")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Dual actions of albumin packaging and tumor targeting enhance the antitumor efficacy and reduce the cardiotoxicity of doxorubicin in vivo
Authors Zheng K, Li R, Zhou X, Hu P, Zhang Y, Huang Y , Chen Z , Huang M
Received 12 March 2015
Accepted for publication 30 April 2015
Published 24 August 2015 Volume 2015:10(1) Pages 5327—5342
DOI https://doi.org/10.2147/IJN.S84478
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Ke Zheng,1 Rui Li,2 Xiaolei Zhou,2 Ping Hu,2 Yaxin Zhang,2 Yunmei Huang,3 Zhuo Chen,2 Mingdong Huang2
1College of Chemistry, Fuzhou University, Fuzhou, People’s Republic of China; 2State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, People’s Republic of China; 3Fujian Academy of Integrative Medicine, Fujian University of Traditional Chinese Medicine, Fuzhou, People’s Republic of China
Abstract: Doxorubicin (DOX) is an effective chemotherapy drug used to treat different types of cancers. However, DOX has severe side effects, especially life-threatening cardiotoxicity. We herein report a new approach to reduce the toxicity of DOX by embedding DOX inside human serum albumin (HSA). HSA is further fused by a molecular biology technique with a tumor-targeting agent, amino-terminal fragment of urokinase (ATF). ATF binds with a high affinity to urokinase receptor, which is a cell-surface receptor overexpressed in many types of tumors. The as-prepared macromolecule complex (ATF–HSA:DOX) was not as cytotoxic as free DOX to cells in vitro, and was mainly localized in cell cytosol in contrast to DOX that was localized in cell nuclei. However, in tumor-bearing mice, ATF–HSA:DOX was demonstrated to have an enhanced tumor-targeting and antitumor efficacy compared with free DOX. More importantly, histopathological examinations of the hearts from the mice treated with ATF–HSA:DOX showed a significantly reduced cardiotoxicity compared with hearts from mice treated with free DOX. These results demonstrate the feasibility of this approach in reducing the cardiotoxicity of DOX while strengthening its antitumor efficacy. Such a tumor-targeted albumin packaging strategy can also be applied to other antitumor drugs.
Keywords: amino-terminal fragment of urokinase, urokinase receptor, drug carrier, human serum albumin, doxorubicin, cytotoxicity
Introduction
Doxorubicin (DOX) is a member of anthracycline compounds and is a US Food and Drug Administration (FDA)-approved chemotherapeutic drug used for effective treatment of a number of cancers, including hematological cancers, some types of solid tumors, and soft-tissue sarcomas.1 The mechanism of action for DOX in tumor cells is not completely understood, but it is generally believed that DOX intercalates into DNA, disrupts the replication and transcription processes, and leads to cell death.1 Despite its effectiveness and broad clinical indications, DOX is also known for its severe side effects, including hepatotoxicity,2 cardiotoxicity, and nephrotoxicity.3 Especially, cardiotoxicity is identified as the most life-threatening adverse effect that may cause the loss of myofibrils and the vacuolization of myocardial cells and, in some cases, congestive heart failure.4
Various approaches to minimize this side effect of DOX and increase its therapeutic window have been attempted. DOX can be co-administrated with cardioprotective drugs such as dexrazoxane and monoHER.5 Chemically modified DOX or other anthracycline compounds also have been explored.6 There are some successful examples with this approach. Epirubicin (4′-hydroxyl derivative of doxorubicin, Farmorubicin®)7 and pirarubicin (4′-tetrahydropyranyl-doxorubicin, Theprubicin®)1 have been approved for clinical use. In addition, DOX is modified into a (6-maleimidocaproyl) hydrazone derivative (DOXO-EMCH),8 which can bind to circulating serum albumin after intravenous administration by covalently attaching to albumin residue Cys34. The albumin-bound form of DOXO-EMCH has pronounced acid lability that allows DOX to be released in the acidic environment present in tumor tissues or intracellular endosomes. DOXO-EMCH has been approved for clinical trials and is found to have a two-fold to five-fold increase in the maximum tolerated dose and a lower cardiotoxicity compared with free DOX.8 Another broadly studied approach is to formulate DOX with nanomaterials, such as liposomes,9,10 iron oxide nanoparticles,11 gold nanoparticles,12,13 polymeric micelles,14 or albumin nanoparticles,15 by covalent linking or physical adsorption. These nanomaterials can be further functionalized by coupling with targeting proteins,15 peptides,13 or RNA aptamers.10 Among the nanomaterials, the DOX-loaded liposomes DOXIL® and Lipodox® have been approved by the FDA and used in clinics.16
Human serum albumin (HSA) is the most abundant plasma protein (35–50 g/L or ~0.6 mM) with a molecular weight of 66.5 kDa,17 and is the primary carrier protein in blood circulation, acting as a solubilizing agent for long-chain fatty acids and a detoxifying protein for toxic substances (bilirubin, heavy metal ions, etc) in plasma.18 HSA also binds to various exogenous drugs, eg, antibiotics, anticoagulants, anti-inflammatory agents, and anesthetics,18 and modulates the pharmacokinetic profiles of drugs. Therefore, HSA can be used as a carrier for drug delivery. Moreover, HSA is an important component in nanoparticle albumin-bound (Nab) technology. This technology aggregates albumin into nanoparticles with a size of ~100–200 nm and traps the hydrophobic drugs with the nanoparticles. Nab–paclitaxel (Abraxane®), a Nab formulation of paclitaxel using HSA, has been approved by the FDA to treat metastatic breast cancer,19 non-small-cell lung cancer,20 and pancreatic cancer.21
In this study, we fused HSA with a peptide (amino-terminal fragment of urokinase, ATF) using a molecular biology technique to generate a recombinant protein (ATF–HSA). ATF targets to urokinase-type plasminogen activator receptor (uPAR), a cell-surface receptor that is overexpressed in many types of cancer cells and tumor tissues, but not in most normal tissues.22,23 uPAR is widely advocated as a surface marker for tumors and is an important target for tumor therapy and tumor imaging.24,25 A number of different types of uPAR inhibitors have been developed in the last two decades, including antibodies, peptides, and organic molecules. One peptidyl inhibitor of uPAR conjugated with a radioactive isotope is used in a clinical trial as a positive emission tomography tracer to identify tumors with aggressive phenotype.26 Urokinase-type plasminogen activator (uPA) is an endogenous ligand of uPAR and functions as a natural inhibitor for uPAR, as uPA binds to uPAR with a high affinity (subnanomolar dissociation constant).23 Our crystal structure studies,27,28 together with biochemical studies from others,29 demonstrate that the ATF is the primary receptor-binding region of uPA and accounts for all the binding affinity. ATF has been used as a target agent for uPAR in a number of studies.30,31 Therefore, ATF–HSA possesses dual properties: uPAR targeting by ATF and drug binding by HSA.
Here, we used ATF–HSA as a drug carrier to embed DOX inside HSA molecules through a novel strategy of dilution–incubation–purification (DIP) to form a macromolecule complex, ATF–HSA:DOX. We demonstrated that ATF–HSA:DOX indeed possessed a tumor-targeting capability and an enhanced antitumor effect and at the same time showed a reduced cardiotoxicity compared to free DOX in vivo.
Materials and methods
Materials
A Pichia pastoris yeast strain X-33 (Invitrogen, USA) with plasmid pPICZαA encoding ATF–HSA was constructed, as previously described.30 DOX was purchased from Wuhan DKY Technology Co. Ltd. (Wuhan, People’s Republic of China). Diethylaminoethyl (DEAE) anion exchange resin and Ni-chelating Sepharose Fast Flow resin were purchased from GE Healthcare (Uppsala, Sweden). Other chemicals were purchased either from Sigma-Aldrich (St Louis, MO, USA) or from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, People’s Republic of China). All the experimental procedures were approved by the ethics committee of Fuzhou University and Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. Non-small-cell lung carcinoma cells (H1299) and human embryo lung fibroblasts (HELF) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal calf serum at 37°C in a humidified incubator with 5% CO2 atmosphere. The viability of cells was determined by trypan blue dye exclusion. Cells were maintained in logarithmic phase with viability >95%.
Expression of ATF–HSA with Pichia pastoris strain X-33
The transformed Pichia pastoris strain X-33 integrated with ATF–HSA expression vector was cultured in YPD medium (1% yeast extract, 2% peptone, 2% dextrose) containing 100 μg/mL Zeocin® at 28°C for 2 days before it was transferred into BMGY medium (2% peptone, 1% yeast extract, 100 mM potassium phosphate, pH 6, 1% glycerol). The X-33 strain was further cultured in BMGY medium at 28°C for about 24 hours to an OD600 of 4–5. After being transferred into BMMY medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6, 1% methanol), the cells were induced every 24 hours with methanol (at a final concentration of 1%) over the following 4 days to express the protein ATF–HSA.
Purification and characterization of ATF–HSA
After 4-day induction with 1% methanol, the BMMY medium was harvested by centrifugation at 9,000 g for 20 minutes. After the supernatant was adjusted to pH 7.4 using 1 M Tris-HCl (pH 8.5) and centrifuged one more time, the supernatant was collected and applied to a Ni2+-chelating column that was pre-equilibrated with 20 mM Tris-HCl containing 500 mM NaCl, pH 7.4. The column was then washed by 20 mM Tris-HCl, pH 7.4, 500 mM NaCl containing 5 mM imidazole, followed by the elution of the target protein (ATF–HSA) using the above buffer containing 500 mM imidazole. The fraction containing ATF–HSA from the column was dialyzed overnight against 20 mM Tris-HCl, 50 mM NaCl, pH 8.0, at 4°C by a dialysis membrane with molecular weight cutoff of 8–10 kDa. The molecular weight of the target protein was confirmed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) in a Bio-Rad Biologic system (Bio-Rad Laboratories Inc., Hercules, CA, USA), and the concentration of the target protein was measured by a GE Nanovue® spectrophotometer (Cambridge, UK).
