")
Back to Journals » International Journal of Nanomedicine » Volume 17
Drug Release from Gelsolin-Targeted Phase-Transition Nanoparticles Triggered by Low-Intensity Focused Ultrasound
Authors Qin H, Teng R, Liu Y, Li J, Yu M
Received 26 September 2021
Accepted for publication 26 December 2021
Published 6 January 2022 Volume 2022:17 Pages 61—71
DOI https://doi.org/10.2147/IJN.S341421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Farooq A. Shiekh
Haocheng Qin,1 Rong Teng,1 Yan Liu,1 Juan Li,2,* Ming Yu1,*
1Department of Ultrasound, The Affiliated Lianyungang Hospital of Xuzhou Medical University, Lianyungang, 222002, People’s Republic of China; 2Department of Oncology, The Second People’s Hospital of Lianyungang, Lianyungang, 222023, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ming Yu
Department of Ultrasound, The Affiliated Lianyungang Hospital of Xuzhou Medical University, 182 Northern Tongguan Road, Lianyungang, 222002, People’s Republic of China
Tel/Fax +86 18961326101
Email [email protected]
Juan Li
Department of Oncology, The Second People’s Hospital of Lianyungang, 161 Southern Xingfu Road, Lianyungang, 222023, People’s Republic of China
Tel/Fax +86 18936641336
Email [email protected]
Purpose: Current strategies for tumour-induced sentinel lymph node detection and metastasis therapy have limitations. It is essential to identify and provide warnings earlier for tumour metastasis to carry out effective clinical interventions. In addition, traditional cancer chemotherapy encounters drastic limitations due to the nonspecific delivery of antitumour drugs and severe side effects. We aimed to exploit the potential of gelsolin (GSN) monoclonal antibody as a targeting agent and perfluorohexane (PFH) as a phase-transition agent to maximize the cytotoxic effect of poly(lactic-co-glycolic acid) (PLGA) nanoparticle-based drug controllable release systems for Hca-F cells.
Methods: We co-encapsulated PFH and doxorubicin (DOX) into PLGA nanoparticles (NPs) and further conjugated GSN monoclonal antibody onto the surface of NPs to form GSN-targeted phase transition polymer NPs (GSN-PLGA-PFH-DOX) for both imaging and therapy of tumours and metastatic lymph nodes. To promote and trigger drug release on demand, low-intensity focused ultrasound (LIFU) was applied to achieve a controllable release of the encapsulated drug.
Results: GSN-PLGA-PFH-DOX NPs exhibited characteristics such as a narrow size distribution and smooth surface. GSN-PLGA-PFH-DOX NPs could also specifically bind to Hca-F cells and increase the ultrasound contrast agent (UCA) image contrast intensity. GSN-PLGA-PFH-DOX NPs enable GSN-mediated targeting and biotherapeutic effects as well as LIFU-responsive drug release, resulting in synergistic cytotoxic effects in GSN-overexpressing cells in vitro.
Conclusion: Our work might provide a strategy for the imaging and chemotherapy of primary tumours and their metastases.
Keywords: poly(lactic-co-glycolic acid), gelsolin, phase transition, controllable drug release
Introduction
Traditional cancer chemotherapy encounters drastic limitations due to the nonspecific delivery of antitumour drugs and severe side effects.1–3 Therefore, the design and fabrication of novel drug delivery systems are promising and rapidly developing areas. Microbubbles, known to be UCAs, have played important roles in the diagnosis, differential diagnosis and treatment of benign and malignant tumours. In addition, they have been used to load therapeutic agents for efficient tumour chemotherapy.4–6 However, these gas-filled microbubbles have a limited capacity for loading therapeutic agents, short circulation time, and large micrometre size, making it difficult to achieve effective drug concentration at the tumour site, which hampers their wide application in drug delivery. It has been reported that only particles with a diameter of <700 nm are permitted to pass through the vascular endothelial space of tumours.7 To address these problems, different kinds of nanoscale drug-delivery systems with selective targeting and controlled release properties have been designed and fabricated to minimize systemic cytotoxicity.8–10 The stability of nanodroplets is increased, and they can accumulate in tumour tissues by passive targeting due to their nanoscale size.
