Back to Journals » Drug Design, Development and Therapy » Volume 10
Druggability analysis and classification of protein tyrosine phosphatase active sites
Authors Ghattas M ![]() , Raslan N, Sadeq A, Al Sorkhy M
, Raslan N, Sadeq A, Al Sorkhy M ![]() , Atatreh N
, Atatreh N
Received 26 April 2016
Accepted for publication 9 June 2016
Published 30 September 2016 Volume 2016:10 Pages 3197—3209
DOI https://doi.org/10.2147/DDDT.S111443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Mohammad A Ghattas, Noor Raslan, Asil Sadeq, Mohammad Al Sorkhy, Noor Atatreh
College of Pharmacy, Al Ain University of Science and Technology, Al Ain, UAE
Abstract: Protein tyrosine phosphatases (PTP) play important roles in the pathogenesis of many diseases. The fact that no PTP inhibitors have reached the market so far has raised many questions about their druggability. In this study, the active sites of 17 PTPs were characterized and assessed for its ability to bind drug-like molecules. Consequently, PTPs were classified according to their druggability scores into four main categories. Only four members showed intermediate to very druggable pocket; interestingly, the rest of them exhibited poor druggability. Particularly focusing on PTP1B, we also demonstrated the influence of several factors on the druggability of PTP active site. For instance, the open conformation showed better druggability than the closed conformation, while the tight-bound water molecules appeared to have minimal effect on the PTP1B druggability. Finally, the allosteric site of PTP1B was found to exhibit superior druggability compared to the catalytic pocket. This analysis can prove useful in the discovery of new PTP inhibitors by assisting researchers in predicting hit rates from high throughput or virtual screening and saving unnecessary cost, time, and efforts via prioritizing PTP targets according to their predicted druggability.
Keywords: PTP1B, oral bioavailability, drug-like inhibitors, drug design, active site, allosteric site, MPtpB, CD45, SHP2, YopH
Introduction
Protein phosphorylation and dephosphorylation are key regulatory mechanisms in living organisms; aberrant protein phosphorylation is highly correlated with many human diseases ranging from diabetes to many forms of cancers.1 These two reactions are catalyzed by two major enzymes categories – protein kinases that catalyze the phosphorylation process and phosphatases that are responsible for the dephosphorylation process.2 Although the two processes are of even importance as molecular switches, the scientific world paid more attention toward the kinases; it was not until recently when protein phosphatases started to gain proper attention in the medicinal chemistry field.3,4 Protein phosphatases have been grouped into two major categories – serine/threonine phosphatases and tyrosine phosphatases.5 The latter group, that is protein tyrosine phosphatases (PTPs), has gained considerable amount of attention for their pivotal non-redundant role in many cellular pathways and in the pathogenesis of many diseases.6 Thus, the growing number of human diseases associated with aberrant PTP function has brought them in the spotlight as a key drug target.
The first bona fide piece of evidence came with the PTP1B knockout mouse which clearly demonstrated that PTP1B acts as a negative regulator of insulin signaling.7,8 Since then, a large list of key PTPs associated with different human abnormalities has emerged as possible drug targets. The strong relationship between PTP and different human diseases made them hot targets for pharmaceutical companies trying to design suitable inhibitors. The design of PTP inhibitors has been rendered by the difficulty in identifying cell permeable compound with sufficient selectivity; so far, no PTP inhibitors have reached the market.9–11 Figure 1 displays examples of different PTP inhibitors that have inhibitory potency ranging between low and medium nanomolar levels.11 Obviously, the shown compounds carry multiple charges on their structures, so as to become capable of binding with the PTP positively charged pocket; this can partially explain the cell permeability issues generally accompanied with such inhibitors.
 | Figure 1 Examples of potent PTP inhibitors. |
SiteMap12 has been able to help researchers in assessing the active site of important targets, for example, the bromodomain acetyl-lysine proteins were found to have significantly different druggability, with a median druggability score (Dscore) ranging from 0.38 to 1.08.13 In another study, enoyl-acyl carrier protein reductases were all predicted to be druggable by SiteMap, with average Dscore ranging from 1.05 to 1.30; nonetheless, the size of their active site varied significantly, with the average pocket size ranging from 101 to 284.14 In this study, we assessed the active site of PTPs, studying their druggability and other physical characteristics using SiteMap,12 and then analyzed/classified them according to the resultant scores. In this work, we also studied the effect of different structural aspects on the PTP druggability, that is, loop flexibility, solvation effect, and allosteric site, which can be proven valuable for designing new PTP inhibitors.
Methods
Selection and preparation of PTP crystal structures
The PTPs considered in this study are known to be involved in many diseases.3,5 All PTP crystal structures were obtained from the Protein Data Bank.15 All PTP structures considered in the study were solved by X-crystallography and have no mutated residues in their active site; otherwise, they were discarded. PTP structures with covalently bound inhibitors were not considered in the final list. Our resultant PTP crystal structure library finally contained a total number of 116 crystal structures that belong to 17 PTP enzymes.
Preparation of PTP crystal structures
The crystal structures obtained from PDB were categorized according to the PTP member they belong to and then subcategorized according to whether or not they have a ligand molecule in their active site. Subsequently, each crystal structure was processed manually to keep one protein chain. All water molecules were removed (where applicable); all other solvent molecules and co-crystallized ligands were also removed. Afterward, each crystal structure was processed automatically via the protein preparation wizard in the MOE software.16 This wizard prepares a PTP crystal structure by checking the protein part for any missing atoms, residues, or loops; accordingly, it conducts all required corrections. Next, the MOE protein preparation wizard assigns the protonation state of each ionizable side chain (using the Protonate 3D algorithm17).
Druggability assessment of PTPs
The prepared protein crystal structures were processed by the SiteMap module12 in Maestro,18 so the druggability of the catalytic pocket can be assessed. The assessment was conducted using contributions from the cavity size, the enclosure, and the degree of hydrophobicity. The pockets were then scored by SiteScore and Dscore according to Equations (1) and (2), respectively.