Preparation and purification of ATF–HSA:DOX
An aliquot of 200 μL solution of DOX (20 mM in dimethylsulfoxide) was added dropwise into 400 mL buffer (20 mM Tris-HCl, 50 mM NaCl, pH 8.0) containing 67.2 mg purified ATF–HSA with the protein and DOX at a molar ratio of 1:5. The final concentration of DOX was 10 μM. The mixture was kept stirring in the dark for 12 hours and then applied to a DEAE column pre-equilibrated with 20 mM Tris-HCl, 50 mM NaCl, pH 8.0. The final product, ATF–HSA:DOX, was eluted with 20 mM Tris-HCl, 300 mM NaCl, pH 8.0. The fraction of ATF–HSA:DOX from DEAE column was dialyzed overnight against phosphate-buffered saline (PBS) in the dark at 4°C by a dialysis membrane with molecular weight cutoff of 8–10 kDa and further concentrated by ultrafiltration.
Ultraviolet-visible and fluorescence spectra of ATF–HSA:DOX
The ultraviolet-visible absorption or fluorescence spectra of ATF–HSA:DOX were measured on a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) or a Varian Cary Eclipse instrument (Agilent Technologies, Santa Clara, CA, USA).
Stoichiometric ratio measurement
The stoichiometric ratio was obtained by measuring the ratio of the concentrations of ATF–HSA and DOX in the ATF–HSA:DOX complex. DOX was quantified using its characteristic maximal absorption at 490 nm. A standard curve covering concentration ranging from 1–30 μM was established (Figure S1). The concentration of ATF–HSA was measured by a BCA Protein Assay Kit (BioTek Corporation, Beijing, People’s Republic of China). The presence of DOX did not affect such protein assay because the protein was assayed at 562 nm, quite different from the DOX absorption maximum (490 nm).32
Cellular uptake of ATF–HSA:DOX
H1299 and HELF cells were used in current study. Aliquots of 200 μL of the suspended cells at a concentration of 2×105 cells/mL were placed in each well of 96-well Costar® plates to a confluence of about 70%; 20 μL of ATF–HSA:DOX (50 μM in PBS), HSA:DOX (50 μM in PBS), or DOX (50 μM in PBS) was added into the wells containing 180 μL medium, and final concentrations of ATF–HSA:DOX, HSA:DOX, and DOX were 5 μM. The cells were incubated with ATF–HSA:DOX, HSA:DOX, or DOX for various periods of time (0 hours, 4 hours, 8 hours, 16 hours, or 24 hours). After being washed by PBS, the cells were then lysed by a buffer of 2% SDS (w/v) and 0.1 M NaOH, and the fluorescence of cell lysates was measured by a BioTek microplate reader using λex=490 nm and λem=590 nm. The protein concentrations of cell lysates were determined by the BCA Protein Assay Kit (BioTek Corporation, Beijing, People’s Republic of China). Each experiment was repeated three times with four replicates at each time point.
Cellular localization of ATF–HSA:DOX
The suspended H1299 cells (5×104 cells) were plated onto confocal chamber slides at 37°C and incubated for 18–24 hours. Then ATF–HSA:DOX (50 μM in PBS) or DOX (50 μM in PBS) was added onto confocal chamber slides, and the cells were incubated with ATF–HSA:DOX (at a final concentration of 5 μM) or DOX (at a final concentration of 5 μM) for 2 hours and 12 hours, respectively. After being washed in PBS to remove unbound ATF–HSA:DOX or DOX, cells were fixed by a phosphate buffer containing 4% paraformaldehyde. The cells were then stained with 1 mL DNA fluorescent dye, DAPI (4′,6-diamidino-2-phenylindole, 5 μg/mL), for 15 minutes and were washed in PBS thoroughly before being mounted for direct confocal microscopic observation. The Olympus FluoView™ FV1000 laser scanning confocal microscope (Olympus Corp., Tokyo, Japan) was coupled to an inverted microscope with a 60× differential interference contrast oil immersion objective. The fluorescence of cell specimens in the confocal chamber slides was excited by a diode laser light (λ=405 nm for DAPI) or an argon–ion laser light (λ=488 nm for DOX), while the emitted fluorescence was filtered with barrier filters (450/30 nm and 590/30 nm band pass, respectively). All parameters, including the laser line intensity, photometric gain, settings of photo-multiplier tube and filter attenuation, were kept constant throughout the entire imaging experiment. The cellular localization of ATF–HSA:DOX or DOX in HELF cells was measured using the same method. All images were analyzed by Olympus Fluoview v2.1 software.
In vitro cytotoxicity of ATF–HSA:DOX
The cytotoxicity experiment in vitro was carried out using the electric cell-substrate impedance sensing (ECIS) technique, which is a noninvasive method to measure cell attachment of adherent cells growing in culture medium.33 The array plates (8W10E) used in this study have a pattern of microelectrode printed at the bottom of the plates for detecting the current flow through the media. In our experiments, 6×104 cells (H1299) were seeded into wells and the electric impedance value in each well was monitored at 16,000 Hz with 60 s sampling intervals. After cells reached a confluence of about 70%, ATF–HSA:DOX (500 μM in PBS) or DOX (500 μM in PBS) was added to the wells to achieve a final concentration of 50 μM. Meanwhile, the electric impedance data was collected for 12 hours. When the data collection stopped, the cells were photographed to record cell density and morphology.
Flow cytometry analysis of ATF–HSA:DOX
Induction of apoptosis was measured using Annexin V–FITC (fluorescein isothiocyanate)/PI (propidium iodide) Apoptosis Assay Kit (MultiSciences Biotech Co. Ltd., Hangzhou, People’s Republic of China). The stock solution of ATF–HSA:DOX was 100 μM in PBS and the stock solution of DOX was 100 μM in PBS. H1299 cells (5×104) were incubated with ATF–HSA:DOX (at a final concentration of 10 μM) or DOX (at a final concentration of 10 μM) for 12 hours before they were harvested by centrifugation at 100 g for 5 minutes, and washed by Hanks balanced saline solution. The cells were then resuspended in 200 μL of staining buffer and stained by Annexin V–FITC (2 μL) and PI (4 μL) for 5 minutes in the dark, followed by an analysis on a flow cytometer (Accuri C6; BD, Franklin Lakes, NJ, USA). The percentage of live, early apoptotic, and late apoptotic or necrotic cells was determined. Each experiment was repeated three times with four replicates at each time point.
Establishment of hepatoma-22 (H22) tumor-bearing mice model
Male Kunming mice (purchased from Shanghai SLAC Laboratory Animal Co. Ltd., Shanghai, People’s Republic of China), 4 weeks old and weighing 18–22 g, were maintained in a temperature-regulated environment and were allowed free access to water and food throughout the whole course of the experiment. All animal procedures were conducted in accordance with the recommendations of the institutional animal care and use committee. The mouse hepatocellular carcinoma cell line (H22) was purchased from Shanghai Institute of Cell Biology (Chinese Academy of Sciences, Shanghai, People’s Republic of China) and propagated in the form of ascites from the mouse abdominal cavity. To establish the tumor-bearing model, each mouse was injected subcutaneously 200 μL H22 cells (2.0×106) on the back. Typically, the tumor reaches a volume around 70 mm3 in 5–7 days after the inoculation.
In vivo fluorescence imaging using ATF–HSA:DOX as a probe
The H22 tumor-bearing Kunming mice were randomly divided into three groups (five mice per group) with the equivalent average starting tumor size (~70 mm3) and body weight (~23 g). ATF–HSA:DOX (600 μM in saline) or DOX (600 μM in saline) was injected into mice via caudal veins at a dosage of 5 μmol/kg. At different time points (3 hours, 6 hours, 12 hours, 24 hours, and 48 hours) after the injection, five mice per group were anaesthetized with isoflurane and imaged on a IVIS Lumina II animal imaging system (Caliper Life Sciences, Inc. Hopkinton, MA, USA) with the green fluorescent protein filter for excitation (445–490 nm). The fluorescence of ATF–HSA:DOX and DOX can be excited at 445–490 nm (Figure S2). The imaging results were analyzed with Living Imaging Software (IVIS Imaging Systems).
Antitumor effect and cardiotoxicity of ATF–HSA:DOX on H22 tumor-bearing mice
H22 tumor-bearing mice (established using the method in the section Establishment of H22 tumor-bearing mice model) were randomly divided into three groups (eight mice in each group) with equivalent average starting tumor size (~70 mm3) and body weight (~23 g). Two treatment groups were respectively injected with ATF–HSA:DOX (600 μM in saline) or DOX (600 μM in saline) intravenously via the caudal vein at a dosage of 5 μmol/kg. The control group received saline of an equivalent volume. The subcutaneous tumor sizes were estimated by the formula (W2 × L)/2, where L = tumor length (the longest diameter of the tumor) and W = tumor width (diameter perpendicular to the length), and were recorded daily throughout the experiment. Antitumor effects of both ATF–HSA:DOX and DOX against H22 tumor implanted in mice were evaluated by a tumor growth-inhibition analysis. After 6 days, the mice were sacrificed. Their hearts were removed, immersed in a buffered solution of 4% paraformaldehyde and embedded in paraffin. The specimens were sectioned serially at a 4 mm thickness and stained by hematoxylin and eosin. The histological sections were evaluated on an optical microscopy.
Statistics
All data represent group means and standard errors of the mean. Data were analyzed by two-way or three-way analyses of variance. Individual group means were compared by the Newman–Keuls multiple-range test. GB-STAT software (Dynamic Microsystems, Inc., Silver Spring, MD, USA) was used for all statistical analyses.
Results
Embedding DOX into ATF–HSA by the DIP method
The tumor-targeting drug carrier used here (recombinant protein ATF–HSA, Figure 1) was expressed in the yeast cells transformed with ATF–HSA expression vector and purified over a Ni2+-chelated Sepharose Fast Flow column. The ATF–HSA was purified to a high purity with a single band at a molecular weight of ~84 kDa on reducing SDS-PAGE (Figure S3A).
