Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Dual GIP–GLP1-Receptor Agonists In The Treatment Of Type 2 Diabetes: A Short Review On Emerging Data And Therapeutic Potential
Authors Bastin M ![]() , Andreelli F
, Andreelli F
Received 12 May 2019
Accepted for publication 17 August 2019
Published 30 September 2019 Volume 2019:12 Pages 1973—1985
DOI https://doi.org/10.2147/DMSO.S191438
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Konstantinos Tziomalos
Marie Bastin, Fabrizio Andreelli
Diabetology-Metabolism Department, Sorbonne Université, ICAN, Assistance Publique Hôpitaux de Paris, Pitié-Salpêtrière Hospital, Paris, F-75013, France
Correspondence: Fabrizio Andreelli
Diabetology-Metabolism Department, Pitié-Salpêtrière Hospital, 47-83 Boulevard de l’Hôpital, Paris cedex 13, 75951, France
Email [email protected]
Abstract: The need for efficient and safe therapy to improve such metabolic diseases as obesity and type 2 diabetes mellitus is currently unmet. The development of dual GIPR–GLP1R coagonists that bind to either one or the other receptor (sequence-mixed dual agonists) has emerged as an innovative therapeutic strategy for obesity and type 2 diabetes. Combined activation of both receptors may act synergistically providing additive effects on glucose and body weight in comparison of GLP1 analogues alone. Preclinical studies have confirmed that GIPR–GLP1R coagonists improve several hallmarks of metabolic syndrome, such as obesity, hyperglycemia, and dyslipidemia. These metabolic benefits have been translated from mice to nonhuman primates and humans. Recent clinical trials have shown that coagonists induce significant benefits on body weight, fasting, and postprandial glucose levels, insulin sensitivity, and total cholesterol. Combined GIP- and GLP1R activators have the potential to become a treatment option for patients with type 2 diabetes.
Keywords: incretins, type 2 diabetes, coagonists, GLP1, GIP, body weight
Introduction
Obesity and type 2 diabetes (T2D) remain epidemic problems. Both diseases are known to reduce quality of life and lead to serious complications, such as cardiovascular events and microangiopathic alterations in eyes, kidneys, and peripheral nerves. Prospective studies, such as the UK Prospective Diabetes Study (UKPDS) demonstrated that T2D is a progressive disease that worsens over time, whatever the pharmacological management used to reduce daily glucose excursion.1 Therefore, T2D needs progressive therapeutic intensification targeting β-cells, as described in worldwide diabetes-management guidelines.2 Nevertheless, it has been found that despite eight available families of hypoglycemic compounds (biguanides, sulfonylureas, glinides, α-glucosidase inhibitors, DPPIV inhibitors, GLP1 analogues, thiazolidinediones, and SGLT2 inhibitors), <50% of patients with T2D (in France and other countries) reached an HbA1c level <7% (a target generally accepted as sufficient in most cases to reduce the risk of developing specific complications of this disease).3 This suggests strongly that the therapeutic management of T2D remains partly efficient and that innovation in the management of this disease is urgently needed.
Because T2D is a multifaceted disease, an emerging pharmacotherapy is the development of dual gut hormones to target multiple signaling pathways in a coordinated manner with a single molecular entity. This strategy may lead to superior metabolic action with fewer side effects compared to monotherapies. Until recently (and in contrast to GLP1-receptor [GLP1R] agonists), GIP had not been considered a suitable candidate for the treatment of T2D. This assumption was based on the observation that GIPR–/– mice are resistant to weight gain during an obesogenic diet, suggesting that GIPR antagonists (rather than agonists, as for GLP1R) could provide therapeutic benefit in this disease.4 In addition, incretin response to GIP is blunted in patients with T2D, limiting the rationale for its use as a hypoglycemic compound. Challenging these observations is the demonstration that GIPR agonists improve glucose tolerance and reduce body weight in preclinical studies (two well-known beneficial effects of GLP1R agonists). These new preclinical results raise the question of the possibility of cumulating metabolic benefits by a simultaneous activation of GIPRs and GLP1Rs. In this context, new dual-incretin peptides that display agonist activity on GLP1R and GIPR have been developed recently and demonstrated interesting beneficial effects on glucose homeostasis in preclinical evaluations and randomized clinical studies in humans.