SiteScore = 0.0733n1/2 + 0.6688e − 0.20p | (1) |
Dscore = 0.094n1/2 + 0.60e − 0.324p | (2) |
where n is the number of site points, e is the enclosure score, and p is the hydrophilic score. As shown in the equations, both scoring functions use the same terms but with different coefficients so that highly polar active sites are more penalized by the Dscore function than SiteScore. Thus, a polar pocket may score low in Dscore, although a high score can be achieved when the other algorithm, SiteScore, is employed. Such a pocket is more likely to encourage the binding of highly polar ligands that do not display druglikeness.12 Another parameter calculated by SiteMap is the size of the active site; the number of points that constitute an examined pocket is a measure of the size of the site. Approximately, two to three site points correspond to each atom of the bound ligand, including hydrogen atoms.12
Analyzing scores obtained by SiteMap
SiteMap has been validated by a set of 538 complexes and was able to accurately identify the ligand binding site in 86% of the cases.12 Pockets with a SiteScore value of ≥0.8 are identified as a possible binding pocket by SiteMap and those with a SiteScore of ≥1.0 are classified as binding sites with particular importance, tight-binding sites.12 A slightly different classification system used by Vidler et al13 will be used in this work where the Dscore function is employed to assess the PTP binding pocket for their druggability. This system categorizes proteins into four main classes: very druggable (Dscore ≥1.0), druggable (Dscore =0.8–1.0), intermediate (Dscore =0.7–0.8), and difficult (Dscore ≤0.7).
Results and discussion
Druggability assessment and binding site analysis
The term “druggable” was defined previously by Hopkins and Groom19 as a protein that is able, or predicted to be able, to bind drug-like molecules. This concept is considered in this study to assess the catalytic pocket of different members from the PTP family. The fact that no PTP inhibitors have reached the market, so far, highlights the difficulty that medicinal chemists face to design new PTP inhibitors with adequate cell permeability.10 In order to attain the full picture, the catalytic pocket of PTP enzymes was studied and assessed for druggability.
Searching the protein data bank resulted in obtaining 116 crystal structures that belong to 17 members of the PTP family, all of which were assessed for their druggability using SiteMap.12 Interestingly, the results show that PTPs had a very wide range of median Dscores (0.25–1.20; Table 1). The mycobacterial phosphatase, MPtpB, showed the highest Dscore value among all tested PTPs, while JSP-1 exhibited the poorest druggability. As shown in Figure 2, PTPs are not well distributed into these four classes, as only one member was found under the “very druggable” category (MPtpB: Dscore =1.20) and one member under the “druggable” category (GLEPP-1: Dscore 0.89). Only two PTPs were found in the next category “intermediate” (ie, SHP-1 and PTPε), with Dscore values ranging between 0.78 and 0.71. The rest of the PTP members, including the mostly targeted enzyme PTP1B, were classified as “difficult”.
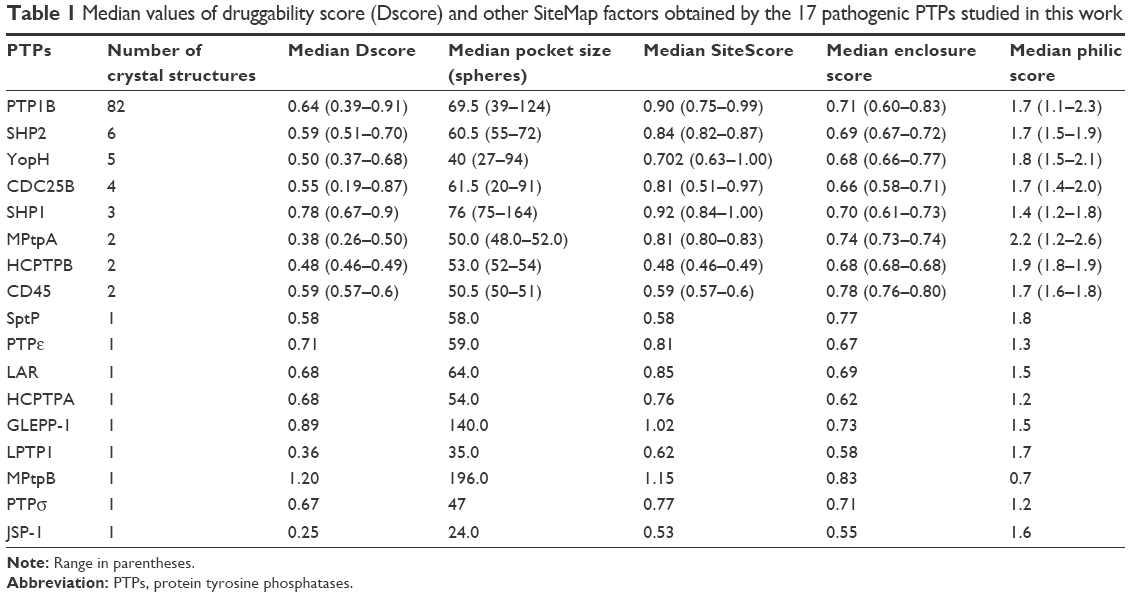 | Table 1 Median values of druggability score (Dscore) and other SiteMap factors obtained by the 17 pathogenic PTPs studied in this work |
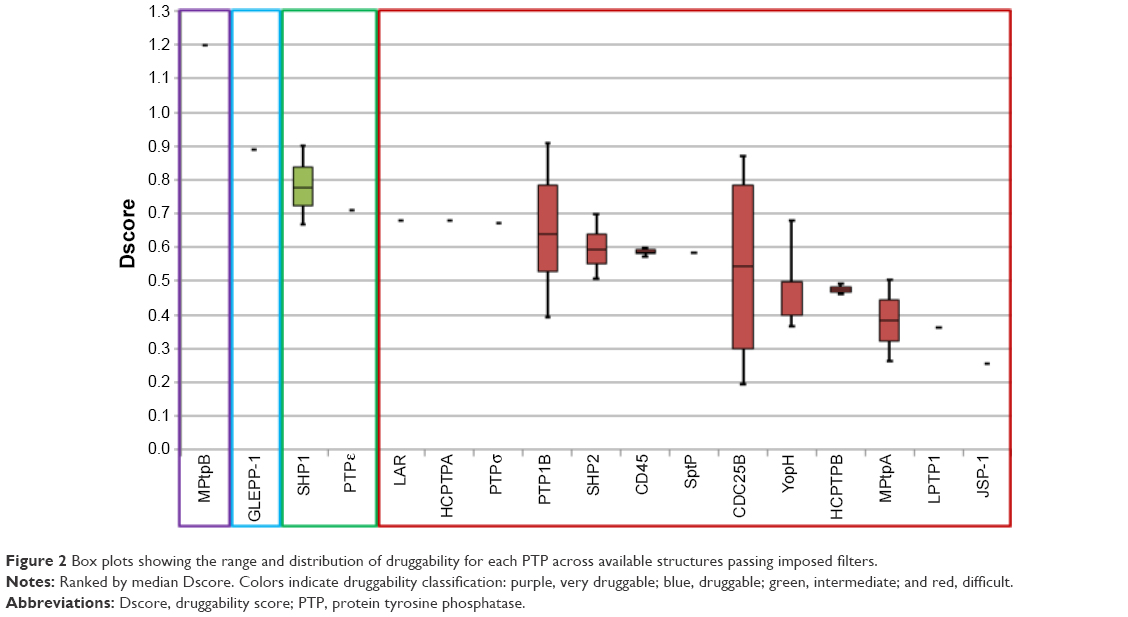 | Figure 2 Box plots showing the range and distribution of druggability for each PTP across available structures passing imposed filters. |
Looking at a representative example of each class, differences in the active site of the PTPs can be readily detected. As shown in Figure 3A and Table 1, MPtpB showed a large (pocket size 196 spheres) and well-defined cavity (enclosure score 0.83) with a low philic score (0.7). A similar pocket was also shown by GLEPP-1 (Figure 3B) but with a smaller pocket size (140 spheres), a less well-defined cavity (enclosure score 0.73), and higher philic score (1.5), in comparison with MPtpB. Although the “moderately druggable” class representative PTPε showed a less hydrophilic pocket (philic score 1.3) than GLEPP-1, its active site was observed to have a less well-defined cavity (enclosure 0.67) with a significantly smaller pocket size (59 spheres) (Figure 3C). The least druggable member among the PTP family, JSP-1, exhibited a very shallow cavity (enclosure 0.55) with a very small pocket size (=24 spheres), in addition to high hydrophilic nature (philic score =1.6), as shown in Figure 3D.
