Back to Journals » Drug Design, Development and Therapy » Volume 11
Drugs in development for toxoplasmosis: advances, challenges, and current status
Authors Alday PH, Doggett JS
Received 6 July 2016
Accepted for publication 23 November 2016
Published 25 January 2017 Volume 2017:11 Pages 273—293
DOI https://doi.org/10.2147/DDDT.S60973
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr James Janetka
P Holland Alday,1 Joseph Stone Doggett1,2
1Division of Infectious Diseases, Oregon Health & Science University, 2Portland Veterans Affairs Medical Center, Portland, OR, USA
Abstract: Toxoplasma gondii causes fatal and debilitating brain and eye diseases. Medicines that are currently used to treat toxoplasmosis commonly have toxic side effects and require prolonged courses that range from weeks to more than a year. The need for long treatment durations and the risk of relapsing disease are in part due to the lack of efficacy against T. gondii tissue cysts. The challenges for developing a more effective treatment for toxoplasmosis include decreasing toxicity, achieving therapeutic concentrations in the brain and eye, shortening duration, eliminating tissue cysts from the host, safety in pregnancy, and creating a formulation that is inexpensive and practical for use in resource-poor areas of the world. Over the last decade, significant progress has been made in identifying and developing new compounds for the treatment of toxoplasmosis. Unlike clinically used medicines that were repurposed for toxoplasmosis, these compounds have been optimized for efficacy against toxoplasmosis during preclinical development. Medicines with enhanced efficacy as well as features that address the unique aspects of toxoplasmosis have the potential to greatly improve toxoplasmosis therapy. This review discusses the facets of toxoplasmosis that are pertinent to drug design and the advances, challenges, and current status of preclinical drug research for toxoplasmosis.
Keywords: Toxoplasma gondii, therapeutics, preclinical medicine, experimental medicine, mechanism of action, Apicomplexa
Toxoplasma gondii
Toxoplasma gondii is a protozoan parasite that belongs to the phylum Apicomplexa. Apicomplexa also includes the medically important genera, Plasmodium, Babesia, and Cryptosporidium. The eponymous organelle of Apicomplexa, the apical complex, is used to invade the host cell. Biological similarities among apicomplexans are the basis for shared susceptibility to drugs such as the antifolate drugs pyrimethamine and sulfonamides and the anti-respiratory drug atovaquone. The current treatments for T. gondii and Babesia microti are drugs that were used as anti-malarials prior to being repurposed. However, an examination of parasite genomes, routes of infection, life cycle stages, hosts, and disease manifestations reveals diversity in the underlying biology of apicomplexan pathogens. Drugs that are specifically designed to optimize the efficacy against T. gondii hold potential for improving the treatment of toxoplasmosis.
The unique pathogenesis of T. gondii also presents challenges for drug therapy. Unlike many apicomplexans, T. gondii crosses the blood–brain barrier and establishes persistent infection in a drug-resistant bradyzoite stage. An ideal medicine for toxoplasmosis would achieve therapeutic, systemic, brain and eye concentrations to be effective in the organs where the majority of disease occurs and would be active against both the acute replicating tachyzoite and latent bradyzoite stages of the parasite. New drugs should also prioritize having fewer, milder side effects, a significant problem with the current first-line drugs. Current research into new drugs developed specifically for toxoplasmosis has led to promising preclinical compounds. This review discusses the aspects of toxoplasmosis that are germane to drug development and ongoing preclinical drug research.
T. gondii is a remarkably successful parasite that is broadly distributed throughout the world and is capable of infecting both mammals and birds. Up to one-third of the human population is estimated to have been infected.1 The great majority of human T. gondii infection occurs either by ingestion of oocysts that are generated in the felid intestine and spread throughout the environment via feces or ingestion of T. gondii tissue cysts in undercooked meat. Congenital infection occurs through vertical transmission when a previously uninfected mother is infected during pregnancy. Otherwise, uncommon means of transmission include transplantation of infected organs, blood transfusion, or inhalation of oocyst-contaminated dust.2 The resiliency of T. gondii oocysts in the environment contributes to the high rates of T. gondii infection in humans, and the risk of toxoplasmosis outbreaks, as evidenced by large waterborne outbreaks of T. gondii infection from oocyst-contaminated drinking water in Canada and Brazil.3,4 Although preventive measures focused on hygiene and sanitary meat production may have reduced the prevalence of human T. gondii infection, these measures will not reduce the overall burden of human T. gondii infection enough to decrease the need for better anti-Toxoplasma therapies in the near future.
Prevalence of T. gondii infection and disease
The seroprevalence of T. gondii antibodies varies significantly worldwide as rates of human infection are influenced by climate, the consumption of undercooked meat, hygiene, and exposure to cats.5 Direct comparisons of seroprevalence studies are limited by heterogeneous methodologies, but have been important in identifying specific high prevalence populations. For example, seropositivity for T. gondii antibodies in Brazil ranges from 20% to >90% among different groups.6 In the US, the seroprevalence among people aged 12–49 years has declined from 14.1% to 6.7% between 1994 and 2010.7 However, prevalence in the US was reported to be 29.9% in people aged >70 years and 25.1% in US residents born outside of the US.7 In studies of pregnant women and women of child bearing age, seroprevalence in Europe, Asia, and Africa ranges from 20% to 60%.5
Although studies of seroprevalence provide valuable insight into T. gondii transmission and the underlying risk for the development of toxoplasmosis in a population, the worldwide incidence of disease caused by T. gondii is less understood. In addition to the risk of acquisition of T. gondii infection, the development of symptomatic toxoplasmosis is influenced by the prevalence of immunosuppressive conditions such as AIDS and may be influenced by differences in the virulence of T. gondii strains found all over the world. The hypothesis that certain strains of T. gondii are more virulent is most evident in Brazil where non-archetypal strains are associated with symptomatic ocular toxoplasmosis8 and in French Guiana with severe disseminated disease in the immunocompetent.9 By comparison, 1%–2% of persons infected with T. gondii develop eye disease in the US, whereas eye disease can approach up to 20% of seropositive persons in highly endemic areas of Brazil.10 The broad distribution of T. gondii disease in populations with limited medical resources indicates that a new anti-T. gondii drug should be orally bioavailable, have a chemically stable formulation, and be inexpensive to produce.
Toxoplasmosis
T. gondii infection in immunocompetent hosts rarely requires drug therapy. Over 80% of primary T. gondii infections in immunocompetent hosts are asymptomatic.11 Symptomatic infection typically consists of self-limited bilateral nontender cervical lymphadenopathy, which may be accompanied by fevers and myalgias. Following primary infection, T. gondii establishes latent infection, converting to the quiescent bradyzoite form within tissue cysts. Although the host immune system is capable of controlling most T. gondii primary infections, ocular cysts may be reactivated and lead to vision loss from retinal scarring. In the US, 21,000 persons per year are estimated to develop ocular lesions and 4,800 persons per year develop symptomatic ocular lesions.10 The effect of latent T. gondii brain infection on human behavior and mental health in immunocompetent individuals is an active area of research, but a causal relationship between latent infection and psychiatric disease or changes in human behavior has not been established.12–14 Unlike T. gondii in most healthy people, infection in the immunocompromised is often debilitating or fatal.
Severe manifestations of T. gondii most often occur when tissue cysts reactivate in the setting of immunosuppression due to AIDS or medical immunosuppression. The most frequent severe clinical presentation is Toxoplasma encephalitis, which typically consists of multiple discrete brain lesions. Ocular and pulmonary diseases are the most common extra-cerebral sites of infection; however, disease involving other organs has been reported as well as sepsis due to disseminated disease.15 In individuals with AIDS and a CD4 T-cell count <100 cells/μL, the incidence of encephalitis is 28% in seropositive patients not taking prophylaxis.16 In allogeneic stem cell transplant patients, the 6-month incidence of symptomatic toxoplasmosis has been reported to be as high as 6% in a prospective multicenter study of seropositive patients, although prior retrospective studies have reported a lower incidence.17–20 Of note, patients who developed symptomatic toxoplasmosis in this study were not taking standard prophylaxis. Toxoplasmosis has been reported in autologous stem cell transplant patients as well, although the risk for disease is lower.21 Despite the relatively low prevalence of T. gondii infection in the US, in 2008, there were ~3,585 toxoplasmosis-related hospitalizations.22 The cost of illness in the US caused by T. gondii has been estimated to be ~$3 billion and an annual loss of 11,000 quality-adjusted life years.23 Severe toxoplasmosis in the US is reduced from the height of the AIDS epidemic prior to highly active antiretroviral therapy, but remains a significant source of morbidity and mortality in the US and worldwide.
Severe toxoplasmosis also results from congenital infection and may occur rarely in healthy individuals. Congenital toxoplasmosis ranges from subclinical to severe disease that consists primarily of brain and eye manifestations, but may also include extracranial pathology in up to half of neonates.24 Although rare, severe toxoplasmosis in the immunocompetent has been reported, and in a notable cluster of cases in French Guiana, it seems to be due to virulent, non-archetypal strains.9,25–27 Unlike typical toxoplasmosis, patients in these series presented with disseminated infection involving multiple organs, frequently had pulmonary involvement, and required intensive care.
Current medicines for toxoplasmosis
First-line therapy consists of the combination of pyrimethamine and sulfadiazine with leucovorin added to prevent hematologic toxicity. In observational studies and controlled trials for Toxoplasma encephalitis, this regimen has been found to have high rates of toxic side effects leading to discontinuation of therapy. A review of 115 patients with Toxoplasma encephalitis found toxicity in 62% of patients and severe side effects requiring a change in therapy in 44% of patients.28 Similarly, in treatment trials of Toxoplasma encephalitis, toxicity led to discontinuation of pyrimethamine-sulfadiazine in one-third of patients.29,30 Sulfadiazine may be replaced with clindamycin if the patient has an allergy to sulfa drugs; however, the clindamycin-containing regimen is less effective in preventing relapse and had similar rates of toxicity.29 Trimethoprim-sulfamethoxazole has been shown to have efficacy similar to pyrimethamine-sulfadiazine and may be used as an alternative if patients do not have sulfa allergy and pyrimethamine is not tolerated or is not available.31–33 Atovaquone or azithromycin may be used as alternate therapy in combination with pyrimethamine or sulfadiazine for the treatment and prophylaxis of toxoplasmosis when first-line therapy is contraindicated. However, use of these alternate therapies is supported by less clinical data, and these regimens have similar rates of patient intolerance.34–36 In addition to frequent side effects, pyrimethamine and sulfadiazine are associated with rare severe reactions that may be fatal, including agranulocytosis, Stevens–Johnson syndrome, toxic epidermal necrolysis, and hepatic necrosis. Drugs that are less toxic would greatly improve the care of patients with toxoplasmosis.
The need for nontoxic medicines is further emphasized by the prolonged courses of therapy required for treatment and suppression of infection. Initial treatment duration for Toxoplasma encephalitis is at least 6 weeks followed by secondary suppression until sufficient immune reconstitution; duration for congenital infection is at least 1 year; ocular infection is 4–6 weeks with the consideration of continued suppression to prevent relapse.24,37,38 Prolonged courses are required, in part because current clinical medicines are unable to eliminate the tissue-cyst stage of T. gondii. A promising aspect of several experimental compounds is activity against T. gondii tissue cysts that are established in mice 5 weeks prior to treatment.39,40 Previous studies have shown that atovaquone possesses activity in experimental models in vivo against tissue cysts; however, clinical studies of ocular infection and encephalitis have not shown an advantage in preventing relapse, which may be related to subtherapeutic drug concentrations in infected tissue compartments or translational limitations of toxoplasmosis animal models.34,38,41
Challenges for anti-T. gondii drugs
The biology of T. gondii bradyzoite containing tissue cysts has been poorly understood and is an active area of research that will be important for the development of drugs that eliminate T. gondii infection from the host. Tissue cysts are made up of bradyzoites surrounded by a thick glycan-rich cyst wall. Animal models have provided important insights into drugs that have the potential to cure latent T. gondii infection. However, virulence and cyst formation vary depending on the T. gondii strain and the host species and strain. It is unclear which animal model most resembles human infection. In addition to uncertainty regarding the most appropriate translational animal models, the key biological questions of the metabolic pathways that are essential for bradyzoite survival in the tissue cyst and which of these are viable drug targets remain mostly unanswered. McPhillie et al42 made recent progress in understanding bradyzoite biology, demonstrating that the cyst-forming EGS strain possesses a mutation in the transcription factor Apetala 2, a known repressor of bradyzoite genes. Provocative studies of the ME49 T. gondii strain in CBA/J mice have shown heterogeneous levels of replication among bradyzoites within cysts and variation between cysts.43 Future studies probing the biology and drug susceptibility of latent T. gondii infection should provide more effective strategies for eliminating T. gondii tissue cysts from the host, preventing relapsing disease and potentially shortening the required duration of drug administration.
