Back to Journals » International Journal of Nanomedicine » Volume 17
Dual pH-Responsive and Tumor-Targeted Nanoparticle-Mediated Anti-Angiogenesis siRNA Delivery for Tumor Treatment
Authors Zhang X, Qin B, Wang M, Feng J, Zhang C, Zhu C, He S, Liu H ![]() , Wang Y
, Wang Y ![]() , Averick SE, Vo NTN, Huang L, Liu W, Wang Z
, Averick SE, Vo NTN, Huang L, Liu W, Wang Z
Received 22 October 2021
Accepted for publication 18 February 2022
Published 5 March 2022 Volume 2022:17 Pages 953—967
DOI https://doi.org/10.2147/IJN.S340926
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Xiangyang Zhang,1 Bin Qin,1 Min Wang,1 Junyi Feng,1 Chenglin Zhang,1 Chengshen Zhu,2 Suqin He,2 Hao Liu,2 Yaohe Wang,1 Saadyah E Averick,3 Nga TN Vo,4 Lei Huang,5 Wentao Liu,2 Zhimin Wang1
1Sino-British Research Centre for Molecular Oncology, National Centre for International Research in Cell and Gene Therapy, School of Basic Medical Sciences, Academy of Medical Sciences, Zhengzhou University, Zhengzhou, Henan Province, People’s Republic of China; 2School of Material Science and Engineering, Zhengzhou University, Zhengzhou, Henan Province, People’s Republic of China; 3Neuroscience Institute, Allegheny Health Network, Allegheny General Hospital, Pittsburgh, PA, USA; 4School of Engineering, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK; 5Inflammations Immunity Research Theme, Translational and Clinical Research Institute, FMS, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK
Correspondence: Zhimin Wang; Wentao Liu, Email [email protected]; [email protected]
Purpose: In order to overcome the biological barriers at all levels and enhance the delivery efficiency of siRNA, we have prepared a multifunctional siRNA delivery system (CHCE/siRNA nanoparticles) through self-assembly of the carboxymethyl chitosan modified with histidine, cholesterol, and anti-EGFR antibody (CHCE).
Methods: The morphology of CHCE/siRNA NPs was detected by dynamic light scattering and scanning electron microscope. In vitro, we assessed the tumor-targeting, cellular uptake, and endosomal escape by flow cytometry and confocal laser scanning microscopy, confirming the CHCE/siRNA NPs functions in gene silencing and cell killing ability. In vivo, we examined the biodistribution of the CHCE/siRNA NPs by the IVIS imaging system and confirmed the therapeutic effect of NPs in the nude-mouse tumor model.
Results: The CHCE/siRNA NPs exhibited nanosized spherical with narrow size distribution. In vitro, the CHCE/siRNA NPs incorporated a dual capability of tumor targeting and pH response that could facilitate cellular bind, cellular uptake, and endosomal escape. The CHCE/siRNA NPs could effectively silence the vascular endothelial growth factor A (VEGFA) to cause cell apoptosis and inhibit proliferation. In vivo, the CHCE/siRNA NPs could target tumor sites to knock down VEGFA and achieve a better anti-tumor effect.
Conclusion: We successfully prepared a novel siRNA delivery system with the double capability of tumor targeting and pH response, which can break through the biological barriers to penetrate deep into tumors and achieve better therapeutic tumor effects, providing a new ideal delivery platform for siRNA.
Keywords: multifunctional carboxymethyl chitosan, targeting delivery, endosomal escape, gene silencing, anti-tumor therapy
Introduction
RNA interference (RNAi) is an evolutionarily conserved biological mechanism regulating gene expression through small interfering RNA (siRNA) after transcription.1 RNAi has attracted much attention in biomedical fields, as siRNAs are highly potent in silencing the expression of specific target genes in human cells.2,3 siRNA-based therapeutics have been proposed to treat various diseases, including inherited disorders, viral infections, and cancers.4 The approval of the small interfering RNA (siRNA) drug Patisiran is a milestone in the clinical translation of Nobel Prize achievements after a 20-year wait, which provides a successful model for the research of nano-delivery systems.5 However, siRNA has poor therapeutic efficacy due to low enzymatic stability, poor pharmacokinetics, and inability to cross through biological barriers in vivo6,7 and thus requires careful consider with carrier groups to enhance the stability of siRNA, improve effective therapeutic dose in target tissues and promote efficient cell uptake.
Nanocarriers are a promising platform for siRNA delivery. Over the past few decades, various novel materials, including liposomes, natural and synthetic polymers, peptides, and inorganic, have been produced for siRNA delivery.8–12 Although these delivery systems improved the bioavailability of siRNA in vivo, they have seen limited efficacy in translation to clinical settings.13–15 Many factors can affect the effective delivery of nanocarriers in vivo.16 It is postulated that the very features that promote siRNA binding are the factors that lead to these materials’ limited clinical success. For example, a positive charge is required to bind siRNA and promote the cellular internalization of the NPs. However, the positively charged surfaces also promote NPs binding to various opsonizing proteins, leading to the rapid clearance by macrophages and monocytes.17 The non-specific distribution of nanocarriers is also an important issue facing in vivo delivery. Previous studies have shown that the utilization rate of existing drugs is only 0.7%, and more than 99% of drugs are wasted in the body, producing strong adverse effects and low therapeutic efficacy.18 Therefore, improving the targeted delivery ability of nanocarriers is of great significance for improving the interference ability of siRNA. The surface properties of the NPs, such as polarity, particle sizes, and morphology also directly impact the performance of NPs in vivo.19–21 Furthermore, the drug-loading/release rate of NPs influences the cumulative concentration of the drug at the targeted tissue, eg, tumor, thus affecting the therapeutic efficacy.22 Therefore, designing optimal NPs for breaking through the biological barriers at all levels has been an intricate engineering process.
In this manuscript, we report a novel siRNA delivery system targeting epidermal growth factor receptor (EGFR) expressing cells using carboxymethyl chitosan as the backbone polymer, modified with functional groups including histidine, cholesterol, and anti-EGFR antibody (CHCE/siRNA NPs) (Figure 1A). The CHCE/siRNA NPs incorporate a tumor-targeting mechanism (Figure 1B), pH responsiveness (Figure 1C), and cargo siRNA stability, which can overcome most biological barriers and achieve higher delivery efficiency. In summary, the CHCE/siRNA NPs silenced the VEGFA gene in tumors to inhibit intratumor vasculature growth and inhibited tumor growth.
