Back to Journals » Journal of Pain Research » Volume 13
Dual-Acting Peripherally Restricted Delta/Kappa Opioid (CAV1001) Produces Antinociception in Animal Models of Sub-Acute and Chronic Pain
Authors Hartrick CT, Poulin D, Molenaar R, Hartrick A
Received 16 May 2020
Accepted for publication 20 August 2020
Published 6 October 2020 Volume 2020:13 Pages 2461—2474
DOI https://doi.org/10.2147/JPR.S262303
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Craig T Hartrick,1 Dominic Poulin,2 Rebekka Molenaar,3 Allison Hartrick1
1Caventure Drug Discovery, Bloomfield Hills, MI, USA; 2Charles River Laboratories, Montreal, Canada; 3American Preclinical Services, Minneapolis, MN, USA
Correspondence: Craig T Hartrick Email [email protected]
Background: The development of highly efficacious alternatives to mu-opioid analgesics represents an urgent unmet medical and public health need. In the presence of inflammation both delta- and kappa-opioid agonists, acting on peripheral sensory neurons, mediate analgesia. The dual-acting, peripherally restricted kappa/delta-opioid agonist, CAV1001, was tested in four rodent pain models.
Methods: Experiment 1 – Formalin testing in mice. Three doses (1– 10 mg/kg) of CAV1001 or ICI204448 at 30 minutes were tested after formalin injection. Spontaneous nocifensive responses were video recorded. Experiment 2 – Complete Freund’s Adjuvant (CFA)-induced arthritis. CFA was injected into the ankle joint of rats. Joint compression thresholds (JCT) were measured. CAV1001 was compared to celecoxib. Experiment 3 – Spinal nerve ligation (SNL) in rats. Paw compression thresholds (PCT) were measured. CAV1001 was compared to gabapentin. Experiment 4 – MMRT-1 bone cancer implantation into the rat tibia. Weight-bearing was assessed. CAV1001 was compared to morphine.
Results: In Phase 2 of the formalin model, CAV1001 (1 mg/kg) significantly reduced pain behaviors to a degree comparable to the peripherally restricted kappa-opioid agonist, ICI204448 (10 mg/kg). CAV1001 (10 mg/kg) effectively eliminated pain behaviors associated with phase 2. In the CFA-induced arthritis model, a significant increase in JCTs, similar to the comparator celecoxib, was observed with CAV1001 at 1 mg/kg at 2 hours; CAV1001 (10 mg/kg) was effective at 1 hour. In the SNL model, both the comparator gabapentin and CAV1001 (5 mg/kg) significantly reduced PCT at 2 hours, but at 4 hours, the CAV1001 thresholds improved to baseline. CAV1001 10 mg/kg significantly improved weight bearing at 4-hour post-dosing compared to baseline following MMRT-1 implantation.
Conclusion: CAV1001 demonstrated efficacy in several different preclinical pain models. Time- and dose-dependent differences in the efficacy of CAV1001 amongst these rodent pain models parallel the degree of underlying inflammation.
Keywords: inflammatory pain, CFA-inflammatory arthritis, neuropathic pain, bone cancer pain, spinal nerve ligation, formalin model
Introduction
Trauma or other injuries to peripheral tissues initiate an inflammatory response. This process provides both protective and restorative roles in recovery, but also induces sensitization of nociceptors in the peripheral tissues.1 The resultant hyperalgesia following sensitization is proportionate to the degree of inflammation.2 When the inflammation is prolonged or persistent, chronic pain results, which can be severe and debilitating.3
The pharmacologic treatment of moderate-to-severe pain has historically been dominated by mu-opioid analgesics. Unfortunately, centrally-acting mu-opioid agonists not only induce unwanted and even life-threatening adverse effects acutely, but they also precipitate tolerance and altered pain pathways within a week of use.4 Moreover, chronic administration of opioids can even make pain worse in some cases.5 Consequently, the use of mu-opioid analgesics for persistent and chronic pain has become the subject of debate. Moreover, some pain states, such as neuropathic pain, are well known to be resistant to mu-opioids unless given concurrently with adjunctive medications.6 Proinflammatory mechanisms induced by mu-opioids, leading to glial activation, can contribute to the worsening of pain and the induction of hyperalgesia with continued use.7 The development of efficacious analgesics devoid of central mu-opioid adverse effects such as sedation, respiratory depression and risk for addiction, and that are not proinflammatory, is an urgent unmet medical need.
The pathophysiology of chronic pain includes a number of alterations in the pain pathway itself. Some of these changes reduce the effectiveness of traditional mu-opioid analgesics. It is well known that neuropathic pain is characteristically difficult to treat with mu-opioid analgesics resulting clinically in the use of polypharmacy. Because of the phenotypic shift in neuropathic pain away from mu-opioid dominated pain pathways, the use of co-analgesics has been one approach. Exploiting non-opioid and non-mu opioid pathways has been the subject of extensive research in this often devastatingly intractable pain condition.8 Kappa-opioid receptor agonists are active in neuropathic pain states, such as postherpetic neuralgia.9 Moreover, neuropathic pains, including diabetic neuropathy, also are associated with a significant degree of inflammatory response in the peripheral tissues.10,11 Bone cancer pain and pain resulting from metastases to bone are often very severe and also characteristically difficult to treat. One contributing factor is that while the severity necessitates strong analgesics, such as mu-opioid agonists, the mu-opioid receptors (MOR) are actually downregulated in bone cancer, making traditional opioids less effective.12 Consequently, alternatives to mu-opioid agonists that exploit changes in the pain pathway in association with bone cancer are needed.
Mu, delta, and kappa opioid receptors (MOR, DOR, KOR, respectively), each encoded by separate and unique genes, are expressed widely throughout the central and peripheral nervous systems and other tissues including the gastrointestinal and immune systems.13 Endogenous opioid peptides differentially interact with these receptors following volumetric release into synaptic and extrasynaptic spaces.14 Moreover, each of these receptors has a distinct anatomic expression pattern.15,16 MOR and DOR are co-expressed in unmyelinated pain fibers in peripheral tissues and both KOR and DOR are expressed in peripheral sensory neurons.17,18
Phenotypic shifting in opioid receptor distribution, trafficking, and function occurs following a variety of pathological states, including peripheral tissue injury.16 Inflammatory injuries cause an increase in axonal transport of MORs from the DRG of sensory neurons into the periphery.19–21 Additionally, extrasynaptic MORs in immune cells are important mediators of inflammation-induced pain.22 KORs are also constitutively present in peripheral sensory neurons.23 In contrast, constitutively present, but normally quiescent, DORs become activated in the presence of inflammation.24 This DOR activation is associated with allosteric facilitation of KORs.24 A dual-acting delta/kappa-opioid agonist that is restricted to the periphery would take advantage of this synergy while avoiding unwanted dysphoria, hallucinations, and other central effects associated with kappa and delta opioid agonists.
The hexahydrodiimidazodiazepine heterocyclic peptidomimetic, CAV1001 (Caventure Drug Discovery, Bloomfield Hills, MI, USA) is a peripherally restricted dual-acting delta/kappa opioid agonist. Both the opioid receptor selectivity and peripheral restriction of this novel diazaheterocyclic have been previously described by Eans et al.25 These investigators identified this peptidomimetic, formerly known as Compound 2065–14, as an analgesic in mice that acts peripherally. In the warm water (55° C) tail-withdrawal assay, morphine at 10mg/kg i.p. and CAV1001 administered at either 10 mg/kg i.p. or the equivalent oral dose (30 mg/kg p.o.) were equianalgesic. However, in the acetic acid writhing assay CAV1001 at a dose of 1 mg/kg i.p. was as effective as morphine 10 mg/kg i.p. Efficient absorption without penetration across the blood-brain barrier (BBB) was confirmed by comparing blood levels to brain levels following oral administration (30 mg/kg p.o.). While blood levels were readily demonstrated, only negligible amounts were observed in brain samples at 30, 60, and 90 minutes after dosing. Impermeability to the BBB was confirmed by pretreatment with the BBB impermeable opioid antagonist, naloxone methiodide. Antagonist-induced antinociception was only noted when treated on the same side of the BBB as CAV1001 administration (central: CAV1001 30 nmol i.c.v./naloxone methiodide 30 nmol i.c.v; peripheral: CAV1001 10 mg/kg i.p./naloxone methiodide 30mg/kg s.c.). Further, neither reward nor aversion effects were noted as assessed by conditioned place preference (CPP) (30 nmol i.c.v.). Additionally, in contrast to morphine, there was no acute analgesic tolerance when 5 nmol i.c.v. was followed by 0.3–100 nmol i.c.v. dosing. There was also a lack of effect on locomotor function or balance on rotarod testing (30 nmol i.c.v.). Peripherally, due to negligible mu-opioid activity, no appreciable slowing of gastrointestinal transit was observed following charcoal ingestion.