Drug resistance in T. gondii is suspected to contribute to treatment failures that have occurred in ~10% of patients during initial therapy and 10%–20% of patients who relapse during suppressive therapy. However, the contribution of drug resistance to treatment failure is difficult to quantify because T. gondii is not routinely recovered from patients with toxoplasmosis. Moreover, treatment failures of toxoplasmosis during clinical studies have been difficult to characterize due to diagnostic limitations, drug intolerance, nonadherence, variable drug absorption, and the complex morbidities of patients.29,30,32,34,44 High-level sulfonamide resistance has been found in clinical isolates without known sulfonamide exposure and after sulfadoxine exposure.45,46 The resistant T. gondii strain in which there was no drug exposure possessed a mutation in the dihydropteroate synthase (DHPS) gene known to confer resistance in both T. gondii and Plasmodium falciparum and demonstrated cross-resistance to several sulfonamides including sulfadiazine and sulfamethoxazole.43 However, genetic mutations in DHPS do not account for sulfonamide resistance in all clinical isolates, and research into alternate mechanisms is ongoing.45,47,48
The range of sulfonamide susceptibility seems to be greater than that of pyrimethamine or atovaquone. Genetic evidence of atovaquone resistance has not been found in clinical isolates, but has been obtained in vitro with chemical mutagenesis.49 Variations in atovaquone susceptibility among different T. gondii genotypes have been observed but subsequent experiments did not find significant differences in genotype susceptibility.45,50 Similarly, type I T. gondii strains have been shown to be less susceptible to pyrimethamine in vitro compared to type II and type III strains; however, subsequent studies found that decreased susceptibility to pyrimethamine correlated with the replication rate rather than genotype and decreased susceptibility was not great enough to be considered significant in pyrimethamine resistance.45,51 Differences in in vitro susceptibility for pyrimethamine and atovaquone found in these studies likely reflect the differences in methodologies, and the translational significance of these findings is not clear. However, given that sulfonamides are part of first-line therapy, variations in sulfonamide susceptibility, whether preexisting or occurring after drug exposure are concerning and further accentuate the need for new drugs for toxoplasmosis.
Preclinical testing of anti-toxoplasma compounds
Well-established in vitro assays and animal models exist for preclinical anti-T. gondii drug development, but there is no standard testing cascade. Most often, anti-Toxoplasma compounds are initially evaluated in vitro in tachyzoite replication assays. Common assays use T. gondii that expresses β-galactosidase or fluorescent proteins, or plaque formation or [3H] uracil uptake in non-transgenic T. gondii. T. gondii requires a host cell to replicate; therefore, it is also important to assess the effects of the compounds on the viability of the host cells while assessing the degree of compound inhibition. Although T. gondii inhibition assays may be performed on different strains of T. gondii, rates of replication vary among different strains, and the type I RH strain is often used initially because it is well adapted to in vitro culture and replicates relatively quickly and reliably.
The RH strain is typically fatal in mice within 10 days providing a model that reflects fulminate, disseminated infection that is not controlled by the host immune system. This strain is useful to evaluate acute infection; however, this model does not evaluate activity against brain tissue cysts, and type I strains are not the dominant strain in most of the areas of the world. Type II strains are more prominent in North America and Europe, are less virulent in mice, and establish tissue cysts in the mouse brain. Susceptibility to infection with type II strains differs among mouse strains, and the degree to which tissue cysts in the mouse brain resemble latent human T. gondii infection is unknown. Recent experiments have indicated that the ongoing replication of bradyzoites occurs within the established brain tissue cysts as long as 8 weeks after infection drawing into question the presumption that latent bradyzoite infection is metabolically dormant.43,52 Models of latent infection that started treatment with experimental compounds after 5 weeks of sublethal infection with the ME49 strain have demonstrated brain cyst reduction by experimental compounds.39,53 In addition to acute and latent murine models of infection, congenital and ocular toxoplasmosis models as well as other animal species have been evaluated, but the majority of experimental compounds are initially tested in models wherein mice are infected orally or by intraperitoneal injection, and parasite burden in tissues or survival is measured as outcome.54
Future testing of preclinical compounds against genetically diverse, non-archetypal strains of T. gondii both in vitro and in vivo should prove insightful in that virulence and pathogenesis vary between strains. Recently characterized parasite strains, such as the EGS strain that forms thick-walled cysts spontaneously in tissue culture allows for the study of inhibitors against cysts in vitro.42 In addition, this strain has been genetically modified to express stage-specific fluorescence to further characterize the activity against bradyzoites and tissue cysts.55 Beyond improvements in testing cascades, new methods of target identification are likely to contribute to drug discovery. Future investigation of drug targets in Toxoplasma has recently been advanced by a genome-wide screen using CRISPR/Cas9 to identify essential genes.56 Using the phenotypic data from this screen, which is available through www.toxodb.org, researchers are able to investigate fitness-conferring proteins for potential drug targets.57
Basic research in T. gondii biology and drug screening has identified a diverse array of drug targets. Over 20 preclinical drug development projects have been described in publications over the past decade. The research studies selected in this review reflect the advances, challenges, and current status of drug development for toxoplasmosis. Table 1 presents the classes of compounds in alphabetical order as they appear in the text and with the chemical structures.
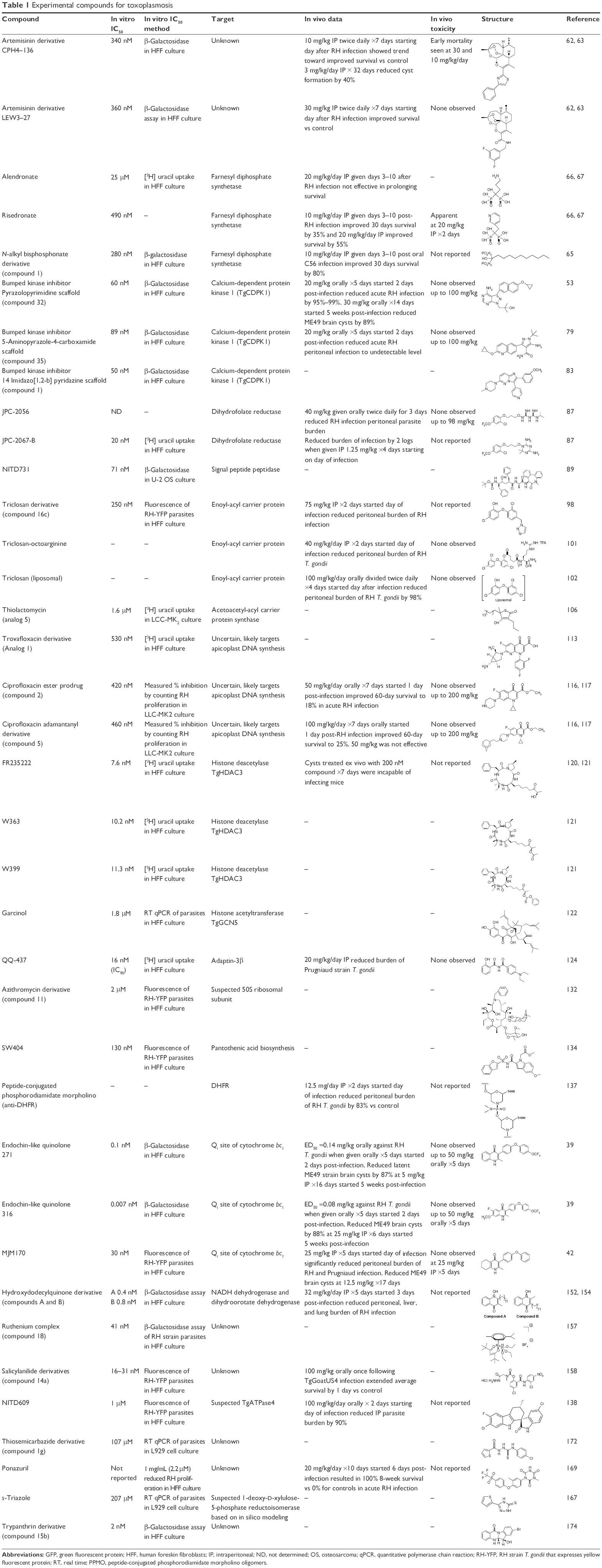 | Table 1 Experimental compounds for toxoplasmosis |
Artemisinin derivatives
Derivatives of artemisinin, an endoperoxidase, are the backbone of first-line treatment for malaria. Artemisinin and its derivatives also have in vitro efficacy against T. gondii.58–60 Anti-parasitic activity of these compounds requires a 1,2,4-trioxane moiety and is enhanced by substitutions at the C-10 position.61
Schultz et al62 tested two artemisinin derivatives, CPH4–136 and LEW3–27, in murine models of acute and chronic infection. CPH4–136 and LEW3–27 contain thiazole and carboxamide substitutions at their respective C-10 positions and were previously found to have in vitro activity against tachyzoites (340 and 360 nM, respectively).63 Mice were infected intraperitoneally (IP) with RH strain tachyzoites and then treated with 3–50 mg/kg of either compound IP for 7 days. Both the compounds prolonged survival to a modest extent. Approximately 20% of infected mice treated with LEW3–27 at 30 mg/kg/day were alive at day 20, and all the mice treated with CPH4–136 at 10 mg/kg/day were dead by day 18. All the infected mice treated with atovaquone (10 mg/kg/day) survived until day 20, and the infected, untreated mice died by 12 days post-infection. In addition, CPH4–136 at doses of 30 and 10 mg/kg was associated with early mortality in the treatment group compared to the control group treated with vehicle alone, raising questions regarding toxicity at these doses. However, in a murine model of infection with the ME49 strain, a lower 3 mg/kg dose of CPH4–136 reduced cyst burden by ~40% when given for 32 days. Neither LEW3–27 nor artemether reduced the cyst burden.
Bisphosphonates: farnysyl diphosphate inhibitors
Bisphosphonates are used for the treatment of osteoporosis and other diseases of bone resorption. Nitrogen-containing bisphosphonates, such as, alendronate or risedronate, inhibit farnesyl pyrophosphate synthase in the mevalonate pathway in osteoclasts.64 Many parasitic protozoa, including T. gondii, possess a mevalonate pathway for the synthesis of sterols and polyisoprenoids. A key branch of this pathway is the synthesis of farnesyl diphosphate, a precursor to ubiquinone, sterols, and prenylated proteins. Synthesis of farnesyl diphosphate in T. gondii is catalyzed by the bifunctional enzyme farnesyl diphosphate/geranylgeranyl-diphosphate synthase65 and is inhibited by nitrogen-containing bisphosphonates.66
In an early study,66 risedronate was shown to have an in vitro IC50 of 490 nM against T. gondii. In vivo experiments67 demonstrated that risedronate (10 mg/kg/day) improved 30-day survival by 35% in Swiss Webster mice infected orally with 10 cysts of the C56 strain of T. gondii. An amount of 20 mg/kg/day improved 30-day survival by 55%. Later work67 further improved the potency of bisphosphonate derivatives. Compound 1 in the series described in Ling et al is an n-alkyl derivative that has an in vitro IC50 of 280 nM. When administered to mice infected with 5 cysts of C56 T. gondii at 10 mg/kg IP daily for 10 days, compound 1 improved 30-day survival to 80%.