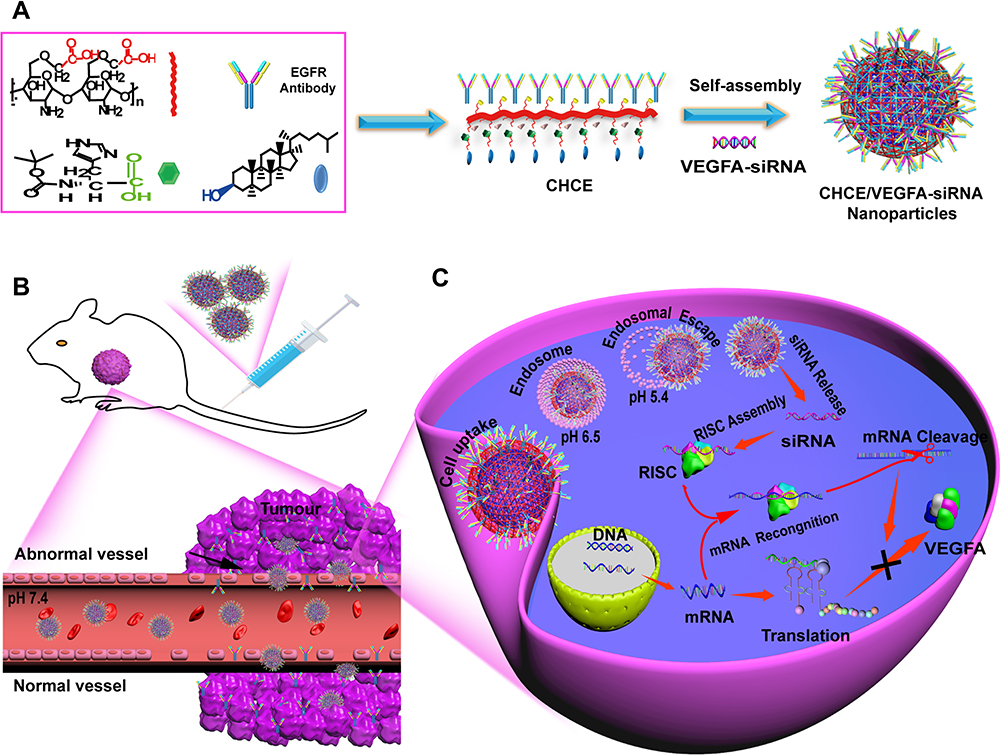 |
Figure 1 The preparation and siRNA delivery of CHCE/siRNA NPs. (A) Synthesis of CHCE/siRNA nanoparticle. Nanocarrier backbone carboxymethyl chitosan (red), cholesterol (blue), and helper histidine (Green). The CHCE forms stable NPs through self-assembly. (B) The CHCE/siRNA NPs target delivery to the tumor. (C) The endosomal escape and RNAi-induced gene silencing. Abbreviation: RISC, RNA-induced silencing complex. |
Materials and Methods
Materials
Breast cancer MDA-MB-231 cells and malignant melanoma SK-MEL-28 cells were purchased from American Type Culture Collection (ATCC). MDA-MB-231 and SK-MEL-28 cells were cultured in DMEM supplemented with 10% FBS and 100 units mL−1 penicillin and streptomycin. All cells were incubated at 37 °C with 5% CO2. The C57BL/6 mice and CD1 nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Ethical approval was obtained before the research from the Zhengzhou University Animal Ethics Committee (ZZU-LAC2020070703). The siRNAs and the FITC-siRNA were purchased from Shanghai GenePharma Co., Ltd. The siRNA sequences of VEGFA are shown as follows: sense 5’-GCAGCUACUGCCAUCCAAUTT-3; antisense 5’-AUUGGAUGGCAGUAGCUGCTT-3. The negative control siRNAs sequences are shown as follows: sense 5’-UUCUCCGAACGUGUCACGUTT-3; antisense 5’-ACGUGACACGUUCGGAGAATT-3.
Synthesis of CHCE/siRNA NPs
The CHCE was synthesized as per the method described in the Supplementary Methods section. The infrared spectra of CHCE were shown in Supplementary Figure 1. 10 mg CHCE was dissolved in 5 mL HEPES buffer (20 mM, pH 7.4). Then, a probe-type ultrasonic processor is used for ultrasonic treatment at 100 W for 2 minutes, and the ultrasonic treatment step is repeated three times. To prevent the sample solution from accumulating heat during ultrasonic treatment, the pulse function is 2s on and 2s off. Mix a certain concentration of CHCE solution and siRNA solution according to a certain mass ratio (8:1) at 4°C for 30 min to synthesize the multifunctional CHCE/siRNA nanoparticles.The siRNA encapsulation efficiency and siRNA loading content of the CHCE/siRNA NPs were detected in Supplementary Table 1.
Characterization of CHCE/siRNA NPs
The particle size and Zeta potential of CHCE/siRNA NPs were determined by dynamic light scattering (DLS) using a Zetasizer Nano ZS90 (Malvern Instruments, U.K). The CHCE/siRNA NPs were diluted in phosphate buffer solution, the concentration of CHCE/siRNA NPs was 1 mg/mL, the test temperature was 25°C.
The morphology of CHCE/siRNA NPs was measured by scanning electron microscope (SEM). Drop 2 microliters of the solution on the silicon wafer, let it dry naturally, then spray gold for 10 sec and observe it under a scanning electron microscope.
The CHCE/siRNA NPs were incubated in 50% mouse serum at 37°C. Collect 5 μL aliquots at different time points (5 min, 3 h, 6 h, 12 h, 24 h) and probe on a 1% agarose gel with nucleic acid dye. The fluorescence imaging system obtained the gel images, and the dissociated siRNA is positioned and the pure siRNA band (the second lane). The siRNA dissociation was analyzed by ImageJ image processing software.
Targeted Adhesion of CHCE/siRNA NPs
1×105 MDA-MB-231 cells with low expression of EGFR (EGFR-) and SK-MEL-28 cells with high expression of EGFR (EGFR+) cells23 were inoculated with PBS, pure siRNA, CHC/siRNA, and CHCE/siRNA (the siRNA were modified with FITC, and the siRNA concentration in each group was 1.6 μg/mL) were incubated with MDA-MB-231 cells and SK-MEL-28 cells for 1 h at room temperature. Collect the cells and the binding efficiency of CHCE/siRNA NPs to cells was detected by flow cytometry. The cells treated with PBS were used as blank control. The inhibitory effect of Panitumumab on the binding between CHCE/siRNA NPs and SK-MEL-28 cells were detected according to the following method: The SK-MEL-28 cells were cultured with different concentrations of Panitumumab and a fixed concentration of CHCE/siRNA NPs for 1 h at room temperature (the siRNA was modified with FITC, the siRNA concentration was 1.6 μg/mL, the Panitumumab concentration were 2.4 μg/mL, 1.2 μg/mL, 0.6 μg/mL, 0.3 μg/mL, 0 μg/mL). The inhibitory effects of Panitumumab were detected by flow cytometry. For laser scanning confocal microscope detection, fix with 4% paraformaldehyde for 5 min, then stained with DAPI (Invitrogen) for 10 minutes. The inhibitory effect of Panitumumab was detected by a laser scanning confocal microscope.
The Cellular Uptake of CHCE/siRNA NPs
1×105 SK-MEL-28 cells were cultured with CHCE/siRNA NPs (the siRNA concentration was 1.6 μg/mL) for a different time (0 min, 15 min, 30 min, 1 h, 2 h, 4 h), then collected the cells and flow cytometry detected the cellular uptake efficiency of CHCE/siRNA NPs. The cells untreated were used as blank control. For laser scanning confocal microscope detection, fix with 4% paraformaldehyde for 5 min and stain with DAPI for 10 minutes. Then laser scanning confocal microscope detected the cellular uptake efficiency of CHCE/siRNA NPs.