The purpose of this work is to evaluate CAV1001 in a range of subacute and chronic pain models in rodents. Specifically, we now report CAV1001 compared to a peripherally restricted kappa opioid receptor (KOR) agonist to assess the degree of synergy offered by the delta opioid receptor (DOR) agonist activity under inflammatory conditions without central nervous system involvement. The efficacy of CAV1001 was then assessed in preclinical pain models associated with varying degrees of inflammatory response. Although these experiments were not designed to measure the inflammatory response, the variable extent to which the inflammatory cascade is elicited in these models has been the subject of extensive previous study. With a dual-acting DOR/KOR agonist, greater analgesic activity might be anticipated in conditions associated with greater degrees of inflammation.
Methods
Experiment 1: Formalin Model
Subcutaneous plantar injection of formalin causes a bi-phasic nocifensive behavioral response in rodents. The early acute phase (Phase 1) lasts for about 5–10 minutes, following which an interphase occurs without any discernible nociceptive reactions. A late phase (phase 2) nociceptive reaction then ensues continuing from about 20 minutes after formalin injection and continues until about 60 minutes after injection. Phase 2 of the formalin model arises as a consequence of the induction of the inflammatory response. The late phase (phase 2), in particular, is considered as a pharmacodynamic surrogate of central sensitization. CAV1001 (MW 486.3; 98.6% purity; Torrey Pines Institute for Molecular Studies, Port St. Lucie, FL, USA) was compared to a peripherally restricted kappa agonist, ICI204448 (MW 465.4; >98% purity; Sigma, Natick, MA, USA) in the formalin model in the mouse at the Charles River Laboratories in Montreal, CA.
Following IACUC approval in accordance with the International Association for the Study of Pain Guidelines for the Use of Animals in Research (Charles River Montreal Institutional Animal Care and Use Committee; CR-MTL IACUC#: 5,900,579; 03/10/17) and acclimation for a minimum of 6 days, individually housed male mice (C57BL6; Charles River Ltd., Canada) weighing 20–30 g were randomly pretreated with inert vehicle (phosphate buffered saline; PBS), ICI204448 1mg/kg, ICI204448 3mg/kg, ICI204448 10mg/kg, CAV1001 1mg/kg, CAV1001 3mg/kg, or CAV1001 10 mg/kg administered IP (intraperitoneal) 30 minutes prior to formalin injection (Table 1). Food and water were provided ad libitum throughout the study; a light cycle of 12-hour light and 12-hour dark was maintained; nesting, hiding and chewing materials were provided for psychological and environmental enrichment.
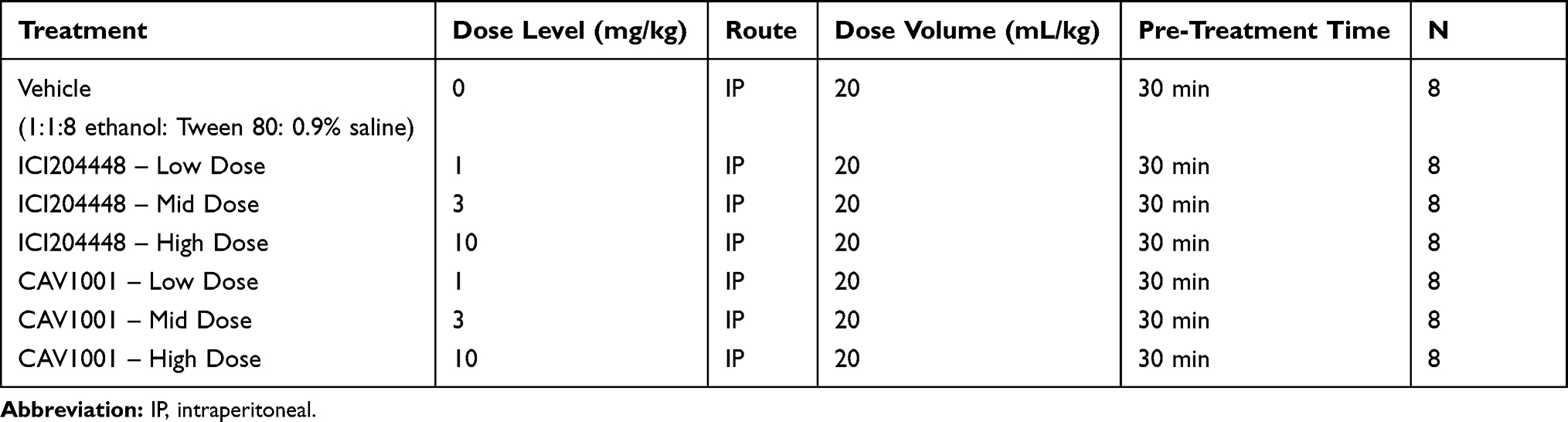 |
Table 1 Formalin Study Design |
Animals were next acclimated to an observation chamber for 15 minutes immediately prior to formalin injection. All animals received an intraplantar subcutaneous injection of 30 μL of freshly prepared room temperature 5% formalin in PBS into the left hind paw, then immediately placed into an observation chamber where formalin-evoked spontaneous nociceptive behaviors in the mice were continuously recorded for 40 min using a commercial camcorder.
Scoring from the recorded video files was done off-line using a PC by an observer who had been validated to score such nociceptive behavior in rodents and was blinded as to study group assignment. The total time spent in each 5-minute bin was recorded in seconds using a stop-watch for the following nociceptive behaviors: biting and licking of the formalin-injected paw. The cumulative total of behaviors recorded in bins 20–25, 25–30, and 30–35 minutes were defined as the sum of phase 2 behaviors. Percentage maximal possible effects (%MPE) during phase 2 were calculated as previously described:26
%MPE = [sum of phase 2 behaviors with drug]/[sum of phase 2 behaviors with control] x 100
Curves were fit using simple linear regression. Statistical analyses were performed using Graphpad Prism 8.42 for Mac (San Diego, CA).
Study size determination, eligibility, model validity: A minimum of eight animals per group was required to achieve meaningful differences between groups at a significance level of p<0.05. Animals without an interphase (ie between phase 1 and phase 2) demonstrating reduced behaviors were excluded as model failures.
Experiment 2: CFA Model of Inflammatory Arthritis
This experiment evaluated the efficacy of a single intraperitoneal injection of CAV1001 on hyperalgesic nociceptive behaviors in the CFA (Complete Freund’s Adjuvant) Model of Inflammatory Arthritis Pain in rats at American Preclinical Services, Minneapolis, MN, USA.
Following IACUC approval in accordance with the International Association for the Study of Pain Guidelines for the Use of Animals in Research (APS IACUC# CDD003-PH15; 02/02/18) and a minimum 5-day acclimation period, inflammatory arthritis pain was induced in male (175–200 g), Sprague-Dawley (Envigo; Indianapolis, IN) rats by intracapsular injection of 50 μL of 100% complete Freund’s adjuvant (CFA) into the tibiotarsal joint of the left hind leg. Animals were anesthetized (isoflurane 1–4%) prior to CFA injection.
Ipsilateral and contralateral joint compression thresholds (JCTs) were determined using a digital Randall-Selitto device (dRS; IITC Life Sciences; Woodland Hills, CA) prior to CFA injection and 14 days post-CFA (Day 0), prior to study article administration. At that time 50 animals that met inclusion criteria, ie animals exhibiting at least a 25% decrease in Day 0 pre-dosing baseline (BL) JCT compared to pre-injury baseline JCT, were assigned to 5 groups with 10 animals per group.
Treatment group assignment was based on Day 0 pre-dosing JCTs such that group means of the ipsilateral JCTs were approximately equal. All animals were ranked by ipsilateral JCT and randomly assigned within stratified subgroups according to the total number of treatment groups in the study. Animals were administered a single dose of the CAV1001 (1, 5, or 10 mg/kg IP), the anti-inflammatory/cyclooxygenase-2 inhibitor celecoxib (30 mg/kg PO [oral gavage]: active control; internal validity; 98% purity; Toronto Research Chemicals, North York, Canada) or vehicle on Day 0 (Table 2). Dosing sequence was based on the animal number so that the distribution of treatments across a given set of animals was not predictable.