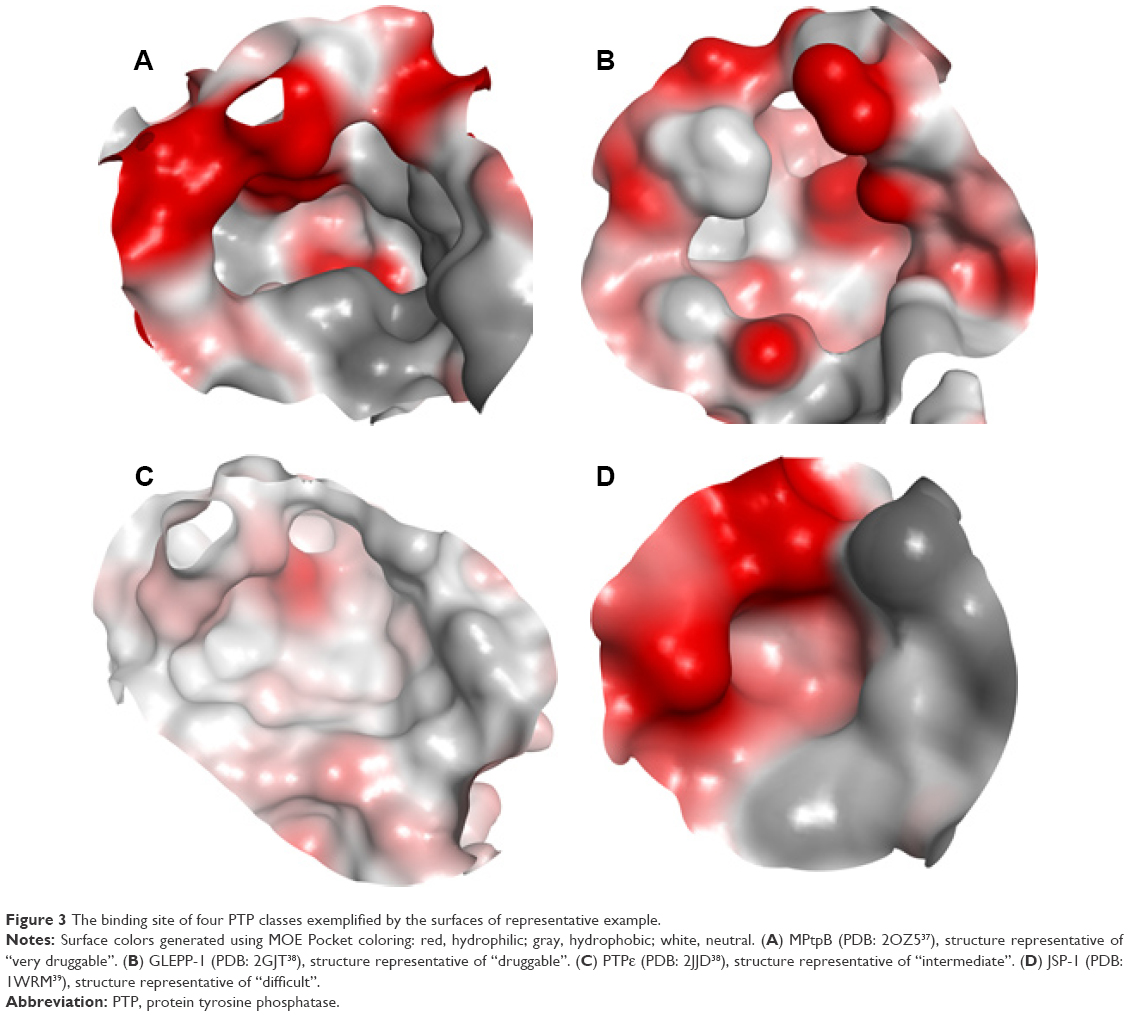 | Figure 3 The binding site of four PTP classes exemplified by the surfaces of representative example. |
It is worth noting that some of the PTP members were classified as difficult targets, although they demonstrated reasonably well-defined cavities in their active pocket. For instance, PTP1B and CDC25B have a medium pocket size (60–70 spheres) with a comparable enclosure score (~0.7) to the intermediate druggable member PTPε (Table 1), which led all of them to score well by SiteScore (0.81–0.9). However, the former PTPs seem to have 31% more polar pockets (philic factor =1.7) than PTPε, which prevented them from obtaining good druggability scores (median Dscore ~0.6). In other words, such PTPs have binding pockets that are good enough to bind ligand molecules but are not good enough to particularly bind ligands with high druglikeness. Classifying PTP1B via SiteMap as difficult to target by drug-like molecules may further explain why PTP1B inhibitors usually suffer from cell permeability problems.9
Since Dscore is calculated based on size, enclosure, and hydrophilicity of a tested pocket, it would be interesting to see which of these three factors has the greatest correlation with the obtained Dscore values of the assessed PTPs. Figure 4A shows that the pocket size factor has a direct and a proportional correlation with the PTP druggability scores. In contrast, pocket enclosure demonstrates a proportional correlation with Dscore; however, this correlation looks less significant compared to pocket size. Figure 4B illustrates a strong correlation between the hydrophilicity and druggability factors; it can be readily detected that when the pocket hydrophilicity increases, the druggability decreases. Hence it can be concluded that the most influential factors on the PTP druggability are the size and the hydrophilicity of their catalytic pocket.