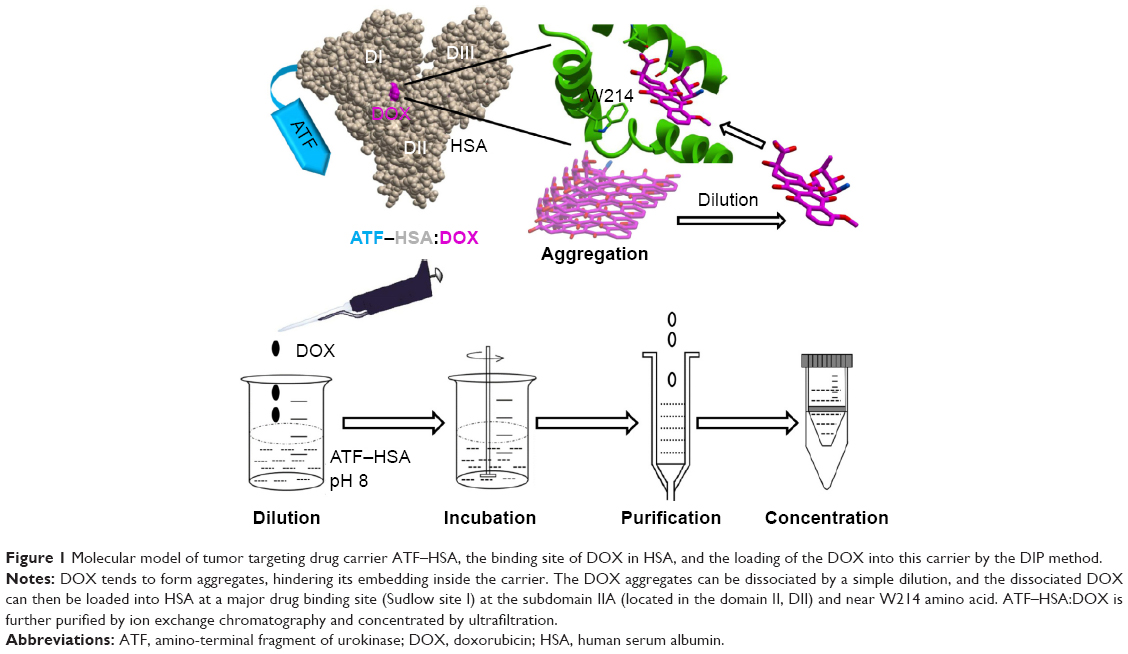 | Figure 1 Molecular model of tumor targeting drug carrier ATF–HSA, the binding site of DOX in HSA, and the loading of the DOX into this carrier by the DIP method. |
Albumin is composed of three domains (DI, DII, DIII) and each domain has two subdomains (A and B). HSA has multiple ligand-binding sites and accommodates a number of endogenous metabolites and exogenous drugs. The HSA in the ATF–HSA fusion protein is quite likely to maintain the same ligand-binding capability as free HSA, as ATF is linked to HSA through a spacer (four glycine residues) and unlikely to perturb the structure of HSA. It was reported that DOX can bind to HSA with a binding constant K =1.1×104 M−1.34 Meanwhile, ATF–HSA has the ability to bind to the cancer cell-surface receptor uPAR.30 A major challenge to embed DOX into ATF–HSA is that DOX has a strong tendency to aggregate in aqueous solution at high concentrations due to its hydrophobicity (>20 μM, Figure S4, see also Fülöp et al35). We developed a unique method (DIP, ie, dilution–incubation–purification) to solve this problem (Figure 1). In this method, DOX was diluted to a concentration below its aggregation point (~20 μM, Figure S4) and mixed with ATF–HSA. After incubation for 12 hours, the ATF–HSA:DOX complex was separated from unbound DOX by anion–ion exchange chromatography to a high purity.
Properties of the embedded DOX (ATF–HSA:DOX)
The ultraviolet-visible spectra of ATF–HSA:DOX complex showed the characteristic absorption peak of DOX at 490 nm (Figure S3B), demonstrating the presence of DOX in the complex. It is interesting to note that the ATF–HSA:DOX complex showed fine structures in its major absorption peak compared with DOX itself. It was inferred that the DOX was buried inside the HSA molecule and lost its rotational freedom. This result further supported the presence of DOX in the complex.
The fluorescent spectra measurement also supported the direct and specific interaction of DOX with ATF–HSA. We observed that the fluorescence of tryptophan in ATF–HSA:DOX complex was blue-shifted and slightly weaker compared with that of ATF–HSA under equal conditions (Figure S3C). HSA contains only one tryptophan residue (W214), which is located near a drug-binding site (named Sudlow drug binding site I) in subdomain IIA (Figure 1) of HSA. Thus, this result demonstrated that DOX was buried in the Sudlow site I of HSA, consistent with a previous study.34 A molecular model of ATF-HSA:DOX was generated and is shown in Figure 1. We also observed the presence of ATF–HSA greatly quenched the fluorescence of DOX (ex/em 490/590 nm) by ~three-and-one-half-fold (Figure S3D). This is likely due to the destabilization of the excited electron state of DOX by surrounding residues of ATF–HSA.
Next, we measured the stoichiometric ratio of ATF–HSA and DOX in the complex. The results showed only one molecule of DOX bound to one ATF–HSA molecule. It is interesting to note that HSA has seven binding sites for fatty acids or drug molecules but only one binding site is used for DOX, as found in our study. It should be pointed out that DOX most likely does not bind to ATF, because there is no binding pocket in ATF.27
Stability of ATF–HSA:DOX
DOX is not covalently attached to ATF–HSA in the ATF–HSA:DOX. To test if DOX would leak out of the binding site of ATF–HSA, we stored ATF–HSA:DOX in PBS at 4°C for a month and observed no visible color change or precipitation. Furthermore, the stoichiometric ratio of ATF–HSA and DOX was measured after storage and was found to be 1:1. Thus, it appears that ATF–HSA:DOX is quite stable. ATF–HSA:DOX showed an enhanced antitumor efficacy in vivo compared with free DOX at equal concentrations (see the ATF-HSA:DOX shows a higher antitumor efficacy than DOX in vivo section), providing additional support on the stability of ATF–HSA:DOX in blood circulation.
At low pH value (pH 4.5), DOX was partly released from the ATF–HSA:DOX based on the fluorescence measurements (Figure S5). Such release is likely due to the deformation of HSA structure at such a low pH value.36 The complete release of DOX requires stringent conditions, eg, the denaturing of ATF–HSA:DOX complex by 1% SDS (Figure S5).
To test the stability of ATF–HSA:DOX in cell culture medium (RPMI-1640) containing 10% fetal calf serum (FCS), ATF–HSA:DOX was incubated with culture medium for 24 hours. If DOX was competed off from ATF–HSA by BSA or small molecules in the FCS, the fluorescence of DOX would be enhanced. This was not the case from our results (Figure S5), which showed that the fluorescence emission spectra had almost no change, demonstrating the stability of ATF–HSA:DOX in FCS (Figure S6).
ATF–HSA:DOX reduces the cellular uptake of DOX
As the DOX is buried inside the ATF–HSA:DOX, will it be able to enter the cells? We carried out the cellular uptake experiments of ATF–HSA:DOX in both H1299 and HELF cell lines with free DOX as a control. H1299, a lung cancer cell line, has a large amount of uPAR expressed on the cell surface, while fibroblast HELF cell line has little uPAR expression on the cell surface.37 In our experiment, cells were incubated with DOX, HSA:DOX, or ATF–HSA:DOX (all at a final concentration of 5 μM) for different time periods (0 hours, 4 hours, 8 hours, 16 hours, and 24 hours) followed by washing to remove the unbound DOX. The taken DOX was quantified by measuring the fluorescence of DOX. We noticed that the amount of ATF–HSA:DOX was higher than that of HSA:DOX in H1299 at any time periods, whereas there was almost no difference in the amount of ATF–HSA:DOX and DOX in HELF (Figure 2), presumably due to the higher level of uPAR expression on the cell surface of H1299 compared with HELF.
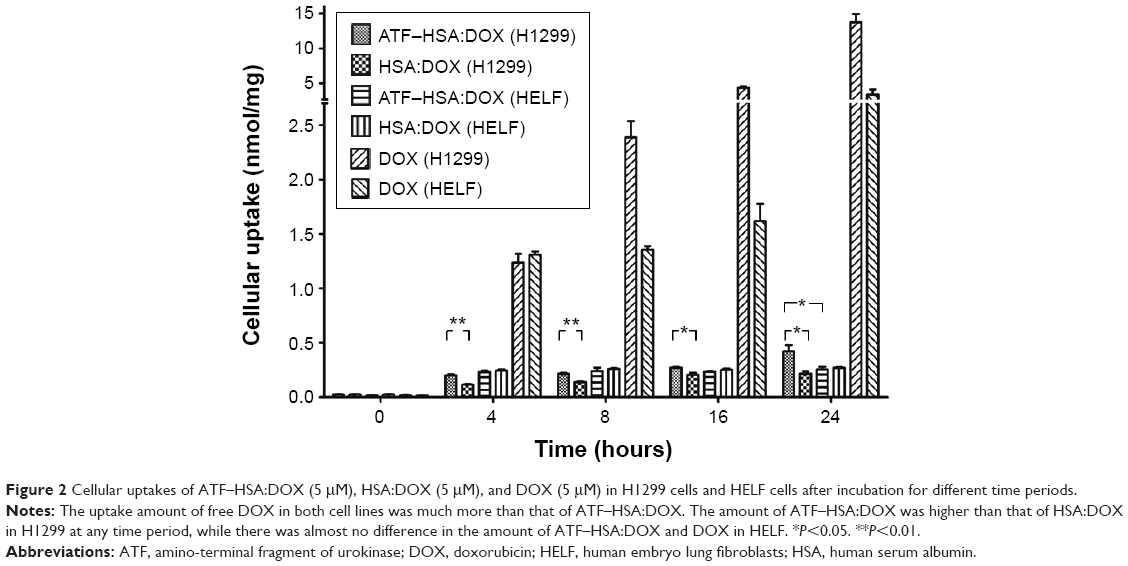 | Figure 2 Cellular uptakes of ATF–HSA:DOX (5 μM), HSA:DOX (5 μM), and DOX (5 μM) in H1299 cells and HELF cells after incubation for different time periods. |
It is interesting to note that both H1299 and HELF cell lines absorbed much more free DOX than ATF–HSA:DOX. The uptake amount of DOX, after an 8-hour incubation, was more than ten-fold higher than that of ATF–HSA:DOX for both cell lines (Figure 2), suggesting that the cellular uptake mechanism of ATF–HSA:DOX is different from that of free DOX.