Some synthetic polymers, which are composed of one or two monomers and demonstrate controllable degradation rates, have been widely used for drug delivery applications.11–13 PLGA is a degradable polyester polymer that is randomly polymerized by two monomers, lactic acid and glycolic acid. It has been certified by the FDA in the United States and has been formally included in the United States Pharmacopoeia as a medicinal excipient. It has good biocompatibility, encapsulation and film-forming properties and ultimately metabolizes into water and carbon dioxide in vivo and is thus called a “good environmental material”. It is widely used in pharmaceuticals, medical engineering materials and modern industrial fields.14,15 PLGA NPs can protect the biological activity of drugs and enhance their tolerance to external conditions (acids, alkalis, enzymes, water, heat, etc.) and prolong the half-lives of biological drugs in the body, which has important clinical significance for long-term drug users. Meanwhile, to enhance the accuracy of drug delivery to tumours, controlled drug-delivery systems have been functionalized with a variety of tumour-specific targeting ligands—such as aptamers, peptides, antibodies and growth factors—to selectively target molecules and receptors that are overexpressed on the surface of tumour cells.16–19
The lymphatic system plays an important role in the spread of cancer cells. Tumour metastasis to the lymphatic system is a universal sign of tumour progression and is closely related to the prognosis of patients. Once tumour cells invade the lymphatic system, they can take root and seed into other regions of the body by these invaded lymphatic nodes.20 Studies have revealed that GSN plays important roles in cell adhesion, migration and invasion. Differential proteomics has revealed that GSN is closely associated with the lymphatic metastasis of tumours and is mainly located in the cytoplasm and cytoskeleton of the mouse hepatocarcinoma cell line Hca-P with low metastatic potential.21–23 Meanwhile, it also appears in the cell membrane of the mouse hepatocarcinoma cell line Hca-F, which has a high metastatic potential.24,25 It is a kind of skeleton-regulating protein that regulates polymerization and depolymerization of cell actin in a Ca2+-dependent manner and thus affects cell morphology and motility.26 Abbeele et al27 demonstrated that the downregulation of gelsolin tends to decrease the invasion and migration of many tumour cells, such as cervical HeLa cells and MDA-MB-231 cells, in human breast cancer. Hence, it may be an important membrane antigen in the early stages of the invasion and metastasis of tumour cells and is expected to become a molecular target for early identification and prediction of lymphatic metastasis. Nevertheless, the existing imaging diagnosis mainly depends on the morphology of tumours and characteristics of blood perfusion but is unable to provide specifications of the biological behaviours of tumours, which leads to obvious limitations in the early identification of tumour metastasis. Therefore, designing a molecular targeted contrast agent for specific imaging of tumour antigens will be helpful for early detection, accurate diagnosis and precise treatment of tumour metastasis.