The Endosomal Escape of CHCE/siRNA NPs
1×105 SK-MEL-28 cells were cultured with CHCE/siRNA NPs (the siRNA concentration was 1.6 μg/mL) for a different time (30 min, 1 h, 2 h, and 4 h). The endosomes were labeled with LysoTracker-Red (Invitrogen) for 5 min. Then fix with 4% paraformaldehyde for 5 min and stained with DAPI for 10 min. Finally, the intracellular delivery and endosomal escape of CHCE/siRNA NPs in SK-MEL-28 cells were detected by laser scanning confocal microscope. The corresponding colocalization fluorescence intensity profiles and the corresponding colocalization ratios of siRNA (green) and endosomes (red) were analyzed by ZEN 3.0 software.
Gene Silencing of CHCE/siRNA NPs
2×105 SK-MEL-28 cells were cultured with PBS, CHCE/nc-siRNA, siRNA, and CHC/siRNA, CHCE/siRNA, PEI/siRNA (the siRNA concentration in each group was 1.6 μg/mL) for 48 h, respectively. Then the cells were collected to extract the total RNA and protein, respectively. The gene silencing of CHCE/siRNA NPs was detected by qPCR and Western blot. The detailed qPCR and Western blot scheme are in the Supplementary Material.
Cell Proliferation and Apoptosis Detection
For Cell proliferation, 1×104 SK-MEL-28 cells were seeded into 96-well microtiter plates and cultured for 24 hours for attachment. The PBS, CHCE/nc-siRNA, siRNA, and CHC/siRNA, CHCE/siRNA, PEI/siRNA NPs (the siRNA concentration in each group was 1.6 μg/mL) were added into different wells in triplicate. After 48 h of incubation, the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)- 2-(4-sulfophenyl)- 2H-tetrazolium) solution (Sigma) was added to each well and the plates were incubated at 37°C for an additional 1 h. The optical densities were recorded at 490 nm using a microplate reader. Cell viability was calculated as a percentage of PBS.
For Apoptosis detection, 2×105 SK-MEL-28 cells were cultured with PBS, CHCE/nc-siRNA, siRNA, and CHC/siRNA, CHCE/siRNA, PEI/siRNA (the siRNA concentration in each group was 1.6 μg/mL) for 48h in 5% CO2 at 37°C, respectively. The cells were collected and 5 μL AnnexinV- FITC solution and 5 μL PI dyeing solution (Nanjing Vazyme Biotech Co., Ltd) were added to react for 30 min at room temperature away from light. 400 μL of binding buffer was then added and detected by flow cytometry within 1 h. The negative control was used without Annexin V-FITC and PI.
Pharmacokinetic Study
The siRNA, CHC/siRNA, and CHCE/siRNA nanoparticles (The siRNA was labeled with FITC) were injected into the body at the same siRNA dose per nude mouse through the tail vein. Blood samples were collected from the venous plexus of the naked eye of the mouse at the 0 h, 4 h, 8 h, 12 h, 16 h, 20 h, and 24 h time points and placed in sample tubes pretreated with sodium heparin-centrifuge at 2000 rpm for 10 mins at 4°C. An equal amount of plasma samples was collected. FITC-labeled siRNA at a standard concentration was used as a control. Pharmacokinetic studies were performed by measuring the blood siRNA concentration in nude mice using a fluorescence optical protractor.
The Biodistribution and Antitumor of CHCE/siRNA NPs
Harvest SK-MEL-28 cells and dilute the cells with serum-free MEM medium at the appropriate concentration 5×107 cells/mL; Inject 1×107 cells in 200 μL subcutaneously into 5–6 weeks old Nude mice. For in vivo biodistribution studies, the siRNA, CHC/siRNA, and CHCE/siRNA (the siRNA in each group was 8 μg, total volume of 250 µL) were administrated into mice bearing tumors (400–600 mm3) via tail vein injection. Images were taken on an IVIS imaging system at 1 h, 10 h, and 24 h post-injection. The tumors and other major organs were collected for ex vivo imaging. Then the tissue sections were prepared and were observed by optical fluorescence microscopy. For antitumor effect studies, when the tumor sizes reach 100 mm3, regroup the animals; The PBS, siRNA, CHC/siRNA, PEI/siRNA, and CHCE/siRNA were administrated into mice bearing tumors (100 mm3) via tail vein injection. Intravenous injections were repeated 4 times, 8 μg siRNA each time, a total of 32 μg siRNA. The volume and weight of nude mice were measured monitored once every three days. The tumor volume was measured by the formula V = LW2/2. L and W represent the length and width of the tumor, respectively. The tumors and main organs were collected, including the heart, liver, spleen, lung, and kidney. The tissue sections were obtained by paraffin embedding, and hematoxylin-eosin stain (H&E) and Immunohistochemistry (IHC) were performed. Observe with a fluorescence microscope.
Potential Immune Responses of CHCE/siRNA NPs
The PBS, siRNA, CHC/siRNA, PEI/siRNA, and CHCE/siRNA were administrated into C57BL/6 mice via tail vein injection. Then the mice were sacrificed, and blood samples were collected after 24h and 48h intravenous administrations. The plasma was isolated for cytokine ELISA measurements of IL-1, IL-6, IFN-γ, and TNF-α (MultiSciences (Lianke) Biotech Co., Ltd).
Statistical Analyses
All experiments were replicated over three times, and the resulting data were represented mean ± s.e.m. The resulting data were statistically analyzed by independent samples t-test. The values are significantly different (p < 0.05). All analyses were done by using GraphPad Prism software (version 5).
Results
Preparation and Characterization of CHCE/siRNA NPs
We first synthesized the CHCE, and the infrared spectroscopy signature indicated that the CHCE has been successfully synthesized (Supplementary Figure 1). The CHCE/siRNA NPs were formed through CHCE and siRNA by self-assembly (Figure 2A). The lower critical micelle concentration of CHCE (0.1 mg/mL) indicated the CHCE owned excellent self-assembling ability to form spheroids (Figure 2B). Analyzing the mechanism of self-assembly into a ball may be that the amino acid cholesteryl ester can form a hydrophobic domain through the hydrophobic force and promote the long-chain carboxymethyl chitosan polymer in the stretched state to curl to form a hydrophilic shell.24 Scanning electron microscope results confirmed the formation of stable spherical particles with a high degree of dispersion and a narrow particle size distribution (Figure 2C and D). The above results confirmed that we have successfully engineered a novel nanoparticle delivery system for siRNA with a uniform and stable physical structure.