ATF–HSA:DOX distributes mainly in cytoplasm, DOX in nuclei
It is important to study the subcellular localization of ATF–HSA:DOX, which helps to understand the molecular mechanism of DOX-induced cytotoxicity. We incubated DOX or ATF–HSA:DOX (both at 5 μM) with either H1299 or HELF cells in confocal chamber slides for two different time periods (2 hours and 12 hours). After the cells were fixed, a cell nucleus dye DAPI was applied. Cells were imaged by a laser scanning microscope under identical experimental conditions. ATF–HSA:DOX and DOX were found inside both H1299 and HELF cells after 2-hour incubation (Figure 3, red color). As expected, DOX was observed to localize in cell nuclei (Figure 3, blue color; magenta color is the mix of blue and red). Surprisingly, ATF–HSA:DOX was found to distribute mainly in cytoplasm, but not in nuclei at all. Such distribution pattern remained almost the same after 12-hour incubation for both DOX and ATF–HSA:DOX in both cell lines, except that a trace amount of ATF–HSA:DOX was observed to enter the nuclei in H1299 cells. Therefore, we concluded that DOX embedded into ATF–HSA had a subcellular distribution different from that of DOX.
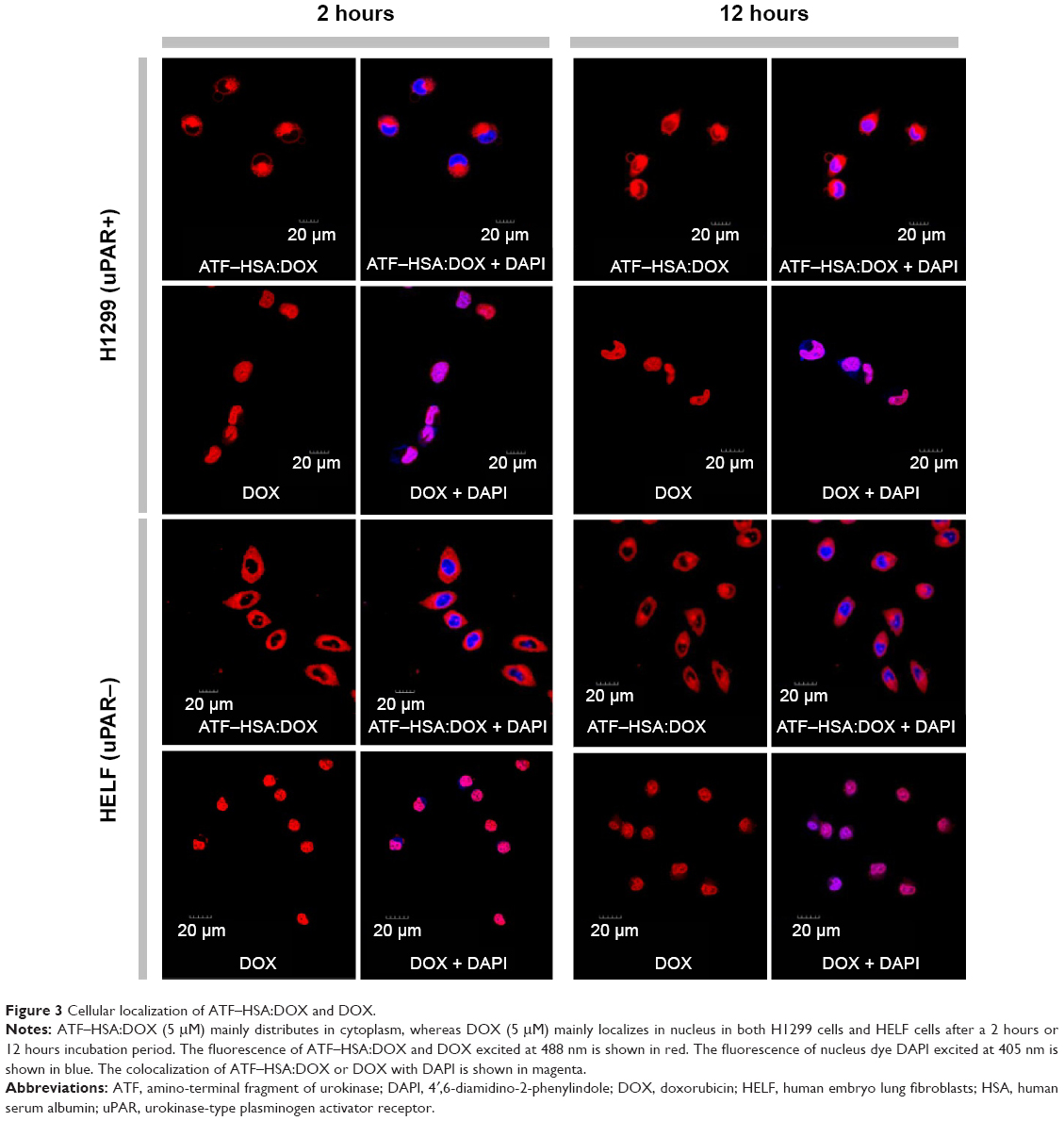 | Figure 3 Cellular localization of ATF–HSA:DOX and DOX. |
This difference in cellular localization between ATF–HSA:DOX and DOX suggests that the whole ATF–HSA:DOX complex enters the cells before DOX is dissociated out of the ATF–HSA:DOX complex. Once inside the cells, DOX may release from the complex because the reducing environment of cytosol can loosen up the tertiary structure of HSA.
This difference in cellular localization between ATF–HSA:DOX and DOX also helps to answer a question involving FCS. Ten percent FCS was included in all of the in vitro experiments to facilitate cell culture. FCS contains bovine serum albumin (BSA). Will BSA affect the in vitro experiments by binding to DOX in the control experiment or even extract DOX out of ATF–HSA:DOX complex? We think this effect is small. If DOX would be in complex with BSA and enter cells, it would be quite likely to localize in cytoplasm, similar to ATF–HSA:DOX, which was not the case observed. This result here provides support that DOX is likely not in complex with BSA in the settings of our in vitro experiments. On another hand, BSA binding to DOX is weaker than HSA (the binding constant KDOX-HSA =1.1×104 M−1, KDOX-BSA =7.8×103 M−1).34
Lower cytotoxicity of ATF–HSA:DOX compared to DOX in vitro
To compare the cytotoxicities of ATF–HSA:DOX and free DOX on H1299 cells, we used a sensitive, noninvasive and in situ method named electric cell-substrate impedance sensing (ECIS) in our experiments. The cells were grown on the gold electrodes attached at the bottom of the ECIS plates. The electric impedance values measured are proportional to the number of live cells attached to the plate, while cells detaching from the gold electrodes will not contribute to impedance values. The results of ECIS for H1299 cells incubated with or without 50 μM ATF–HSA:DOX or DOX were shown in Figure 4. In the absence of DOX or ATF–HSA:DOX, H1299 cells continued to grow, leading to the rise of the impedance (Figure 4). In the presence of DOX, the cells started to die at the fifth hour, and after 12 hours only a few cells were alive and a large quantity of cell debris was visible (Figure 4). ATF–HSA:DOX showed some toxicity, but not as cytotoxic as DOX (Figure 4) at the same dosage (50 μM). This difference between DOX and ATF–HSA:DOX is in line with the different subcellular localization and the different amounts of cellular uptake stated in the section ATF-HSA:DOX reduces the cellular uptake of DOX and ATF-HSA:DOX distributes mainly in cytoplasm, DOX in nuclei.
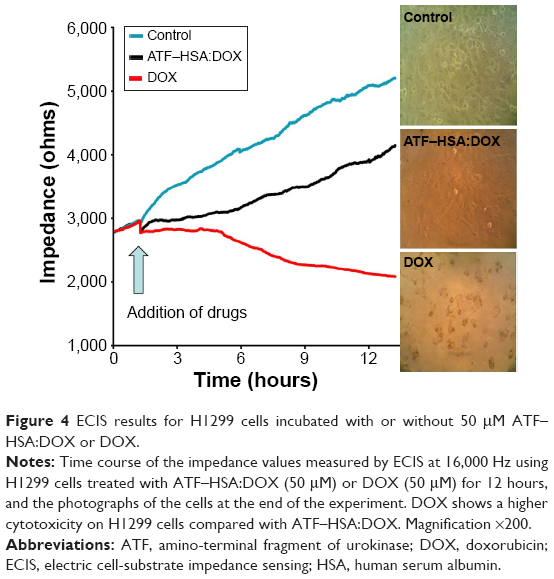 | Figure 4 ECIS results for H1299 cells incubated with or without 50 μM ATF–HSA:DOX or DOX. |
Mechanism of cell death induced by ATF–HSA:DOX
Apoptosis and necrosis are two major types of cell death. Does the different subcellular localization of ATF–HSA:DOX affect the mechanism of cell death? We incubated H1299 with ATF–HSA:DOX (10 μM) or DOX (10 μM) for 12 hours and stained the cells with FITC-label Annexin V and PI for a flow cytometry analysis. The Annexin V is commonly used to measure the extracellular exposure of phosphotidylserine, a sign of apoptosis. PI is a probe for nucleic acid and measures the number of necrotic cells. High Annexin V but low PI staining is an indication of early apoptosis of cells, whereas strong staining of both Annexin V and PI indicates the cells are in the stage of late apoptosis or necrosis. Figure 5 shows that the number of cells treated with DOX in both early apoptosis and necrosis quarters was higher than that treated with ATF–HSA:DOX, demonstrating that DOX was more toxic than ATF–HSA:DOX. The early apoptotic cells after the treatment with DOX were four-fold more than those treated with ATF–HSA:DOX (Figure 5B). Interestingly, the number of early apoptotic cells after the treatment with ATF–HSA:DOX (10 μM) was ~three-fold less than cells of the late apoptotic or necrosis (Figure 5A and B) after a 12-hour incubation, which was different from the case of DOX (10 μM). This difference suggests that ATF–HSA:DOX may induce cell death by a mechanism different from that of DOX.