Despite the great efforts to design and produce novel drug delivery systems, drug efficacy in treating tumours remains limited by the relatively small amount of drug delivery and release in available systems. In addition, at present, most of the developed PLGA contrast agents are nanoscale, and their imaging ability does not seem very optimistic in in vivo experiments. Recent studies have demonstrated that PFH holds great potential in imaging-based approaches via a “liquid-to-gas” phase transition in the tumour region.28,29 In contrast to conventional UCA microbubbles, PFH droplets are generally enclosed in liposomes, PLGA, or albumin shells30,31 to enhance their stability and avoid air embolism at the beginning of administration. Furthermore, the small dimensions of PFH-containing NPs enhance their ability to permeate the internal regions of a tumour and elude capture by the mononuclear phagocyte system to further prolong their circulation in the bloodstream.30 Meanwhile, when PFH-containing NPs are triggered by ultrasound (US) to enter the gaseous phase, they possess significantly augmented US contrast due to their volume expansion. Therefore, to develop a controllable drug delivery system with the simultaneous ability to realize a molecular imaging diagnosis of early metastasis of tumours, we fabricated drug-encapsulated GSN-targeted phase-transition PLGA nanoparticles using the double emulsion-solvent evaporation technique and carbodiimide method. In the NPs we produced, the phase-transition material PFH and an anticancer drug (DOX) were selected to be incorporated in the PLGA shell. In addition, we investigated the influence of different volumes of PFH on drug encapsulation efficiency. PFH in the core of NPs can undergo a liquid-gas phase transition upon activation by US energy, called acoustic droplet vaporization. When converged in tumour sites, these NPs convert into microbubbles under LIFU irradiation. These acoustic microbubbles oscillate and collapse by the ultrasound-targeted microbubble destruction (UTMD) effect.6,32,33 During this process, the anticancer drug encapsulated in bubbles is released in tumour cells and possesses significantly augmented US contrast.
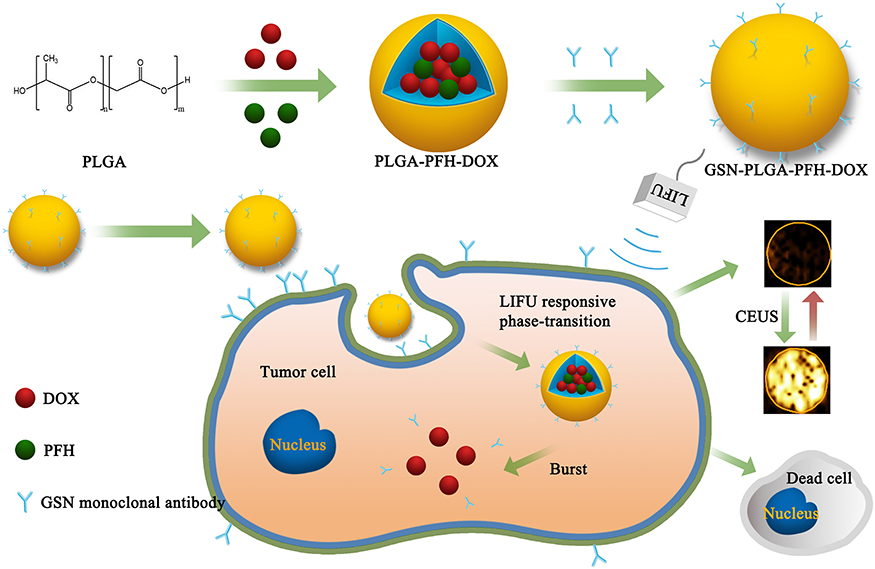 |
Figure 1 Schematic representation of the preparation process of phase-transition NPs. |
Experimental Section
Materials and Instruments
PLGA (lactide: glycolide = 50:50, MW = 12,000 Da) was purchased from Jinan Daigang Biological Material Limited Company (Shandong, China). PFH was purchased from J&K Scientific Ltd. (Beijing, China). DOX was obtained from Beijing Yihe Biological Co. Ltd. (Beijing, China). GSN monoclonal antibodies, poly(vinyl alcohol) (PVA, 99% MW`=30,000–70,000), 2-(N-morpholino)ethanesulfonic acid (MES), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 1,1-dioctadecyl-3,3,3,3-tetrame-thylindocarbocyanine perchlorate (DiI), 4,6-diamino-2-phenylindole (DAPI), and coumarin-6 were purchased from Sigma–Aldrich (St. Louis, MO, USA). The use of a mouse ascitic hepatocarcinoma cell line with high lymphatic metastasis (Hca-F) was approved by the ethics committee of the Affiliated Lianyungang Hospital of Xuzhou Medical University (LW-20180611012). All chemicals used in this work were analytical grade and used without further processing.