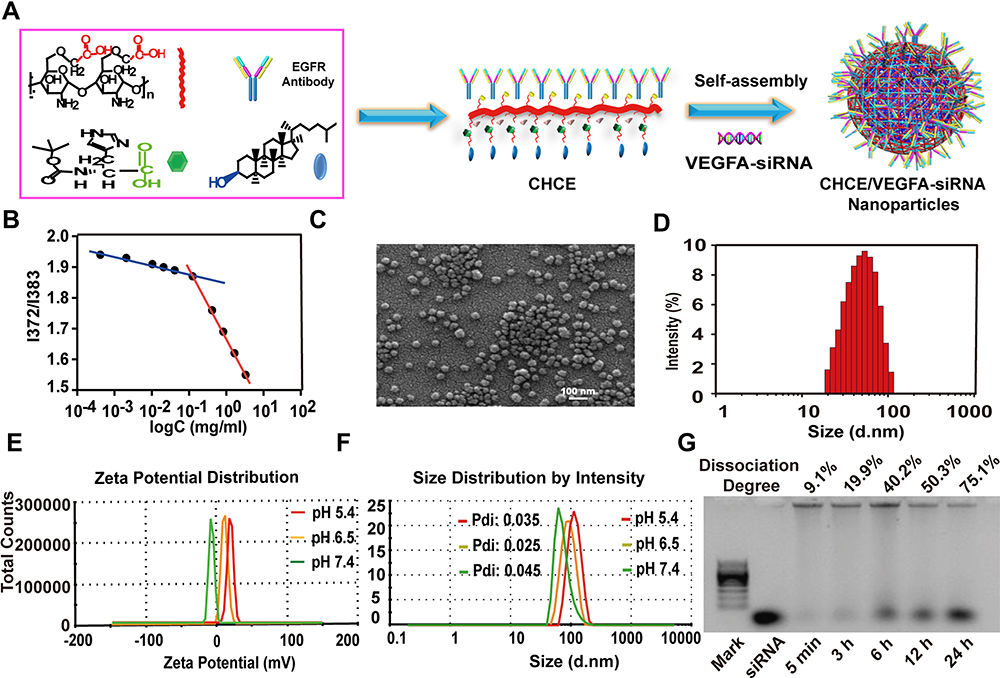 |
Figure 2 Preparation and characterization of CHCE/siRNA NPs. (A) Synthesis of CHCE/siRNA nanoparticle. (B) The relationship between the intensity ratio (I372/I385) from excitation spectra of pyrene and the logarithm of the concentration of CHCE. (C) SEM image of CHCE/siRNA NPs in distilled water. (D) The size distribution of CHCE/siRNA NPs in SEM image. (E) The zeta potential distribution of CHCE/siRNA NPs in PBS buffers with different pH. (F) The size distribution of CHCE/siRNA NPs in PBS buffers with different pH. (G) The stability of CHCE/siRNA NPs in 50% serum at 37°C was detected by the electrophoresis gel. The dissociated siRNA is positioned against the mark band and the pure siRNA band. |
PH-Responsive Behaviors of CHCE/siRNA NPs
The Zeta potential of CHCE/siRNA NPs was measured in different pH buffer solutions (7.4, 6.5, and 5.4). The results indicated that in neutral (or basic conditions, pH > 7), the Zeta potential maintained an anionic surface, once the pH reverses toward acidic conditions (pH≤6.5), amine groups on histidine were protonated leading to the positively charged surface and the lower the pH value, the higher the positive charge on the surface (Figure 2E). Although the particle size of CHCE/siRNA NPs increased with the pH decreasing, the polydispersity index (PDI) showed that the CHCE/siRNA NPs had good stability at different pH levels (Figure 2F). Besides, the results of the electrophoresis gel showed that the release of the cargo siRNA from CHCE/siRNA NPs was slow with better stability (Figure 2G). All the above characterization of CHCE/siRNA NPs indicated that our novel siRNA delivery vehicle has a stable physical structure, pH-responsive particle charges.
Tumor Adhesion of CHCE/siRNA NPs in vitro
An active targeting strategy of modifying with EGFR monoclonal antibody was adopted to improve the tumor adhesion ability of CHCE/siRNA NPs.25 The results showed that the CHCE/siRNA NPs target SK-MEL-28 cells (high expression of EGFR) efficiency with 90% of the cells labeled by the fluorescence, while the MDA-MB-231 cells (low expression of EGFR) was only about 5% (Figure 3A and B). To further confirm that the targeting specificity of CHCE/siRNA NPs was via EGFR, the Panitumumab, which bonded to EGFR and prevented EGFR monoclonal antibody from EGFR,26 was used to incubate sk28 cells with CHCE/siRNA NPs. The results indicated that the Panitumumab could well inhibit the binding between CHCE/siRNA NPs and SK-MEL-28 cells, a dose-dependent inhibition of CHCE/siRNA NPs binding by SK-MEK28 cells, and that the Panitumumab could well inhibit the binding between CHCE/siRNA NPs and SK-MEL-28 cells (Figure 3C). The laser confocal microscope images also showed an apparent reduction of the binding of CHCE/siRNA NPs to SK-MEL-28 cells with an increase of Panitumumab concentration (Figure 3D). Taking these findings into account, we concluded that the CHCE/siRNA NPs could specifically target and bind to the tumor cells.
 |
Figure 3 EGFR-mediated CHCE/siRNA specific binding to tumor cells. (A) Flow cytometry detected the binding efficiency of CHCE/siRNA. (B) The binding efficiency of CHCE/siRNA. *P<0.05 (two-tailed t-test). NS: no significant difference. (C) Flow cytometry and (D) Laser scanning confocal microscope detected the inhibitory effect of panitumumab on the binding between CHCE/siRNA NPs and SK-MEL-28 cells. Scale bar, 10 µm. |
Cellular Uptake and Endosomal Escape of CHCE/siRNA NPs
For the cellular uptake of CHCE/siRNA NPs, the results showed that compared with the simple siRNA, both CHC/siRNA NPs and CHCE/siRNA NPs could enter the cytoplasm, and the most CHCE/siRNA NPs entered the cytoplasm (Supplementary Figure 2). The cellular uptake of CHCE/siRNA NPs was concentration-dependent, and the cell uptake efficiency reached the maximum when the siRNA concentration was 1.6ug/mL (Supplementary Figure 3). The cellular uptake of CHCE/siRNA NPs was also time-dependent, and the longer the time, the higher the cell uptake efficiency (Figure 4A). Interestingly, the CHCE/siRNA NPs are only attached to the cell surface at 30 minutes of incubation. As the time prolonged, more CHCE/siRNA NPs were internalized via endocytosis and accumulated in the intracellular vesicles (Figure 4B). For the endosomal escape of CHCE/siRNA NPs, the results showed the CHCE/siRNA NPs bound to the cell surface at 30 min, accumulated in the endosomes at 1h-2h, and separated from the endosomes again at 4h (Figure 4C). The corresponding colocalization fluorescence intensity profiles and the corresponding colocalization ratio indicated that the CHCE/siRNA NPs have successfully escaped from the endosomes (Figure 4D and E).