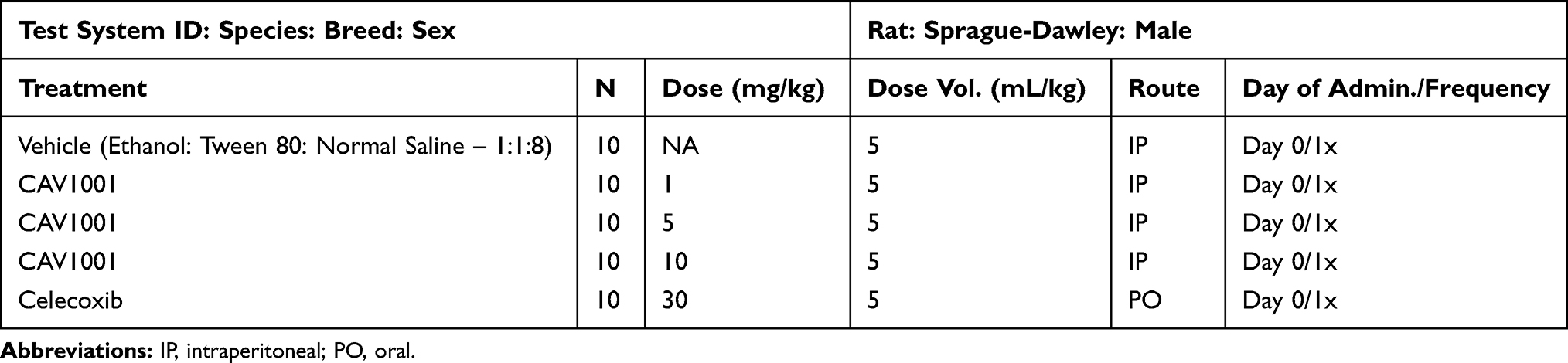 |
Table 2 Arthritis (CFA Ankle): Study Design |
Animals were allowed to acclimate to the testing room for a minimum of 15 minutes before mechanical hyperalgesia testing. For testing, animals were placed in a restraint sling that suspended the animal, leaving the hind limbs exposed for testing. The stimulus was applied to the ankle joint by a blunt tip and pressure was applied gradually over approximately 10 seconds. JCT values were recorded at the first observed nocifensive behavior (vocalization, struggle, or withdrawal). One reading per joint was taken at each time point, and a maximum stimulus cut-off of 500 g was used to prevent injury. The mean and standard error of the mean (SEM) were determined for ipsilateral and contralateral joints for each treatment group at each time point. Bilateral JCTs were determined 1, 2, and 4 hours after post-dosing. All behavioral evaluations were performed in a blinded manner, with all experimenters involved in the study being unaware of the group assignment of any animal they were testing. One staff member prepared the dose solutions, coded the syringes of solutions, and created the blind (treatment key).
Study size was calculated using the Massachusetts General Hospital on-line power calculator (http://hedwig.mgh.harvard.edu/sample_size/size.html). To detect a JCT difference of 68 g with a power of 80% and a standard deviation of 51, 10 animals per group were required. Based upon prior experience it was anticipated that approximately 10% of animals would fail to reach inclusion criteria. Statistical analyses were conducted using Prism 8.42 for Mac (Graphpad, San Diego, CA, USA). The level of significance was set at α = 0.05. The development of the pain model was assessed by t-test with internal validation with active control and the effect of CAV1001 assessed using one-way ANOVA with Dunnett’s test for multiple comparisons.
Experiment 3: Spinal Nerve Ligation
This study evaluated the efficacy of a single intraperitoneal injection of CA1001 and the active comparator, the anticonvulsant gabapentin, in the spinal nerve ligation (SNL) model for neuropathic pain in the rat at American Preclinical Services, Minneapolis, MN, USA.
Following IACUC approval (APS IACUC# CDD002-PH01; 01/26/18) and acclimation, neuropathy was induced by spinal nerve ligation (SNL) under isoflurane anesthesia (up to 5%) in male (75–100 g), Sprague-Dawley rats (Envigo; Indianapolis, IN) by surgically ligating the 5th and 6th lumbar spinal nerves (L5 and L6). Mechanical sensitivity was assessed via paw compression thresholds (PCTs) using a dRS device fitted with a cone-shaped tip. Thresholds were determined prior to surgery and 15 days post-surgery, prior to study article administration.
Animals meeting inclusion criteria were assigned to 5 groups with 10 animals per group. Only animals that exhibited at least a 25% decrease in PCT from pre-injury BL to pre-dosing BL, or a 1.5 ratio of contralateral/ipsilateral thresholds were included. Included study animals were administered a single dose of CAV1001 (1, 5, or 10 mg/kg IP), the gabapentin (100 mg/kg IP; active control; internal validity; >98% purity; Spectrum Chemical Manufacturing Corporation, Gardena, CA, USA), or vehicle on Day 0 (15 days after SNL surgery). Group assignment was based on Day 0 pre-dosing dRS PCTs such that group means of ipsilateral paw compression thresholds were equal. Animals were ranked by ipsilateral PCT measurement from lowest to highest and treatment group assigned randomly within stratified subgroups according to the total number of treatment groups. Animals were dosed in sequence based on the animal number so that the distribution of treatments across a given set of animals was not predictable. Behavioral assessments were made by investigators who were blind to treatment allocation (Table 3).
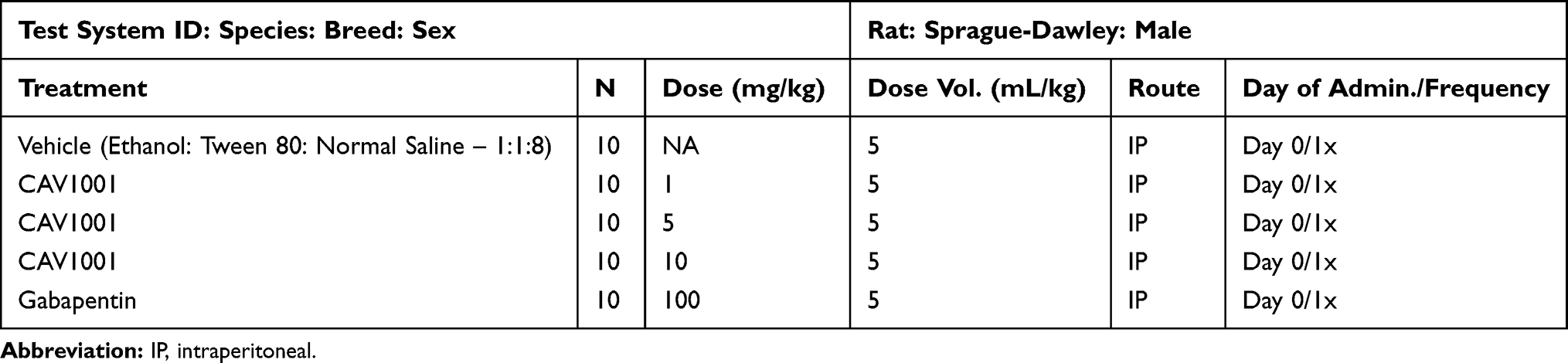 |
Table 3 Neuropathic Pain (SNL): Study Design |
PCTs were assessed at 1, 2, and 4 hours after administration. Animals were given a minimum of 15 minutes to acclimate to the room prior to testing. As in the CFA model previously described, the animals were suspended in a restraint sling for testing. The pressure stimulus was applied gradually over a period of 10 seconds to the plantar surface of the hind paw. PCT values were recorded at the first observed nocifensive behavior (vocalization, struggle, or withdrawal). One reading per paw was taken at each time point, and a maximum stimulus cut-off at 300 g was used to prevent injury. Mean and SEM were determined for ipsilateral and contralateral paws for each group at each time point.
Study size was calculated using the Massachusetts General Hospital on-line power calculator (http://hedwig.mgh.harvard.edu/sample_size/size.html). In order to detect a threshold difference of 4.5 g with a power of 80%, (assuming mean control = 3.42, mean treated = 7.92, standard deviation = 2.1) group size of 10 was required for each treatment. Because approximately 10% of animals undergoing surgery may not meet inclusion criteria, surgery was performed on 55 animals. Statistical analyses were conducted using Prism 8.42 for Mac (Graphpad, San Diego, CA, USA). The level of significance was set at α = 0.05. The development of the pain model was assessed by t-test with internal validation with active control and the effect of CAV1001 assessed using one-way ANOVA with Dunnett’s test for multiple comparisons.