Bumped kinase inhibitors (BKIs)
BKIs inhibit the T. gondii calcium-dependent protein kinase 1 (TgCDPK1), a member of the serine/threonine protein kinase family.68,69 TgCDPK1 regulates the calcium-dependent pathway of T. gondii microneme secretion and is required for gliding motility, host-cell invasion, and egress.70 BKIs inhibit T. gondii replication by blocking host-cell invasion and egress.71,72 T. gondii mitogen-activated protein kinase like 1 (TgMAPKL1) has also been suggested as a secondary target for the BKI, 1NM-PP1, as a mutation in TgMAPKL1, was associated with decreased susceptibility to 1NM-PP1 and similar BKIs.73,74
A key structural difference between TgCDPK1 and human kinases occurs at the “gatekeeper residue” in the ATP-binding pocket. TgCDPK1 contains a small glycine residue at this position, whereas human kinases have larger residues. The additional space afforded by the glycine residue in TgCDPK1 has been exploited for the design of potent and selective ATP-competitive TgCDPK1 inhibitors.68,75 Numerous BKI analogs have been developed around different core scaffolds that have promising in vitro and in vivo activities.53,76–79
Potent TgCDPK1 inhibitors have been synthesized from pyrazolopyrimidine (PP) and 5-aminopyrazole-4-carboxamide (AC) scaffolds.53,71,72,74,79,80–82 The BKI scaffold binds in the ATP-binding pocket of TgCDPK1 and the addition of a 6-alkoxy-2-naphthyl group at the C-3 position, and a 4-piperidinylmethylene group at the N-1 position of the PP scaffold further increases selectivity.75,76 These BKIs were >15,000-fold more active against TgCDPK1 compared to the human kinases SRC and ABL, with no inhibition at 20 μM. BKI 1294, which was synthesized with N-methylation of the 4-piperidinyl-methylene substituent to improve metabolic stability, had an in vitro IC50 of 140 nM and reduced acute T. gondii infection by 93% when given orally at 30 mg/kg.76 Later, BKI 1294 was found to inhibit the human Ether-à-go-go-Related Gene (hERG) ion channel preventing further advancement due to the risk of cardiotoxicity. Vidadala et al investigated the modifications of PP scaffold that maintained efficacy and selectivity for TgCDPK1 while eliminating hERG liability.53 Compound 32 in their series of BKIs with reduced hERG activity had a >10 μM IC50 against hERG, a 60 nM in vitro IC50 against T. gondii and was effective in vivo. Compound 32 administered orally to mice at 20 mg/kg for 5 days eliminated acute RH strain peritoneal infection. In the latent T. gondii infection model using the ME49 strain, 30 mg/kg of compound 32 given orally reduced the number of brain cysts by 88.7%. Pharmacokinetic studies in noninfected mice demonstrated a brain to plasma concentration ratio of 0.33.53 Lourido et al77 described a series of PP analogs. Compounds 11 and 24 were metabolically stable and had in vitro IC50s of 250 and 610 nM. These compounds increased 30-day survival in mice infected with type II Pru-LUC strain and decreased brain cysts when administered IP prior to infection at 5 mg/kg for 10 days.77
Development of the AC scaffold by Huang et al identified compounds 34 and 35 as lead compounds. Compound 35 had an in vitro IC50 of 89 nM against T. gondii, and in an acute in vivo model using the RH strain, compound 35 reduced infection in the peritoneum below the limits of detection when given orally at 20 mg/kg.79 Compounds 34 and 35 had a brain to plasma concentration ratio of 0.16 and 0.43, respectively.
In addition to the PP and AC scaffolds, Moine et al identified biphenylimidazoazines as T. gondii growth inhibitors by screening a library of compounds.82 Later, a series of 14 imidazo[1,2-b]pyridazines based on the biphenylimidazoazines were found to inhibit TgCDPK1 in enzymatic studies at EC50s of <1 μM and 7 compounds inhibited T. gondii in vitro at IC50s of <1 μM. Compound 1, 16a and 16f demonstrated in vitro IC50s of 50 nM, 100 nM and 70 nM, respectively, with minimal host cell toxicity.83
Dihydrofolate reductase (DHFR) inhibitors
T. gondii, like several other protozoal parasites, has a unique bifunctional DHFR–thymidylate synthase (TS) that contains both catalytic sites on the same protein. DHFR is an extensively studied drug target that is inhibited by pyrimethamine, one of the first-line agents currently used against toxoplasmosis. Clinical use of pyrimethamine is primarily limited by the inhibition of host folate metabolism, which may cause neutropenia. DHFR inhibitors that have greater selectivity for T. gondii84 could potentially be less toxic and more effective.
The dihydrotriazines are a new class of DHFR inhibitors originally developed for use against malaria85,86 that have also been found to be effective against T. gondii. The dihydrotriazine JPC-2067-B87 inhibited in vitro growth of T. gondii with an IC50 of 20 nM. JPC-2067-B has poor oral bioavailability but was effective in a murine model of acute toxoplasmosis when given IP at a dose of 1.25 mg/kg/day for 4 days. Likewise, the orally available prodrug of JPC-2067-B, JPC-2056, was effective in the same murine model at an oral dose of 40 mg/kg twice daily for 3 days.
Endoplasmic reticulum-associated degradation (ERAD) inhibitors
Proteins that are inserted into a membrane or secreted from the cell undergo folding and post-translational modification in the ER. A small fraction of proteins become irreversibly misfolded, and these proteins are recycled by a ubiquitin- and proteasome-mediated process called ERAD.88 Most eukaryotic cells possess an extensive system for detecting misfolded proteins and target them to the ERAD pathway. However, the ERAD system in T. gondii, P. falciparum, and other protozoan parasites is limited relative to other eukaryotes89 rendering these parasites more susceptible to interference with this pathway. Harbut et al89 screened inhibitors known to target various proteins in the ERAD pathway for activity against P. falciparum. The authors determined that inhibitors of signal peptide peptidase (SPP) were the most effective and nontoxic agents based on in vitro IC50s against P. falciparum and human hepatocytes. One of these compounds, NITD731, inhibited T. gondii in human U-2 osteosarcoma (OS) cell culture at an IC50 of 71 nM. No toxicity to host cells was noted at a compound concentration of 10 μM. These results demonstrate that SPP is a potential target for apicomplexan inhibitors.
Fatty acid synthesis inhibitors
Synthesis of fatty acids in T. gondii takes place in the apicoplast.90–92 Consistent with the evolutionary origin of this organelle, iterative elongation of nascent fatty acids is catalyzed by the multienzyme fatty acid synthase II (FAS II) pathway also found in bacteria and plants.91,93,94 This is in contrast to the FAS I pathway found in animals and fungi that consists of a single large multifunctional polypeptide that catalyzes the steps in fatty acid elongation.95
The FAS II enzyme enoyl-acetyl carrier protein reductase (ENR), which catalyzes the last step in fatty acid elongation, has been the focus of a number of drug development efforts. ENR is inhibited by the antibacterial compound triclosan, which inhibits the in vitro growth of T. gondii at low micromolar to nanomolar concentrations.96,97 Triclosan has a very low solubility in water and poor oral bioavailability and a number of triclosan derivatives have been synthesized to increase potency and solubility.98–100 The most promising triclosan derivatives are described by Stec et al;98 compound 16c of this series had an in vitro IC50 of 250 nM compared to 3 μM for triclosan. Compound 16c also demonstrated improved solubility over triclosan, with a computational log P of 3.9 versus 5.5 for triclosan. Compound 16c, when given at an IP dose of 75 mg/kg/day for 2 days starting on the day of infection, decreased T. gondii proliferation in vivo compared to vehicle controls. Doses of ≤50 mg/kg were not effective.
Samuel et al101 demonstrated delivery of fluorescently labeled triclosan-octoarginine conjugates to intracellular and extracellular tachyzoites as well as encysted bradyzoites. Fluorescent triclosan-octoarginine conjugates were observed to localize around but not clearly enter the apicoplast. However, incubation of the RH strain in cell culture with a hydrolyzable triclosan-octoarginine conjugate resulted in concentration-dependent inhibition of T. gondii growth with maximum effect seen at 12 μM. Furthermore, administration of 40 mg/kg/day triclosan-octoarginine conjugate IP to mice infected IP with the RH strain reduced parasite counts over threefold after 5 days of treatment, compared to treatment with vehicle or unconjugated triclosan.
In another approach to improving the drug properties of triclosan, El-Zawawy et al102 created triclosan-loaded liposomes and compared their efficacy against triclosan in a murine model of acute toxoplasmosis. Liposomal triclosan at an oral dose of 100 mg/kg/day reduced the parasite burden by 96% in peritoneal fluid compared to a reduction of 74% with standard triclosan at an oral dose of 150 mg/kg/day. There was no evidence of drug toxicity in uninfected controls treated with the same doses.
The benzimidazoles are a class of compounds known to target bacterial ENRs.103 These compounds inhibit in vitro growth of T. gondii with IC50s in the low micromolar range.104 Based on co-crystallization studies, these compounds do not seem to bind TgENR tightly, raising questions as to their primary mode of action.
Another validated drug target in the FAS II pathway is the enzyme β-ketoacyl-acyl carrier protein synthase, which is inhibited by thiolactomycin, a naturally occurring thiolactone.105 Martins-Duarte et al106 tested eight thiolactomycin analogs against RH strain T. gondii growing in LCC-MK2 cell culture and noted IC50s between 1.6 and 29.4 μM. Electron microscopy of treated parasites revealed swollen mitochondrial, enlarged Golgi complex cisternae, and incomplete separation of dividing daughter cells.
Fluoroquinolone derivatives
The fluoroquinolones are inhibitors of DNA gyrase and DNA topoisomerase IV widely used in human and veterinary medicine as antibacterial agents.107–109 Trovafloxacin was found to have activity against T. gondii;110 however, this drug is no longer used because of the risk of liver failure. Subsequent studies have examined fluoroquinolone derivatives and veterinary fluoroquinolones against T. gondii. Although the mechanism of action of fluoroquinolones against T. gondii is unknown, they are presumed to inhibit DNA synthesis in the apicoplast.
The veterinary fluoroquinolone enrofloxacin was found to have a modest protective effect against vertical transmission of the apicomplexan parasite Neospora caninum in a mouse model.111 When tested against in vitro replication of T. gondii in human foreskin fibroblast (HFF) cell culture, enrofloxacin at 25 μg/mL reduced the number of cells infected by 59% versus an untreated control.112 It was more effective than sulfadiazine at 100 μg/mL, which reduced the number of infected cells by 27%. Enrofloxacin also demonstrated efficacy in a murine model of infection with the ME49 strain, reducing brain cysts in Calomys callosus by 68%, similar to sulfadiazine (79%).
A number of fluoroquinolone derivatives have been synthesized in an effort to create more potent inhibitors of T. gondii.113–115 Khan et al113 evaluated six compounds with in vitro IC50s below that of trovafloxacin. Analogs 1 and 2 of this series both showed IC50s of 530 nM, compared to 2.93 μM for trovafloxacin. Dubar et al116 synthesized several derivatives of ciprofloxacin and noted in vitro IC50s of 420 nM for compound 2, an ester prodrug, and 460 nM for compound 5, an adamantyl derivative. Neither compound was observed to be toxic to host cells at 30 μM. In a murine model of acute infection, compound 2 at 50 mg/kg/day orally for 7 days improved 60-day mortality to 18% versus 0% for ciprofloxacin-treated controls.117 In the same model, compound 5 at 100 mg/kg/day orally for 7 days improved 60-day mortality to 25%.