Synthesis of GSN-PLGA-PFH-DOX NPs
PFH and DOX-containing NPs were prepared using a double emulsion-solvent evaporation technique. One hundred milligrams of PLGA-COOH was fully dissolved in 2 mL of dichloromethane, and 0.2 mL of DOX solution (50 mg/mL, 10 mg of DOX dissolved in 200 μL of deionized water) and 200 μL of PFH were added. First, PLGA, DOX, and PFH with dichloromethane were vibrated acoustically via an ultrasonic oscillation instrument (VCY-500, Shanghai Yanyong Ultrasonic Instrument Co. Ltd.) for 100 s. Then, 5 mL of 4% PVA solution was added to the above solution, on which ultrasonic vibration was performed for 60 s again. Then, 20 mL of 2% isopropanol solution was added and magnetically stirred continuously for 4 h until the dichloromethane evaporated and the surfaces of the NPs solidified. After centrifugation, the supernatant was discarded, and the precipitate was washed with double-distilled water. The centrifugation and washing processes were repeated three times, and PLGA-PFH-DOX NPs were obtained. A carbodiimide-mediated amide bond formation method was used to prepare the GSN-PLGA-PFH-DOX NPs. Briefly, 2 mL each of EDC (200 mmol/L) and NHS (50 mmol/L) were successively added to 1 mL (10 mg/mL) of PLGA-PFH-DOX NP solution, and then shaking incubation was performed for 30 min, followed by multiple centrifugations using double-distilled water to remove the unreacted EDC and NHS to obtain NPs with an activated surface. Then, these NPs were dissolved with MES buffer (PH=8.0). An appropriate amount of GSN monoclonal antibody (final concentration 2.0 mM) was added and allowed to react for 2 h. Finally, the GSN-PLGA-PFH-DOX NPs were washed with double-distilled water three times to remove the unreacted materials, diluted to 10 mL, stored at 4 °C and observed for 7 days. The entire fabrication process was performed in ice water at a temperature of 4 °C. All NP types were prepared using the same procedures described above without adding the corresponding elements.
Characteristics of the NPs and Determination of DOX Loading
First, 200 μL of GSN-PLGA-PFH-DOX NPs was diluted in double-distilled water to 10 mL, and then 1 drop was deposited on a 200-mesh copper grid. When the sample was dry, the morphology of these NPs was studied using a scanning electron microscope (SEM, Hitachi S-3400N, Japan), and the structure was analysed using transmission electron microscopy (TEM, Hitachi H-7600, Japan). In addition, the size distribution and surface zeta potential of each NP type were assessed using a Malvern Zetasizer Nano ZS unit (Malvern Instrument, UK).
Drug loading and encapsulation efficiency were determined using a high-performance liquid chromatography (HPLC) method. The amount of DOX recovered from washed supernatants during preparation (WR) was determined. The drug payload (WNP) was calculated as the difference between the total amount of DOX (WT) and WR. The drug encapsulation efficiency and loading efficiency were calculated by the following equations:
Encapsulation efficiency = WNP/WT × 100%
Loading efficiency = WNP/Wt × 100%
where WT represents the total weight of DOX used in the preparation of the NPs; WNP represents the total drug amount in the NPs; and Wt represents the total weight of NPs.
Connection of GSN Monoclonal Antibody with NPs
DiI-labelled GSN-PLGA-PFH-DOX was collected and diluted to 2 mg/mL with double-distilled water, and then 20 μL of fluorescein isothiocyanate (FITC)-labelled goat-anti-mouse IgG antibody was added and oscillated for 2 h in an ice bath, followed by observations under a laser scanning confocal microscope (LSCM, Olympus FV1200, Japan). Considering PLGA-PFH-DOX as the control, the GSN monoclonal antibody connection rate on the surface of GSN-PLGA-PFH-DOX was detected by flow cytometry.