Experiment 4: Bone Cancer
This study evaluated the efficacy of a single intraperitoneal injection of CAV1001 and the comparator, morphine, a mu-agonist opioid, in the mammary gland carcinoma (MRMT-1) model for osteolytic bone cancer pain in the rat at American Preclinical Services, Minneapolis, MN, USA.
Following IACUC approval (APS IACUC # CDD001-PH12; 01/23/18, amendment approval date: 02/16/18) and acclimation for a minimum of 5 days, under isoflurane anesthesia (up to 5%), bone cancer pain was induced by injection of 3000 MMRT-1 cells into the intramedullary space of the left tibia in male (100–250 g) Sprague-Dawley rats (Envigo; Indianapolis, IN). Weight bearing was assessed prior to inoculation, at BL (Day 0) prior to test substance administration 14–18 days after inoculation, and at 1, 2 and 4 hours after test article administration. Test articles were vehicle, CAV1001, and the active comparator, morphine.
All testing was performed in a blinded manner where experimenters were unaware of group assignment. Hind limb weight bearing scores (WBS) were measured using a Linton Incapacitance Tester (Stoelting Co., Wood Dale, IL). Animals were acclimated to the testing room for a minimum of 15 minutes prior to testing. When the animals were in the correct position in the acrylic testing chamber, the average force exerted individually by each hind paw over a 3-second interval was taken. Three evaluations of force per animal were taken at each time point. The percent WBS for the injured limb was calculated for each evaluation of force as follows:
%WBS = [weight on Left leg/(weight on left leg + weight on right leg)] x 100
The mean of the three values was taken as the %WBS for that time point.
Group assignment was based on Day 0 pre-dosing %WBS scoring so that the group means were approximately equal. Animals were ranked by %WBS from lowest to highest and treatments randomly assigned within stratified sub-groups according to the total number of treatment groups. As in Experiments 2 and 3, successful model creation was defined by both the behavioral response, in this case, a significant decrease in %WBS, and by significant reversal of the behavioral response with the active control, in this case, morphine (purity: USP grade; Hospira, Lake Forest, IL, USA).
Study size was determined using substantial data generated in previous studies and the Massachusetts General Hospital on-line power calculator as before. In order to detect a threshold difference of 30 units (%WBS) with a power of 90% (SD=19), 10 animals per group were required. Given the historic failure rate in this model of 30%, 65 rats were requested (n=10/group with 5 treatment groups, plus 15 additional animals). With 10 animals in the vehicle group completed, the model failed to meet the second criteria (significant reversal with morphine) at any time point. A revised study size calculation using the interim data suggested that detecting a threshold difference of 11.5 units (%WBS) with the power of 90% (using SD=9), with a 30% failure rate, would require 14 animals per group. The IACUC amendment was approved allowing 3 groups to continue: vehicle, active control (morphine) and CAV1001 (high dose; 10 mg/kg). The CAV1001 1 mg/kg and 5 mg/kg groups, being underpowered (n=8 per group), were excluded from analysis (Table 4). Statistical analyses were conducted using Prism 8.42 for Mac (Graphpad, San Diego, CA, USA). The level of significance was set at α = 0.05. The development of the pain model was assessed by t-test. Internal validation with active control and the effect of CAV1001 assessed using one-way ANOVA with Dunnett’s test for multiple comparisons.
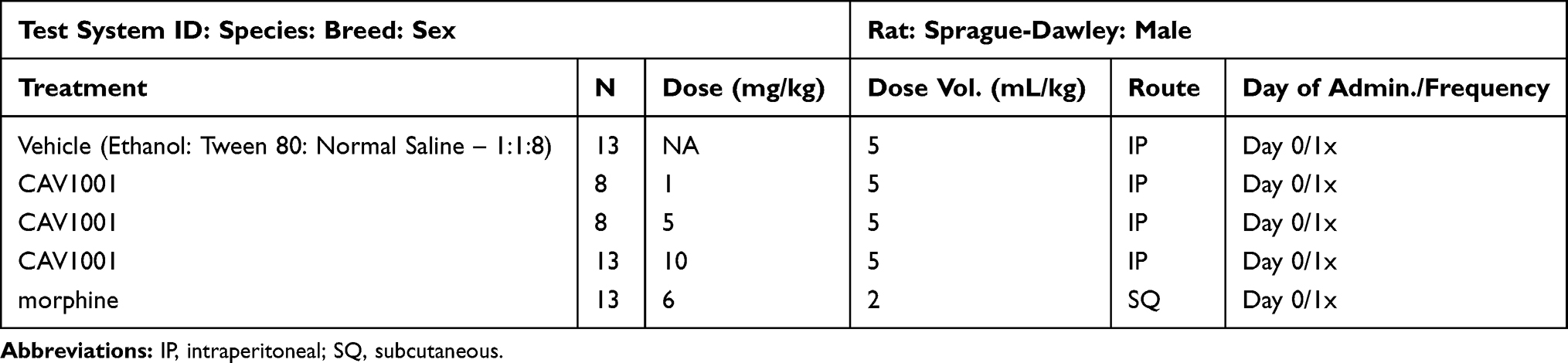 |
Table 4 Bone Cancer Study Design |
Results
Experiment 1: Formalin Testing
Four animals receiving CAV1001 mid-dose, one animal receiving ICI204448 mid-dose and one animal receiving vehicle exhibited an injury resulting in persistent or increased nociceptive behaviors after phase 1, without any discernable interphase. All animals that did exhibit an interphase were included in the linear regression curve generation. However, the failed model mid-dose (3 mg/kg) animals rendered those groupings underpowered and thus were excluded from further statistical analysis. Mid-dose grouping results, including those with failed model development, are provided as supplemental data (Table S1). The results of the remaining groupings are provided in Figure 1.
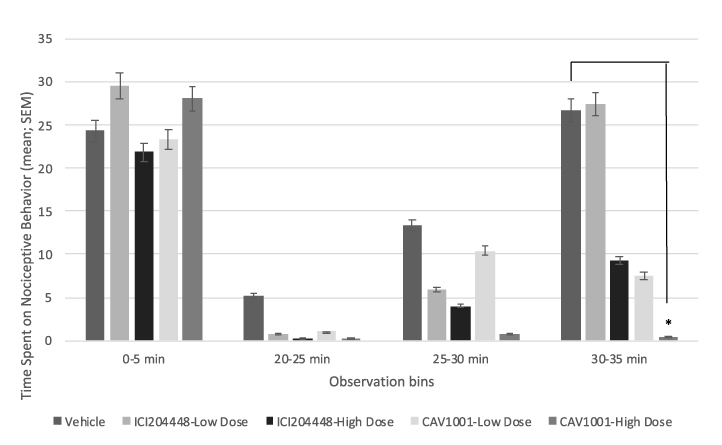 |
Figure 1 Formalin testing. Notes: *CAV1001 10mg/kg v. vehicle (30-35 min), p<0.03; Phase 2 comparisons (cumulative: 20-35 min): CAV1001 10mg/kg v. vehicle, p<0.003; ICI204448 10mg/kg v. vehicle, p<0.05; CAV1001 10mg/kg v. ICI204448 10mg/kg, p<0.01. |
Neither agent (CAV1001 or ICI204448) was effective in reducing the immediate (0–5 minutes) response to formalin injection (phase 1). Time spent in nociceptive behavior with CA1001 1 mg/kg (mean ± SEM: 18.88 ± 8.08 [n=8; range: 0–57]; % inhibition compared to vehicle: 58.29 ± 17.86 [n=8; range: -26.0–100]) was similar to ICI204448 10 mg/kg (13.50 ± 9.37 [n=8; range: 0–77]; % inhibition: 70.17 ± 20.70 [n=8; range: −70.2–100]) in phase 2 (cumulative: 20–35 minutes). CA1001 10 mg/kg was significantly more effective, with 96.96 ± 1.56% inhibition [n=8; range: 89.0–100], than was ICI204448 10 mg/kg (phase 2 mean behavior time ± SEM; 1.38 ± 0.71 [n=8; range: 0–5], p<0.01). Moreover, CA1001 10 mg/kg effectively prevented the development of the phase 2 hyperalgesic response (nociceptive behavior during the 20–35 minute observation period for vehicle: 44.25 ± 18.93 [n=8; range: 1–146], p<0.003).