Histone acetyltransferase/histone deacetylase inhibitors
The epigenetic control of gene expression by post-translational modification of histone proteins is a key process by which many organisms including T. gondii modulate transcription. Acetylation of conserved histone lysine residues by histone acetyltransferases (HATs) creates the post-translation modification, generally increasing transcription of the target gene. Conversely, de-acetylation by histone deacetylases (HDACs) removes the modification, generally decreasing the transcription of the target gene. The multiple stages in the T. gondii life cycle require significant changes in gene expression, and several groups have suggested developing novel therapeutic compounds that target epigenetic modification.118,119
The cyclic tetrapeptide FR235222 causes hyper-acetylation of histone H4 in T. gondii, inhibits growth of the parasite with an in vitro EC50 of 7.6 nM, and is associated with the conversion from the tachyzoite to bradyzoite stage.120 Electron micrographs of bradyzoites formed during treatment with FR235222 show altered morphology and multiple nuclei. T. gondii resistant to FR235222 has T99A and T99I mutations in the HDAC TgHDAC3. Introducing either mutation into the parental strain reproduced the FR235222-resistant phenotype, demonstrating that TgHDAC3 is the target of FR235222. Interestingly, T99 is part of a two amino acid insertion (A98T99) that is unique to the members of the apicomplexan HDAC3 family, making this enzyme an attractive drug target.
Treatment of bradyzoites with FR235222 prevents differentiation into tachyzoites. Pretreated bradyzoites are incapable of infecting either an HFF monolayer in vitro or infecting mice.121 However, host cell toxicity was a concern for FR235222 in that it inhibited HFF cells at an IC50 of 128 nM. Two derivatives of FR235222, W363 and W399, display IC50s against T. gondii equivalent to that of the parent compound but less toxic against HFF cells (HFF IC50: W363 =632 nM and W399 =539 nM).
A recently described HAT inhibitor is garcinol, a polyisoprenylated benzophenone derivative extracted from the kokum fruit (Garcinia indica) that targets the HAT TgGCN5b.122 TgGCN5b is essential for tachyzoite replication.123 T. gondii exposed to garcinol showed decreased transcription of genes regulated by TgGCN5b. Compared to other histone deacetyl transferase inhibitors the in vitro IC50 of garcinol was less potent at 1.8 μM.
N-benzoyl-2-hydroxybenzamides
In an effort to identify novel leads effective against T. gondii, Fomovska et al124 screened a library of 6,811 synthetic compounds. The most promising compound to emerge from this screen, MP-IV-1, was found to have an IC90 of 31 nM against RH strain tachyzoites and no observed toxicity against host cells at 10 μM. A chemically diverse series of MP-IV-1 derivatives were synthesized and evaluated. Of these, the compound QQ-437 was the most potent with an in vitro IC90 of 16 nM and no apparent toxicity to host cells at 250 nM. Treatment with MP-IV-1 at a dose of 50 mg/kg/day IP reduced RH strain peritoneal fluid parasite counts. In an experiment using type II Prugniaud strain, QQ-437 at a dose of 20 mg/kg/day decreased parasite burden compared to MP-IV-1.
Using an insertional mutagenesis library, the authors identified four clones, all with insertions in the gene coding for adaptin-3β. Although little is known about adaptin-3β in T. gondii, the related protein adaptin-1 is known to be involved in sorting proteins from the Golgi complex to rhoptries.125 Rhoptries, along with micronemes and dense granules, are secretory organelles found in T. gondii whose biogenesis requires correct trafficking of proteins from the Golgi complex.126 Electron microscopy of parasites treated with either MP-IV-1 or QQ-437 demonstrated distortion of micronemes, dense granules, and rhoptries along with the absence of acidocalcisome, another secretory organelle. These results demonstrate that the N-benzoyl-2-hydroxybenzamides are potent inhibitors of secretory processes in T. gondii.
Macrolide derivatives
The apicoplast is a plastid organelle present in most of the members of the phylum Apicomplexa acquired from endosymbiosis.127 The T. gondii apicoplast is the site of several essential metabolic pathways, many of which contain potential drug targets.128 Molecules that inhibit protein synthesis in the apicoplast have been shown to be effective agents against T. gondii. Clindamycin inhibits the bacterial 50S ribosomal subunit and is used clinically as an antibacterial and for the treatment of toxoplasmosis. The macrolides erythromycin and azithromycin also target the 50S subunit and are routinely prescribed for the treatment of bacterial infections. Azithromycin prevents death from acute toxoplasmosis in a murine model129 and has several advantageous properties as a drug including high oral bioavailability and a long half-life. Similar to clindamycin, azithromycin causes a delayed death phenotype130 in which parasites are only modestly inhibited during the first round of replication, but all daughter parasites are severely impaired in their ability to replicate further, even after the removal of azithromycin.131
Lee et al132 synthesized a series of erythromycin and azithromycin analogs with the goal of improving antiparasitic efficacy. Compound 11 of their series is an azithromycin derivative in which the ring nitrogen was alkylated with a benzyl substituent. Compound 11 had an IC50 of 2 μM. Compound 11 displayed a similar delayed death phenotype as azithromycin with an IC50 of 5 μM against the first cycle of replication and an IC50 of 500 nM against the second cycle. Interestingly, in contrast to azithromycin, compound 11 significantly reduced the number of parasitophorous vacuoles formed on initial infection, suggesting an alternate mode of action.
Pantothenate synthetase inhibitors
All organisms that require coenzyme A and acyl carrier protein have pathways for synthesizing or acquiring these cofactors from pantothenate (vitamin B5). In plants, fungi, and bacteria, pantothenate is synthesized de novo from pyruvate via a conserved three-enzyme pathway;133 however, animals must acquire pantothenate through their diet or from gastrointestinal bacteria. By searching the T. gondii genome for orthologs of the pantothenate synthesis pathway, Mageed et al134 identified three conserved genes coding for de novo pantothenate biosynthesis in T. gondii. Among these genes is the terminal enzyme in the pathway, pantothenate synthetase that converts pantoate to pantothenate. Consistent with this finding, T. gondii did not exhibit a requirement for exogenous pantothenate when grown in culture.
Mageed et al134 tested a series of pantothenate synthetase inhibitors originally developed for Mycobacterium tuberculosis135 and found that two compounds, SW413 and SW404, inhibited in vitro growth of T. gondii with IC50s at 20 and 130 nM, respectively. Supplementing the growth media with 50 mg/L pantothenate decreased drug susceptibility to SW404 216-fold. In silico modeling placed SW404 in the pantoate-binding site of pantothenate synthetase and predicted binding to two conserved glutamine residues of this enzyme.
These results suggest that de novo pantothenic acid biosynthesis is an attractive target for drug development due to the lack of a homologous pathway in the human host. The rapid metabolism of free pantothenate by host cells to coenzyme A would likely prevent T. gondii from scavenging pantothenate from the host and overcoming pantothenate synthetase inhibition; however, this would need to be verified by in vivo experiments.
Peptide-conjugated phosphorodiamidate morpholino oligomers (PMOs)
PMOs are synthetic oligomers that bind complementary mRNA sequences and interfere with gene expression.136 PMOs are potentially a highly specific means of disrupting the translation of key parasitic proteins. The challenge for developing effective PMOs is delivering the PMO to the target. Using a similar approach to that of Samuel et al,105 Lai et al137 conjugated various PMOs to arginine octomers, creating peptide-conjugated PMOs (PPMOs). The authors demonstrated specific knockdown of T. gondii DHFR, enoyl-acyl carrier protein reductase, and the transcription factor AP2XI-3, a key regulator of bradyzoite differentiation. Knocking down expression of each protein with 3–5 μM of the targeted PMO partially inhibited the replication of RH strain tachyzoites without toxicity to the host HFF cells at 20 μM. Growth of parasites treated with an anti-DHFR PPMO was rescued by supplementation with exogenous folic acid.137 When a dose of 12.5 mg/kg was given IP for 2 days, the anti-DHFR PPMO reduced parasite count by 83% after 96 h in mice infected with RH strain T. gondii compared to treatment with vehicle or an off-target PPMO. PPMOs targeting the 5′ end of the T. gondii cytochrome b gene partially inhibited T. gondii replication at 10 μM.42 In principal, PPMOs could be extended to target many essential genes.137,138
4-(1H)-quinolones
The cytochrome bc1 complex (bc1) is a drug target in several apicomplexan pathogens, including T. gondii, P. falciparum and B. microti. The bc1 reduces cytochrome c as part of the electron transport chain and generates an electrochemical gradient by transferring protons to the intermembrane space. The bc1 also creates ubiquinone for pyrimidine biosynthesis. The bc1 Qo site oxidizes ubiquinol and the bc1 Qi site reduces ubiquinone. Atovaquone is a Qo-site inhibitor that is used as an alternate treatment and prophylactic for toxoplasmosis, and in drug-combination regimens for malaria and mild to moderate babesiosis.37,139 Qi site inhibitors are not currently in clinical use; however, pyridones, which are Qi site inhibitors, were advanced to human studies for malaria, but were found to have cardiotoxicity in rats and activity against human bc1.140 The endochin-like-quinolone (ELQ) series of 4(1H)-quinolone-3-diarylethers are derived from endochin, target the Qi site and have been designed to avoid human bc1 inhibition.141,142
Endochin was initially investigated as an antimalarial drug in an avian model of malaria by Salzer et al in 1948.143 Gingrich and Darrow later found endochin to be active against avian and murine toxoplasmosis in 1951.144 A library of 4(1H)-quinolone-3-diarylethers was made to improve metabolic stability, solubility and the in vivo efficacy of endochin. ELQ-316 and ELQ-271 were found to be highly effective against acute and latent toxoplasmosis.39 The in vitro IC50 values of ELQ-271 and ELQ-316 were 0.1 and 0.007 nM, respectively. The ED50 values of ELQ-271 and ELQ-316 were 0.14 and 0.08 mg/kg when administered orally against acute toxoplasmosis with the RH strain in mice. ELQ-271 and ELQ-316 reduced the cyst burden in a mouse model of latent infection with the ME49 strain by 76%–88%.39 ELQ-271 was found to inhibit the human bc1 at 800 nM, whereas ELQ-316 did not inhibit the human bc1 and was not toxic to human fibroblasts or human hepatocarcinoma cells (HepG2) at 10 μM, the highest concentration tested.141 Despite being highly efficacious, the bioavailability of ELQs was limited by crystallinity and a lack of aqueous solubility, which was viewed as a liability for further clinical development. Improved bioavailability was achieved by the synthesis of ethyl carbonate prodrugs in which the ethyl carbonate promoiety disrupts crystal lattice formation.145 Like atovaquone, ELQ-316 has broad anti-apicomplexan activity and is also highly effective against P. falciparum and B. microti and offers a novel approach to prevent the development of resistance when combined with atovaquone for dual inhibition of the parasite bc1.141,146
The 4-(1H)-quinolone scaffold was also recently investigated by McPhillie et al.42 A screen of 5,6,7,8-tetrahydroquinolin-4-one derivatives identified MJM170 as a lead compound that inhibits RH tachyzoites in vitro with an IC50 of 30 nM and EGS encysted bradyzoites with an IC50 of 4 μM. A 25 mg/kg daily dose given for 5 days IP reduced the acute infection in mice with RH and Prugniaud parasites, and a 12.5 mg/kg dose given for 17 days reduced ME49 brain cysts. Inhibition of Saccharomyces cerevisiae bc1 Qi-site mutants and co-crystallography with bovine bc1 indicate that MJM170 binds to the bc1 Qi site.