In vitro Liquid-Gas Phase Transition and Responsive Drug Release Activated by LIFU
Two millilitres of GSN-PLGA-PFH-DOX NP solution (1 mg/mL) was added to the preformed wells of an agarose gel phantom (3% agarose). Then, a LIFU transducer (1 MHz, acoustic intensity: 1.2 W/cm2, duty cycle: 50%, Chongqing Medical University, P. R. China) was used to irradiate the NPs with a power of 1–8 W and a duration of 1–5 min. B-mode US imaging signals and contrast-enhanced ultrasound (CEUS) imaging signals of the post-formed microbubbles from NPs were observed in real time using a MyLab 90 ultrasound diagnostic system (Esaote, Italy). The average ultrasound intensity was determined by DFY software (Chongqing Medical University, P. R. China), and the change in size of NPs was observed under an optical microscope (OM, Olympus BX51-P, Japan).
To measure the drug release patterns, we collected two types of drug-loaded NPs, divided them into four groups and incubated them in conical flasks with 20 mL of prewarmed PBS (120 rpm/min) at 37 °C. For the GSN-PLGA-DOX+LIFU and GSN-PLGA-PFH-DOX+LIFU groups, LIFU irradiation (8 W, 3 min) was applied to activate drug release at a predetermined time point (8 h). One millilitre of release medium was removed for measurement at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, and 72 h. Then, the same amount of fresh buffer was added to maintain the sink condition. HPLC was used to determine the content of DOX in these samples, calculate the cumulative release rate of the drug, and draw a release curve.
Cellular Uptake of NPs
Hca-F cells in logarithmic phase were seeded into confocal culture dishes with 1640 complete medium (without foetal bovine serum) for 4 h. They were divided into 3 groups: Hca-F cells+ PLGA-PFH-DOX, Hca-F cells+ GSN monoclonal antibody+ GSN-PLGA-PFH-DOX (receptor blockade group), and Hca-F cells+ GSN-PLGA-PFH-DOX. First, 100 μL of GSN monoclonal antibody was added to the receptor blockade group and incubated for 20 min. After that, 100 μL of coumarin-6-labelled corresponding NP sample was added, and the cells were incubated at 37 °C and 5% CO2 for 2 h. Subsequently, cells were fixed with 4% paraformaldehyde and stained fluorescently with DAPI after washing with PBS. Intracellular uptake of NPs was investigated under LSCM. Image-Pro Plus 6.0 software (IPP 6.0, Media Cybernetics, USA) was used to measure the green fluorescence intensity in the field of vision and count the number of cells. The fluorescence intensity of each cell was obtained by dividing the two. Each group took three fields of vision.
Cytotoxicity Assay, Cell Apoptosis and Half Inhibition Concentration
Hca-F cells were seeded into 96-well plates at a density of 1×104 per well and cultured overnight. Free DOX at a concentration of 20 μg·mL−1 and GSN-PLGA-PFH NPs, PLGA-PFH-DOX NPs, and GSN-PLGA-PFH-DOX NPs at a concentration of 300 μg·mL−1 were prepared in double-free medium. To investigate the cytotoxicity of NPs, eleven groups were formed, namely, the empty group, control group, and experimental groups: GSN-PLGA-PFH NPs, PLGA-PFH-DOX NPs, GSN-PLGA-PFH-DOX NPs, DOX, control + LIFU, GSN-PLGA-PFH NPs + LIFU, PLGA-PFH-DOX NPs + LIFU, GSN-PLGA-PFH-DOX NPs + LIFU, and DOX + LIFU groups. Then, 0.1 mL of medium was added to the control group when 0.1 mL of NPs or free DOX was added to the other groups. Then, these cells were incubated for 24 h at 37 °C and 5% CO2. For those groups with US irradiation, LIFU irradiation (8 W, 3 min) was performed after 2 h incubation. Then, the CCK-8 method was used to detect the cell survival rate of each group.