Dose–response curves for nocifensive behaviors and %MPE created using simple linear regression demonstrate similar slopes with CAV1001 being shifted leftward compared to ICI204448 at each dose studied (Figure 2). For phase 2 pain behaviors the dose–response curve for CAV1001 described by y = −2.34x + 26.2 (R2 = 0.79) was nearly parallel to that seen for ICI204448, described by y = −2.72x + 42.3 (R2 = 0.80). For the %MPE a similar relationship was noted with the line for CAV1001, being described by y = −5.16x + 57.8 (R2 = 0.78), and the line for ICI204448 described by y = −6.03x + 93.6 (R2 = 0.80).
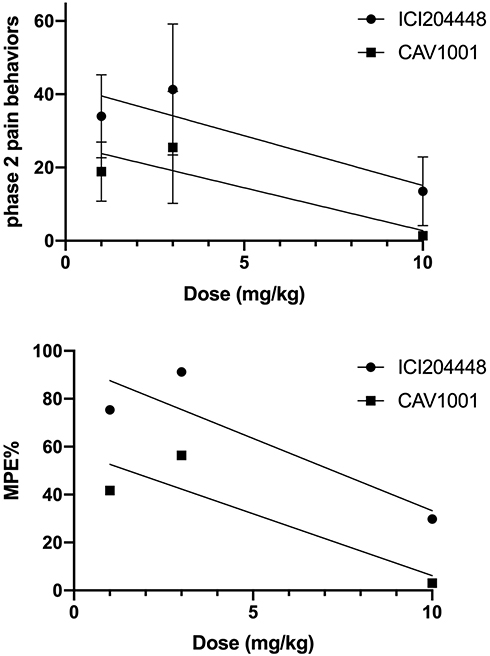 |
Figure 2 Dose-response: CAV1001 versus ICI204448. |
Experiment 2: CFA-Induced Arthritis
Arthritis: Hyperalgesia Development
To verify the development of mechanical hyperalgesia due to CFA-induced rheumatoid arthritis pain, bilateral JCTs prior to CFA injection, and 14 days after CFA-injection at pre-dosing on Day 0 and at 1, 2, and 4-hours post-dosing. Ipsilateral JCTs were compared to contralateral JCTs using an unpaired t-test at each time point. Ipsilateral JCTs were significantly lower at all post-CFA time points (pre-dose BL, 1-h and 4-h: p<0.0001; 2-h: p<0.0002; one-way ANOVA with Dunnett’s Multiple Comparison Test), indicating persistent mechanical hyperalgesia due to CFA injection.
To verify the sensitivity of the assay to detect changes in mechanical hyperalgesia, JCTs of celecoxib-treated animals were compared to vehicle-treated animals at all post-CFA time points (repeated-measures, one-way ANOVA; Dunnett’s Multiple Comparison Test). The JCTs of the celecoxib-treated animals were significantly higher at 2 hours (mean ± SEM [95% CI]: celecoxib 335.1 ± 22.21 [284.9–385.3] n=10; range: 196–434; vehicle 210.5 ± 18.78 [168.0–253.0] n=10; range: 106–323; p=0.0015) and 4 hours post-dose (celecoxib 329.5 ± 32.50 [256.0–403.0] n=10; range: 137–500; vehicle 187.4 ± 12.85 [158.3–216.5] n=10; range: 108–255; p=0.02) (Figure 3).
Efficacy of CAV1001 in CFA-induced Arthritis
Intraperitoneal administration of CAV1001 significantly reversed CFA-induced mechanical hyperalgesia. All three doses of CAV 1001 significantly reversed mechanical hyperalgesia (one-way ANOVA with Dunnett’s Multiple Comparison Test) at the 2-hour (JCT mean ± SEM [95% CI]: vehicle 210.5 ± 18.78 [168.0–253.0] n=10; range: 106–323; CAV1001 1 mg/kg 347.1 ± 28.74 [282.1–412.1] n=10; range: 177–455, p=0.001; CAV1001 5 mg/kg 325.8 ± 19.62 [281.4–370.2] n=10; range: 200–412, p<0.006; CAV1001 10 mg/kg 370.2 ± 28.89 [304.9–435.5] n=10; range: 262–500, p=0.0001) and 4-hour time points (vehicle 187.4 ± 12.85 [158.3–216.5] n=10; range: 108–255; CAV1001 1 mg/kg 318.0 ± 30.82 [248.3–387.7] n=10; range: 151–500, p<0.003; CAV1001 5 mg/kg 306.7 ± 22.04 [256.8–356.6] n=10; range: 213–434, p<0.007; CAV1001 10mg/kg 380.4 ± 32.84 [306.1–454.7] n=10; range: 133–500, <0.0001) while the 10 mg/kg dose also significantly reversed mechanical hyperalgesia at the 1-hour time point (mean ± SEM [95% CI]: vehicle 204.5 ± 20.00 [159.3–249.7] n=10; range: 105–329; CAV1001 10 mg/kg 399.1 ± 36.60 [316.3–481.9] n=10; range: 224–500, p=0.0002) (Figure 3).
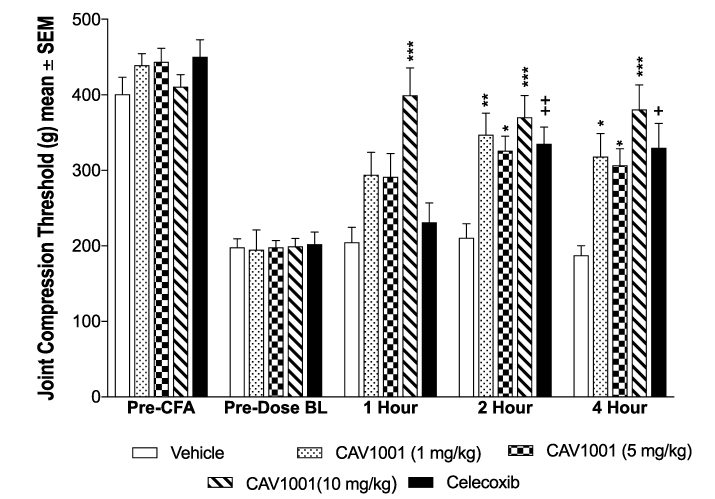 |
Figure 3 Efficacy of CAV1001 in CFA-induced JCT reduction. Notes: +: p<0.02; ++: p<0.002; *: p<0.01; **: p<0.001; ***: p<0.0002 (one-way ANOVA). |
Experiment 3: SNL Results
Neuropathic Pain: Model Development
As in experiment 2, in order to verify the development of mechanical hyperalgesia due to SNL surgery, ipsilateral and contralateral PCTs were assessed prior to surgery, post-SNL surgery prior to dosing (Day 0), and at 1, 2, and 4-hour post-dosing on Day 0. Ipsilateral PCTs were compared to contralateral PCTs using an unpaired t-test at each time point. Ipsilateral PCTs were significantly lower at all post-SNL time points in vehicle-treated animals (pre-dosing BL and 2-h: p<0.001; 1-h and 4-h: p<0.0001, one-way ANOVA), confirming the development of persistent mechanical hyperalgesia due to SNL surgery.
In order to further verify the model, 15 days after SNL surgery, on Day 0, mechanical hyperalgesia (PCTs) in animals receiving the positive control, gabapentin, were compared to vehicle controls (one-way ANOVA, Dunnett’s Multiple Comparison Test). Gabapentin significantly reversed SNL-induced mechanical hyperalgesia at 1 hour (mean ± SEM [95% CI]: gabapentin 140.5 ± 21.26 [92.41–188.6] n=10; range: 65–250; vehicle 72.00 ± 3.815 [63.37–80.63] n=10; range: 50–90, p<0.03), at 2 hours (gabapentin 137.5 ± 17.08 [98.86–176.1] n=10; range: 75–250; vehicle 78.50 ± 5.92 [65.11–91.89] n=10; range: 55–120, p<0.02) and at 4 hours (gabapentin 122.5 ± 16.01 [86.29–158.7] n=10; range: 65–250; vehicle 68.50 ± 3.88 [59.73–77.27] n=10; range: 50–85, p<0.034) (Figure 4).
