:10.1016/j.bj.2017.09.004
53. Singh R, Garcha D, Patel A, Mackay A, Donaldson G, Wedzicha J. The impact of bacterial colonisation on airway inflammation in stable COPD. Eur Respir J. 2012;40(Suppl 56). doi:10.1183/09031936.00213711
54. Singh R, Mackay A, Brill S, et al. Effect of lower airway bacterial colonisation on time to acute exacerbation in patients with COPD. Eur Respir J. 2015;46(suppl 59):PA661.
55. Smith JA, Mayeux PR, Schnellmann RG. Delayed MEK/ERK inhibition by trametinib attenuates systemic inflammatory responses and multi-organ injury in murine sepsis. Crit Care Med. 2016;44(8):e711–e720. doi:10.1097/CCM.0000000000001672
56. Long ME, Gong K-Q, Eddy WE, Liles WC, Manicone AM. Pharmacologic inhibition of MEK1/2 reduces lung inflammation without impairing bacterial clearance in experimental pseudomonas aeruginosa pneumonia. Pneumonia. 2017;9:13. doi:10.1186/s41479-017-0037-y
57. Long ME, Eddy WE, Gong K-Q, et al. MEK1/2 inhibition promotes macrophage reparative properties. J Immunol. 2017;198(2):862–872. doi:10.4049/jimmunol.1601059
58. Droebner K, Pleschka S, Ludwig S, Planz O. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res. 2011;92(2):195–203. doi:10.1016/j.antiviral.2011.08.002
59. Haasbach E, Müller C, Ehrhardt C, et al. The MEK-inhibitor CI-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antiviral Res. 2017;142:178–184. doi:10.1016/j.antiviral.2017.03.024
60. Baturcam E, Vollmer S, Schlüter H, et al. MEK inhibition drives anti-viral defence in RV but not RSV challenged human airway epithelial cells through AKT/p70S6K/4E-BP1 signalling. Cell Commun Signaling. 2019;17(1):78. doi:10.1186/s12964-019-0378-7
61. Pleschka S, Wolff T, Ehrhardt C, et al. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. 2001;3(3):301–305. doi:10.1038/35060098
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.