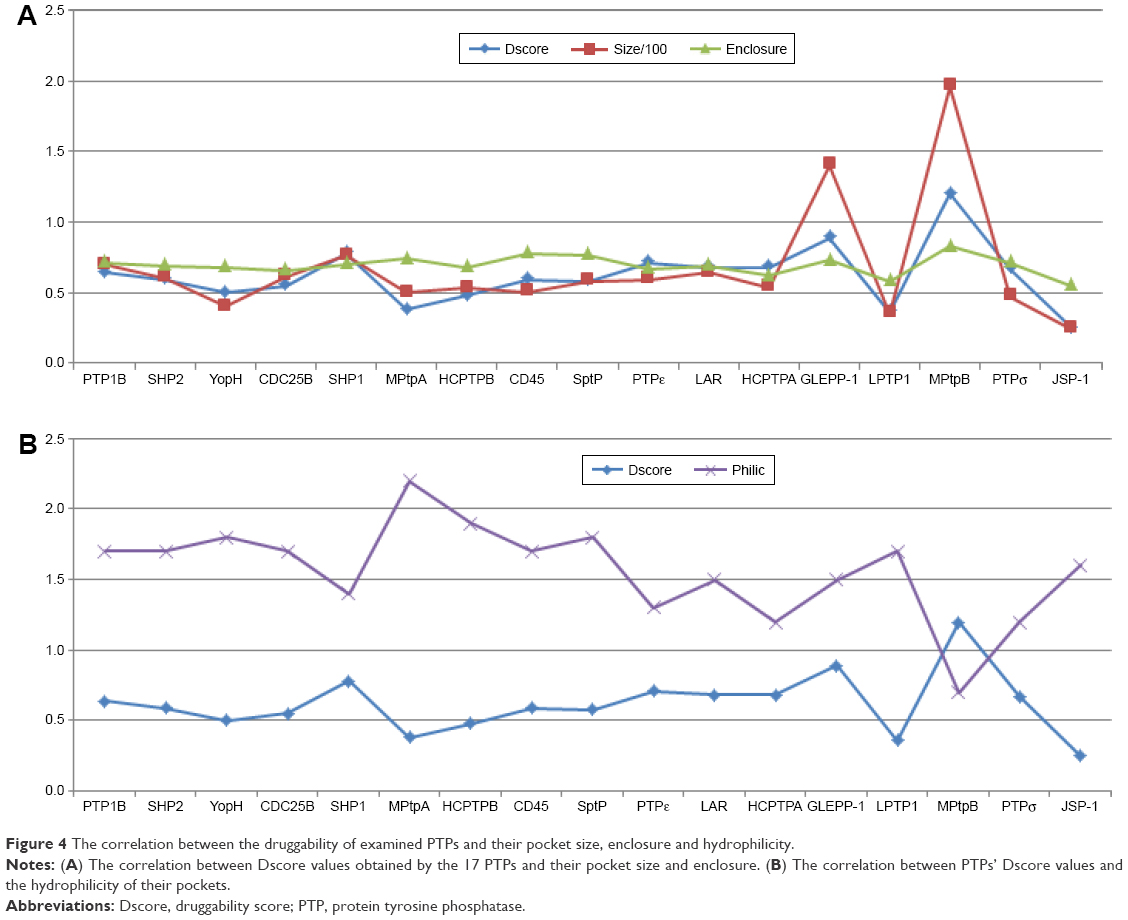 | Figure 4 The correlation between the druggability of examined PTPs and their pocket size, enclosure and hydrophilicity. |
The effect of pocket conformation on PTP druggability
Proteins are known to be flexible enough to change their binding site conformation according to their pocket status – the apo form or ligand-bound form. Moreover, their binding pocket can change its conformation in accordance with the shape and size of bound ligands, so ligands with different physical and chemical characteristics can still bind.20 Thus, it was just sensible to separate those structures in the apo form from those having ligands in their binding pocket, and then reassess them for their druggability. As shown in Table 2, the apo and ligand-bound forms were only seen in five PTPs, four of which exhibited different Dscore and pocket size values when the two forms are compared. Three of these PTPs (ie, PTP1B, YopH, and MPtpA) have their apo form more druggable than the ligand-bound form, and only CDC25B showed the opposite.
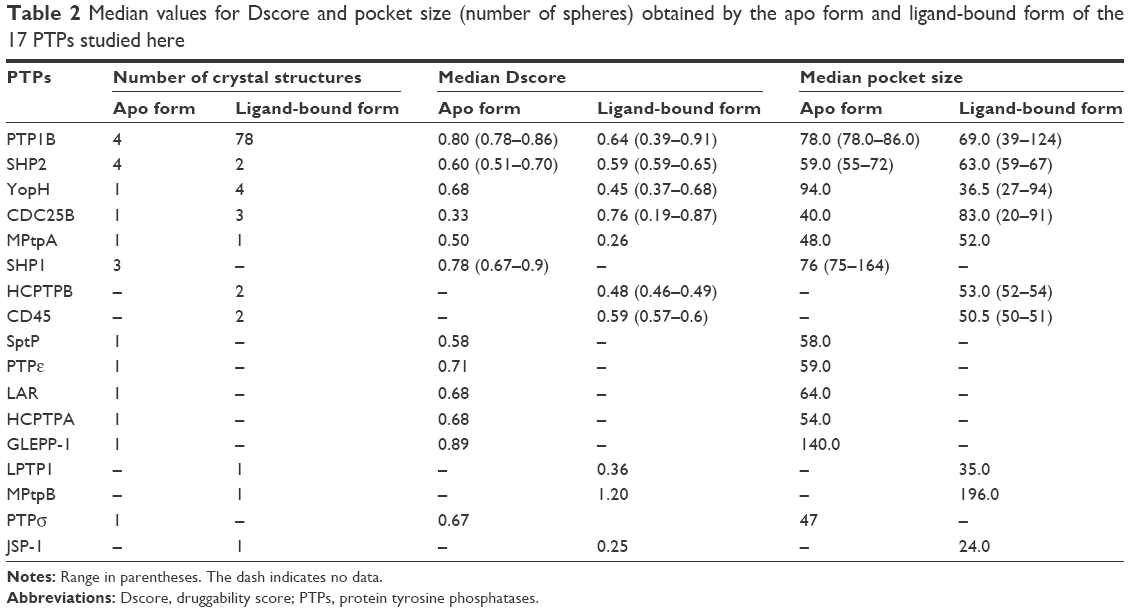 | Table 2 Median values for Dscore and pocket size (number of spheres) obtained by the apo form and ligand-bound form of the 17 PTPs studied here |
Figure 5 shows examples of CDC25B apo protein and ligand-bound forms aligned on each other. The apo form of CDC25B showed as low Dscore as 0.33, while the ligand-bound form has more than double druggability score (median Dscore =0.76; Table 2). The main reason behind having such druggability variation in the CDC25B active site seems to be due to the side chain flexibility of two arginine residues, Arg479 and Arg482 (Figure 5A). In the apo protein form, the two residues adopt certain conformations making the binding pocket really small (=40 spheres), preventing it from having an adequate Dscore value (Figure 5B). However, when a ligand binds to the enzyme, the two arginine residues rotate away, channeling the active site with an adjacent pocket so that to get large enough (pocket size =83 spheres) to bind drug-like molecules (Figure 5C). Consequently, the ligand-bound form of CDC25B may be individually classified as “intermediate” rather than “difficult” (where the apo form is listed in).