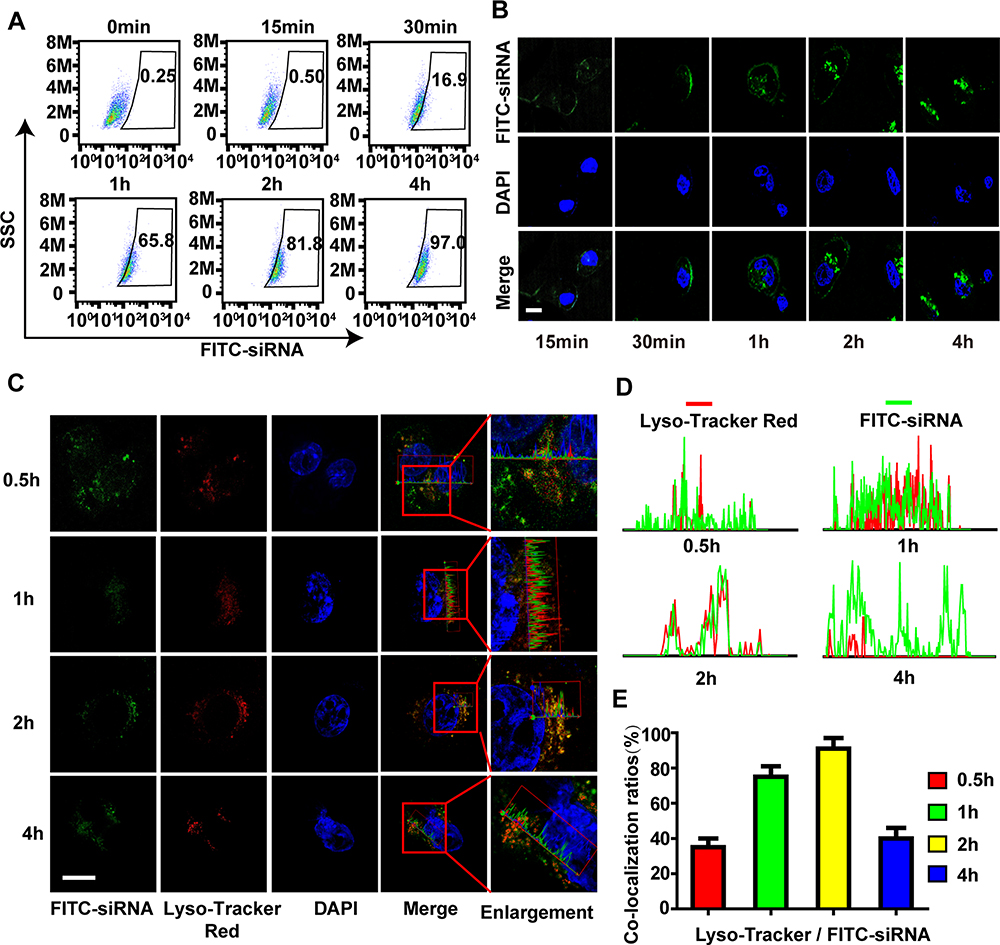 |
Figure 4 The cellular uptake and endosomal escape of CHCE/siRNA NPs. (A) Flow cytometry and (B) Laser scanning confocal microscope detected the cellular uptake efficiency of CHCE/siRNA NPs. Scale bar, 10 µm. (C) A laser scanning confocal microscope detected the endosomal escape of CHCE/siRNA NPs. (the siRNA were modified with FITC, the endosomes were labeled with LysoTracker). Scale bar, 10 µm. (D) The corresponding colocalization fluorescence intensity profiles of siRNA (green) and endosomes (red) in SK-MEL-28 cells after CHCE/siRNA NPs treatment for different times. (E) The corresponding colocalization ratios of siRNA (green) and endosomes (red). |
Gene Silencing of CHCE/siRNA NPs
The qPCR results indicated that, compared with other treatments, the CHCE/siRNA NPs showed a marked downregulation of VEGFA mRNA (Figure 5A). The Western blot results also further verify the silencing efficiency of CHCE/siRNA NPs (Figure 5B). Some researchers have demonstrated that VEGF knockdown suppresses tumor cells proliferation.27 To assess the cytotoxicity of the CHCE/siRNA NPs, the proliferation of SK-MEL-28 cells treated was tested by MTS. The results showed that the CHCE/siRNA NPs had the best anti-proliferation effect on the SK-MEL-28 cells (Figure 5C). Some researchers have also demonstrated that downregulation of VEGF could promote apoptosis.28,29 The apoptosis results showed that CHCE/siRNA NPs could induce cell apoptosis with higher efficacy than other treatment groups (Figure 5D). To summarize, the above results clearly showed that the CHCE/siRNA NPs mediated the effective gene silencing of VEGFA mRNA, which induced the cell apoptosis, and finally had a good anti-proliferation effect.
 |
Figure 5 The gene silence of CHCE/siRNA NPs in vitro. The PBS was the blank control group, the CHCE/nc-siRNA was the negative control group, and the PEI/siRNA was the positive control group. (A) The qPCR detected the VEGFA mRNA expression in tumor cells. (mean ± sem, n = 3). *p < 0.05 (two-tailed t-test). (B) Western blot detected the VEGFA protein expression. GAPDH was used as a reference protein. The band intensity of VEGFA and GAPDH expression was measured with ImageJ. The relative ratio was calculated with the intensity of VEGFA divided by the intensity of GAPDH in the same treatment group. (C) The cell viability of CHCE/siRNA NPs was detected by MTS (mean ± sem, n = 3). * p <0.05, (two-tailed t-test). NS: no significant difference. (D) Cell apoptosis was detected by Annexin V and PI staining. The lower left, lower right, upper right, and upper left quadrants represent cell survival, early apoptosis, late apoptosis, and necrosis. |
Biodistribution of CHCE/siRNA NPs
The fluorescence reflectance imaging showed that CHC/siRNA NPs and CHCE/siRNA NPs maintained high concentrations at 10 and 24 hours post-injection, while most of the naked siRNA was cleared at 10 h post-injection (Figure 6A). The pharmacokinetic results also showed that the CHC/siRNA NPs and CHCE/siRNA NPs could effectively prolong the circulation time of siRNA, and the naked siRNA was fast cleared from the circulation (Figure 6B). The above results indicated that the CHC/siRNA NPs and CHCE/siRNA have good stability and protect siRNA from ribozyme degradation. In this paper, an active targeting strategy was adopted to improve the tumor-targeted delivery capability of CHCE/siRNA NPs. The fluorescence reflectance imaging showed that CHCE/siRNA accumulated in the tumor with a higher concentration than CHC/siRNA (Figure 6A black arrow). The imagines of various organs and tumors further showed that compared with the simple siRNA and CHC/siRNA NPs, the CHCE/siRNA NPs had a higher accumulation in the tumor and organs (Figure 6C and D). Besides, the histology sections also proved that the CHCE/siRNA NPs penetrated the tumor tissue with a higher concentration (Figure 6E and F). Taking these findings into account, we concluded that CHCE/siRNA NPs prolong the circulation of siRNA and actively target the tumor.