Insulin-Secretion Deficiency As The Main Therapeutic Target In Type 2 Diabetes
T2D is a complex disease. A common pathogenic pathway appears to exist for most people with T2D. Since the pioneer work of Jean Vague in 1956, it is generally accepted that people with T2D who are obese and insulin-resistant have a more central distribution of fat excess.5 In comparison with subcutaneous adipose tissue, visceral fat is characterized by immune-cell infiltration, excess of secretion of inflammatory cytokines, and high lipolysis rate.6,7 This led to the concept that visceral fat accumulation not only promotes low-grade inflammation at the systemic level but also favors ectopic fat accumulation in insulin-target tissue, mainly the liver and skeletal muscle. Altogether, these mechanisms lead to the development of a reduction of insulin action in these tissue types, namely the insulin-resistance state. Early in T2D pathophysiology, insulin secretion increases to compensate for the development of insulin resistance. During this period, glucose homeostasis in fasting and fed periods remains in the normal range (prediabetic state). The reduction in insulin-secretion capacity subsequently observed during the evolution of the disease in individuals genetically destined to develop T2D is the main contributor to the rise in glucose levels. In addition, the UKPDS demonstrated that progressive reduction in insulin secretion during follow-up (rather than changes in insulin resistance) explained the deterioration of diabetes control over time.8 This has been replicated in the Belfast diet-intervention study, suggesting that β-cell failure is an important determinant not only for diabetes pathophysiology but also for its evolution.9 As a consequence, restoration of β-cellfunctionality by lifestyle modifications and/or pharmacological management has become an important therapeutic goal in T2D populations. Lifestyle modifications may promote substantial improvement of T2D (especially in early diabetes), as shown in the DIRECT study (Diabetes Remission Clinical Trial).10 The aim of this study was to assess whether intensive weight management (with a target of ≥15 kg weight loss) within routine primary care would achieve remission of T2D. Almost half the cohort with early T2D achieved remission (73% for ≥10 kg loss) at 12 months, and sustained remission at 24 months for more than a third of people with T2D was observed. In another study, Lim et al showed in eleven patients with T2D 8 weeks after an energy-restriction diet a normalization of both β-cell function and hepatic insulin sensitivity, suggesting that in some patients, β-cell dysfunction may be reversible.11 β-Cell dysfunction in T2D populations has been extensively studied. Insulin responses to an intravenous glucose challenge are abnormal in diabetes. First, it has been demonstrated that acute insulin response to glucose load is profoundly reduced or absent in most patients with T2D.12 It is interesting to note that alteration of the first phase of insulin secretion in T2D is specific for glucose, since β-cells can respond acutely to other secretagogues (arginine, isoproterenol, tolbutamide). In addition, the second phase of insulin secretion is also deficient when glucose levels and the degree of insulin resistance are taken into account. Lastly, disruptions to short- and long-term pulsatility of insulin secretion13 and alterations in insulin maturation (and excess of circulating proinsulin levels)14 are commonly observed in T2D.
Alteration of the gut–β-cell axis has been proposed as an important determinant of β-cell dysfunction in T2D. The incretin effect is first defined by increased stimulation of insulin secretion elicited by oral (as opposed to intravenous) administration of glucose under similar plasma-glucose levels.15 This phenomenon is not exclusive to glucose, and is also observed after oral load of lipids and amino acids.16 This observation led to the hypothesis that intestinal factors (termed “incretins”) secreted during the meal potentiated insulin secretion. Structures of the main incretin hormones, GIP and GLP1, were discovered in the 1980s.17,18 GLP1 and GIP are secreted by intestinal L cells (whose abundance increases toward the ileum and the colon) and K cells (present mainly in the duodenum and the upper jejunum), respectively. The development of pharmacological therapy of T2D by incretins focused on GLP1 was based on the observation that administration of GLP1 agonists reduced glucose levels in patients with T2D,19,20 while GIP may worsen glucose levels when administered acutely.21,22 Furthermore, the observation that inhibition of GIP signaling in animal models prevents obesity or ameliorates insulin resistance suggested that GIP agonists may not have therapeutic benefit.23–25
When GIP Is Rehabilitated As A Key Hormone Of Incretin Effect
The concept that GLP1 is a better pharmacological target for diabetes therapy than GIP is probably a misconception, as discussed elsewhere.16 Indeed, some reports have suggested that hyposecretion of GLP1 and hypersecretion of GIP are the usual pattern of patients with T2D during an oral glucose load;26,27 however, other publications and recent meta-analysis have failed to confirm that imbalance in incretin levels is a generalized defect in T2D patients. Therefore, if it is true that the relationship between the dose of oral glucose and the incretin effect is reduced in T2D when compared to healthy subjects, this defect is mainly explained by a reduction in β-cell mass in some patients with T2D, rather than by a reduction in gut-hormone secretion.16,28 Beyond pathogenesis of T2D, the importance of GIP in improve glucosing tolerance has also been underestimated in physiology. In healthy individuals, the contribution of GIP to the incretin effect seemed more important than GLP1. This has been suggested by the demonstration that circulating GIP levels are higher than GLP1 levels in comparisons of oral and intravenous glucose load.16,29 In addition, when GIP and GLP1 are infused in healthy subjects, GIP seems to have the major contribution to the incretin effect after oral glucose administration.30 The importance of GIP has also been demonstrated by a weak reduction in incretin effect when exendin (9–39) amide (a GLP1R antagonist) was infused during an oral glucose load.31 Conversely, it must be mentioned that inhibition of DPP4 increases levels of both active GLP1 and GIP during improvement in glucose levels without deleterious effects of GIP on body weight, suggesting that both incretins may act synergistically during DPP4 therapy.32
The important contribution of GIP to the incretin effect has been also emphasized by the observation that in patients with T2D with reduced incretin effect, the insulinotropic effect of GIP was lost, while that of GLP1 was relatively preserved.16,21 Importantly, the reduction of chronic hyperglycemia in patients with T2D may improve the ability to respond partially to GIP, suggesting that the reduced incretin effect of GIP is the consequence of glucotoxicity and not a primary defect.21,33 Therefore, in contrast to previous studies showing deleterious effects of acute GIP administration on glucose levels in patients with T2D, it could be hypothesized that partial restoration of GIP activity in well-controlled patients could ameliorate β-cell responsiveness and glucose homeostasis. This was confirmed by Højberg et al in eight patients with T2D in which β-cell responsiveness to GIP improved (but did not reach the levels observed in healthy subjects) with 4 weeks of near-normalization of blood glucose using insulin therapy.34 Because GLP1 and GIP may act synergistically on pancreatic β-cells, simultaneous pharmacological activation of both pathways may have additional benefits on β-cell functionality. Indeed, it could be speculated that GLP1 activity promotes a reduction in blood-glucose levels that fosters GIP activity to achieve better metabolic control.