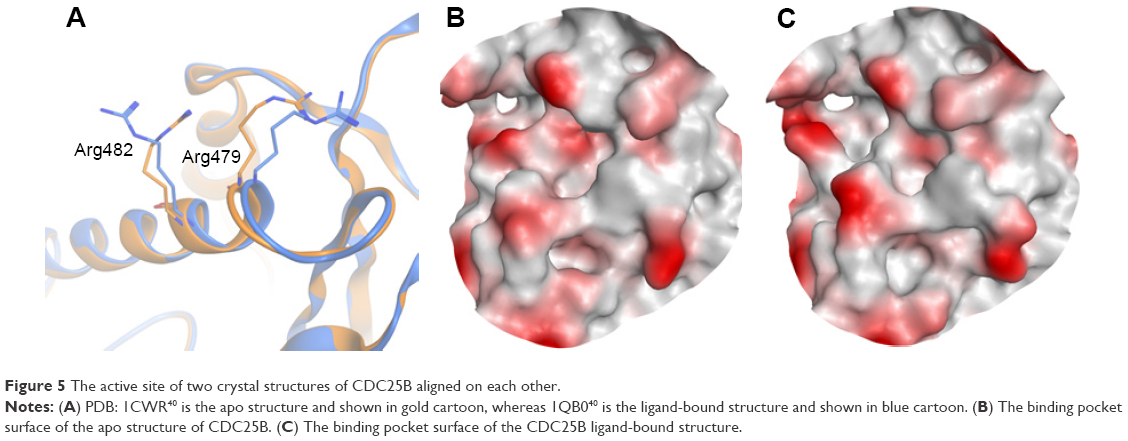 | Figure 5 The active site of two crystal structures of CDC25B aligned on each other. |
The other PTP that had druggability differences between its apo form and ligand-bound form is the YopH. In contrast to CDC25B, the YopH apo structure showed a better druggability score (Dscore =0.68) when compared to its ligand-bound form (median Dscore =0.45). Figure 6 shows all YopH crystal structures aligned on each other. It can be observed that when YopH is in the unbound form, the WPD-loop adopts an open conformation (pocket size =94 spheres). However, binding of a ligand causes the WPD-loop to swing down to adopt a closed conformation,21 making the YopH binding pocket significantly smaller in size (median pocket size =37 spheres) and less able to achieve good druggability score. This means YopH would have even less chance to bind drug-like molecules if the catalytic pocket deviated from the apo conformation, giving emphasis to its druggability issue.
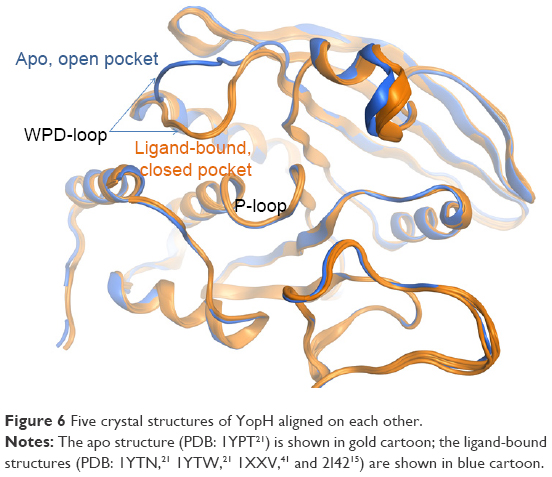 | Figure 6 Five crystal structures of YopH aligned on each other. |
The effect of binding site conformation on PTP druggability
Just like YopH, PTP1B is known to have a flexible loop (WPD-loop) that can adopt two different conformations upon ligand binding, closed or open.9 It was previously observed that the open structure of PTP1B shows a conformational change in the WPD loop (Trp179 to Ser187) where the main chain atoms are moved upward by 5.5 Å and the side chain atoms of Asp181 and Phe182 are shifted by 8 and 12 Å, respectively (Figure 7A), in comparison to the PTP1B closed conformation.22 Among the 82 PTP1B structures used here, the closed conformation was adopted by 64 structures (only one is in the apo form), and the open conformation was only seen in 18 structures, three of which are in the apo form. Thus, similar to YopH, the PTP1B catalytic pocket is more likely to adopt the closed conformation when a ligand binds to. Interestingly, the open conformation was also seen in the ligand-bound form of PTP1B. Hence, it would be interesting to study the druggability influence of the WPD-loop flexibility on the PTP1B binding site.