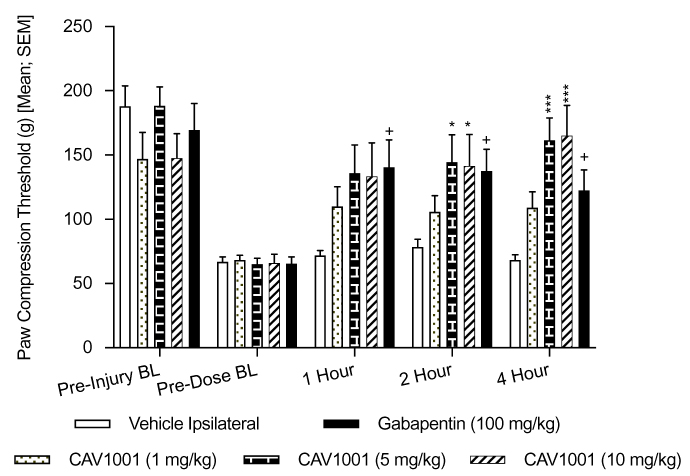 |
Figure 4 Efficacy of CAV1001 in SNL-induced mechanical hyperalgesia. Notes: +: p<0.05; *; p<0.05; ***: p<0.0003 (one-way ANOVA). |
Efficacy of CAV1001 in SNL-Induced Mechanical Hyperalgesia
CAV1001 at the 1 mg/kg dose did not significantly reverse mechanical hyperalgesia at any of the time points tested. Significant reversal of mechanical hyperalgesia was observed at the 2-hour time point for CAV1001 5 mg/kg (mean ± SEM [95% CI]: 144.5 ± 21.23 [96.47–192.5] n=10; range: 65–250, versus vehicle 78.50 ± 5.92 [65.11–91.89] n=10; range: 55–120) and for CAV1001 10 mg/kg (141.5 ± 24.35 [86.42–196.6] n=10; range: 70–250, p<0.035, one-way ANOVA with Dunnett’s Multiple Comparison Test). Significant reversal of mechanical hyperalgesia was also observed at the 4-hour time point for CAV1001 5 mg/kg (mean ± SEM [95% CI]: 161.5 ± 17.27 [122.4–200.6] n=10; range: 85–250, versus vehicle 68.50 ± 3.88 [59.73–77.27] n=10; range: 50–85) and for CAV1001 10 mg/kg (165.0 ± 23.71 [111.4–218.6] n=10; range: 85–250, p=0.0003, one-way ANOVA with Dunnett’s Multiple Comparison Test) (Figure 4).
Experiment 4: Bone Cancer Results
Bone Cancer Pain: Model Development
In order to verify the development of weight bearing incapacitance in this model, ipsilateral and contralateral %WBS were assessed prior to surgery, MMRT-1 implantation prior to dosing (Day 0), and at 1, 2, and 4-hour post-dosing on Day 0. Weight bearing asymmetry, as assessed by %WBS, was confirmed in the vehicle group by comparing pre-implantation BL to pre-injection BL (p<0.001; unpaired, two-tailed t-test). Mean hind limb weight bearing scores pre-dosing Day 0 (BL) were significantly lower than pre-implantation only at the 1-hour time point (mean ± SEM: 24.12 ± 2.849 n=13; range: 7.76–38.28, versus 50.31 ± 1.064 n=13; range: 9.17–66.48) in vehicle-treated animals (p<0.02). Consequently, the model, as defined a priori, failed to develop. The results from the underpowered groupings (CAV1001 1 mg/kg and CAV1001 5 mg/kg) are provided in the supplemental data (Table S2) but were otherwise excluded from further analysis.
CAV1001 Compared to Morphine
On study Day 0%WBS were assessed at BL (prior to dosing) and 1, 2, and 4-hour post-dosing with morphine (6mg/kg SQ) and CAV1001 (10mg/kg IP). Morphine administration significantly increased mean weight bearing scores at 1 hour (mean ± SEM: 46.52 ± 3.38, n=13; range: 24.75–62.02; p<0.001) and at 2 hours (39.56 ± 3.84, n=13; range: 15.27–61.64; p<0.006) compared to the pre-dosing BL (22.26 ± 3.24 n=13; range: 1.64–39.95; one-way ANOVA with Dunnett’s Multiple Comparison Test) (Figure 5).
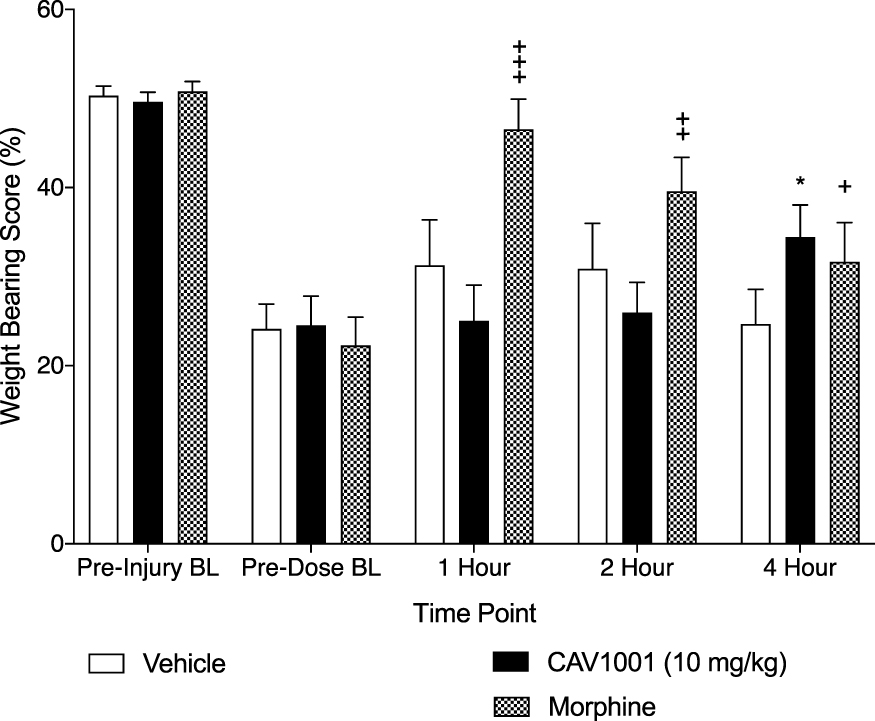 |
Figure 5 Effect of CAV1001 and morphine on pain induced weight bearing. Notes: Morphine compared to BL: +++: p<0.001, ++: p <0.006, +: p = not significant; CAV1001 compared to BL: *: p<0.003 (one-way ANOVA). |
CAV1001 at the 10 mg/kg dose did not significantly reverse weight bearing discrepancy at the 1-hour or 2-hour time points compared to BL. CAV1001 10 mg/kg did significantly impact %WBS at 4 hours compared to pre-dosing BL (mean ± SEM: 34.43 ± 3.58 n=13; range: 7.61–55.54; BL 24.49 ± 3.34 n=13; range: 3.38–37.65, p<0.003, one-way ANOVA with Dunnett’s Multiple Comparison Test) (Figure 5).
Discussion
The development of an efficacious alternative to mu-opioid agonists is a timely scientific challenge.27,28 The lack of clinically available alternatives to opioids contributes to the public health crisis known as the opioid epidemic. However, the inherent differences in mechanism associated with the different chronic pain conditions for which highly efficacious treatments are needed suggest that no single treatment will likely be suitable for all indications.
Despite the disparate mechanisms associated with chronic pain, a number of clinically important pain conditions are known to be associated with significant involvement of the inflammatory response.2 The relative contribution of this inflammatory response in the generation and perpetuation of the resultant pain conditions is variable, but the inflammatory response to tissue injury generally includes the elaboration of cytokines and substances that sensitize peripheral nociceptors, including bradykinin.29 Bradykinin is a mediator of peripheral sensitization in acute and subacute inflammatory pain states and the initiation of hyperalgesia as seen in postoperative pain, posttraumatic pain including burn pain.30
CAV1001 reduced hyperalgesia, as evidenced by both spontaneous and evoked pain behaviors in rodents, in several different pain models associated with differing degrees of inflammation. The hypothesis that the intensity of analgesia might relate to the degree of inflammation in the underlying pain state was consistent with the findings that the lowest doses tested were effective in the models having the highest degree of inflammatory response. Preclinical evidence previously mentioned suggests the potential for peripherally mediated kappa-opioid effects to be enhanced through activation of the delta-opioid receptors by the inflammatory mediator, bradykinin, possibly due to the presence of delta/kappa-opioid heterodimers in peripheral sensory neurons.18,24 Consequently, absent inflammation, the peripheral delta-opioid effects would be less significant, rendering a dual-acting kappa/delta agonist agent effectively a kappa-opioid agonist.