The compound 1-hydroxy-2-dodecyl-4(1H)quinolone (HDQ), a structural analog of ubiquinone, has been identified as an inhibitor of type II NADH dehydrogenase isolated from the yeast Yarrow lipolitica, S. cerevisiae bc1 Qi site and T. gondii dihydroorotate dehydrogenase.147–149 The electron transport chain of T. gondii contains a single-component type II NADH dehydrogenase instead of the multisubunit type I NADH dehydrogenase found in mammals.150,151 Hegewald et al152 investigated HDQ in T. gondii by generating RH-strain mutants resistant to HDQ. Sequencing of genes coding for ubiquinone-binding proteins showed a N302S point mutation in dihydroorotate dehydrogenase. Enzymatic studies with purified wild-type and mutant enzyme confirmed that HDQ inhibited dihydroorotate dehydrogenase. HDQ was shown to inhibit the in vitro growth of RH strain T. gondii with an IC50 of 3.7 nM.153 Later work154 explored the activity of four HDQ derivatives, two of which were more potent than the parent molecule with in vitro IC50s of 0.4 and 0.8 nM (compounds A and B, respectively). The in vivo efficacy of these compounds was examined in murine models of acute and chronic infection. Compounds A and B showed similar efficacy as atovaquone in controlling parasite replication in the peritoneal cavity, but HDQ was less effective than atovaquone. HDQ, compound A, and compound B were effective against parasite replication in liver and lung, but not as effective as atovaquone. In a model of chronic infection with the ME49 strain, compound B showed a trend toward reduced brain cyst burden; however, this result did not reach statistical significance. Similar to the above-mentioned 4-(1H)-quinolones, HDQ derivatives are limited by low aqueous solubility via π-stacking of aromatic rings as Pidathala et al155 describe in their investigation of HDQ derivatives.
Ruthenium complexes
Ruthenium-based complexes have been investigated for various purposes including the treatment of cancer and bacterial infections.156 Barna et al157 examined 18 ruthenium-containing compounds for activity against T. gondii, and they found that two compounds, 16 and 18 of their series, had IC50s of 18.7 and 41.1 nM, respectively. Treated parasites displayed empty or lipid-filled inclusions after 12 h of incubation with compound 16, and by 36 h, the parasite cytoplasm was disorganized. Both compounds 16 and 18 are hydrolytically stable ruthenium phosphite complexes with hydrocarbon exteriors. The authors hypothesize that the hydrocarbon exterior facilitates movement across lipid bilayers while shielding the core of the molecule from nucleophilic attacks and subsequent degradation.
Salicylanilides
Fomovska et al158 synthesized 39 derivatives of the antihelmintic drug niclosamide and examined their in vitro efficacy against RH strain tachyzoites. Six derivative compounds had IC50s below 250 nM, and of these, four were tested further to assess whether they killed the parasites or the parasites resumed replication after removal of the compound (compounds 3i, 3j, 7a, and 14a). Parasites resumed replication after removal of compound 3i. Parasites did not recover after removal of 3j, 7a, or 14a. Two compounds, 14a and 14b, were tested for in vivo efficacy by challenging mice with oral infection of either ME49 or TgGoatUS4 oocysts and then treating orally with 100 mg/kg/day, 25 mg/kg/day, or vehicle control. All the infected mice died, but a mild protective effect was seen on treated animals as they died on average 1 day later. No gross toxicities were noted in animals treated with 100 mg/kg of either compound. The mechanism of action of salicylanilides is unknown despite efforts using random insertional mutagenesis to identify a target.
Spiroindolones
The spiroindolone NITD609 was originally discovered in a drug screen for new antimalarial agents159 and later found to be effective against T. gondii in vitro and in vivo. The target of NITD609 in P. falciparum is a P-type cation-transporting adenosine triphosphatase, PfATP4. In T. gondii, NITD609 has an in vitro IC50 of 1 μM against RH strain tachyzoites. In vivo, NITD609 at an oral dose of 100 mg/kg/day for 5 days decreased the parasite burden by 90% in mice infected IP with RH strain parasites. Although the T. gondii ATP4 has a high degree of sequence homology to PfATP4, further work is needed to definitively identify the target of NITD609 in T. gondii.
Thiosemicarbazones
Hydroxyurea, or hydroxycarbamide, is an antineoplastic drug primarily used in the treatment of polycythemia vera as well as other myeloproliferative disorders. In mammalian cells, it acts by inhibiting ribonucleotide reductase,160 which catalyzes the rate-limiting step in DNA synthesis. Inhibition of ribonucleotide reductase leads to cell cycle arrest at the G1/S juncture. de Melo et al161 demonstrated that incubation of infected Vero cells in 4 mM hydroxyurea for 5 h interfered with intracellular parasite replication. Electron microscopy showed that hydroxyurea caused severe morphologic alterations to intracellular parasites followed by the elimination of parasites from parasitophorous vacuoles.
Hydroxyurea is limited by its relatively low affinity for ribonucleotide reductase as well as its short half-life in humans.160 However, the thiosemicarbazones have higher affinity for ribonucleoside reductase162 and are active against P. falciparum.163
Tenório et al164 synthesized a series of thiosemicarbazones as well as the structurally related 4-thiazolidinones and screened the resulting compounds for activity. Compounds were found to have IC50s ranging from 80 to 500 μM based on microscopy of infected host cells. By comparison, both hydroxyurea and sulfadiazine were found to have IC50s of 100 μM in this assay. The authors did not note any morphologic alterations to the Vero cells at the highest compound concentration of 30 mM. Of all the compounds tested, 4-thiazolidinones with phenyl substituents on the N-3 position showed the lowest IC50s. With this in mind, de Aquino et al165 made an additional set of 4-thiazolidinones with the phenyl group fixed at the N-3 position and a set of thiosemicarbazones with the phenyl at the N-4 position (4-arylthiazolidinones). The two compounds with the lowest IC50, 2i and 2k, were 4-arylthiazolidinones with IC50s of 50 μM against intracellular tachyzoites and 1 mM against the host Vero cells.
Liesen et al166 continued this work by synthesizing additional 4-thiazolidinones and 1,3,4-thiadiazoles from acylthiosemicarbazides with imidazole substituents at their N-1 position. Several members of this series showed equal efficacy to compounds from previous series but with less toxicity toward the host cells. Dzitko et al167 expanded on this series and examined how replacement of the imidazole ring with other heteroaryl rings might alter the activity. The most potent compound in their series 1g had an IC50 of 33.2 μg/mL (107 μM) against RH tachyzoites in L929 fibroblast culture as measured by the incorporation of [3H]uracil. The authors conducted in silico studies in which these molecules were docked against possible target enzymes; however, no clear target was identified.
Triazines
The anticoccidial triazine toltrazuril is widely used in the poultry industry for its activity against Eimeria spp. This drug is not used in humans due to its potential to cause developmental malformations in a rat model.168 The major metabolite of toltrazuril is ponazuril. Mitchell et al169 demonstrated that ponazuril reduced the proliferation of T. gondii grown in African green monkey kidney cells at a concentration of 1 mg/mL (2.2 μM). The authors also found that oral ponazuril (10 mg/kg/day for 10 days) given 6 days after subcutaneous infection with RH parasites reduced mortality at 8 weeks (5/5 control mice dead versus 3/5 treated). With a higher dose (20 mg/kg/day for 10 days) given 6 days post-infection, all treated mice survived to 8 weeks.
Triazole derivatives
Itraconazole, a member of the triazole family of antifungal agents, is known to have in vitro activity against T. gondii tachyzoites.170 However, in vivo studies of its protective effect against mortality and brain cyst formation in a mouse model of infection with the ME49 strain failed to demonstrate efficacy.171 In the same study, the authors showed that the related compound fluconazole reduced 45-day mortality from 50% in untreated controls to 5% when given at 20 mg/kg/day to mice infected IP with ME49. However, fluconazole treatment did not reduce the formation of brain cysts when compared to untreated animals.
The idea of optimizing triazoles for better antiparasitic activity is a particularly attractive one, as these compounds are well tolerated and have high oral bioavailability. Dzitko et al172 synthesized two such compounds, namely 3-(thiophen-2-yl)-1,2,4-triazole-5-thione and 4-ethyl-3-(4-methyl-1,2,3-thiadiazol-5-yl)-1,2,4–5-thione. The first of these, which the authors describe as “s-triazole” showed in vitro activity against the RH strain of T. gondii grown in L929 mouse fibroblast culture. IC50s measured by incorporation of 3H-uracil and quantitative real-time PCR were 42.46 μg/mL (235.6 μM) and 37.33 μg/mL (207.1 μM), respectively. These compared favorably to those obtained for sulfadiazine by the same methods, which were 2,225.25 μg/mL (8.9 mM) by 3H-uracil incorporation and 1,217.62 μg/mL (4.86 mM) by quantitative real-time PCR. Cytotoxic effects on host cells were measured using the MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay and calculated as the concentration needed to cause a 30% maximal effect (CC30). The CC30 for s-triazole was 194.46 μg/mL (1.08 mM) and that of sulfadiazine was 537.06 μg/mL (2.15 mM), demonstrating a better therapeutic index for s-triazole as calculated by IC50/CC30.
The target of s-triazole and other triazole compounds in T. gondii is unclear. Through a literature search, Dzitko et al172 identified six likely enzymes and attempted in silico docking of s-triazole. Out of three potential targets, the authors suggested that Protein Data Bank number 3AU9 (1-deoxy-D-xylulose-5-phosphate reductoisomerase) was the most likely, followed by 4M84 (calmodulin-domain protein kinase 1) and 3MB8 (purine nucleoside phosphorylase).
Tryptanthrin derivatives
Tryptanthrin (indolo[2,1-b]quinazoline-6,12-dione) is a compound found in several natural sources, among them the plant Isatis indigotica. Krivogorsky et al173 examined the activity of tryptanthrin and derivative molecules in vitro. Several compounds, in particular derivatives with halogen substituents at the 8 position, had IC50s under 10 nM. Further work174 investigated the structure-activity relationship of the 6-keto group by replacing it with oximes, hydrazones, or alcohols. The oxime and alcohol derivatives of 8-bromotrypanthrin had excellent activity, with IC50s of 3 nM (for the oxime, compound 6b) and 2 nM (for the alcohol, compound 15b). Cytotoxic effects were not seen at concentrations as high as 100 μM. The target of these compounds was not determined.
Acknowledgment
This work was supported by Career Development Award # BX002440 from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development.
Disclosure
The authors report no conflicts of interest in this work.