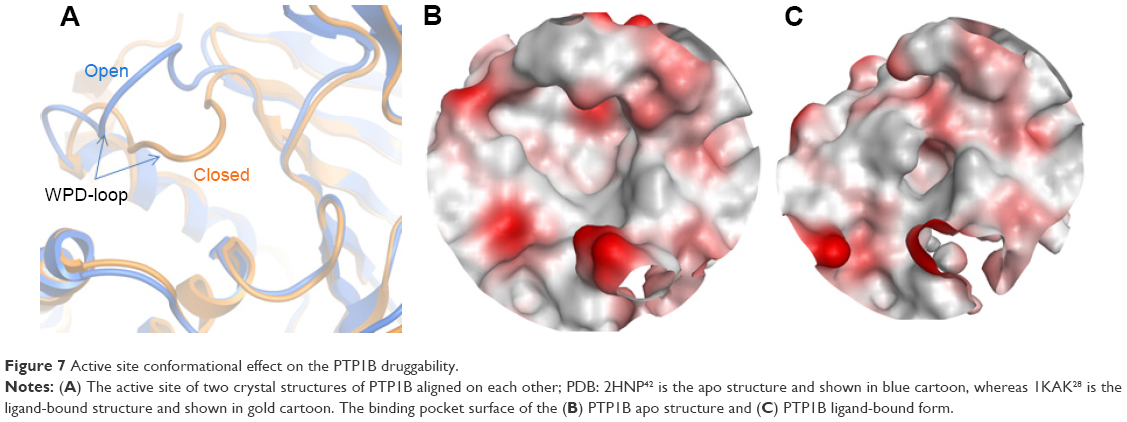 | Figure 7 Active site conformational effect on the PTP1B druggability. |
Table 3 lists the results obtained by SiteMap for the open and closed conformations of PTP1B. It can be observed that the open conformation was able to score a greater druggability score (Dscore =0.79), that almost touched the lower border of the “druggable” class, when compared to the closed conformation (Dscore =0.61, classified as “difficult”). However, the open and closed conformations of PTP1B scored well by SiteScore (~0.9). This means both conformations are equally capable of binding small organic molecules, but the closed conformation lacks the same capability to bind drug-like molecules (the open conformation looks superior in this context). The open structure (Figure 7B) showed a 16% increase in the pocket size and a 15% decrease in the philic score compared to the closed pocket (Figure 7C), although the latter exhibited a 7% greater enclosure than the open conformation. Hence, the pocket size and hydrophilicity seem to be the main responsible factors for the PTP1B open conformation superiority over the closed one.
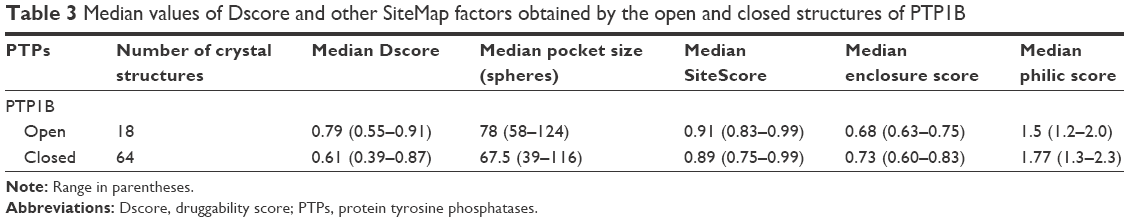 | Table 3 Median values of Dscore and other SiteMap factors obtained by the open and closed structures of PTP1B |
In fact, druggability data obtained here come in line with previous results obtained from our seeding experiments on PTP1B.23 In that study, the open conformation was able to retrieve a larger number of known PTP1B inhibitors (that obey Lipinski’s rule of five24), in comparison with the closed conformation. That was explained at the time by the fact that the PTP1B open conformation has a more spacious binding pocket than the closed conformation, and therefore, a greater range of ligands can be readily accommodated in the active site of the open conformation. It can also be added now that, as mentioned previously, the open conformation demonstrates a relatively less polar surface area than the closed conformation, which should, theoretically, assist and facilitate the binding of less polar ligands (that exhibit greater druglikeness) to the open pocket. Practically, so far, most of the open-conformation-bound inhibitors still possess a significant amount of polarity that prevents them from having a good cell membrane permeability profile (Figure 8). More studies are thus needed in this area to prove the druggability of the PTP1B open conformation.
 | Figure 8 Examples of potent PTP inhibitors belonging to different druggability class along with their experimental potency and calculated TPSA (produced by MOE16). |
The effect of water on PTP druggability
Different-ordered water molecules buried just behind the WPD loop have been observed in the closed PTP1B conformation.25–30 Out of the 82 PTP1B structures considered in this study, the highly ordered water molecules were seen in as much as 43 structures, acting as a hydrogen bonding bridge between the co-crystallized ligand and the corresponding amino acid. Therefore, the ordered water molecules represent an important aspect in the PTP1B drug discovery,31 and hence it was important to study whether these solvent molecules have an influence on the druggability characteristics of the PTP1B active site.
Table 4 shows the Dscore values of the solvated and unsolvated forms of the PTP1B active site. It was found that the solvated PTP1B structures have less druggable catalytic pocket by 7% than the unsolvated structure. This appeared to result mainly from the pocket size and enclosure factors as the unsolvated form showed a bigger pocket and a greater enclosure score by 14% and 6%, respectively, when compared to the solvated form. Surprisingly, although the solvated pocket contains more polar sites (ie, water molecules) than the unsolvated form, the former pocket scored a 7% lower philic score (=1.69; Table 4) when compared to the unsolvated one (philic score =1.82). It seems that these conserved water molecules are covering an even more polar spots in the active site, making the PTP1B pocket less hydrophilic in nature. Overall, it can be concluded that water does not appear to have a great influence on the PTP1B druggability as both forms were not able to escape from the “difficult” class. It is noteworthy that, some of the known PTP1B inhibitors, such as isothiazolidinone, demonstrated high ability to displace all ordered solvent molecules from the PTP1B active site.32,33 Thus, water should be handled with caution when designing new PTP inhibitors.
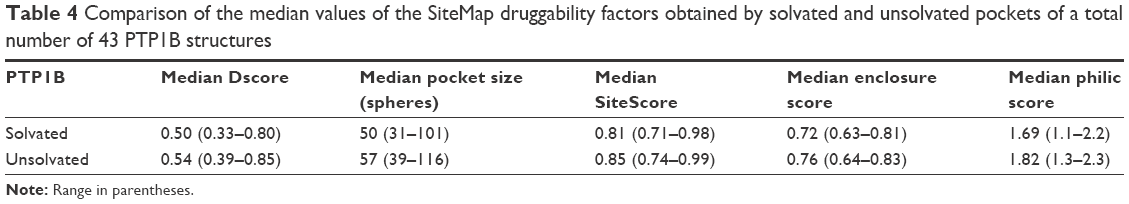 | Table 4 Comparison of the median values of the SiteMap druggability factors obtained by solvated and unsolvated pockets of a total number of 43 PTP1B structures |
Druggability of the PTP1B allosteric pocket
In 2004, Wiesmann et al34 discovered that PTP1B has an allosteric pocket located around 20 Å away from the active site (Figure 9). The allosteric pocket was targeted by several types of molecules that possess drug-like characteristics; however, good inhibition potency was always difficult to achieve. Regardless of its potency issue, it was interesting to assess the allosteric pocket in terms of druggability and compare with the PTP1B catalytic site.
 | Figure 9 The location of catalytic and allosteric pockets on the PTP1B structure along with their co-crystallized ligands. |
Table 5 shows the data obtained from SiteMap for both PTP1B pockets. Obviously, the allosteric site is much more druggable than the catalytic pocket, scoring as high Dscore as 0.97, which is 52% greater than the catalytic pocket Dscore (Dscore =0.64). Although both pockets had comparable SiteScore (which means that both of them can bind organic molecules), they were not able to score similarly in Dscore. As shown in Table 5, the active pocket scored greater philic score (=1.7) by more than threefold compared to the allosteric pocket (philic score =0.5), making polarity the supreme contributor to druggability variations between the two PTP1B pockets.