To investigate the half inhibition concentration (IC50) of NPs, 0.1 mL of GSN-PLGA-PFH-DOX NPs at different concentrations (0.025, 0.05, 0.1, 0.25, 0.5 and 1.0 mg·mL−1) was added to Hca-F cells when the control group was added to 0.1 mL medium. Then, these cells were incubated for 24 h at 37 °C and 5% CO2. LIFU irradiation (8 W, 3 min) was performed after 2 h of incubation. Then, the CCK8 method was used to detect the OD value of each group, and the cell inhibition rate was calculated.
Cell viability rate (%) = (OD1-OD0)/(OD2-OD0) × 100%
Cell inhibition rate (%) = (OD2-OD1)/(OD2-OD0) × 100%
where OD0 represents the OD value of the empty group; OD1 represents the OD value of the experimental group; and OD2 represents the OD value of the control group.
Two-colour flow cytometric analysis was used to investigate cellular apoptosis. The experiment was grouped as above, and Hca-F cells were collected at the end of the incubation. Cells were washed with PBS and stained with reagents from the Apoptosis Assay Kit (Bestbio Co. Ltd, P. R. China) following the manufacturer’s instructions and analysed using a flow cytometer (FACSVantage SE, BD Biosciences, US).
Results and Discussion
Design, Synthesis and Characterization of GSN-PLGA-PFH-DOX
The fabrication of GSN-PLGA-PFH-DOX NPs is elucidated in Figure 1. First, we constructed PLGA NPs encapsulating PFH and DOX. Then, the GSN monoclonal antibody was coupled to the surface of the PLGA-PFH-DOX NPs via the carbodiimide method, forming GSN-PLGA-PFH-DOX NPs for US imaging and chemotherapy applications. GSN receptor-specific endocytosis of NPs is also demonstrated in Figure 1. When GSN-PLGA-PFH-DOX NPs were endocytosed into Hca-F cells, LIFU was applied to trigger the liquid-gas phase transition of PFH, thus achieving an enhanced ultrasound imaging effect and on-demand drug release.
The GSN-PLGA-PFH-DOX NPs appeared as a milky-white mixed suspension. SEM imaging of these NPs (Figure 2A) showed that GSN-PLGA-PFH-DOX NPs exhibited a smooth and uniform spherical morphology. The presence of PFH and DOX in the shells of NPs was demonstrated by the enhanced contrast manifested as dark domains in Figure 2B by TEM. The size distribution of GSN-PLGA-PFH-DOX NPs was between 200 and 300 nm (mean diameter = 285.62± 12.76 nm), and the polydispersity index was PI = 0.141±0.052. The mean diameters of pure PLGA NPs, PLGA-PFH NPs, PLGA-DOX NPs and PLGA-PFH-DOX NPs were 248.35±13.23 nm (PI= 0.133±0.053), 259.17±16.45 nm (PI= 0.136±0.027), 259.46±13.71 nm (PI=0.364±0.075) and 260.51±14.38 nm (PI=0.263±0.043), respectively (Figure 2C and D). These results suggest that the addition of PFH and DOX in the centre does not significantly affect the NP size distributions. The particle sizes of GSN-PLGA-PFH-DOX NPs compared with other NPs were all P<0.05, and the difference was statistically significant. These results indicate that the connection of GSN monoclonal antibody on the surface of the NPs is the main reason for the increase in particle size. Figure 2E shows that when the NPs carry GSN monoclonal antibody, the negative charge decreases gradually and tends to the positive potential (1.68±0.53 mV). This is similar to the results of previous studies,34 which showed that the GSN monoclonal antibody was successfully connected with NPs.