 |
Figure 6 Biodistribution of CHCE/siRNA in vivo. (A) Whole-body NIR fluorescence imaging of mice bearing tumors. (B) Pharmacokinetic studies were performed by measuring the blood siRNA concentration in vivo. (mean ± sem, n = 3). *p < 0.05 (two-tailed t-test). (C) Ex vivo imaging of tumors and other major organs. (D) Quantitative analysis of CHCE/siRNA in tumors and other major organs by the ex vivo fluorescence signals. *P < 0.05 (two-tailed t-test). (E) The tumor penetration of CHCE/siRNA. Green, FITC-labelled siRNA. Scale bar, 50 µm. (F) The green fluorescent signal intensity of CHCE/siRNA. *p < 0.05 (two-tailed t-test). |
Anti-Tumor Effect of CHCE/siRNA NPs
Considering that CHCE/siRNA NPs have better tumor-targeted delivery capabilities and longer circulation time in vivo, we further tested the anti-tumor effect of CHCE/siRNA NPs in vivo (Figure 7A). The results showed that the CHCE/siRNA NPs had a better ability to inhibit tumor growth (Figure 7B and Supplementary Figure 4A). The changes in body weight and tumor mass also showed that the CHCE/siRNA NPs had better anti-tumor effects and low side effects (Figure 7C and D). The anti-tumor effect of CHCE/siRNA NPs was also proved by the tumor histopathology with sparse and disordered tumor structure (Figure 7E). The low side effects were further proved by the organ’s histopathology with the complete tissue structure and the normal cell morphology (Supplementary Figure 4B). The tumor immunohistochemistry showed that the CHCE/siRNA NPs could reduce the expression level of VEGFA protein in tumor tissues (Figure 7E), which was the reason that inhibited tumor growth. Finally, the inflammatory response of CHCE/siRNA NPs was detected after injection 24h and 72h. The results showed that the CHCE/siRNA NPs did not induce any inflammatory response (Figure 7F-I). The above results indicated that the CHCE/siRNA could effectively inhibit tumor growth with low side effects.
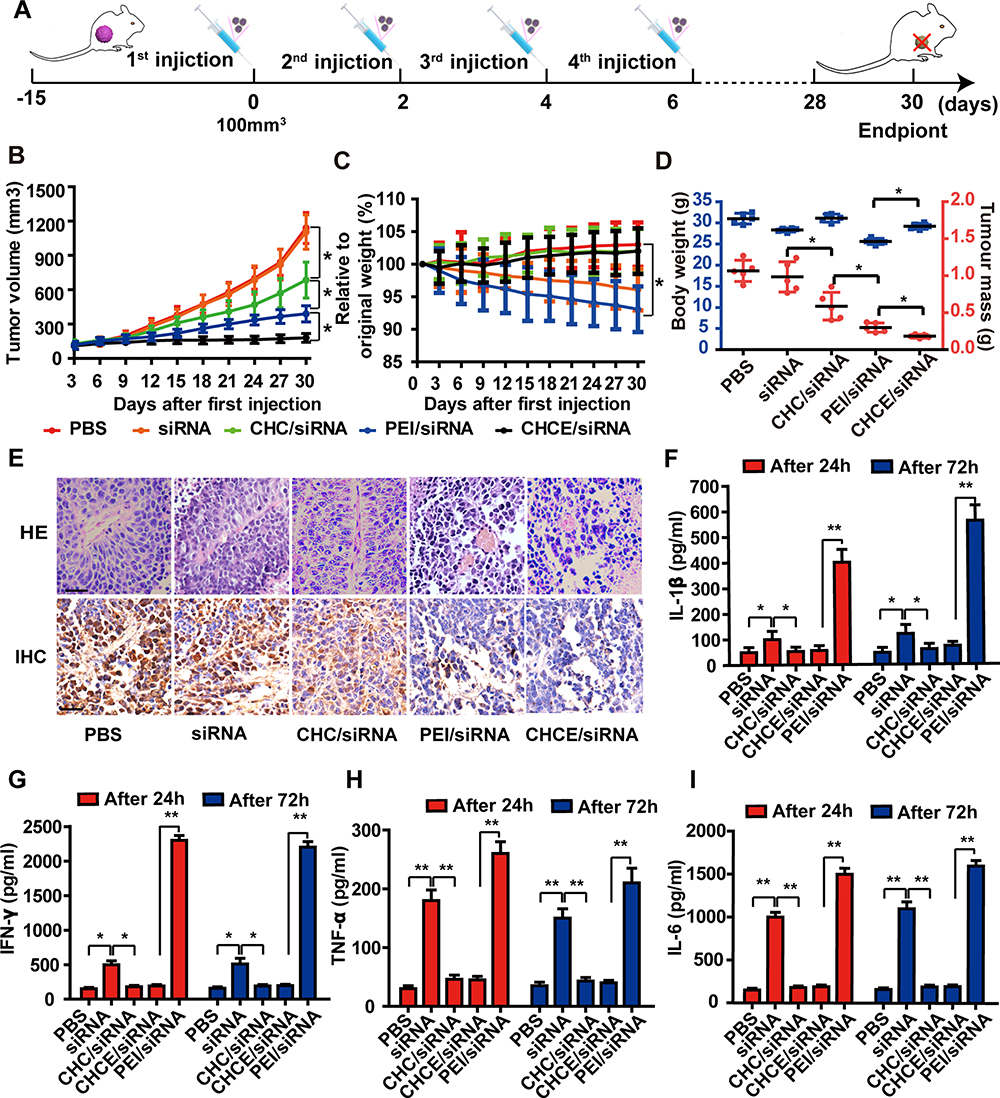 |
Figure 7 The antitumor effect of CHCE/siRNA NPs in vivo. (A) The treatment schemes. (B) The growth curves of tumors with various siRNA agents. The PEI/siRNA was the positive control group. (mean ± s.e.m., n=5). *p < 0.05, (two-tailed t-test). (C) The change in body weight after treatments (mean ± s.e.m., n=5). *p < 0.05, (two-tailed t-test). (D) The tumor volume and the tumor weight at the end of treatment. (mean ± s.e.m., n=5). *p < 0.05, (two-tailed t-test). (E) H&E and IHC showing antitumor effect. Scale bar, 50 µm. (F–I) Potential immune responses after treatments were detected by ELISA (data represent mean ± s.e.m., n = 3), *p < 0.05, **p < 0.01 (two-tailed t-test). |
Discussions
The pH-Responsiveness Promotes CHCE/siRNA NPs Breakthrough Biological Barriers at All Levels
It is crucial to evaluate the effect of the surrounding environment on the particles’ critical characteristics as the surface charge directly affects the delivery efficiency and circulation time in vivo.30 Considering that the high metabolic rate of cancer cells and the hypoxic tumor microenvironment generate a lower pH in the tumor microenvironment comparing normal tissues,31 we incorporated histidine due to its reversible protonation on ionizable amines in the imidazole group, resulting in the pH-responsiveness of the particles’ charges. Therefore, the CHCE/siRNA NPs would present as anionic particles in the blood circulation system and cationic particles in tumor tissue, prolonging circulation time and averting rapid clearance. NPs cannot enter the cells to deliver siRNA via passive diffusion, and cell entry will depend on cellular endo/phagocytic mechanisms.32 Factors affecting the cellular uptake efficiency of NPs include the membrane compatibility, surface charge, cellular mechanisms mediating uptake and microenvironment, etc.33 The hypoxic and low pH in the tumor microenvironment would trigger histidine protonation, inducing a positively charged surface on the CHCE/siRNA NPs, facilitating binding to the negatively charged cell membrane.