GIP may have also therapeutic potential for the management of body-weight excess in patients with T2D. Recent studies have shown that GIP analogues with agonist properties induce a dose-dependent decrease in body weight in diet-induced obesity (DIO) mice by a reduction in food intake without changes in energy expenditure.35 These effects were preserved in DIO GLP1R–/– mice, but lost in DIO GIPR–/– mice, suggesting that the GIPR pathway was the main driver of body-weight loss during GIP administration. Confirmation that GIP-agonism therapy is beneficial for body-weight loss comes from the observation that a long-acting GIP-antagonist analogue does not induce body-weight change in rodents. Restoration of the GIP effect is important not only to improve the incretin effect (leading to a reduction in postprandial glucose levels) but also potentially to reverse the decline in β-cell mass. Indeed, Campbell et al demonstrated in mice with selective ablation of GIPR in β-cells that GIP had cytoprotective effects mediated by Tcf7 gene (encoded TCF1) expression via cAMP-independent and ERK-dependent pathways.36 Beneficial properties of GIP on β-cell physiology and survival have been confirmed in vivo when administration in rodents of a truncated GIP analogue,
Development Of GIPR–GLP1R Co-Agonists
The goal is to generate dual GIPR–GLP1R coagonists that bind to one or the other receptor (sequence-mixed dual agonists) and not to bind simultaneously to different related receptors at the same cell (as for fusion peptides or multimers, Figure 1).39,40 Finan et al investigated the potential of engineering a single-molecule GLP1R–GIPR coagonist.32 They designed a series of peptides tested for their ability to activate human GLP1R, GIPR, and GCGR (the glucagon receptor) in a cell-based reporter-gene assay for cAMP induction. The challenge was to provide balanced activity at GLP1R and GIPR while minimizing activity at GCGR to <1% that of native glucagon. After substitutions of residues in the middle and C-terminal regions of different intermixed peptides, the most interesting synthetic peptide was aminoisobutyric acid at position 2 (important to prevent physiological degradation and inactivation by DPPIV) and position 20 (in order to maximize stabilization of the helix, Figure 2).40 Substitution of a C-terminal residue (Cys40) with Lys40 allowed direct lipidation with a 16-carbon acyl chain (16:0, co-agonist in acylated form for daily administration, also called RG7697, Figure 2). Lastly, 40kDa PEGylation at Cys24 (coagonist in PEGylated form for weekly administration) maintained activity at both incretin receptors and reduced activity at GCGR to <0.02% of native glucagon. Finan et al described the metabolic effects of acute or chronic administration of either acetylated or PEGylated GIP–GLP1 coagonist in rodent models (wild-type DIO mice, GLP1R-knockout), in mice with pharmacologically silenced GIPR signaling, in mice with both incretin receptors silenced, in nonhuman primates, in healthy subjects, and in patients with T2D.32 Both dual-incretin agonists were compared to both GLP1 (liraglutide) and GIP monoagonists. First, the coagonist peptide improved glucose tolerance in DIO wild-type mice, similarly to the GIP analogue or coadministration of both monoagonists. In a mouse model mimicking the dual incretin receptor–knockout mice, the coagonist peptide and both monoagonists, individually or in combination, failed to improve glucose tolerance, suggesting that the GIP–GLP1 co-agonist has in vivo activity at both receptors without off-target activity. Then, it was demonstrated that when compared to a GLP1 monoagonist (exendin4 or liraglutide), the GIP–GLP1 coagonists (acylated or PEGylated forms) had greater efficacy in reducing fat intake, fat mass, circulating cholesterol levels, and decreasing ad libitum–fed blood glucose. Those metabolic effects were observed in wild-type DIO mice, db/db mice, and diabetic ZDF rats. Interestingly, the coagonist did not demonstrate significant influence on energy expenditure, respiratory quotient, or locomotor activity. Changes in insulin-secretory response after administration of both coagonists (acetylated and PEGylated) was dependent on the rodent model. Coagonists increased insulin-secretory response in normoglycemic lean mice, while they reduced it in diabetic ZDF rats, suggesting that coagonists decreased insulin resistance in the latter model (as assessed by a reduction in homeostatic model assessment [HOMA] insulin resistance–index values). Interestingly, in diabetic ZDF rats, both coagonists increased HOMA-β index values, a marker of β-cell functionality in parallel with improved pancreatic islet cytoarchitecture. At this step, the observation that acute administration of dual incretins enhances the insulinotropic effect to a greater extent than liraglutide in cynomolgus monkeys (as observed in rodents) strongly supported the rationale for a clinical study in humans. Acute administration of a PEGylated coagonist increased insulin secretion and reduced blood-glucose levels in healthy nondiabetic subjects, suggesting that preclinical data observed in rodents and nonhuman primates may translate to human subjects.32 A 6-week phase II clinical study assessed safety, efficacy, and pharmacodynamics of escalating doses of a PEGylated coagonist in 53 patients with T2D. The mean decrease from baseline for HbA1c ranged from 0.53% for patients receiving 4 mg coagonist to 1.11% for 30 mg compared to a decrease of 0.16% in the placebo group. In contrast to what is usually observed with GLP1 analogues, only minimal gastrointestinal adverse effects (diarrhea, nausea) were observed in this PEGylated coagonist clinical trial.