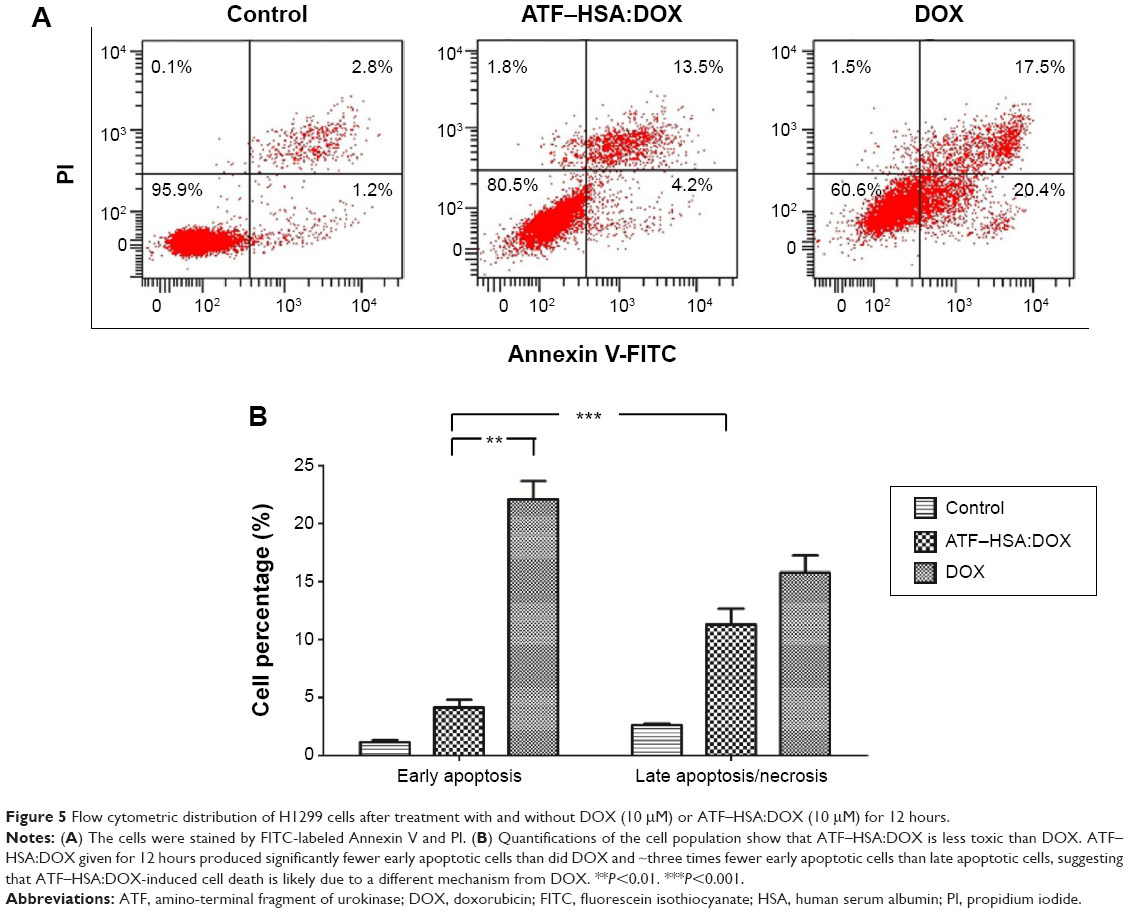 | Figure 5 Flow cytometric distribution of H1299 cells after treatment with and without DOX (10 μM) or ATF–HSA:DOX (10 μM) for 12 hours. |
ATF–HSA:DOX shows an enhanced tumor specificity in vivo compared with free DOX
To further investigate the biodistribution of ATF–HSA:DOX in vivo, we established an H22 tumor-bearing mouse model. The mice were injected with ATF–HSA:DOX or free DOX (5 μmol/kg body weight) through their caudal veins. The mice were imaged using DOX fluorescence at different time points (3 hours, 6 hours, 12 hours, 24 hours, and 48 hours) after the injection on an IVIS Lumina II imaging system. ATF–HSA:DOX was found to accumulate at the tumor sites, and its accumulation was quite fast (Figure 6A), reaching almost a maximal amount in tumors at 3 hours after injection and gradually reducing after the sixth hour. In contrast, DOX did not show clear specific accumulation in tumors. As a result, the observed amount of DOX at the tumor sites was much less than that of ATF–HSA:DOX at each time point we investigated (Figure 6B). Therefore, ATF–HSA:DOX exhibited a specific and much higher amount of tumor retention compared with DOX in tumor-bearing mice. This is quite different from the cellular experimental results shown in the section ATF-HSA:DOX reduces the cellular uptake of DOX.
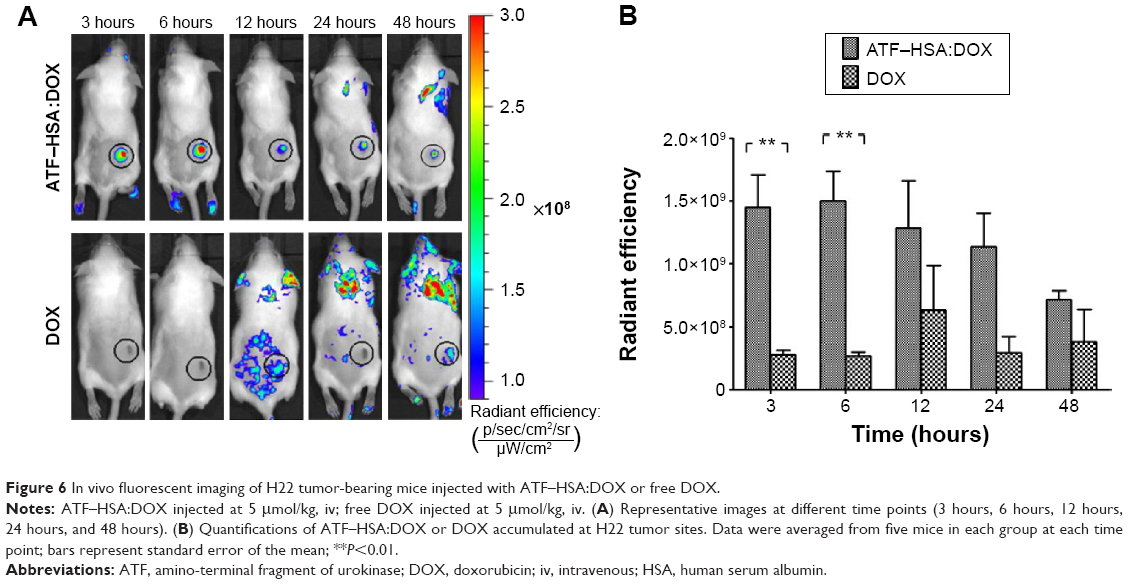 | Figure 6 In vivo fluorescent imaging of H22 tumor-bearing mice injected with ATF–HSA:DOX or free DOX. |
ATF–HSA:DOX shows a higher antitumor efficacy than DOX in vivo
To investigate the antitumor efficacy of ATF–HSA:DOX in H22 tumor-bearing mice, we measured the tumor volumes of mice (eight per group) for 6 continuous days after one bolus injection of ATF–HSA:DOX (5 μmol/kg), DOX (5 μmol/kg or 2.9 mg/kg), or saline (as a control) through the caudal vein. The DOX treatment inhibited the tumor growth rate (Figure 7A) compared with the untreated group. In contrast, ATF–HSA:DOX treatment further reduced the tumor growth rate compared to the group treated with DOX (~60% of the tumor size from the DOX-treated group at day 6). These data showed that ATF–HSA:DOX had an enhanced antitumor efficacy compared with DOX.