Moreover, as a component of the cell membrane, cholesterol also makes nanocarriers more biocompatible with the cell membrane, facilitating cellular absorption.34 The pH-responsiveness protonation can promote the lysosomal escape of CHCE/siRNA NPs. As a critical biological barrier with extremely low pH, lysosomes can rapidly degrade nanocarriers and strongly efflux. Previous studies have found that histidine with reversible protonation elicits the proton sponge effect, which causes the lysosome to swell, rupture, and finally release the NPs.35 The pH-responsive protonation could also endow the CHCE/siRNA NPs with better release ability, and its pH sensitivity mainly comes from the protonation of the imidazole and amine groups of histidine. The greater the degree of protonation, the greater the charge on the internal groups, resulting in increased electrostatic repulsion between groups and greater swelling.36 The hydrophobic effect of cholesterol can also promote the swelling transition of nanocarriers, which can be completed in a narrow pH range.37 In this study, we did not specifically design experiments to measure how siRNA is released from CHCE/siRNA NPs, but studies by other researchers have demonstrated that changes in pH can cause siRNA to be released from NPs.38 Because our design resulted in a marked change in the pH of the nanoparticles in the endosome, we speculate that the persistence of their low pH also contributed to their release from the nanoparticles. However, the specific release mechanism needs further study. In conclusion, the pH-responsiveness promotes CHCE/siRNA NPs breakthrough biological barriers at all levels and promotes releasing of cargo siRNA into the cytosol of the target cells.
Tumor Targeting Can Increase the Use Rate of siRNA in Tumors
Previous studies have shown that the utilization rate of existing drugs is only 0.7%, and more than 99% of drugs are wasted in the body, producing strong adverse effects and low therapeutic efficacy.18 To improve the tumor-targeting delivery, an active targeting strategy of modifying the EGFR monoclonal antibody was adopted.25 The surface modification with EGFR monoclonal antibody enhances the specific adhesion of CHCE/siRNA NPs and triggers receptor-mediated endocytosis in conjunction with corresponding receptor processes to increase cell uptake efficiency.39 The experiments also have confirmed that CHCE/siRNA NPs had the better tumor adhesion and tumor targeting delivery ability. However, although the active targeting strategy is a promising approach for the delivery in vivo, targeted modification still faces many problems so far.40 The main problem is that most of the so-called specific antigens in tumor tissues are not completely specific and slightly expressed in other normal tissues. Although scientists have worked hard to explore a variety of tumor targets, these targets are only tumor-related rather than tumor-specific.41–43
The second problem is that some studies have shown that targeted modified NPs would bind to various protein factors in the blood circulation system and form a “protein halo” on the surface of the nanocarrier complex.44 This “protein halo” could prevent the NPs from specifically binding to tumor receptors, resulting in the loss of targeting effect of the NPs.45 In addition, the heterogeneity of tumor cells leads to the expression of different tumor-specific receptors on the tumor surface, which also brings severe challenges to the targeted modification of NPs.46 Although nanocarrier targeted modification still faces many problems in its fabrication and in vivo application, fortunately, the successful clinical application of monoclonal antibody-drug conjugates provides a good demonstration for nanocarrier targeted modification.47 We believe that nanocarriers will gain efficiently targeted delivery capabilities with the resolution of various problems.
Knockdown of VEGFA Can Effectively Inhibit the Growth of Blood Vessels and Tumor Growth
The abnormal tumor vessels supply oxygen and nutrients and remove waste products and promote abnormal tumor microenvironment by impairing perfusion.48 The tumor microenvironments own hypoxia, low pH, and high interstitial pressure.49,50 This specific tumor microenvironment not only reduces the delivery efficiency of various therapeutic drugs but also inhibits the activity of the delivered payloads, resulting in poor therapeutic effects and even drug resistance.51 Among the various factors supporting tumor microenvironment growth, the protein VEGFA is crucial for promoting abnormal blood vessels’ rapid and disordered growth and the unlimited growth of tumor cells.52 Many experiments have proved that as long as the expression of the VEGFA gene is reduced in tumors, it can inhibit angiogenesis growth.53,54 Moreover, the VEGFA can also potentiate the metastasis of metastatic cancer cells in the blood vessels to distant organs.55 Therefore, silencing the expression of VEGFA by siRNA can potentially inhibit tumor angiogenesis and thus change tumor microenvironment and inhibit tumor growth. The experiment results indicated that CHCE/siRNA NPs could effectively knock down the expression of VEGFA and inhibit tumor growth, which also powerfully proved that the CHCE/siRNA NPs could breakthrough biological barriers at all levels and promote the siRNA to perform corresponding biological functions. Although CHCE/siRNA NPs had an excellent inhibitory effect on tumor growth, the tumor was not eliminated. Therefore, instead of single siRNA delivery, our focus is shifted to simultaneous delivery of multiple siRNA of oncogenes, bringing synergistic effects and increasing the opportunity to overcome cancer completely. In addition, RNA interference technology can also be combined with radiotherapy and chemotherapy for standard anti-tumor treatment to complete cancer treatment.
Conclusions
In short, we optimized and designed a novel delivery system that could simultaneously accommodate all the requirements of breaking through all biological barriers and could target delivery siRNA to the tumor site and achieve a better therapeutic effect. We believe that the delivery system will provide exciting opportunities for RNAi technology.
Ethics Approval
We certify that all applicable institutional and governmental regulations concerning the ethical use of animals were followed during the course of this research. The experiments were approved by the Animal Care and Use Committee of Zhengzhou University Health Science Center.
Acknowledgments
The authors are sincerely grateful for financial support from the National Natural Science Foundation of China (Grant No. U1504527) and the Science and Technology Research Key Project of the Henan Science and Technology Commission (Grant No. CXJD2020001).
Disclosure
The authors report a patent siRNA Delivery System Compound as well as Preparation Method and Application thereof pending to Zhimin Wang, Chengshen Zhu, Wentao Liu, Xiangyang Zhang, Suqin He, Yaohe Wang, Hao Liu, Miaoming Huang, Chenglin Zhang, Bin Qin, and Lei Huang. The authors report no other potential conflicts of interest for this work.
References
1. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. doi:10.1038/35888
2. Quon K, Kassner PD. RNA interference screening for the discovery of oncology targets. Expert Opin Ther Targets. 2009;13(9):1027–1035.
3. Freeman EC, Weiland LM, Meng WS. Modeling the proton sponge hypothesis: examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J Biomater Sci Polym Ed. 2013;24(4):398–416.
4. Dykxhoorn DM, Palliser D, Lieberman J. The silent treatment: siRNAs as small molecule drugs. Gene Ther. 2006;13(6):541–552.
5. Heidi L. Gene-silencing technology gets first drug approval after 20-year wait. Nature. 2018;1:45.
6. Haupenthal J, Baehr C, Kiermayer S, Zeuzem S, Piiper A. Inhibition of RNAse A family enzymes prevents degradation and loss of silencing activity of siRNAs in serum. Biochem Pharmacol. 2006;71(5):702–710.
7. Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28(4):181–188.
8. Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12(11):967–977.