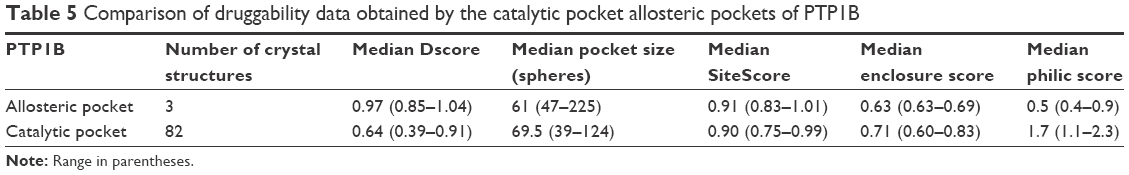 | Table 5 Comparison of druggability data obtained by the catalytic pocket allosteric pockets of PTP1B |
It is worth noting that this pocket seems to be not the only allosteric site on the PTP1B structure. A recently discovered non-competitive inhibitor, MSI-1436 (Figure 8), showed an inhibition activity in the low micromolar level with a unique binding to PTP1B.35 This binding was found to partially involve the aforementioned allosteric site, in addition to a distinct pocket located within the 20 amino acids at the C-terminus of the catalytic domain.35 MSI-1436 is a promising PTP1B inhibitor as it successfully passed Phase I clinical trials where it showed good pharmacodynamic and pharmacokinetic profiles with minimal side effect.36 However, MSI-1436 was not advanced further to Phase II trials due to limited financial resources. In summary, combining the successful story of MSI-1436 along with the druggability findings obtained in this study, allosteric pocket can be considered as a viable target for medicinal chemists.
PTP inhibitors
In this study, 17 different PTPs showed a wide range of druggability with greater tendency of being undruggable. In order to get some insight about this finding, we list a number of previously identified potent inhibitors of different PTP enzymes, belonging to different druggability categories, along with their calculated total polar surface area (TPSA). As shown in Figure 8, the inhibitors of the two druggable members, MPtpB and GLEPP-1, have less polar characteristics (TPSA =129–274 Å2) than the inhibitors of PTPs belonging to the “difficult” category. The latter group of inhibitors scored TPSA values greater than 320 Å2 with only one exception to Cdc25b inhibitor (TPSA =150 Å2). On the other hand, the allosteric inhibitor of PTP1B (TPSA =272 Å2) showed lower polarity nature than the competitive inhibitors of the same enzyme which had TPSA values in a much higher range 353–564 Å2, shedding light on the importance of targeting the PTP1B allosteric site rather than active site. Therefore, druggability data obtained in this study come in line with the inhibitor nature of the corresponding PTP enzyme, although more extensive analyses for known inhibitors are required to get the full picture about PTP druggability.
Conclusion
Druggability assessment of 17 PTPs has revealed that only two of the tested PTPs seem to be druggable. MPtpB and GLEPP-1 were found to be the only druggable among the PTP family as they showed large hydrophobic active site. The rest of the PTPs were ranging from intermediate to undruggable because of their hydrophilic and/or shallow catalytic pocket. Focusing on PTP1B, several factors affecting the active site druggability were studied. Our results showed that the open conformation of the PTP active site has a better druggability than the closed form, while the tight bound water molecule has negligible effect on the PTP1B druggability. Finally, the PTP1B allosteric site demonstrated a significantly improved druggability compared to the catalytic pocket, which suggests that allosteric modulation may become an important pillar for the future of PTP drug discovery. The outcomes of these findings can shed more light in the search for new PTP inhibitors and can assist researchers to avoid extra coast and effort.
Acknowledgment
This work was funded by a grant from the Deanship of Scientific Research and Graduate Studies at Al Ain University of Science and Technology, Al Ain, UAE.
Disclosure
The abstract of this paper was presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutic, November 5–9, 2015, Boston, USA, as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in journal Molecular Cancer Therapeutics: doi:10.1158/1535-7163.TARG-15-B44. The authors report no other conflicts of interest in this work.