References
Tenter AM, Heckeroth AR, Weiss LM. Toxoplasma gondii: from animals to humans. Int J Parasitol. 2000;30(12–13):1217–1258. | ||
Jones JL, Akstein RB, Hlavsa MC, Lopez AS, Wilson M, Holland GN. Follow-up of the 1977 Georgia outbreak of toxoplasmosis. Am J Trop Med Hyg. 2016;94(6):1299–1300. | ||
Vaudaux JD, Muccioli C, James ER, et al. Identification of an atypical strain of toxoplasma gondii as the cause of a waterborne outbreak of toxoplasmosis in Santa Isabel do Ivai, Brazil. J Infect Dis. 2010; 202(8):1226–1233. | ||
Bowie WR, King AS, Werker DH, et al. Outbreak of toxoplasmosis associated with municipal drinking water. The BC Toxoplasma Investigation Team. Lancet. 1997;350(9072):173–177. | ||
Pappas G, Roussos N, Falagas ME. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol. 2009;39(12):1385–1394. | ||
Dubey JP, Lago EG, Gennari SM, Su C, Jones JL. Toxoplasmosis in humans and animals in Brazil: high prevalence, high burden of disease, and epidemiology. Parasitology. 2012;139(11):1375–1424. | ||
Jones JL, Kruszon-Moran D, Rivera HN, Price C, Wilkins PP. Toxoplasma gondii seroprevalence in the United States 2009–2010 and comparison with the past two decades. Am J Trop Med Hyg. 2014;90(6):1135–1139. | ||
Khan A, Jordan C, Muccioli C, et al. Genetic Divergence of Toxoplasma gondii Strains Associated with Ocular Toxoplasmosis, Brazil. Emerging Infectious Diseases. 2006;12(6):942–949. | ||
Demar M, Hommel D, Djossou F, et al. Acute toxoplasmoses in immunocompetent patients hospitalized in an intensive care unit in French Guiana. Clin Microbiol Infect. 2012;18(7):E221–E231. | ||
Jones JL, Holland GN. Annual burden of ocular toxoplasmosis in the US. Am J Trop Med Hyg. 2010;82(3):464–465. | ||
Robert-Gangneux F, Darde ML. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin Microbiol Rev. 2012;25(2):264–296. | ||
Coccaro EF, Lee R, Groer MW, Can A, Coussons-Read M, Postolache TT. Toxoplasma gondii infection: relationship with aggression in psychiatric subjects. J Clin Psychiatry. 2016;77(3):334–341. | ||
Sugden K, Moffitt TE, Pinto L, Poulton R, Williams BS, Caspi A. Is Toxoplasma gondii infection related to brain and behavior impairments in humans? Evidence from a population-representative birth cohort. PLoS One. 2016;11(2):e0148435. | ||
Sutterland AL, Fond G, Kuin A, et al. Beyond the association. Toxoplasma gondii in schizophrenia, bipolar disorder, and addiction: systematic review and meta-analysis. Acta Psychiatr Scand. 2015;132(3):161–179. | ||
Rabaud C, May T, Amiel C, et al. Extracerebral toxoplasmosis in patients infected with HIV. A French National Survey. Medicine (Baltimore). 1994;73(6):306–314. | ||
Grant IH, Gold JW, Rosenblum M, Niedzwiecki D, Armstrong D. Toxoplasma gondii serology in HIV-infected patients: the development of central nervous system toxoplasmosis in AIDS. AIDS. 1990;4(6):519–521. | ||
Mele A, Paterson PJ, Prentice HG, Leoni P, Kibbler CC. Toxoplasmosis in bone marrow transplantation: a report of two cases and systematic review of the literature. Bone Marrow Transplant. 2002;29:691–698. | ||
Martino R, Maertens J, Bretagne S, et al. Toxoplasmosis after hematopoietic stem cell transplantation. Clin Infect Dis. 2000;31:1188–1195. | ||
Martino R, Bretagne S, Einsele H, et al. Early detection of Toxoplasma infection by molecular monitoring of Toxoplasma gondii in peripheral blood samples after allogeneic stem cell transplantation. Clin Infect Dis. 2005;40(1):67–78. | ||
Martino R, Bretagne S, Rovira M, et al. Toxoplasmosis after hematopoietic stem transplantation. Report of a 5-year survey from the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2000;25:1111–1114. | ||
Gajurel K, Dhakal R, Montoya JG. Toxoplasma prophylaxis in haematopoietic cell transplant recipients: a review of the literature and recommendations. Curr Opin Infect Dis. 2015;28(4):283–292. | ||
Jones JL, Roberts JM. Toxoplasmosis hospitalizations in the United States, 2008, and trends, 1993–2008. Clin Infect Dis. 2012;54(7):e58–e61. | ||
Hoffmann S, Batz MB, Morris JG Jr. Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J Food Prot. 2012;75(7):1292–1302. | ||
McLeod R, Boyer K, Karrison T, et al. Outcome of treatment for congenital toxoplasmosis, 1981–2004: the National Collaborative Chicago-Based, Congenital Toxoplasmosis Study. Clin Infect Dis. 2006;42(10):1383–1394. | ||
Carme B, Demar M, Ajzenberg D, Darde ML. Severe acquired toxoplasmosis caused by wild cycle of Toxoplasma gondii, French Guiana. Emerg Infect Dis. 2009;15(4):656–658. | ||
Khan A, Ajzenberg D, Mercier A, et al. Geographic separation of domestic and wild strains of Toxoplasma gondii in French Guiana correlates with a monomorphic version of chromosome1a. PLoS Negl Trop Dis. 2014;8(9):e3182. | ||
Demar M, Ajzenberg D, Maubon D, et al. Fatal outbreak of human toxoplasmosis along the Maroni River: epidemiological, clinical, and parasitological aspects. Clin Infect Dis. 2007;45(7):e88–e95. | ||
Porter SB, Sande MA. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N Engl J Med. 1992;327(23):1643–1648. | ||
Katlama C, De Wit S, O’Doherty E, Van Glabeke M, Clumeck N. Pyrimethamine-clindamycin vs. pyrimethamine-sulfadiazine as acute and long-term therapy for toxoplasmic encephalitis in patients with AIDS. Clin Infect Dis. 1996;22(2):268–275. | ||
Dannemann B, McCutchan JA, Israelski D, et al. Treatment of toxoplasmic encephalitis in patients with AIDS. A randomized trial comparing pyrimethamine plus clindamycin to pyrimethamine plus sulfadiazine. The California Collaborative Treatment Group. Ann Intern Med. 1992;116(1):33–43. | ||
Beraud G, Pierre-Francois S, Foltzer A, et al. Cotrimoxazole for treatment of cerebral toxoplasmosis: an observational cohort study during 1994–2006. Am J Trop Med Hyg. 2009;80(4):583–587. | ||
Yan J, Huang B, Liu G, et al. Meta-analysis of prevention and treatment of toxoplasmic encephalitis in HIV-infected patients. Acta Trop. 2013;127(3):236–244. | ||
Torre D, Casari S, Speranza F, et al. Randomized trial of trimethoprim-sulfamethoxazole versus pyrimethamine-sulfadiazine for therapy of toxoplasmic encephalitis in patients with AIDS. Italian Collaborative Study Group. Antimicrob Agents Chemother. 1998;42(6):1346–1349. | ||
Torres RA, Weinberg W, Stansell J, et al. Atovaquone for salvage treatment and suppression of toxoplasmic encephalitis in patients with AIDS. Atovaquone/Toxoplasmic Encephalitis Study Group. Clin Infect Dis. 1997;24(3):422–429. | ||
Chirgwin K, Hafner R, Leport C, et al. Randomized phase II trial of atovaquone with pyrimethamine or sulfadiazine for treatment of toxoplasmic encephalitis in patients with acquired immunodeficiency syndrome: ACTG 237/ANRS 039 Study. AIDS Clinical Trials Group 237/Agence Nationale de Recherche sur le SIDA, Essai 039. Clin Infect Dis. 2002;34(9):1243–1250. | ||
Jacobson JM, Hafner R, Remington J, et al. Dose-escalation, phase I/II study of azithromycin and pyrimethamine for the treatment of toxoplasmic encephalitis in AIDS. AIDS. 2001;15(5):583–589. | ||
Panel on Opportunistic Infections in HIV-Infected Adults and Adolescents. Guidelines for the prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from the Centers for Disease Control and Prevention, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/adult_oi.pdf. Accessed July 17, 2016. | ||
Harrell M, Carvounis PE. Current treatment of toxoplasma retinochoroiditis: an evidence-based review. J Ophthalmol. 2014;2014:273506. | ||
Doggett JS, Nilsen A, Forquer I, et al. Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc Natl Acad Sci U S A. 2012;109(39):15936–15941. | ||
Benmerzouga I, Checkley LA, Ferdig MT, Arrizabalaga G, Wek RC, Sullivan WJ Jr. Guanabenz repurposed as an antiparasitic with activity against acute and latent toxoplasmosis. Antimicrob Agents Chemother. 2015;59(11):6939–6945. | ||
Araujo FG, Huskinson-Mark J, Gutteridge WE, Remington JS. In vitro and in vivo activities of the hydroxynaphthoquinone 566C80 against the cyst form of Toxoplasma gondii. Antimicrob Agents Chemother. 1992;36(2):326–330. | ||
McPhillie M, Zhou Y, El Bissati K, et al. New paradigms for understanding and step changes in treating active and chronic, persistent apicomplexan infections. Nature Publishing Group. 2016;6:29179. | ||
Watts E, Zhao Y, Dhara A, Eller B, Patwardhan A, Sinai AP. Novel approaches reveal that Toxoplasma gondii bradyzoites within tissue cysts are dynamic and replicating entities in vivo. MBio. 2015;6(5):e01155–e01115. | ||
Gajurel K, Gomez CA, Dhakal R, Vogel H, Montoya JG. Failure of primary atovaquone prophylaxis for prevention of toxoplasmosis in hematopoietic cell transplant recipients. Transpl Infect Dis. 2016;18(3):446–452. | ||
Meneceur P, Bouldouyre MA, Aubert D, et al. In vitro susceptibility of various genotypic strains of Toxoplasma gondii to pyrimethamine, sulfadiazine, and atovaquone. Antimicrob Agents Chemother. 2008;52(4):1269–1277. | ||
Aspinall TV, Joynson DH, Guy E, Hyde JE, Sims PF. The molecular basis of sulfonamide resistance in Toxoplasma gondii and implications for the clinical management of toxoplasmosis. J Infect Dis. 2002;185(11):1637–1643. | ||
Doliwa C, Xia D, Escotte-Binet S, et al. Identification of differentially expressed proteins in sulfadiazine resistant and sensitive strains of Toxoplasma gondii using difference-gel electrophoresis (DIGE). Int J Parasitol Drugs Drug Resist. 2013;3:35–44. | ||
Doliwa C, Escotte-Binet S, Aubert D, et al. Sulfadiazine resistance in Toxoplasma gondii: no involvement of overexpression or polymorphisms in genes of therapeutic targets and ABC transporters. Parasite. 2013;20:19. | ||
McFadden DC, Tomavo S, Berry EA, Boothroyd JC. Characterization of cytochrome b from Toxoplasma gondii and Q(o) domain mutations as a mechanism of atovaquone-resistance. Mol Biochem Parasitol. 2000;108(1):1–12. | ||
Araujo FG, Huskinson J, Remington JS. Remarkable in vitro and in vivo activities of the hydroxynaphthoquinone 566C80 against tachyzoites and tissue cysts of Toxoplasma gondii. Antimicrob Agents Chemother. 1991;35(2):293–299. | ||
Reynolds MG, Oh J, Roos DS. In vitro generation of novel pyrimethamine resistance mutations in the Toxoplasma gondii dihydrofolate reductase. Antimicrob Agents Chemother. 2001;45(4):1271–1277. | ||
Kim K. A bradyzoite is a bradyzoite is a bradyzoite? Trends Parasitol. 2015;31(12):610–612. | ||
Vidadala RS, Rivas KL, Ojo KK, et al. Development of an orally available and central nervous system (CNS)-penetrant Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitor with minimal human ether-a-go-go-related gene (hERG) activity for the treatment of toxoplasmosis. J Med Chem. 2016;59(13):6531–6546. | ||
Lüder CGK, Reichard U, Gross U. Chapter 7 – Toxoplasma Animal Models and Therapeutics. Toxoplasma Gondii (Second Edition). Boston: Academic Press; 2014:217–255. | ||