The second phase of the formalin test was used to compare the effect of a peripherally restricted kappa-opioid agonist, ICI204448, on an equimolar basis to the dual-acting kappa/delta-opioid agonist, CAV1001, in reducing the hyperalgesia resulting from this intensely inflammatory stimulus.31 While the peripherally restricted kappa agonist, at high dose, did significantly reduce the hyperalgesic response, the dual-acting delta/kappa agent produced similar efficacy at low dose, suggesting an order of magnitude difference in potency in this assay. At the higher dose, CAV1001 completely abolished the second phase of the formalin testing. This supports the concept that, in the presence of peripheral inflammation, the peripheral effect of the dual-acting kappa/delta-opioid agonist is superior to action at the kappa-opioid receptors alone.
Both subacute and chronic pain states often involve the inflammatory response to varying degrees, resulting in peripheral sensitization and perpetuation of pain. The peripheral sensitization of nociceptors due to neuroinflammatory responses has been documented in preclinical studies and in a number of chronic pain states including radiculitis, complex regional pain syndrome (CRPS), fibromyalgia, diabetic peripheral neuropathy, rheumatoid arthritis, and osteoarthritis.1,32 Accordingly, CAV1001 was subsequently tested in additional pain models having differing inflammatory components. Consistent with the hypothesis that increasing involvement with the inflammatory response would lead to a greater effect of CAV1001, increasing effect would be predicted in a primarily inflammatory pain model of arthritis, as compared to a neuropathic pain model where the inflammatory component is present but less pronounced.
The inflammatory arthritis model was effectively created as evidenced by hyperalgesia induced in vehicle-treated animals comparing ipsilateral and contralateral effects before and after injury.33 The model was further effectively confirmed using an active anti-inflammatory analgesic as a control, the cyclooxygenase-2 inhibitor, celecoxib. Testing various doses of CAV1001 in this model revealed a time and dose-dependent reduction in pain behavior. In CFA-induced inflammatory ankle arthritis, the lowest dose tested (CAV1001 1 mg/kg IP) was as effective as the standard comparator (celecoxib 30 mg/kg PO) at 2 hours and 4 hours postdosing. Celecoxib did not produce significant improvement at the 1-hour time point; however, at the highest dose tested (10 mg/kg) CAV1001 did significantly reduce pain behaviors at the 1-hour time point, the earliest time tested.
The SNL model is a reliable, reproducible, and durable for the induction of peripheral neuropathy in rodents. The model is also associated with an upregulation in inflammatory mediators and downregulation in factors inhibiting inflammation.34,35 While the neuroimmune response in peripheral tissues associated with neuropathic pain involves inflammatory mediators and glial cells, the pain relief provided by delta-opioid agonists in neuropathic pain is not entirely dependent upon action at the glial cells.36,37 Neuropathic pain is also associated with a phenotypic shift away from mu-opioid dominated pain pathways to noradrenergic pathways.38 Alpha-2 noradrenergic agonists have been successfully used clinically as co-analgesics in neuropathic pain states.39 Gabapentin is an inhibitor of the α2-δ2 subunit of the voltage-gated calcium channel. It has been shown to activate descending noradrenergic pathways and thus reduce hyperalgesia in rodents following peripheral nerve injury.40 In the third experiment presented, SNL was effectively created, as evidenced by hyperalgesia induced in vehicle-treated animals comparing ipsilateral and contralateral effects before and after injury. The model was confirmed using gabapentin as the active control. As with the previous experiment, testing various doses of CAV1001 revealed a time and dose-dependent reduction in pain behavior. In the SNL model, the lowest dose tested (CAV1001 1 mg/kg IP) was less effective than the standard comparator (gabapentin 100 mg/kg IP) at the 1-hour time point. However, moderate (5mg/kg IP) and high (10 mg/kg IP) dose were as effective as the active control at 2 hours and continued to increase in efficacy at 4 hours postdosing.
The elaboration of peripheral nerve sensitizing substances seen in bone cancer, including bradykinin, suggests the possibility of peripheral delta-opioid receptor agonism as an approach to improve treatment effectiveness in this recalcitrant pain state.41 In this study, both the severity of pain and the relative resistance to mu-opioids may have contributed to the difficulties in the development of the cancer pain model as defined (ie morphine reversal). Moreover, the rodent model not only faces the challenges in the ability of morphine to validate the model, but the induction of the model itself is also associated with a high failure rate. In this study, difficulties in inducing the model led to an increase in study size. Even after amendment, the study was apparently still underpowered. The model only met the dual criteria defined a priori for the significant discrepancy in weight bearing and its reversal by active comparator, morphine, at the 1-hour time point. However, comparisons to baseline showed improvement in weight bearing following morphine at 1 and 2 hours with the waning of effect at 4 hours. In contrast, CAV1001 at 10 mg/kg (high dose) demonstrated an increasing effect over time with significant effect compared to baseline at 4 hours. In this single-dose study, it is difficult to distinguish between a pharmacodynamic effect, perhaps due to relatively less inflammation compared to the other pain models studied, and a pharmacokinetic effect, where the peak effect of CAV1001 at the peripheral target after a single initial dose may occur at or after 4 hours. Multiple-dose longer-term studies are planned.
Limitations of the current studies include the short-term, single dosing designs. Additional multiple dosing studies are planned given the subacute and chronic indications suggested by the mechanism of action. Longer term studies, as well as studies examining other difficult to treat pain syndromes having significant inflammatory components, are also planned.
A number of other recalcitrant chronic pain states evoke an inflammatory response. They include several common chronic visceral pain states, such as those arising from inflammatory bowel disease, interstitial cystitis and pancreatitis, which may be amenable to peripheral kappa/delta agonists. Kappa-opioid agonists have previously been reported to be useful in various preclinical and clinical visceral pain models.23,42–46 Similarly, peripheral burn injury is associated with a dramatic inflammatory component.47 Finally, although clinically the chronic widespread pain in fibromyalgia is related to a generalized increase in sensitivity to painful stimuli, and not considered to be primarily inflammatory, the condition may be in part a response to chronic low-grade inflammation, making it amenable to analgesics that take advantage of this persistent inflammatory response.48 Specifically, as a consequence of this chronic inflammation, delta-opioid receptor up-regulation has been reported in fibromyalgia making a dual-acting kappa/delta-opioid agonist potentially useful.49
Acknowledgments
The authors wish to acknowledge and thank Adel Nefzi, PhD, Torrey Pines Institute for Molecular Studies in Port St. Lucie, Florida, USA, for preparation of the CAV1001 used in these studies as well as Thomas Van Valkenburg, BS and Jim Pomonis, PhD of American Preclinical Services for assistance in conduct and data collection in the neuropathic and cancer pain models, respectively.
Disclosure
The authors declare this study received funding from Caventure Drug Discovery, Inc. Craig Hartrick reports stock ownership for Caventure Drug Discovery, outside the submitted work; In addition, Dr Craig Hartrick has a patent US: 16/066,184 pending, a patent PCT: PCT/US2018/038936 pending. CH and AH own stock in Caventure. Study conduct, data collection, analysis, and interpretation of the data were performed by researchers at Charles River (Formalin experiment) and American Preclinical Services. CH and AH were involved in study conception, study design, the decision to publish, and manuscript preparation.
References
1. Totsch SK, Sorge RE. Immune system involvement in specific pain conditions. Mol Pain. 2017;13:1–17.
2. Stein C, Clark JD, Oh U, et al. Peripheral mechanisms of pain and analgesia. Brain Res Rev. 2009;60:90–113.
3. Morlion B, Coluzzi F, Aldington D, et al. Pain chronification: what should a non-pain medicine specialist know? Curr Med Res Opin. 2018;34(7):1169–1178.
4. Barry DT, Marshall BDL, Becker WC, et al. Duration of opioid prescriptions predicts incident nonmedical use of prescription opioids among U.S. veterans receiving medical care. Drug Alcohol Depend. 2018;191:348–354.
5. Merighi S, Gessi S, Varani K, et al. Morphine mediates a proinflammatory phenotype via μ-opioid receptor-PKCɛ-Akt-ERK1/2 signaling pathway in activated microglial cells. Biochem Pharmacol. 2013;86(4):487–496.