 |
Figure 1 Differences between coagonist (chimera) and peptide-fusion structures. Notes: The coagonist is achieved by mixing amino-acid sequences from different peptides or proteins (A). Peptide fusions resulted from the fusion of multiple hormones into a single molecule (B). Reprinted by permission from Springer Nature Customer Service Centre GmbH: [Springer Nature], [Nature Reviews Endocrinology], Clemmensen C, Finan B, Müller TD, DiMarchi RD, Tschöp MH, Hofmann SM. Emerging hormonal-based combination pharmacotherapies for the treatment of metabolic diseases. Nat Rev Endocrinol. 2019;15(2):90-104, (COPYRIGHT 2019).4 |
 |
Figure 2 Structure and first steps of molecular signaling through GIPR and GLP1R of GIPR–GLP1R dual agonists of RG7697–NNCOO90-274640 and LY3298176.39 Notes: Adapted from Molecular Metabolism, Vol 18, Coskun T, Sloop KW, Loghin C, et al, LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept, Pages 3-14, Copyright (2018), with permission from Elsevier.39 Adapted from Cell Metabolism, Vol 24/edition 1, Tschöp MH, Finan B, Clemmensen C, et al, Unimolecular Polypharmacy for Treatment of Diabetes and Obesity, Pages 51-62, Copyright (2016), with permission from Elsevier.40 For LY3298176, primary structure based on GIP amino-acid sequence. For RG7697–NNCOO90-2746, primary structure based on mixture of amino-acid sequences for GLP1 and GIP. For both coagonists, aminoisobutyric acid (Aib) at position 2 is important to prevent physiological degradation and inactivation by DPP4 at position 20 (RG7697) or 13 (LYS3298176) to maximize stabilization of helix. RG7697 characterized by substitution of C-terminal residue (Cys40) with Lys40, with direct lipidation with a 16-carbon acyl chain (16:0) for daily subcutaneous administration. LYS3298176 includes a C20 fatty-diacid moiety that allowed once-weekly subcutaneous administration. LY3298176 displayed higher affinity for GIPR and minimal activity on glucagon receptors. RG7697–NNCOO90-2746 had balanced affinity for both incretin receptors, and its activity on glucagon receptors was <0.02% of native glucagon. |
Clinical Trials
Recent review of the development of GIPR–GLP1R coagonists showed that some had been discontinued (Table 1). We focus our analysis of clinical trials on two compounds still in development: RG7697/NNC0090-2746 and LY3298176.
 |
Table 1 All GLP1R/GIPR Coagonists Developed To The End Of 2018 |
RG7697–NNC0090-2746
An acetylated form of a dual-incretin agonist (RG7697–NNC0090-2746; Novo Nordisk), described earlier, has been administered subcutaneously in 51 healthy volunteers in a double-blind, placebo-controlled study with escalating doses of 0.03–5 mg.41 The pharmacodynamic profile confirmed that RG7697–NNC0090-2746 can be administered once daily. In addition to analysis of adverse events and pharmacokinetic studies, a meal-tolerance test (MTT) was performed at the same time on day −1 (baseline) and day 1. For doses ≥1.8 mg, RG7697–NNC0090-2746 reduced both glucose and insulin levels during the MTT, with mild gastrointestinal adverse events (nausea and vomiting). After evaluation in healthy individuals, 56 patients with T2D received once-daily subcutaneous injections of RG7697–NNC0090-2746 (0.25–2.5 mg) or placebo for 14 days in a randomized, double-blind, dose-escalation study (Table 2).42 Patients in the study were predominantly men (52%), aged 32–65 years, and had body-mass index (BMI) values of 26.8–41.6 kg/m2. HbA1c levels at baseline were 6.93–8.51 among the different groups of patients. Several MTT and gastric-emptying tests were performed during the study. Mean HbA1c at baseline was 7.7% The higher dose of RG7697–NNC0090-2746 (2.5 mg) was associated with a significant decrease in HbA1c level (0.67% vs 0.21% for placebo), reduction in body weight (3 kg vs 0.9 kg), decrease in fasting and postprandial glucose levels, and an improvement in insulin resistance. As observed in the study including healthy individuals,41 the most frequent adverse events with RG7697–NNC0090-2746 were diarrhea and nausea, and the number of hypoglycemia events wase not different across dose or placebo groups. In a recent 12-week, randomized, placebo-controlled, double-blind phase IIA trial, 108 patients with T2D inadequately controlled with metformin received 1.8 mg RG7697–NNC0090-2746 or placebo subcutaneously once daily or liraglutide 1.8 mg (as an open-label reference arm, Table 2).43 At baseline, clinical characteristics of the patients randomized to three groups (54.6% female) were comparable, with mean age 54.8 years, mean duration of diabetes 8 years, mean HbA1c 8.3%, and mean BMI 33 kg/m2. At the end of follow-up, change from baseline in HbA1c was statistically significant (−0.96%, p<0.001) when compared to placebo. If postprandial glucose levels were reduced, insulin AUC during the MTT was reduced while C-peptide AUC remained unchanged. Percentage change in body weight with RG7697–NNC0090-2746 treatment from baseline was significant at week 8 of follow-up, but not at the end of the study (−1.80% at