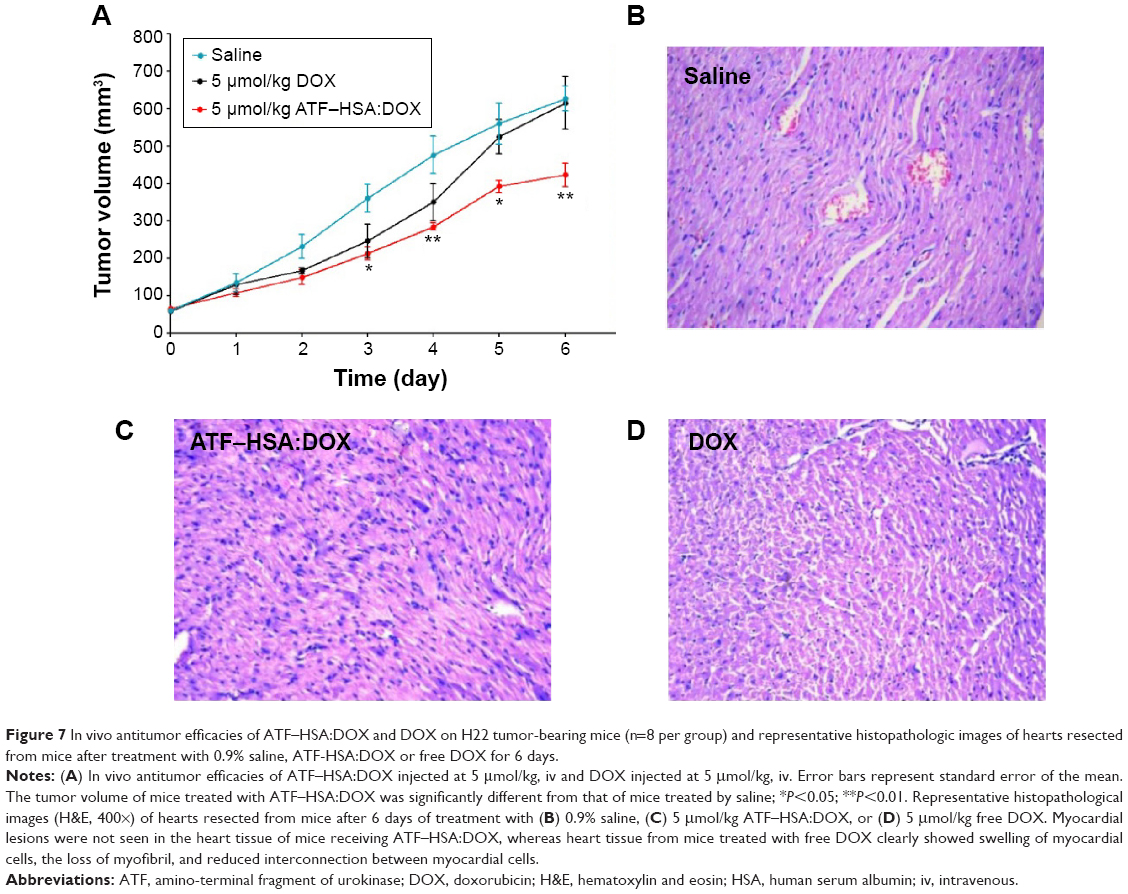 | Figure 7 In vivo antitumor efficacies of ATF–HSA:DOX and DOX on H22 tumor-bearing mice (n=8 per group) and representative histopathologic images of hearts resected from mice after treatment with 0.9% saline, ATF-HSA:DOX or free DOX for 6 days. |
ATF–HSA:DOX reduces cardiotoxicity compared to DOX in vivo
One of the main side effects of DOX is cardiac toxicity. To investigate the cardiotoxicity of ATF–HSA:DOX in vivo, we carried out the histological assessments of hearts from mice after injection with saline, ATF–HSA:DOX (5 μmol/kg), or DOX (5 μmol/kg) for 6 days. Compared with that from saline-treated group (Figure 7B), the histopathological slices of hearts from DOX-treated mice showed an extensive swelling of myocardial cells, reduced interconnection between myocardial cells and the loss of myofibril (Figure 7D). In comparison, no severe myocardial lesions were observed in hearts from ATF–HSA:DOX-treated group (Figure 7C). This observation demonstrated that ATF–HSA:DOX significantly reduced cardiactoxicity and simultaneously exhibited an enhanced antitumor efficacy compared with DOX.
Discussion
ATF–HSA as a molecular drug carrier
HSA is a natural protein in human blood serum and is used as a delivery carrier for many endogenous molecules or drug molecules.18 ATF renders the tumor targeting capability of this drug carrier. A challenge here is how to successfully load DOX into HSA. DOX has a strong tendency to aggregate in aqueous solution at high concentrations due to its hydrophobicity. We developed the DIP method to cope with this challenge.
As a method to embed drug molecules, the DIP method is different from Nab technology.38 ATF–HSA:DOX formed by the DIP method has following characteristics: 1) DOX is buried inside HSA, forming a noncovalent macromolecule complex with a 1:1 stoichiometry; 2) HSA is most likely to maintain its native conformation; 3) the immune reaction from the human body toward ATF–HSA is likely to be low because both components (ATF and HSA) have human sequences and native tertiary conformations. In Nab technology, where the HSA is denatured to some degree and intertwined into nanoparticles (Figure 8A). An advantage of Nab technology is that it can load many more drug molecules than the DIP method, because the native HSA in the DIP method has limited numbers of drug binding sites, whereas the Nab technology uses the denatured HSA nanoparticles to formulate drug molecules.
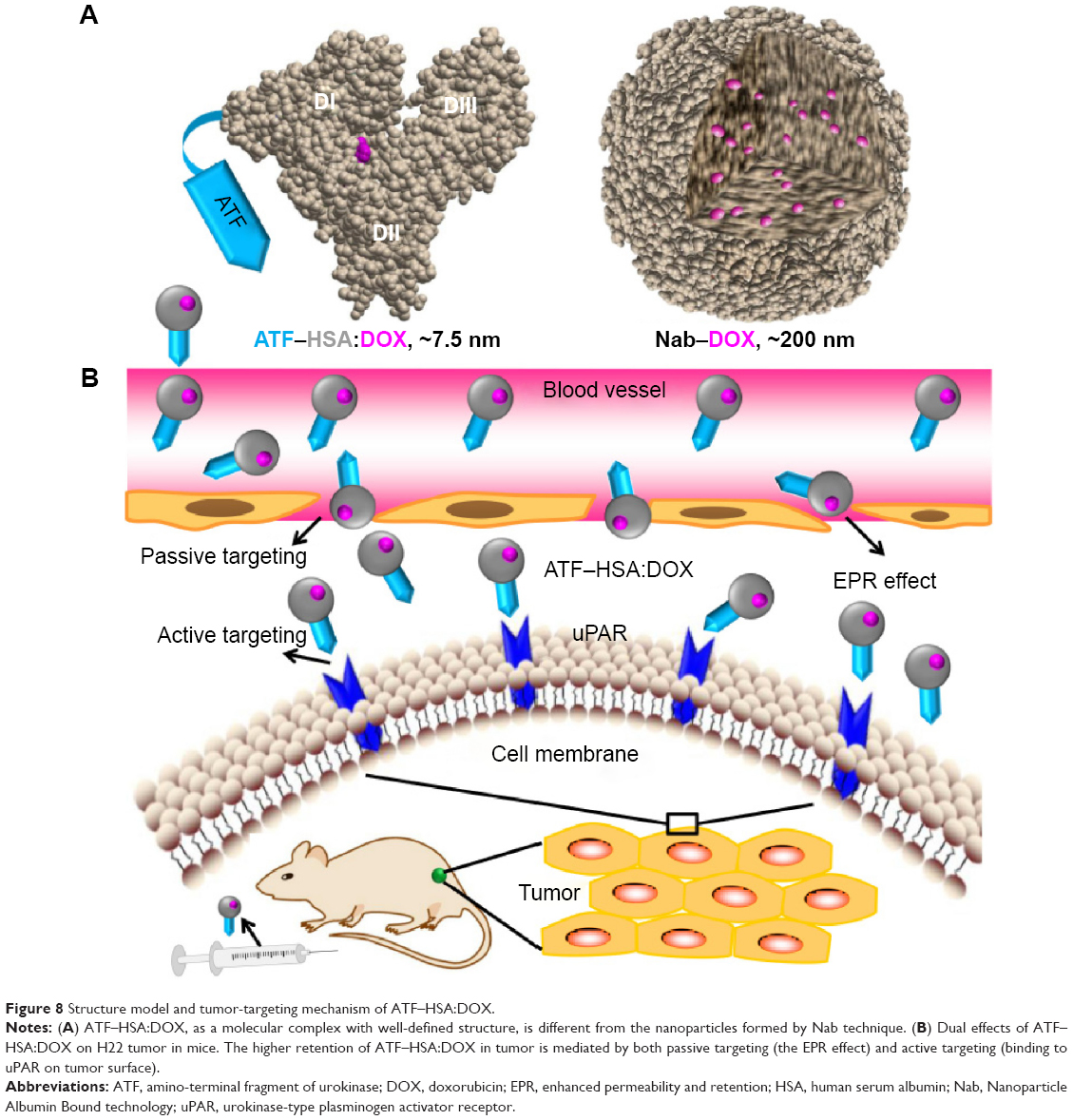 | Figure 8 Structure model and tumor-targeting mechanism of ATF–HSA:DOX. |
An additional potential advantage of albumin-packaged DOX by the DIP method is that it overcomes the multidrug resistance induced by DOX. Multidrug-resistance protein 1, also known as permeability glycoprotein (P-gp),39 is an important adenosine-triphosphate–binding-cassette transporter in the cell membrane that pumps a wide variety of foreign substances, including DOX, out of cells. Crystal structure study reveals that P-gp has a large binding cavity that binds to drugs through hydrophobic interactions.40 HSA is not a substrate of P-gp and buries the hydrophobic DOX inside its own Sudlow site I. Thus ATF–HSA:DOX may affect the recognition of DOX by P-gp and avoid the activation of P-gp in response to DOX. Such a potential advantage needs to be further tested experimentally.
Different mechanism of cellular uptake between ATF–HSA:DOX and DOX
We have noticed that the cellular uptakes of the complex ATF–HSA:DOX in both H1299 cells and HELF cells are significantly lower than that of free DOX (Figure 2). The exact mechanism of the cellular uptake is yet unknown. One possibility for the difference in cellular uptake between ATF–HSA:DOX and free DOX is their different cellular entry routes. DOX is hydrophobic in chemical nature and is demonstrated to traverse the cell membrane mainly by diffusion.41 In contrast, ATF–HSA:DOX is a macromolecule complex and likely to enter cells through endocytosis as the most important mechanism. ATF in the ATF–HSA carrier has a high affinity for uPAR, which is overexpressed on surfaces of many types of tumors and thus may facilitate the entry of the complex ATF–HSA:DOX into tumor cells by receptor-mediated endocytosis.42 In the endocytosis process, the invaginated area on the cell membrane engulfs macromolecules, forming membrane-bound vesicles or endosomes, leading to the transport of macromolecules into cells.43 Such a difference in cellular entry routes may account for the difference of cellular uptake between ATF–HSA:DOX and free DOX.