6. McNicol ED, Midbari A, Eisenberg E. Opioids for neuropathic pain. Cochrane Database Syst Rev. 2013;2013(8):CD006146.
7. Knezevic NN, Yekkirala A, Yaksh TL. Basic/translational development of forthcoming opioid- and nonopioid-targeted pain therapeutics. Anesth Analg. 2017;125(5):1714–1732.
8. Bee LA, Bannister K, Rahman W, Dickenson AH. Mu-opioid and noradrenergic α(2)-adrenoceptor contributions to the effects of tapentadol on spinal electrophysiological measures of nociception in nerve-injured rats. Pain. 2011;152(1):131–139.
9. Wallace MS, Moulin D, Clark AJ, et al. A Phase II, multicenter, randomized, double-blind, placebo-controlled crossover study of CJC-1008–a long-acting, parenteral opioid analgesic–in the treatment of postherpetic neuralgia. J Opioid Manag. 2006;2(3):167–173.
10. Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–187.
11. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–188.
12. Yamamoto J, Kawamata T, Niiyama Y, Omote K, Namiki A. Down-regulation of mu opioid receptor expression within distinct subpopulations of dorsal root ganglion neurons in a murine model of bone cancer pain. Neuroscience. 2008;151(3):843–853.
13. Corder G, Castro DC, Bruchas MR, Scherrer G. Endogenous and exogenous opioids in pain. Annu Rev Neurosci. 2018;41:453–473.
14. Banghart MR, Sabatini BL. Photoactivatable neuropeptides for spatiotemporally precise delivery of opioids in neural tissue. Neuron. 2012;73:249–259.
15. Mansour A, Fox CA, Burke S, et al. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol. 1994;350:412–438.
16. Gendron L, Mittal N, Beaudry H, Walwyn W. Recent advances on the δ opioid receptor: from trafficking to function. Br J Pharmacol. 2015;172:403–419.
17. Chen S-R, Pan H-L. Removing TRPV1-expressing primary afferent neurons potentiates the spinal analgesic effect of δ-opioid agonists on mechano-nociception. Neuropharmacology. 2008;55(2):215–222.
18. Berg KA, Rowan MP, Gupta A, et al. Allosteric interactions between δ and κ opioid receptors in peripheral sensory neurons. Mol Pharmacol. 2012;81:264–272.
19. Puehler W, Zollner C, Brack A, et al. Rapid upregulation of the mu opioid receptor mRNA in dorsal root ganglia in response to peripheral inflammation depends on neuronal conduction. Neuroscience. 2004;129(2):473–479.
20. Mousa SA, Cheppudira BP, Shaqura M, et al. Nerve growth factor governs the enhanced ability of opioids to suppress inflammatory pain. Brain. 2007;130:502–513.
21. Ni J, Gao Y, Gong S, Guo S, Hisamitsu T, Jiang X. Regulation of μ-opioid type 1 receptors by microRNA134 in dorsal root ganglion neurons following peripheral inflammation. Eur J Pain. 2013;17(3):313–323.
22. Celik MO, Labuz D, Henning K, et al. Leukocyte opioid receptors mediate analgesia via Ca(2+)-regulated release of opioid peptides. Brain Behav Immun. 2016;57:227–242.
23. Riviere PJM. Peripheral kappa-opioid agonists for visceral pain. Br J Pharmacol. 2004;141:1331–1334.
24. Brackley AD, Gomez R, Akopian AN, Henry MA, Jeske NA. GRK2 constitutively governs peripheral delta opioid receptor activity. Cell Rep. 2016;16(10):2686–2698.
25. Eans SO, Ganno ML, Mizrachi E, et al. Parallel synthesis of hexahydrodiimidazodiazepines heterocyclic peptidomimetics and their in vitro and in vivo activities at mu (MOR), delta (DOR), and kappa (KOR) opioid receptors. J Med Chem. 2015;58:4905–4917.
26. Yoon MH, Yaksh TL. Evaluation of interaction between gabapentin and ibuprofen in the formalin test in rats. Anesthesiology. 1999;91(4):1006–1013.
27. Soelberg CD, Brown RE, Du Vivier D, Meyer JE, Ramachandran BK. The US opioid crisis: current federal and state legal issues. Anesth Analg. 2017;125(5):1675–1681.
28. Ostling PS, Davidson KS, Anyama BO, et al. America’s opioid epidemic: a comprehensive review and look into the rising crisis. Curr Pain Headache Rep. 2018;22(5):32.
29. Petho G, Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev. 2012;92:1699–1775.
30. George J, Pulickal SJ, Singh A, et al. Locally mediated analgesic effect of bradykinin type 2 receptor antagonist HOE 140 during acute inflammatory pain in rats. J Burn Care Res. 2014;35(6):e391–8.
31. Parada CA, Tambeli CH, Cunha FQ, Ferreira SH. The major role of peripheral release of histamine and 5-hydroxytryptamine in formalin-induced nociception. Neuroscience. 2001;102(4):937–944.
32. Jiang Y-L, He X-F, Shen Y-F, et al. Analgesic roles of peripheral intrinsic met-enkephalin and dynorphin A in long-lasting inflammatory pain induced by complete Freund’s adjuvant in rats. Exp Ther Med. 2015;9:2344–2348.
33. Hawkins P, Armstrong R, Boden T, et al. Applying refinement to the use of mice and rats in rheumatoid arthritis research. Inflammopharmacol. 2015;23:131–150.
34. Zhu X, Eisenach JC. Cyclooxygenase-1 in the spinal cord is altered after peripheral nerve injury. Anesthesiology. 2003;99(5):1175–1179.
35. Sosanya NM, Kumar R, Clifford JL, et al. Identifying plasma derived extracellular vesicle (EV) contained biomarkers in the development of chronic neuropathic pain. J Pain. 2020;21(1–2):82–96.
36. Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokine. J Neuroimmunol. 2010;229:26–50.
37. Mika J, Popiolek-Barczyk K, Rojewska E. Delta-opioid receptor analgesia is independent of microglial activation in a rat model of neuropathic pain. PLoS One. 2014;9(8):e104420.
38. Schroder W, Vry JD, Tzschentke TM. Differential contribution of opioid and noradrenergic mechanisms of tapentadol in rats models of nociceptive and neuropathic pain. Eur J Pain. 2010;14(8):814–821.
39. Hartrick CT. Noradrenergic reuptake inhibition in the treatment of pain. Expert Opin Investig Drugs. 2012;21(12):1827–1834.
40. Tanabe M, Takasu K, Takeuchi Y, Ono H. Pain relief by gabapentin and pregabalin via supraspinal mechanisms after peripheral nerve injury. J Neurosci Res. 2008;86(15):3258–3264.
41. Mantyh P. Bone cancer pain: from mechanism to therapy. Curr Opin Support Palliat Care. 2014;8(2):83–90.
42. Habtezion A. Inflammation in acute and chronic pancreatitis. Curr Opin Gastroenterol. 2015;31(5):395–399.
43. Craft RM, Henley SR, Haaseth RC, Hruby VJ, Porreca F. Opioid antinociception in a rat model of visceral pain: systemic versus local drug administration. J Pharmacol Exp Ther. 1995;275(3):1535–1542.
44. Kannampalli P, Sengupta JN. Role of principal inotropic and metabotropic receptors in visceral pain. J Neurogastroenterol Motil. 2015;21(2):147–158.
45. Mangel AW, Bornstein JD, Hamm LR, et al. Clinical trial: asimadoline in the treatment of patients with irritable bowel syndrome. Aliment Pharmacol Ther. 2008;28(2):239–249.
46. Sengupta JN, Snider A, Su X, Gebhart GF. Effects of kappa opioids in the inflamed rat colon. Pain. 1999;79(2–3):175–185.
47. Fowler M, Clifford JL, Garza TH, et al. A rat model of full thickness thermal injury characterized by thermal hyperalgesia, mechanical allodynia, pronociceptive peptide release and tramadol analgesia. Burns. 2014;40:759–771.
48. Mastrangelo F, Frydas I, Ronconi G, et al. Low-grade chronic inflammation mediated by mast cells in fibromyalgia: role of IL-37. J Biol Regul Homeost Agents. 2018;32(2):195–198.
49. Salemi S, Aeschlimann A, Wollina U, et al. Up-regulation of delta-opioid receptors and kappa-opioid receptors in the skin of fibromyalgia patients. Arthritis Rheum. 2007;56(7):2464–2466.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.