References
Hunter T. Signaling – 2000 and beyond. Cell. 2000;100(1):113–127. | ||
Gurzov EN, Stanley WJ, Brodnicki TC, Thomas HE. Protein tyrosine phosphatases: molecular switches in metabolism and diabetes. Trends Endocrinol Metabol. 2015;26(1):30–39. | ||
van Huijsduijnen RH, Bombrun A, Swinnen D. Selecting protein tyrosine phosphatases as drug targets. Drug Discov Today. 2002;7(19):1013–1019. | ||
Tautz L, Mustelin T. Strategies for developing protein tyrosine phosphatase inhibitors. Methods. 2007;42(3):250–260. | ||
Bialy L, Waldmann H. Inhibitors of protein tyrosine phosphatases: next-generation drugs? Angew Chem Int Ed Engl. 2005;44(25):3814–3839. | ||
Lee H, Yi JS, Lawan A, Min K, Bennett AM. Mining the function of protein tyrosine phosphatases in health and disease. Semin Cell Dev Biol. 2015;37:66–72. | ||
Elchebly M, Payette P, Michaliszyn E, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544–1548. | ||
Klaman LD, Boss O, Peroni OD, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20(15):5479–5489. | ||
Combs AP. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J Med Chem. 2010;53(6):2333–2344. | ||
He RJ, Yu ZH, Zhang RY, Zhang ZY. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin. 2014;35(10):1227–1246. | ||
He R, Zeng LF, He Y, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013;280(2):731–750. | ||
Schrödinger LLC. SiteMap 3.2. Portland: Schrödinger, LLC. Available from: http://www.schrodinger.com/ | ||
Vidler LR, Brown N, Knapp S, Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem. 2012;55(17):7346–7359. | ||
Ghattas MA, Mansour RA, Atatreh N, Bryce RA. Analysis of enoyl-acyl carrier protein reductase structure and interactions yields an efficient virtual screening approach and suggests a potential allosteric site. Chem Biol Drug Des. 2016;87(1):131–142. | ||
RCSB. Protein Data Bank. 2015. Available from: http://www.pdb.org/. Accessed December 15, 2015. | ||
Chemical Computing Group. Molecular Operating Environment. Montreal, Canada: Chemical Computing Group; 2015. Available from: http://www.chemcomp.com | ||
Chemical Computing Group. Molecular Operating Environment (MOE), Manual Version 2015.09. Montreal, Canada: Chemical Computing Group; 2015. Available from: http://www.chemcomp.com | ||
Schrödinger LLC. Maestro 9.9.013. Portland: Schrödinger, LLC. Available from: http://www.schrodinger.com/ | ||
Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–730. | ||
Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol. 2002;6(4):447–452. | ||
Stuckey JA, Schubert HL, Fauman EB, Zhang ZY, Dixon JE, Saper MA. Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature. 1994;370(6490):571–575. | ||
Jia Z, Barford D, Flint AJ, Tonks NK. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science. 1995;268(5218):1754–1758. | ||
Ghattas MA, Atatreh N, Bichenkova EV, Bryce RA. Protein tyrosine phosphatases: ligand interaction analysis and optimisation of virtual screening. J Mol Graph Model. 2014;52:114–123. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. | ||
Andersen HS, Iversen LF, Jeppesen CB, et al. 2-(Oxalylamino)-benzoic acid is a general, competitive inhibitor of protein-tyrosine phosphatases. J Biol Chem. 2000;275(10):7101–7108. | ||
Iversen LF, Andersen HS, Branner S, et al. Structure-based design of a low molecular weight, nonphosphorus, nonpeptide, and highly selective inhibitor of protein-tyrosine phosphatase 1B. J Biol Chem. 2000;275(14):10300–10307. | ||
Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6(6):1401–1412. | ||
Jia Z, Ye Q, Dinaut AN, et al. Structure of protein tyrosine phosphatase 1B in complex with inhibitors bearing two phosphotyrosine mimetics. J Med Chem. 2001;44(26):4584–4594. | ||
Iversen LF, Andersen HS, Moller KB, et al. Steric hindrance as a basis for structure-based design of selective inhibitors of protein-tyrosine phosphatases. Biochemistry. 2001;40(49):14812–14820. | ||
Scapin G, Patel SB, Becker JW, et al. The structural basis for the selectivity of benzotriazole inhibitors of PTP1B. Biochemistry. 2003;42(39):11451–11459. | ||
Cheng T, Li Q, Zhou Z, Wang Y, Bryant SH. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012;14(1):133–141. | ||
Ala PJ, Gonneville L, Hillman MC, et al. Structural basis for inhibition of protein-tyrosine phosphatase 1B by isothiazolidinone heterocyclic phosphonate mimetics. J Biol Chem. 2006;281(43):32784–32795. | ||
Ala PJ, Gonneville L, Hillman M, et al. Structural insights into the design of nonpeptidic isothiazolidinone-containing inhibitors of protein-tyrosine phosphatase 1B. J Biol Chem. 2006;281(49):38013–38021. | ||
Wiesmann C, Barr KJ, Kung J, et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat Struct Mol Biol. 2004;11(8):730–737. | ||
Krishnan N, Koveal D, Miller DH, et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat Chem Biol. 2014;10(7):558–566. | ||
He R, Zeng LF, He Y, Zhang ZY. Recent advances in PTP1B inhibitor development for the treatment of type 2 diabetes and obesity, Chapter 6. New Therapeutic Strategies for Type 2 Diabetes: Small Molecule Approaches (RSC Drug Discovery). Cambridge, UK: The Royal Society of Chemistry; 2012:142–176. | ||
Grundner C, Perrin D, Hooft van Huijsduijnen R, et al. Structural basis for selective inhibition of Mycobacterium tuberculosis protein tyrosine phosphatase PtpB. Structure. 2007;15(4):499–509. | ||
Barr AJ, Ugochukwu E, Lee WH, et al. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136(2):352–363. | ||
Yokota T, Nara Y, Kashima A, et al. Crystal structure of human dual specificity phosphatase, JNK stimulatory phosphatase-1, at 1.5 Å resolution. Proteins. 2007;66(2):272–278. | ||
Reynolds RA, Yem AW, Wolfe CL, Deibel MR Jr, Chidester CG, Watenpaugh KD. Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J Mol Biol. 1999;293(3):559–568. | ||
Ivanov MI, Stuckey JA, Schubert HL, Saper MA, Bliska JB. Two substrate-targeting sites in the Yersinia protein tyrosine phosphatase co-operate to promote bacterial virulence. Mol Microbiol. 2005;55(5):1346–1356. | ||
Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263(5152):1397–1404. | ||
Zeng L-F, Xu J, He Y, et al. A facile hydroxyindole carboxylic acid based focused library approach for potent and selective inhibitors of Mycobacterium protein tyrosine phosphatase B. ChemMedChem. 2013;8(6):904–908. | ||
Gobert RP, van den Eijnden M, Szyndralewiez C, et al. GLEPP1/protein-tyrosine phosphatase φ inhibitors block chemotaxis in vitro and in vivo and improve murine ulcerative colitis. J Biol Chem. 2009;284(17):11385–11395. | ||
Park H, Li M, Choi J, Cho H, Ham SW. Structure-based virtual screening approach to identify novel classes of Cdc25B phosphatase inhibitors. Bioorg Med Chem Lett. 2009;19(15):4372–4375. | ||
Rawls KA, Therese Lang P, Takeuchi J, et al. Fragment-based discovery of selective inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorg Med Chem Lett. 2009;19(24):6851–6854. | ||
Bahta M, Lountos GT, Dyas B, et al. Utilization of nitrophenylphosphates and oxime-based ligation for the development of nanomolar affinity inhibitors of the Yersinia pestis outer protein H (YopH) phosphatase. J Med Chem. 2011;54(8):2933–2943. | ||
Shen K, Keng Y-F, Wu L, Guo X-L, Lawrence DS, Zhang Z-Y. Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure. J Biol Chem. 2001;276:47311–47319. | ||
Szczepankiewicz BG, Liu G, Hajduk PJ, et al. Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J Am Chem Soc. 2003;125(14):4087–4096. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.