Cell-death mechanism
DOX has a strong affinity to the nucleus and is typically found to be located in the cell nucleus.44 DOX intercalates into DNA, causing DNA damage and disrupting the replication and transcription processes. Meanwhile, DOX is able to inhibit topoisomerase II, leading to cell death.1 In our study, we found that ATF–HSA:DOX complex mainly distributed in cytoplasm, which was different from DOX. In addition, ATF–HSA:DOX was observed to induce less early apoptosis compared with DOX after a 12-hour incubation (Figure 5), indicating a different cell death mechanism upon DOX encapsulation inside HSA.
It has been reported recently that cytosolic DOX can generate reactive oxygen species (ROS). The ROS can cause oxidative damage of mitochondria45 and increase the permeabilization of the lysosomal membrane, which causes spillage of catabolic enzymes from the lysosomal lumen into cytosol,46 ultimately leading to cell death. Whether the ATF–HSA:DOX uses the ROS to cause cell death requires further study.
Enhanced antitumor efficacy of ATF–HSA:DOX in mice
Although the antitumor efficacy of ATF–HSA:DOX complex on H1299 cells was inferior to free DOX (Figures 4 and 5), ATF–HSA:DOX complex indeed exhibited an enhanced antitumor efficacy compared with DOX in vivo (Figure 7A). This enhanced antitumor efficacy of ATF–HSA:DOX in vivo is likely due to the higher amount of ATF–HSA:DOX complex than DOX at the tumor sites (Figure 6). Two effects here may contribute to the in vivo tumor-specific accumulation: one is the active targeting of ATF–HSA:DOX to tumor cells by binding to uPAR on the tumor cell surface; the other one is the passive targeting effect, or the enhanced permeability and retention effect, which also increases the retention of ATF–HSA:DOX in tumor (Figure 8B). The enhanced permeability and retention effect is the unique anatomical–pathophysiological nature of tumors,47 due to the permeable nature of tumor blood vessels and the defective lymphatic drain in most solid tumors. It is reported that macromolecules (larger than 40 kDa), including HSA, can be selectively enriched in tumors.48
Conclusion
In this study, we loaded DOX inside ATF–HSA by the DIP method to generate the macromolecule ATF–HSA:DOX with a 1:1 stoichiometric ratio. This albumin-embedded DOX exhibited a lower cellular uptake and reduced cytotoxicity compared with DOX in vitro. In addition, ATF–HSA:DOX showed a different subcellular localization and a different cell death mechanism compared with DOX. However, in vivo, ATF–HSA:DOX showed a stronger antitumor potency and a lower cardiotoxicity compared with DOX. Our work demonstrates that ATF–HSA is an effective drug carrier that may also be used for other drugs to improve the therapeutic window.
Acknowledgments
This work is supported by the Chinese Academy of Sciences/State Administration of Foreign Experts Affairs (CAS/SAFEA) International Partnership Program for Creative Research Teams, the National Natural Science Foundation of China (21171167, U1405229, and 31370737), the CAS Cross-Disciplinary and Collaborative Research Team Program, and the Strategic Priority Research Program of the CAS (XDA09030307).
Disclosure
The authors report no conflicts of interest in this work.
References
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229. | ||
Kalender Y, Yel M, Kalender S. Doxorubicin hepatotoxicity and hepatic free radical metabolism in rats. The effects of vitamin E and catechin. Toxicology. 2005;209(1):39–45. | ||
Yilmaz S, Atessahin A, Sahna E, Karahan I, Ozer S. Protective effect of lycopene on adriamycin-induced cardiotoxicity and nephrotoxicity. Toxicology. 2006;218(2–3):164–171. | ||
Monsuez JJ, Charniot JC, Vignat N, Artigou JY. Cardiac side-effects of cancer chemotherapy. Int J Cardiol. 2010;144(1):3–15. | ||
Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–1225. | ||
Preobrazhenskaya MN, Tevyashova AN, Olsufyeva EN, Huang K, Huang H. Second generation drugs-derivatives of natural antitumor anthracycline antibiotics daunorubicin, doxorubicin and carminomycin. J Med Sci. 2006;26(4):119. | ||
Petrioli R, Fiaschi AI, Francini E, Pascucci A, Francini G. The role of doxorubicin and epirubicin in the treatment of patients with metastatic hormone-refractory prostate cancer. Cancer Treat Rev. 2008;34(8):710–718. | ||
Kratz F. DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs. 2007;16(6):855–866. | ||
Kaminskas LM, McLeod VM, Kelly BD, et al. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomedicine. 2012;8(1):103–111. | ||
Baek SE, Lee KH, Park YS, et al. RNA aptamer-conjugated liposome as an efficient anticancer drug delivery vehicle targeting cancer cells in vivo. J Control Release. 2014;196:234–242. | ||
Maeng JH, Lee DH, Jung KH, et al. Multifunctional doxorubicin loaded superparamagnetic iron oxide nanoparticles for chemotherapy and magnetic resonance imaging in liver cancer. Biomaterials. 2010;31(18):4995–5006. | ||
Minati L, Antonini V, Torrengo S, et al. Sustained in vitro release and cell uptake of doxorubicin adsorbed onto gold nanoparticles and covered by a polyelectrolyte complex layer. Int J Pharm. 2012;438(1–2):45–52. | ||
Park H, Tsutsumi H, Mihara H. Cell-selective intracellular drug delivery using doxorubicin and α-helical peptides conjugated to gold nanoparticles. Biomaterials. 2014;35(10):3480–3487. | ||