week 8 and −1.67% at week 12, respectively, when compared to placebo). A decrease in total cholesterol from baseline was noted for the NNC0090-2746 group (8% relative to placebo), without significant changes in other lipid parameters. Concerning adverse events, gastrointestinal disorders (nausea, vomiting, and diarrhea) were the most frequently reported. Heart rate was significantly increased with RG7697–NNC0090-2746 (5.6 beats/minute compared to placebo) Antidrug antibodies developed in 16 patients (43%) exposed to RG7697–NNC0090-2746. Interestingly, changes in HbA1c and body weight during RG7697–NNC0090-2746 therapy were more important in patients with baseline HbA1c < 8.5% suggesting a restoration in GIP function in parallel with reduction of hyperglycemia. Taken together, these data support the concept that the GIPR–GLP1R coagonist RG7697–NNC0090-2746 is an efficient therapy for the management of glucose levels in patients with T2D. What is worthy of note on RG7697–NNC0090-2746 is that this compound induced substantial reduction in HbA1c in phase II clinical studies, mainly by a reduction in body weight and in insulin resistance, rather than by stimulating insulin secretion.
LY3298176
LY3298176 (tirzepatide; Eli Lilly), is a 39–amino acid synthetic peptide classified as a dual GIPR–GLP1R agonist.39,44 Its structure is based on the GIP amino-acid sequence and a C20 fatty-diacid moiety allowed once-weekly subcutaneous administration (Figure 2).39,44 LY3298176 displayed a greater affinity to GIPRs relative to GLP1Rs expressed in vitro.39,45 Preclinical studies have shown that acute administration of this compound improves both glucose-dependent insulin secretion and glucose tolerance in mice.45 Compared to a GLP1R agonist, administration of LY3298176 to mice decreased body weight and food intake significantly. In a following step, 142 human subjects (healthy and patients with T2D) were included in a placebo-controlled, double-blind study (phase I–IIB) and randomly allocated to LY3298176 (0.25–15 mg), dulaglutide, and placebo groups for 29 days (Table 2).39 In healthy subjects, LY3298176 compared to placebo decreased glucose levels in fasting state and during the oral glucose-tolerance test (OGTT) without significant changes in insulin levels in either situation. Body-weight loss induced by LY3298176 was greater than that observed for dulaglutide 1.5 mg (4.52 and 4.05 kg from baseline for 4.5 mg and 10 mg LY3298176, respectively, and 1.3 kg from baseline for dulaglutide). In patients with T2D, LY3298176 reduced HbA1c significantly from baseline compared to placebo (0.84% at 10 mg and 0.58% at 15 mg), as well as fasting glucose and fasting insulin levels. Glucose excursion during the OGTT was significantly decreased in parallel with increased insulin response. From baseline to day 29, reduction in body weight was less important in patients with T2D than healthy individuals (maximal decrease from baseline 2.62 kg at 10 mg LY3298176). Gastrointestinal adverse events (nausea, vomiting, diarrhea, decreased appetite, abdominal distension) were less frequently reported in patients with T2D than healthy subjects, and there was no cases of severe hypoglycemia or acute pancreatitis. No relevant changes in systolic or diastolic blood pressure were reported, but as observed with dulaglutide, an increase in pulse rate was detected for all doses of LY3298176 in both healthy subjects and patients with T2D (9.80 bpm for 15 mg vs 3.81 bpm for placebo). In 2018, Frias et al reported the results of an international, multicentric, randomised, double-blind phase IIB study of LY3298176 in patients with T2D insufficiently controlled with diet and exercise alone or with stable metformin therapy (Table 2).44 A total of 318 patients were randomly assigned for 26 weeks to one of six treatment groups: LY3298176 1 mg, 5 mg, 10 mg, or 15 mg (once-weekly subcutaneous injection), dulaglutide 1.5 mg, or placebo. Participants (aged 18–75 years, 53%, men, 47% women) had T2D for a mean duration of 9 years and had HbA1c at baseline of 7.0%–10.5% and BMI of 23–50 kg/m2. The primary efficacy outcome was change in HbA1c. Mean changes from baseline in HbA1c were −1.06% (for 1 mg), −1.73% (for 5 mg), −1.89% (for 10mg), and −1.94% (for 15 mg) compared with −0.06% (for placebo). At 26 weeks, 33%–90% of patients treated with LY3298176 had achieved the HbA1c target of <7% (vs 52% with dulaglutide and 12% with placebo) and 15%–82% the HbA1c target of at least 6.5% (vs 39% with dulaglutide and 2% with placebo). Therefore, each dose of LY3298176 reduced HbA1c levels in a dose-dependent manner without a plateau effect. All doses of LY3298176 reduced the concentration of fasting plasma glucose from baseline to week 26 relative to placebo in a dose-dependent manner, and treatment with the 5 mg, 10 mg, and 15 mg LY3298176 reduced fasting plasma glucose more than dulaglutide. The dual agonist improved HOMA2 insulin resistance values and reduced insulin concentrations, whereas dulaglutide did not have significant