Paredes-Santos TC, Tomita T, Yan Fen M, et al. Development of dual fluorescent stage specific reporter strain of Toxoplasma gondii to follow tachyzoite and bradyzoite development in vitro and in vivo. Microbes Infect. 2016;18(1):39–47. | ||
Sidik SM, Huet D, Ganesan SM, et al. A genome-wide CRISPR screen in toxoplasma identifies essential apicomplexan genes. Cell. 2016;166(6):1423–1435.e12. | ||
Gajria B, Bahl A, Brestelli J, et al. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 2008;36(Database issue): D553–D556. | ||
Nagamune K, Beatty WL, Sibley LD. Artemisinin induces calcium-dependent protein secretion in the protozoan parasite Toxoplasma gondii. Eukaryotic Cell. 2007;6:2147–2156. | ||
Nagamune K, Moreno SNJ, Sibley LD. Artemisinin-resistant mutants of Toxoplasma gondii have altered calcium homeostasis. Antimicrob Agents Chemother. 2007;51:3816–3823. | ||
Holfels E, McAuley J, Mack D, Milhous WK, McLeod R. In vitro effects of artemisinin ether, cycloguanil hydrochloride (alone and in combination with sulfadiazine), quinine sulfate, mefloquine, primaquine phosphate, trifluoperazine hydrochloride, and verapamil on Toxoplasma gondii. Antimicrob Agents Chemother. 1994;38:1392–1396. | ||
D’Angelo JG, Bordón C, Posner GH, Yolken R, Jones-Brando L. Artemisinin derivatives inhibit Toxoplasma gondii in vitro at multiple steps in the lytic cycle. J Antimicrob Chemother. 2009;63:146–150. | ||
Schultz TL, Hencken CP, Woodard LE, et al. A thiazole derivative of artemisinin moderately reduces Toxoplasma gondii cyst burden in infected mice. J Parasitol. 2014;100:516–521. | ||
Hencken CP, Jones-Brando L, Bordón C, et al. Thiazole, oxadiazole, and carboxamide derivatives of artemisinin are highly selective and potent inhibitors of Toxoplasma gondii. J Med Chem. 2010;53:3594–3601. | ||
Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83(9):1032–1045. | ||
Ling Y, Li Z-H, Miranda K, Oldfield E, Moreno SNJ. The farnesyl-diphosphate/geranylgeranyl-diphosphate synthase of Toxoplasma gondii is a bifunctional enzyme and a molecular target of bisphosphonates. J Biol Chem. 2007;282:30804–30816. | ||
Martin MB, Grimley JS, Lewis JC, et al. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: a potential route to chemotherapy. J Med Chem. 2001;44:909–916. | ||
Yardley V, Khan AA, Martin MB, et al. In vivo activities of farnesyl pyrophosphate synthase inhibitors against Leishmania donovani and Toxoplasma gondii. Antimicrob Agents Chemother. 2002;46:929–931. | ||
Johnson SM, Murphy RC, Geiger JA, et al. Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-toxoplasma activity. J Med Chem. 2012;55(5):2416–2426. | ||
Winzer P, Muller J, Aguado-Martinez A, et al. In vitro and in vivo effects of the bumped kinase inhibitor 1294 in the related cyst-forming apicomplexans Toxoplasma gondii and Neospora caninum. Antimicrob Agents Chemother. 2015;59(10):6361–6374. | ||
Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature. 2010;465(7296):359–362. | ||
Ojo KK, Larson ET, Keyloun KR, et al. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 2010;17(5):602–607. | ||
Murphy RC, Ojo KK, Larson ET, et al. Discovery of potent and selective inhibitors of calcium-dependent protein kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett. 2010;1(7):331–335. | ||
Sugi T, Kobayashi K, Takemae H, et al. Identification of mutations in TgMAPK1 of Toxoplasma gondii conferring resistance to 1NM-PP1. Int J Parasitol Drugs Drug Resist. 2013;3:93–101. | ||
Sugi T, Kawazu S, Horimoto T, Kato K. A single mutation in the gatekeeper residue in TgMAPKL-1 restores the inhibitory effect of a bumped kinase inhibitor on the cell cycle. Int J Parasitol Drugs Drug Resist. 2015;5(1):1–8. | ||
Larson ET, Ojo KK, Murphy RC, et al. Multiple determinants for selective inhibition of apicomplexan calcium-dependent protein kinase CDPK1. J Med Chem. 2012;55(6):2803–2810. | ||
Doggett JS, Ojo KK, Fan E, Maly DJ, Van Voorhis WC. Bumped kinase inhibitor 1294 treats established Toxoplasma gondii infection. Antimicrob Agents Chemother. 2014;58(6):3547–3549. | ||
Lourido S, Zhang C, Lopez MS, et al. Optimizing small molecule inhibitors of calcium-dependent protein kinase 1 to prevent infection by Toxoplasma gondii. J Med Chem. 2013;56(7):3068–3077. | ||
Sugi T, Kato K, Kobayashi K, et al. 1NM-PP1 treatment of mice infected with Toxoplasma gondii. J Vet Med Sci. 2011;73(10):1377–1379. | ||
Huang W, Ojo KK, Zhang Z, et al. SAR studies of 5-aminopyrazole-4-carboxamide analogues as potent and selective inhibitors of CDPK1. ACS Med Chem Lett. 2015;6(12):1184–1189. | ||
Zhang Z, Ojo KK, Johnson SM, et al. Benzoylbenzimidazole-based selective inhibitors targeting Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase-1. Bioorg Med Chem Lett. 2012;22(16):5264–5267. | ||
Zhang Z, Ojo KK, Vidadala R, et al. Potent and selective inhibitors of CDPK1 from T. gondii and C. parvum based on a 5-aminopyrazole-4-carboxamide scaffold. ACS Med Chem Lett. 2013;5(1):40–44. | ||
Moine E, Denevault-Sabourin C, Debierre-Grockiego F, et al. A small-molecule cell-based screen led to the identification of biphenylimidazoazines with highly potent and broad-spectrum anti-apicomplexan activity. Eur J Med Chem. 2015;89:386–400. | ||
Moine E, Dimier-Poisson I, Enguehard-Gueiffier C, et al. Development of new highly potent imidazo[1,2-b]pyridazines targeting Toxoplasma gondii calcium-dependent protein kinase 1. Eur J Med Chem. 2015;105:80–105. | ||
Pelphrey PM, Popov VM, Joska TM, et al. Highly efficient ligands for dihydrofolate reductase from Cryptosporidium hominis and Toxoplasma gondii inspired by structural analysis. J Med Chem. 2007;50:940–950. | ||
Canfield CJ, Milhous WK, Ager AL, et al. PS-15: a potent, orally active antimalarial from a new class of folic acid antagonists. Am J Trop Med Hyg. 1993;49:121–126. | ||
Fidock DA, Wellems TE. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc Natl Acad Sci U S A. 1997;94:10931–10936. | ||
Mui EJ, Schiehser GA, Milhous WK, et al. Novel triazine JPC-2067-B inhibits Toxoplasma gondii in vitro and in vivo. PLoS Negl Trop Dis. 2008;2(3):e190. | ||
Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. | ||
Harbut MB, Patel BA, Yeung BKS, et al. Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proc Natl Acad Sci U S A. 2012;109:21486–21491. | ||
Jelenska J, Crawford MJ, Harb OS, et al. Subcellular localization of acetyl-CoA carboxylase in the apicomplexan parasite Toxoplasma gondii. Proc Natl Acad Sci U S A. 2001;98:2723–2728. | ||
Roberts F, Roberts CW, Johnson JJ, et al. Evidence for the shikimate pathway in apicomplexan parasites. Nature. 1998;393:801–805. | ||
Waller RF, Keeling PJ, Donald RG, et al. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc Natl Acad Sci U S A. 1998;95:12352–12357. | ||
Magnuson K, Jackowski S, Rock CO, Cronan JE. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol Rev. 1993;57:522–542. | ||
McFadden GI, Roos DS. Apicomplexan plastids as drug targets. Trends Microbiol. 1999;7:328–333. | ||
Wakil SJ, Stoops JK, Joshi VC. Fatty acid synthesis and its regulation. Annu Rev Biochem. 1983;52:537–579. | ||
Martins-Duarte ES, Carias M, Vommaro R, Surolia N, de Souza W. Apicoplast fatty acid synthesis is essential for pellicle formation at the end of cytokinesis in Toxoplasma gondii. J Cell Sci. 2016;129(17):3320–3331. | ||
McLeod R, Muench SP, Rafferty JB, et al. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of apicomplexan Fab I. Int J Parasitol. 2001;31:109–113. | ||
Stec J, Fomovska A, Afanador GA, et al. Modification of triclosan scaffold in search of improved inhibitors for enoyl-acyl carrier protein (ACP) reductase in Toxoplasma gondii. ChemMedChem. 2013;8:1138–1160. | ||
Cheng G, Muench SP, Zhou Y, et al. Design, synthesis, and biological activity of diaryl ether inhibitors of Toxoplasma gondii enoyl reductase. Bioorg Med Chem Lett. 2013;23:2035–2043. | ||
Muench SP, Stec J, Zhou Y, et al. Development of a triclosan scaffold which allows for adaptations on both the A- and B-ring for transport peptides. Bioorg Med Chem Lett. 2013;23:3551–3555. | ||
Samuel BU, Hearn B, Mack D, et al. Delivery of antimicrobials into parasites. Proc Natl Acad Sci U S A. 2003;100:14281–14286. | ||
El-Zawawy LA, El-Said D, Mossallam SF, Ramadan HS, Younis SS. Triclosan and triclosan-loaded liposomal nanoparticles in the treatment of acute experimental toxoplasmosis. Exp Parasitol. 2015;149:54–64. | ||
Hevener KE, Mehboob S, Su P-C, et al. Discovery of a novel and potent class of F. tularensis enoyl-reductase (FabI) inhibitors by molecular shape and electrostatic matching. J Med Chem. 2012;55:268–279. | ||
Wilkinson C, McPhillie MJ, Zhou Y, et al. The benzimidazole based drugs show good activity against T. gondii but poor activity against its proposed enoyl reductase enzyme target. Bioorg Med Chem Lett. 2014;24:911–916. | ||
Jackowski S, Murphy CM, Cronan JE, Rock CO. Acetoacetyl-acyl carrier protein synthase. A target for the antibiotic thiolactomycin. J Biol Chem. 1989;264:7624–7629. | ||
Martins-Duarte ÉS, Jones SM, Gilbert IH, Atella GC, de Souza W, Vommaro RC. Thiolactomycin analogues as potential anti-Toxoplasma gondii agents. Parasitol Int. 2009;58:411–415. | ||
Drlica K, Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev. 1997;61:377–392. | ||
Pan XS, Fisher LM. Targeting of DNA gyrase in Streptococcus pneumoniae by sparfloxacin: selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob Agents Chemother. 1997;41:471–474. | ||
Martinez M, McDermott P, Walker R. Pharmacology of the fluoroquinolones: a perspective for the use in domestic animals. Vet J. 2006;172:10–28. | ||
Khan AA, Slifer T, Araujo FG, Remington JS. Trovafloxacin is active against Toxoplasma gondii. Antimicrob Agents Chemother. 1996;40:1855–1859. | ||
Gottstein B, Razmi GR, Ammann P, Sager H, Müller N. Toltrazuril treatment to control diaplacental Neospora caninum transmission in experimentally infected pregnant mice. Parasitology. 1999;130:41–48. | ||
Barbosa BF, Gomes AO, Ferro EAV, Napolitano DR, Mineo JR, Silva NM. Enrofloxacin is able to control Toxoplasma gondii infection in both in vitro and in vivo experimental models. Vet Parasitol. 2012;187:44–52. | ||
Khan AA, Araujo FG, Brighty KE, Gootz TD, Remington JS. Anti-Toxoplasma gondii activities and structure-activity relationships of novel fluoroquinolones related to trovafloxacin. Antimicrob Agents Chemother. 1999;43:1783–1787. | ||
Anquetin G, Rouquayrol M, Mahmoudi N, et al. Synthesis of new fluoroquinolones and evaluation of their in vitro activity on Toxoplasma gondii and Plasmodium spp. Bioorg Med Chem Lett. 2004;14:2773–2776. | ||
Anquetin G, Greiner J, Mahmoudi N, et al. Design, synthesis and activity against Toxoplasma gondii, Plasmodium spp., and Mycobacterium tuberculosis of new 6-fluoroquinolones. Eur J Med Chem. 2006;41:1478–1493. | ||