9. Santo D, Mendonça PV, Lima MS, Cordeiro RA. Poly(ethylene glycol)- block-poly(2-aminoethyl methacrylate hydrochloride)-Based Polyplexes as Serum-Tolerant Nanosystems for Enhanced Gene Delivery. Int J Med. 2019;16(5):2129–2141.
10. Shrivats AR, Hsu E, Averick S, et al. Cationic Nanogel-mediated Runx2 and Osterix siRNA Delivery Decreases Mineralization in MC3T3 Cells. Clin Orthop Relat Res. 2015;473(6):2139–2149. doi:10.1007/s11999-014-4073-0
11. Siegwart DJ, Whitehead KA, Nuhn L, et al. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc Natl Acad Sci U S A. 2011;108(32):12996–13001. doi:10.1073/pnas.1106379108
12. Averick SE, Paredes E, Dey SK, et al. Autotransfecting short interfering RNA through facile covalent polymer escorts. J Am Chem Soc. 2013;135(34):12508–12511. doi:10.1021/ja404520j
13. Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat Nanotechnol. 2007;2(8):469–478.
14. von Roemeling C, Jiang W, Chan CK, Weissman IL, Kim BYS. Breaking Down the Barriers to Precision Cancer Nanomedicine. Trends Biotechnol. 2017;35(2):159–171.
15. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664.
16. Arvizo RR, Miranda OR, Moyano DF, et al. Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PLoS One. 2011;6(9):e24374.
17. Yue ZG, Wei W, Lv PP, et al. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules. 2011;12(7):2440–2446.
18. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Materials. 2016;1(5):16014.
19. Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515.
20. Dash PR, Read ML, Barrett LB, Wolfert MA, Seymour LW. Factors affecting blood clearance and in vivo distribution of polyelectrolyte complexes for gene delivery. Gene Ther. 1999;6(4):643–650.
21. Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823.
22. Riviere K, Kieler-Ferguson HM, Jerger K, Szoka FC. Anti-tumor activity of liposome encapsulated fluoroorotic acid as a single agent and in combination with liposome irinotecan. J Controlled Release. 2011;153(3):288–296.
23. Subik K, Lee JF, Baxter L, et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer (Auckl). 2010;4:35–41.
24. Akiyoshi K, Deguchi S, Moriguchi N, Yamaguchi S, Sunamoto J. Self-aggregates of hydrophobized polysaccharides in water. Formation and characteristics of nanoparticles. Macromolecules. 1993;26(12):3062–3068.
25. Mamot C, Drummond DC, Greiser U, Hong K, Park JW. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res. 2003;63(12):3154–3161.
26. Martinelli E, Palma RD, Orditura M, Vita FD, Ciardiello F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin Exp Immunol. 2010;158(1):96.
27. Park JH, Seo JH, Jeon HY, et al. Lentivirus-mediated VEGF knockdown suppresses gastric cancer cell proliferation and tumor growth in vitro and in vivo. Angewandte Chem Int Edition. 2020;13:1331–1341.
28. Ge YL, Zhang X, Zhang JY, Hou L, Tian RH. The mechanisms on apoptosis by inhibiting VEGF expression in human breast cancer cells. Int Immunopharmacol. 2009;9(4):389–395.
29. Zhao J, Zhang ZR, Zhao N. VEGF Silencing Inhibits Human Osteosarcoma Angiogenesis and Promotes Cell Apoptosis via PI3K/AKT Signaling Pathway. Cell Biochem Biophys. 2015;73(2):519.
30. He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666.
31. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2013;125(23):5591–5596.
32. Sriraman SK, Aryasomayajula B, Torchilin VP. Barriers to drug delivery in solid tumors. Tissue Barriers. 2014;2:e29528.
33. Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9(9–10):1521–1532.
34. Zhang J, Fan H, Levorse DA, Crocker LS. Interaction of cholesterol-conjugated ionizable amino lipids with biomembranes: lipid polymorphism, structure-activity relationship, and implications for siRNA delivery. Langmuir. 2011;27(15):9473–9483.
35. Boussif O. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92(16):7297–7301.
36. Pradny M, Kopeeek J. Hydrogels for site-specific oral delivery poly [(acrylic acid)-co-(butyl acrylate)] cross-linked with 4, 4bis (methacrsloylamino) azobenzene. MakromolekuelareChemie Macromolecular Chem Phvs. 1990;191(8):1887–1897.
37. Siegel RA, Firestone BA. pH-dependent equilibrium swelling properties of hydrophobic polyelectrolyte copolymer gels. Macromolecules. 1988;21:3254–3259.
38. Duan S, Yuan W, Fei W, Jin T. Polyspermine Imidazole‐4,5‐imine, a Chemically Dynamic and Biologically Responsive Carrier System for Intracellular Delivery of siRNA. Angewandte Chem Int Edition. 2021;51(32):7938–7941.
39. Henriksen L, Grandal MV, Knudsen SL, van Deurs B, Grøvdal LM. Internalization mechanisms of the epidermal growth factor receptor after activation with different ligands. PLoS One. 2013;8(3):e58148.
40. Kumari P, Ghosh B, Biswas S. Nanocarriers for cancer-targeted drug delivery. J Drug Target. 2016;24(3):179–191.
41. Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290.
42. Petrul HM, Schatz CA, Kopitz CC, et al. Therapeutic mechanism and efficacy of the antibody-drug conjugate BAY 79-4620 targeting human carbonic anhydrase 9. Mol Cancer Ther. 2012;11(2):340–349.
43. Fabricius EM, Wildner GP, Kruse-Boitschenko U, Hoffmeister B, Goodman SL, Raguse JD. Immunohistochemical analysis of integrins αvβ3, αvβ5 and α5β1, and their ligands, fibrinogen, fibronectin, osteopontin and vitronectin, in frozen sections of human oral head and neck squamous cell carcinomas. Exp Ther Med. 2011;2(1):9–19.
44. Tenzer S, Docter D, Kuharev J, et al. Rapid formation of plasma protein Corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013;8(10):772–781.
45. Salvati A, Pitek AS, Monopoli MP, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule Corona adsorbs on the surface. Nat Nanotechnol. 2013;8(2):137–143.
46. Pribluda A, de la Cruz CC, Jackson EL. Intratumoral Heterogeneity: from Diversity Comes Resistance. Clin Cancer Res. 2015;21(13):2916–2923.
47. Abdollahpour-Alitappeh M, Lotfinia M, Gharibi T, et al. Antibody-drug conjugates (ADCs) for cancer therapy: strategies, challenges, and successes. J Cell Physiol. 2019;234(5):5628–5642.
48. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–622.
49. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–4200.
50. Heldin C-H, Rubin K, Pietras K, Östman A. High interstitial fluid pressure — an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813.
51. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–1454.
52. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31(17):2205–2218.
53. Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S, Muramatsu T. A small interfering RNA targeting vascular endothelial growth factor as cancer therapeutics. Cancer Res. 2004;64(10):3365–3370.
54. Jia RB, Zhang P, Zhou YX, et al. VEGF-targeted RNA interference suppresses angiogenesis and tumor growth of retinoblastoma. Ophthalmic Res. 2007;39(2):108–115.
55. Hiratsuka S, Goel S, Kamoun WS, et al. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci U S A. 2011;108(9):3725–3730.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.