effects on these measures, suggesting a possible insulin-sensitizing effect of LY3298176 potentially secondary to visceral fat reduction, since mean waist circumference decreased with the 5 mg, 10 mg, and 15 mg doses of LY3298176 compared with placebo and dulaglutide. Glucagon concentrations adjusted by fasting glucose decreased at 5 mg, 10 mg, and 15 mg LY3298176 compared with dulaglutide. Reduction in mean body weight was 0.9–11.3 kg for LY3298176 (0.9 kg for 1 mg, 4.8 kg for 5 mg, 8.7 kg for 10 mg, and 11.3 kg for 15 mg vs 0.4 kg for placebo and 2.7 kg for dulaglutide), 14%–71% of those treated with LY3298176 achieved the weight-loss target of at least 5% (vs 22% with dulaglutide and none with placebo), and 6%–39% achieved the weight-loss target of at least 10% (vs 9% with dulaglutide and none with placebo). Additionally, LY3298176 promoted reduction in total cholesterol (0∙2–0.3 mmol/L for LY3298176 vs 0.3 mmol/L for placebo and 0.2 mmol/L for dulaglutide) and triglyceride levels (from 0–0.8 mmol/L for LY3298176 vs 0.3 mmol/L for placebo and 0.3 mmol/L for dulaglutide). The most common dose-dependent adverse events were gastrointestinal (nausea, diarrhea, and vomiting) and decreased appetite (3.8% for 1 mg LY3298176, 20.0% for 5 mg LY3298176, 25.5% for 10 mg LY3298176, 18.9% for 15 mg LY3298176, 5.6% for dulaglutide, and 2.0% for placebo). There were no reports of severe hypoglycemic episodes. The incidence of cardiovascular adverse events did not differ among the groups. Lipase levels increased from 2.0% in placebo, 1 mg LY3298176, and dulaglutide to 7.8% in 10 mg LY3298176 and 3.8% in 15 mg LY3298176, and two participants treated with 5 mg LY3298176 had clinical pancreatitis. In summary, this phase IIB study established a wide dose range of the dual GIPR–GLP1R agonist LY3298176, that showed clinically meaningful and superior HbA1c control with greater weight loss. Results suggest that LY3298176 improved β-cell function, and noninferiority of LY3298176 versus dulaglutide was established. The authors seemed to consider that adverse events with LY3298176 treatment versus dulaglutide were similar, except for an increased frequency of discontinuation of study treatment for digestive adverse events with 15 mg LY3298176.
Comparison Of RG7697–NNC0090-2746 And LY3298176
If we compare the results of RG7697–NNC0090-2746 and LY3298176 phase II clinical trials,43,44 it seems that LY3298176 is more efficacious in reduction of body weight and HbA1c (Table 2). A potential explanation of this could be differences in study duration (12 weeks for RG7697–NNC0090-2746 and 26 weeks for LY3298176). However, this seems not to be a sufficient explanation, since superior efficiency of LY3298176 in reduction of body weight and HbA1c had already been observed at 12 weeks (a time comparable with available data for both studies). In addition, populations randomized in both studies had similar characteristics at baseline (LY3298176 and for RG7697/NNC0090-2746, respectively): mean age 58.6 vs 54.8 years, male 53% vs 54%, duration of diabetes 9 vs 8.3 years, HbA1c 8.1% vs 8.3%, and BMI (32.4 vs 33 kg/m2). Basically, the results of both clinical trials can be interpreted considering what we know about differences in structure of both compounds. First, LY3298176 and RG7697–NNC0090-2746 share common structural modifications, such as presence of aminoisobutyric acid at position 2 (to prevent physiological degradation and inactivation by DPPDPPIV) and position 20 (RG7697–NNC0090-2746) or 13 (LYS3298176) to maximize stabilization of the helix. In contrast, the structure of LY3298176 is based on the GIP amino-acid sequence and a C20 fatty-diacid moiety (for once-weekly subcutaneous administration) while RG7697–NNC0090-2746 is the result of substitutions of residues in the middle and C-terminal regions of different intermixed peptides. As a consequence, LY3298176 displayed greater affinity to GIPRs relative to GLP1Rs expressed in vitro, while similar affinity for GIPRs/GLP1Rs was observed for RG7697–NNC0090-2746 (Figure 2).4,45 As such, it could be speculated that peptide-based GIPR agonists reduce body weight and improve glucose tolerance mainly by activation of GIP signaling (as for LY3298176), rather than activation of both GIP and GLP1 pathways (as for RG7697–NNC0090-2746). Supporting this speculation is the demonstration that various peptide-based GIPR agonists decrease body weight in DIO mice proportionally to their potency on GIP receptors.38 Additionally, the weight-lowering effect of GIPR agonists was preserved in GLP1R–/– mice, suggesting that systemic GIPR agonism is sufficient to promote body-weight loss. Importantly, these differences in incretin-receptor affinity of both compounds could also explain (almost in part) differences in effects on various organs, as summarized in Figure 3. Further research is needed to better understand the relative contribution of GIP/GLP1 pathways in the regulation of appetite and body weight, an essential step for the design of future compounds.