Dubar F, Wintjens R, Martins-Duarte ÉS, et al. Ester prodrugs of ciprofloxacin as DNA-gyrase inhibitors: synthesis, antiparasitic evaluation and docking studies. Med Chem Commun. 2011;2:430–436. | ||
Martins-Duarte ÉS, Dubar F, Lawton P, et al. Ciprofloxacin Derivatives Affect Parasite Cell Division and Increase the Survival of Mice Infected with Toxoplasma gondii. PLoS ONE. 2015;10:e0125705–0125723. | ||
Andrews KT, Haque A, Jones MK. HDAC inhibitors in parasitic diseases. Immunol Cell Biol. 2011;90:66–77. | ||
Vanagas L, Jeffers V, Bogado SS, Dalmasso MC, Sullivan WJ Jr, Angel SO. Toxoplasma histone acetylation remodelers as novel drug targets. Expert Rev Anti Infect Ther. 2014;10:1189–1201. | ||
Bougdour A, Maubon D, Baldacci P, et al. Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J Exp Med. 2009;206:953–966. | ||
Maubon D, Bougdour A, Wong YS, et al. Activity of the histone deacetylase inhibitor FR235222 on Toxoplasma gondii: inhibition of stage conversion of the parasite cyst form and study of new derivative compounds. Antimicrob Agents Chemother. 2010;54:4843–4850. | ||
Jeffers V, Gao H, Checkley LA, Liu Y, Ferdig MT, Sullivan WJ Jr. Garcinol inhibits GCN5-mediated lysine acetyltransferase activity and prevents replication of the parasite Toxoplasma gondii. Antimicrob Agents Chemother. 2016;60:2164–2170. | ||
Wang J, Dixon SE, Ting L-M, et al. Lysine acetyltransferase GCN5b interacts with AP2 factors and is required for Toxoplasma gondii proliferation. PLoS Pathog. 2014;10:e1003830. | ||
Fomovska A, Huang Q, El Bissati K, et al. Novel N-benzoyl-2-hydroxybenzamide disrupts unique parasite secretory pathway. Antimicrob Agents Chemother. 2012;56:2666–2682. | ||
Ngô HM, Yang M, Paprotka K, Pypaert M, Hoppe H, Joiner KA. AP-1 in Toxoplasma gondii mediates biogenesis of the rhoptry secretory organelle from a post-Golgi compartment. J Biol Chem. 2003;278:5343–5352. | ||
Breinich MS, Ferguson DJP, Foth BJ, et al. A dynamin is required for the biogenesis of secretory organelles in Toxoplasma gondii. Curr Biol. 2009;19:277–286. | ||
Köhler S, Delwiche CF, Denny PW, et al. A plastid of probable green algal origin in Apicomplexan parasites. Science. 1997;275:1485–1489. | ||
Seeber F. Biosynthetic pathways of plastid-derived organelles as potential drug targets against parasitic apicomplexa. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3(2):99–109. | ||
Araujo FG, Guptill DR, Remington JS. Azithromycin, a macrolide antibiotic with potent activity against Toxoplasma gondii. Antimicrob Agents Chemother. 1988;32:755–757. | ||
Pfefferkorn ER, Nothnagel RF, Borotz SE. Parasiticidal effect of clindamycin on Toxoplasma gondii grown in cultured cells and selection of a drug-resistant mutant. Antimicrob Agents Chemother. 1992;36:1091–1096. | ||
Fichera ME, Roos DS. A plastid organelle as a drug target in apicomplexan parasites. Nature. 1997;390:407–409. | ||
Lee Y, Choi JY, Fu H, et al. Chemistry and biology of macrolide antiparasitic agents. J Med Chem. 2011;54:2792–2804. | ||
Webb ME, Smith AG, Abell C. Biosynthesis of pantothenate. Nat Prod Rep. 2004;21:695–721. | ||
Mageed SN, Cunningham F, Hung AW, et al. Pantothenic acid biosynthesis in the parasite Toxoplasma gondii: a target for chemotherapy. Antimicrob Agents Chemother. 2014;58:6345–6353. | ||
Hung AW, Silvestre HL, Wen S, Ciulli A, Blundell TL, Abell C. Application of fragment growing and fragment linking to the discovery of inhibitors of Mycobacterium tuberculosis pantothenate synthetase. Angew Chem Int. 2009;48:8452–8456. | ||
Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7(3):187–195. | ||
Lai B-S, Witola WH, El Bissati K, et al. Molecular target validation, antimicrobial delivery, and potential treatment of Toxoplasma gondii infections. Proc Natl Acad Sci U S A. 2012;109:14182–14187. | ||
Zhou Y, Fomovska A, Muench S, Lai BS, Mui E, McLeod R. Spiroindolone That Inhibits PfATPase4 Is a Potent, Cidal Inhibitor of Toxoplasma gondii Tachyzoites In Vitro and In Vivo. Antimicrob. Agents Chemother. 2014;58:1789–1792. | ||
Vannier E, Krause PJ. Human babesiosis. N Engl J Med. 2012;366(25):2397–2407. | ||
Capper MJ, O’Neill PM, Fisher N, et al. Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc Natl Acad Sci U S A. 2015;112(3):755–760. | ||
Nilsen A, Miley GP, Forquer IP, et al. Discovery, synthesis, and optimization of antimalarial 4(1H)-quinolone-3-diarylethers. J Med Chem. 2014;57(9):3818–3834. | ||
Alday PH, Bruzual I, Nilsen A, et al. Genetic Evidence for Cytochrome b Qi Site Inhibition by 4(1H)-quinolone-3-diarylethers and Antimycin in Toxoplasma gondii. Antimicrob Agents Chemother. 2016. | ||
Salzer WT, Andersag H. A new type of compound active against avian malaria. Chem Ber. 1948;81:12–19. | ||
Gingrich W, Darrow E. The effect of endochin on experimental toxoplasmosis. Am J Trop Med Hyg. 1951;31:12–17. | ||
Miley GP, Pou S, Winter R, et al. ELQ-300 prodrugs for enhanced delivery and single-dose cure of malaria. Antimicrob Agents Chemother. 2015;59(9):5555–5560. | ||
Lawres LA, Garg A, Kumar V, et al. Radical cure of experimental babesiosis in immunodeficient mice using a combination of an endochin-like quinolone and atovaquone. J Exp Med. 2016 Epub June 6. | ||
Vallieres C, Fisher N, Antoine T, et al. HDQ, a potent inhibitor of Plasmodium falciparum proliferation, binds to the quinone reduction site of the cytochrome bc1 complex. Antimicrob Agents Chemother. 2012;56(7):3739–3747. | ||
Eschemann A, Galkin A, Oettmeier W, Brandt U, Kerscher S. HDQ (1-hydroxy-2-dodecyl-4(1H)quinolone), a high affinity inhibitor for mitochondrial alternative NADH dehydrogenase: evidence for a ping-pong mechanism. J Biol Chem. 2005;280:3138–3142. | ||
Hegewald J, Gross U, Bohne W. Identification of dihydroorotate dehydrogenase as a relevant drug target for 1-hydroxyquinolones in Toxoplasma gondii. Mol Biochem Parasitol. 2013;190(1):6–15. | ||
Vercesi AE, Rodrigues CO, Uyemura SA, Zhong L, Moreno SN. Respiration and oxidative phosphorylation in the apicomplexan parasite Toxoplasma gondii. J Biol Chem. 1998;273:31040–31047. | ||
Melo AMP, Bandeiras TM, Teixeira M. New insights into type II NAD(P)H:quinone oxidoreductases. Microbiol Mol Biol Rev. 2004;68:603–616. | ||
Hegewald J, Groβ U, Bohne W. Identification of dihydroorotate dehydrogenase as a relevant drug target for 1-hydroxyquinolones in Toxoplasma gondii. Mol Biochem Parasitol. 2013;190:6–15. | ||
Saleh A, Friesen J, Baumeister S, Groβ U, Bohne W. Growth inhibition of Toxoplasma gondii and Plasmodium falciparum by nanomolar concentrations of 1-hydroxy-2-dodecyl-4(1H)quinolone, a high-affinity inhibitor of alternative (type II) NADH dehydrogenases. Antimicrob Agents Chemother. 2007;51:1217–1222. | ||
Bajohr LL, Ma L, Platte C, et al. In vitro and in vivo activities of 1-hydroxy-2-alkyl-4(1H)quinolone derivatives against Toxoplasma gondii. Antimicrob Agents Chemother. 2010;54:517–521. | ||
Pidathala C, Amewu R, Pacorel B, et al. Identification, design and biological evaluation of bisaryl quinolones targeting Plasmodium falciparum type II NADH: quinone oxidoreductase (PfNDH2). J Med Chem. 2012;55:1831–1843. | ||
Beckford FA, Thessing J, Shaloski M, et al. Synthesis and characterization of mixed-ligand diimine-piperonal thiosemicarbazone complexes of ruthenium(II): biophysical investigations and biological evaluation as anticancer and antibacterial agents. J Mol Struct. 2011;992:39–47. | ||
Barna F, Debache K, Vock CA, Kuster T, Hemphill A. In vitro effects of novel ruthenium complexes in Neospora caninum and Toxoplasma gondii tachyzoites. Antimicrob Agents Chemother. 2013;57:5747–5754. | ||
Fomovska A, Wood RD, Mui E, et al. Salicylanilide inhibitors of Toxoplasma gondii. J Med Chem. 2012;55:8375–8391. | ||
Rottmann M, McNamara C, Yeung BKS, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. | ||
Gwilt PR, Tracewell WG. Pharmacokinetics and pharmacodynamics of hydroxyurea. Clin Pharmacokinet. 1998;34:347–358. | ||
de Melo EJ, Mayerhoffer RO, de Souza W. Hydroxyurea inhibits intracellular Toxoplasma gondii multiplication. FEMS Microbiol Lett. 2000;185:79–82. | ||
Liu MC, Lin TS, Sartorelli AC. Synthesis and antitumor activity of amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J Med Chem. 1992;35:3672–3677. | ||
Walcourt A, Loyevsky M, Lovejoy DB, Gordeuk VR, Richardson DR. Novel aroylhydrazone and thiosemicarbazone iron chelators with anti-malarial activity against chloroquine-resistant and -sensitive parasites. Int J Biochem Cell Biol. 2004;36:401–407. | ||
Tenório RP, Carvalho CS, Pessanha CS, et al. Synthesis of thiosemicarbazone and 4-thiazolidinone derivatives and their in vitro anti-Toxoplasma gondii activity. Bioorg Med Chem Lett. 2005;15:2575–2578. | ||
de Aquino TM, Liesen AP, da Silva REA, et al. Synthesis, anti-Toxoplasma gondii and antimicrobial activities of benzaldehyde 4-phenyl-3-thiosemicarbazones and 2-[(phenylmethylene)hydrazono]-4-oxo-3-phenyl-5-thiazolidineacetic acids. Bioorg Med Chem. 2008;16:446–456. | ||
Liesen AP, de Aquino TM, Carvalho CS, et al. Synthesis and evaluation of anti-Toxoplasma gondii and antimicrobial activities of thiosemicarbazides, 4-thiazolidinones and 1,3,4-thiadiazoles. Eur J Med Chem. 2010;45:3685–3691. | ||
Dzitko K, Paneth A, Plech T, et al. 1,4-Disubstituted thiosemicarbazide derivatives are potent inhibitors of Toxoplasma gondii proliferation. Molecules. 2014;19:9926–9943. | ||
Woodward KN. Toxicological Effects of Veterinary Medical Products in Humans. Vol 2. Cambridge, UK: The Royal Society of Chemistry; 2013. | ||
Mitchell SM, Zajac AM, Davis WL, Lindsay DS. Efficacy of ponazuril in vitro and in preventing and treating Toxoplasma gondii infections in mice. J Parasitol. 2004;90:639–642. | ||
Martins-Duarte ÉDS, de Souza W, Vommaro RC. Itraconazole affects Toxoplasma gondii endodyogeny. FEMS Microbiol Lett. 2008;282:290–298. | ||
Martins-Duarte ÉS, Lemgruber L, de Souza W, Vommaro RC. Toxoplasma gondii: fluconazole and itraconazole activity against toxoplasmosis in a murine model. Exp Parasitol. 2010;124:466–469. | ||
Dzitko K, Paneth A, Plech T, Paweczyk J, Wgliska L, Paneth P. Triazole-based compound as a candidate to develop novel medicines to treat toxoplasmosis. Antimicrob Agents Chemother. 2014;58:7583–7585. | ||
Krivogorsky B, Grundt P, Yolken R, Jones-Brando L. Inhibition of Toxoplasma gondii by indirubin and tryptanthrin analogs. Antimicrob Agents Chemother. 2008;52:4466–4469. | ||
Krivogorsky B, Nelson AC, Douglas KA, Grundt P. Tryptanthrin derivatives as Toxoplasma gondii inhibitors – structure-activity-relationship of the 6-position. Bioorg Med Chem Lett. 2013;23:1032–1035. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.