Perche F, Patel NR, Torchilin VP. Accumulation and toxicity of antibody-targeted doxorubicin-loaded PEG-PE micelles in ovarian cancer cell spheroid model. J Control Release. 2012;164(1):95–102. | ||
Bae S, Ma K, Kim TH, et al. Doxorubicin-loaded human serum albumin nanoparticles surface-modified with TNF-related apoptosis-inducing ligand and transferrin for targeting multiple tumor types. Biomaterials. 2012;33(5):1536–1546. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–182. | ||
Kratz F. A clinical update of using albumin as a drug vehicle – a commentary. J Control Release. 2014;190:331–336. | ||
Bharali DJ, Khalil M, Gurbuz M, Simone TM, Mousa SA. Nanoparticles and cancer therapy: a concise review with emphasis on dendrimers. Int J Nanomedicine. 2009;4:1–7. | ||
Socinski MA, Langer CJ, Okamoto I, et al. Safety and efficacy of weekly nab®-paclitaxel in combination with carboplatin as first-line therapy in elderly patients with advanced non-small-cell lung cancer. Ann Oncol. 2013;24(2):314–321. | ||
Ma WW, Hidalgo M. The winning formulation: the development of paclitaxel in pancreatic cancer. Clin Cancer Res. 2013;19(20):5572–5579. | ||
Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3(12):932–943. | ||
Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11(1):23–36. | ||
Yang L, Sajja HK, Cao Z, et al. uPAR-targeted optical imaging contrasts as theranostic agents for tumor margin detection. Theranostics. 2013;4(1):106–118. | ||
Persson M, Kjaer A. Urokinase-type plasminogen activator receptor (uPAR) as a promising new imaging target: potential clinical applications. Clin Physiol Funct Imaging. 2013;33(5):329–337. | ||
Persson M, Madsen J, Østergaard S, et al. Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: implications for visualizing cancer invasion. J Nucl Med. 2012;53(1):138–145. | ||
Huai Q, Mazar AP, Kuo A, et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311(5761):656–659. | ||
Huai Q, Zhou A, Lin L, et al. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat Struct Mol Biol. 2008;15(4):422–423. | ||
Ploug M, Ellis V. Structure-function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS Lett. 1994;349(2):163–168. | ||
Li R, Zheng K, Hu P, et al. A novel tumor targeting drug carrier for optical imaging and therapy. Theranostics. 2014;4(6):642–659. | ||
Yang L, Peng XH, Wang YA, et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res. 2009;15(14):4722–4732. | ||
Oommen OP, Garousi J, Sloff M, Varghese OP. Tailored doxorubicin-hyaluronan conjugate as a potent anticancer glyco-drug: an alternative to prodrug approach. Macromol Biosci. 2014;14(3):327–333. | ||
Grabarek JB, Zyzyńska K, Saiz N, et al. Differential plasticity of epiblast and primitive endoderm precursors within the ICM of the early mouse embryo. Development. 2012;139(1):129–139. | ||
Agudelo D, Bourassa P, Bruneau J, Bérubé G, Asselin E, Tajmir-Riahi HA. Probing the binding sites of antibiotic drugs doxorubicin and N-(trifluoroacetyl) doxorubicin with human and bovine serum albumins. PLoS One. 2012;7(8):e43814. | ||
Fülöp Z, Gref R, Loftsson T. A permeation method for detection of self-aggregation of doxorubicin in aqueous environment. Int J Pharm. 2013;454(1):559–561. | ||
Peters T Jr. All About Albumin: Biochemistry, Genetics, and Medical Applications. San Diego, USA: Academic Press; 1996. | ||
Abi-Habib RJ, Singh R, Liu S, Bugge TH, Leppla SH, Frankel AE. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol Cancer Ther. 2006;5(10):2556–2562. | ||
Yardley DA. nab-Paclitaxel mechanisms of action and delivery. J Control Release. 2013;170(3):365–372. | ||
Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75(1):13–33. | ||
Aller SG, Yu J, Ward A, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–1722. | ||
Huang Y, Sadée W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Lett. 2006;239(2):168–182. | ||
O’Halloran TV, Ahn R, Hankins P, Swindell E, Mazar AP. The many spaces of uPAR: delivery of theranostic agents and nanobins to multiple tumor compartments through a single target. Theranostics. 2013;3(7):496–506. | ||
Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–758. | ||
Ho JA, Fan NC, Jou AF, Wu LC, Sun TP. Monitoring the subcellular localization of doxorubicin in CHO-K1 using MEKC-LIF: liposomal carrier for enhanced drug delivery. Talanta. 2012;99:683–688. | ||
Han M, Vakili MR, Soleymani Abyaneh H, Molavi O, Lai R, Lavasanifar A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol Pharm. 2014;11(8):2640–2649. | ||
Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32(1):37–43. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. |
Supplementary materials
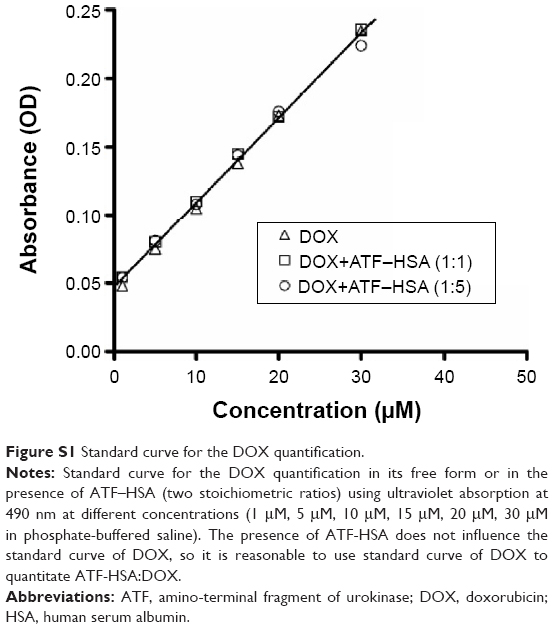 | Figure S1 Standard curve for the DOX quantification. |
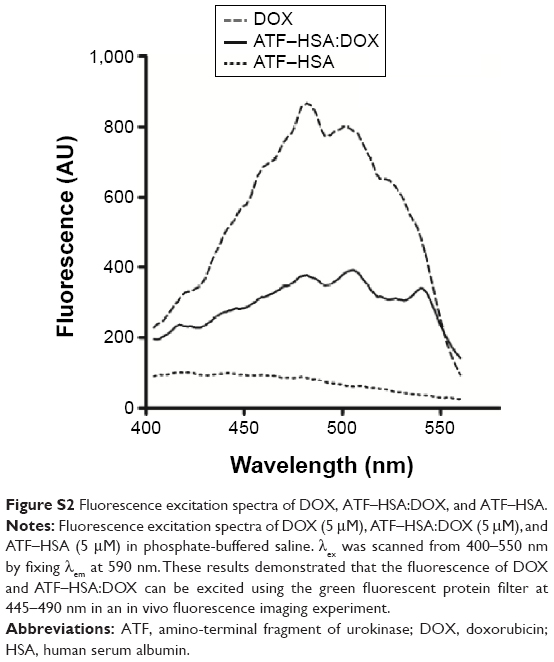 | Figure S2 Fluorescence excitation spectra of DOX, ATF–HSA:DOX, and ATF–HSA. |
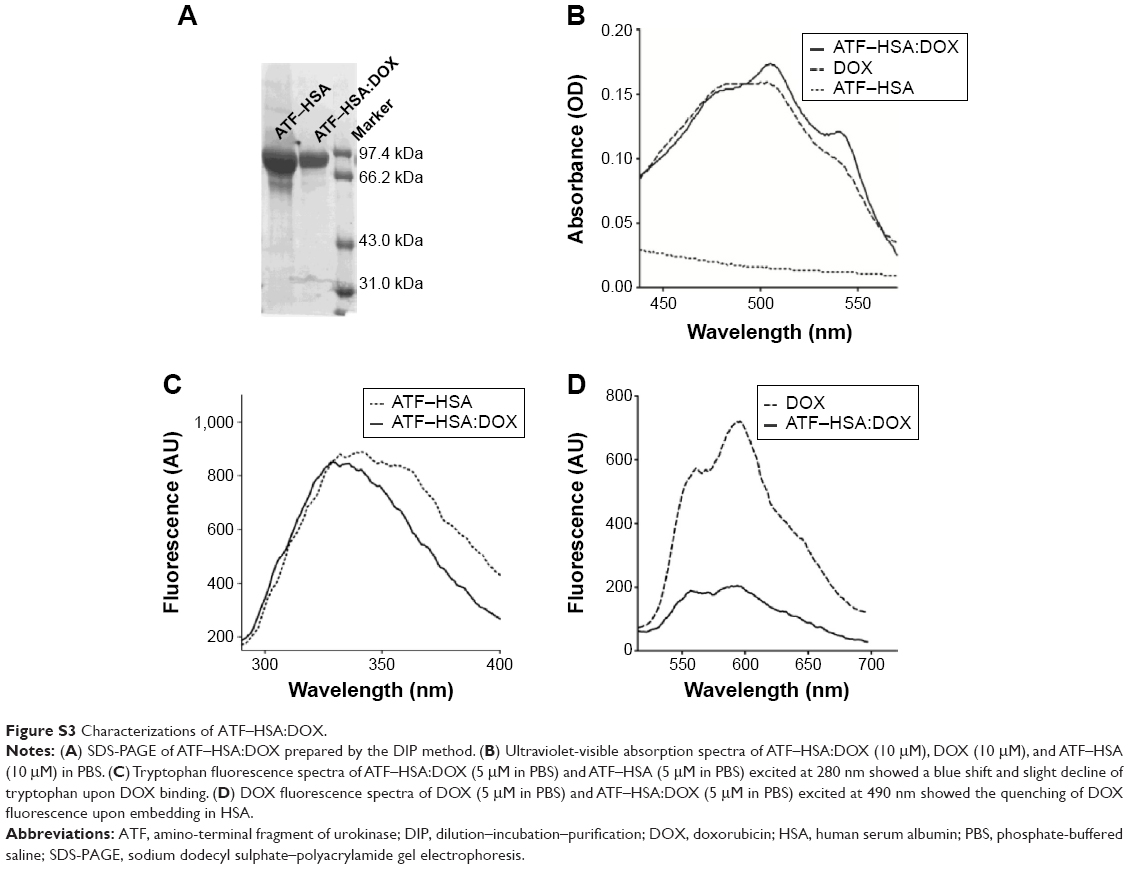 | Figure S3 Characterizations of ATF–HSA:DOX. |
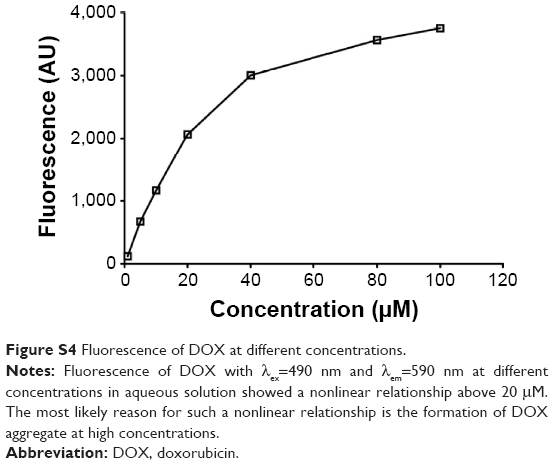 | Figure S4 Fluorescence of DOX at different concentrations. |
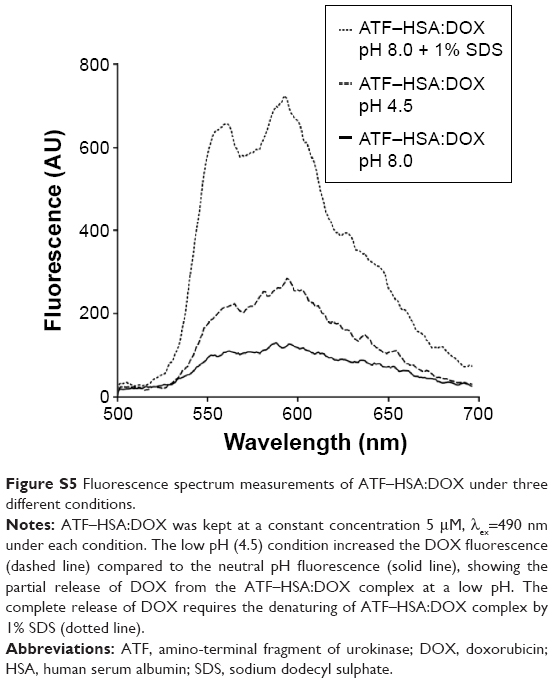 | Figure S5 Fluorescence spectrum measurements of ATF–HSA:DOX under three different conditions. |
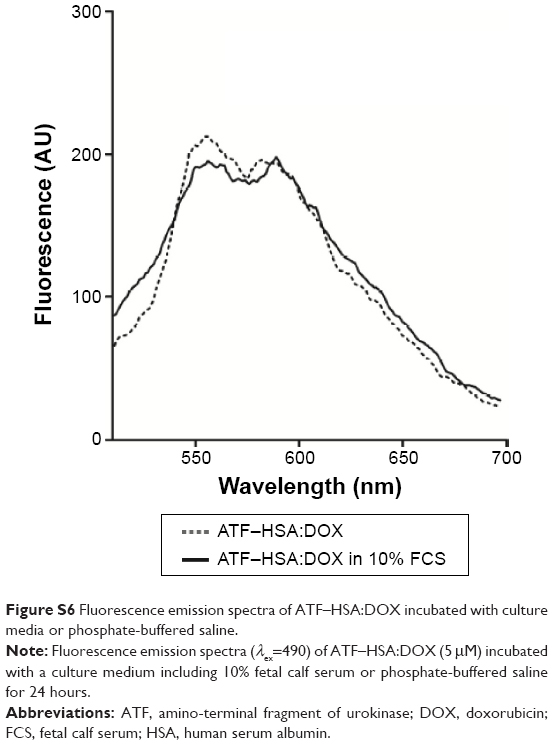 | Figure S6 Fluorescence emission spectra of ATF–HSA:DOX incubated with culture media or phosphate-buffered saline. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.