 |
Table 2 Main results of NNCOO90-2746 and LY3298176 clinical trials39,42–44 in patients with T2D |
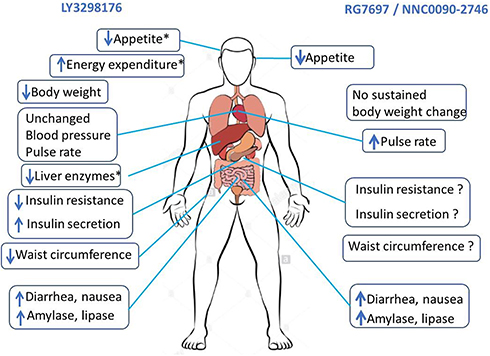 |
Figure 3 Comparative effects of RG7697/NNCOO90-2746 and LY3298176 from clinical trials,40, 43–45 except for where asterisks indicate (demonstrated only in rodents). 45 |
Conclusion
Combined GIPR and GLP1R activation has emerged as a promising therapeutic option in the treatment of chronic metabolic diseases, such as T2D. Based on clinical trials, GIPR–GLP1R dual agonists demonstrated clinical improvement in glycemic control and body weight in patients with T2D, with acceptable safety and tolerability profiles.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Reaven GM. HOMA-beta in the UKPDS and ADOPT. Is the natural history of type 2 diabetes characterised by a progressive and inexorable loss of insulin secretory function? Maybe? Maybe not? Diab Vasc Dis Res. 2009;6(2):133–138. doi:10.1177/1479164109336038
2. American Diabetes Association. Pharmacologic approaches to glycemic treatment. Diabetes Care. 2017;40 Suppl 1:
3. Fosse-Edorh S, Fagot-Campagna A, Detournay B, et al. Impact of socio-economic position on health and quality of care in adults with type 2 diabetes in France: the entred 2007 study. Diabet Med. 2015;32(11):1438–1444. doi:10.1111/dme.12783
4. Clemmensen C, Finan B, Müller TD, DiMarchi RD, Tschöp MH, Hofmann SM. Emerging hormonal-based combination pharmacotherapies for the treatment of metabolic diseases. Nat Rev Endocrinol. 2019;15(2):90–104. doi:10.1038/s41574-018-0118-x
5. Vague J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr. 1956;4(1):20–34. doi:10.1093/ajcn/4.1.20
6. Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol. 2017;13(11):633–643. doi:10.1038/nrendo.2017.90
7. Kumari M, Heeren J, Scheja L. Regulation of immunometabolism in adipose tissue. Semin Immunopathol. 2018;40(2):189–202. doi:10.1007/s00281-017-0668-3
8. Matthews DR, Cull CA, Stratton IM, Holman RR, Turner RC. UKPDS 26: Sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabet Med. 1998;15(4):297–303. doi:10.1002/(SICI)1096-9136(199804)15:4<297::AID-DIA572>3.0.CO;2-W
9. Levy J, Atkinson AB, Bell PM, McCance DR, Hadden DR. Beta-cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: the 10-year follow-up of the Belfast Diet Study. Diabet Med. 1998;15(4):290–296. doi:10.1002/(SICI)1096-9136(199804)15:4<290::AID-DIA570>3.0.CO;2-M
10. Lean MEJ, Leslie WS, Barnes AC, et al. Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2019;7(5):344–355. doi:10.1016/S2213-8587(19)30068-3
11. Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalization of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54(10):2506–2514. doi:10.1007/s00125-011-2204-7
12. Brunzell JD, Robertson RP, Lerner RL, et al. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab. 1976;42(2):222–229. doi:10.1210/jcem-42-2-222
13. O’Rahilly S, Turner RC. Matthews DR Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med. 1988;318(19):1225–1230. doi:10.1056/NEJM198805123181902
14. Pimenta W, Korytkowski M, Mitrakou A, et al. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose-tolerant individuals with a first-degree NIDDM relative. JAMA. 1995;273(23):1855–1861.
15. Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29(1):46–52. doi:10.1007/bf02427280
16. Nauck MA, Meier JJ. The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016;4(6):525–536. doi:10.1016/S2213-8587(15)00482-9
17. Jörnvall H, Carlquist M, Kwauk S, et al. Amino acid sequence and heterogeneity of gastric inhibitory polypeptide (GIP). FEBS Lett. 1981;123(2):205–210. doi:10.1016/0014-5793(81)80288-8
18. Lund PK, Goodman RH, Montminy MR, Dee PC. Habener JF Anglerfish islet pre-proglucagon II. Nucleotide and corresponding amino acid sequence of the cDNA. J Biol Chem. 1983;258(5):3280–3284.
19. Gutniak M, Orskov C, Holst JJ, Ahrén B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. 1992;326(20):1316–1322. doi:10.1056/NEJM199205143262003
20. Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7-36) amide in type 2 (noninsulin-dependent) diabetic patients. J Clin Endocrinol Metab. 1996;81(1):327–332. doi:10.1210/jcem.81.1.8550773
21. Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia. 2002;45(8):1111–1119. doi:10.1007/s00125-002-0878-6
22. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91(1):301–307. doi:10.1172/JCI116186
23. Gault VA, O’Harte FP, Harriott P, Flatt PR. Characterization of the cellular and metabolic effects of a novel enzyme-resistant antagonist of glucose-dependent insulinotropic polypeptide. Biochem Biophys Res Commun. 2002;290(5):1420–1426. doi:10.1006/bbrc.2002.6364
24. Gault VA, Irwin N, Green BD, et al. Chemical ablation of gastric inhibitory polypeptide receptor action by daily (Pro3)GIP administration improves glucose tolerance and ameliorates insulin resistance and abnormalities of islet structure in obesity-related diabetes. Diabetes. 2005;54(8):2436–2446. doi:10.2337/diabetes.54.8.2436
25. Miyawaki K, Yamada Y, Ban N, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. 2002;8(7):738–742. doi:10.1038/nm727
26. Jones IR, Owens DR, Luzio S, Williams S, Hayes TM. The glucose dependent insulinotropic polypeptide response to oral glucose and mixed meals is increased in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1989;32(9):668–677. doi:10.1007/bf00274255
27. Toft-Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab. 2001;86(8):3717–3723. doi:10.1210/jcem.86.8.7750
28. Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia. 2011;54(1):10–18. doi:10.1007/s00125-010-1896-4
29. Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept. 2003;114(2–3):115–121. doi:10.1016/s0167-0115(03)00111-3
30. Nauck MA, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7-36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. 1993;76(4):912–917. doi:10.1210/jcem.76.4.8473405
31. Edwards CM, Todd JF, Mahmoudi M, et al. Glucagon-like peptide 1 has a physiological role in the control of postprandial glucose in humans: studies with the antagonist exendin 9-39. Diabetes. 1999;48(1):86–93.
32. Finan B, Ma T, Ottaway N, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5(209):209ra151. doi:10.1126/scitranslmed.3007218
33. Kjems LL, Holst JJ, Vølund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. 2003;52(2):380–386. doi:10.2337/diabetes.52.2.380
34. Højberg PV, Vilsbøll T, Rabøl R, et al. Madsbad S Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia. 2009;52(2):199–207. doi:10.1007/s00125-008-1195-5
35. Mroz PA, Finan B, Gelfanov V, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab. 2019;20:51–62. doi:10.1016/j.molmet.2018.12.001
36. Campbell JE, Ussher JR, Mulvihill EE, et al. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat Med. 2016;22(1):84–90. doi:10.1038/nm.3997
37. Widenmaier SB, Kim SJ, Yang GK, et al. A GIP receptor agonist exhibits beta-cell anti-apoptotic actions in rat models of diabetes resulting in improved beta-cell function and glycemic control. PLoS One. 2010;5(3):e9590. doi:10.1371/journal.pone.0009590
38. Hasib A, Ng MT, Gault VA, et al. An enzymatically stable GIP/xenin hybrid peptide restores GIP sensitivity, enhances beta cell function and improves glucose homeostasis in high-fat-fed mice. Diabetologia. 2017;60(3):541–552. doi:10.1007/s00125-016-4186-y
39. Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab;2018. 3–14. doi:10.1016/j.molmet.2018.09.009
40. Tschöp MH, Finan B, Clemmensen C, et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 2016;24(1):51–62. doi:10.1016/j.cmet.2016.06.021
41. Portron A, Jadidi S, Sarkar N, DiMarchi R, Schmitt C. Pharmacodynamics, pharmacokinetics, safety and tolerability of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 after single subcutaneous administration in healthy subjects. Diabetes Obes Metab. 2017;19(10):1446–1453. doi:10.1111/dom.13025
42. Schmitt C, Portron A, Jadidi S, Sarkar N, DiMarchi R. Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetes Obes Metab. 2017;19(10):1436–1445. doi:10.1111/dom.13024
43. Frias JP, Bastyr EJ
44. Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392(10160):2180–2193. doi:10.1016/S0140-6736(18)32260-8
45. Bokvist K, Brown R, Coskun T, et al. LY3298176, a novel long-acting GIP/GLP-1 coagonist, shows enhanced activity on weight loss and energy utilization whilst maintaining its efficacy for glycaemic control. Diabetologia. 2017;60(suppl 1):S399.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.