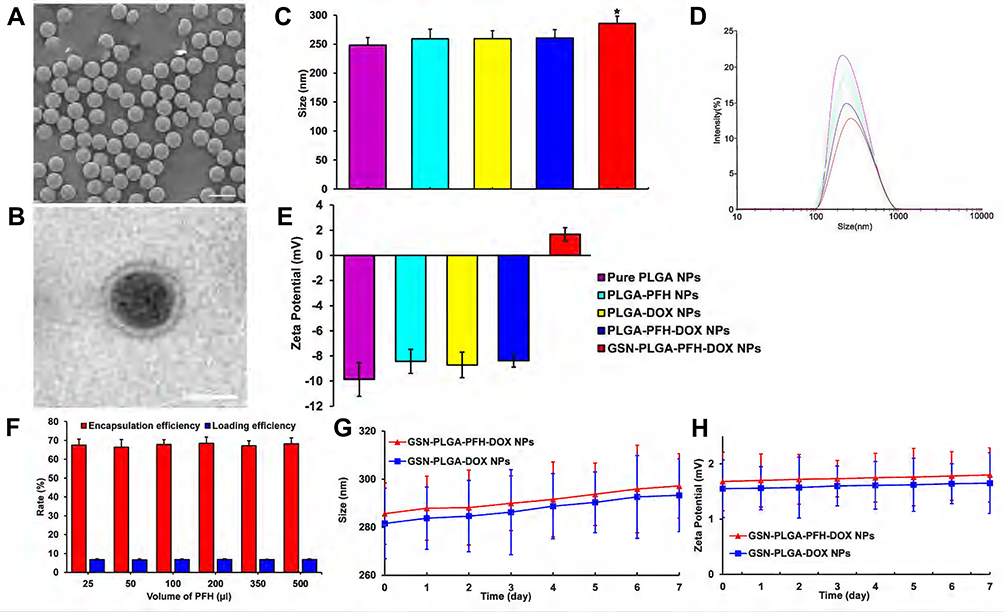 |
Figure 2 (A) SEM image of GSN-PLGA-PFH-DOX NPs (Scale bar: 500 nm). (B) TEM image of GSN-PLGA-PFH-DOX NPs (Scale bar: 200 nm). (C) sizes of the different PLGA NPs (*P<0.05 vs the other groups). (D) Size distribution of the different PLGA NPs. (E) Zeta potential of different PLGA NPs. (F) Different PFH dosages have no significant effect on DOX encapsulation efficiency and drug loading. (G and H) The size (G) and zeta potential (H) of GSN-PLGA-PFH-DOX NPs and GSN-PLGA-DOX NPs over a period of 7 days in a stability test. |
The DOX loading amount and encapsulation efficiency were measured by HPLC. The encapsulation efficiencies of DOX in GSN-PLGA-PFH-DOX with different volumes of PFH (25, 50, 100, 200, 350, 500 μL) were 67.56±3.21%, 66.44±4.06%, 67.92±2.55%, 68.45±3.43%, 67.16±2.66%, and 68.22±3.14%, respectively. The DOX loading efficiencies of these NPs were 6.76±0.32%, 6.64±0.41%, 6.79±0.26%, 6.85±0.34%, 6.72±0.27%, and 6.82±0.31%, respectively. From these results, we can deduce that the addition of PFH to the polymer shell does not significantly affect the DOX encapsulation efficiency or the loading efficiency (Figure 2F). Comparatively, the encapsulation and loading efficiencies of PLGA-PFH-DOX were 68.06±4.63% and 6.81±0.46%, respectively. It can be inferred that the modification of GSN onto the surface of PLGA nanoparticles does not affect the DOX encapsulation or loading efficiency in some respect. The stability of the NPs could be maintained when stored at 4 °C, and the particle size and surface potential of the NPs had no significant change when tested at room temperature within 7 days (Figure 2G and H).
Connection of GSN Monoclonal Antibody
The carbodiimide method, as a classic chemical method for connecting proteins and small peptide molecules, is currently the most popular and widely used method for the production of biochemical conjugates. This method has high target-linking efficiency and mild reaction conditions.35 EDC is a water-soluble crosslinking agent that allows biological coupling between carboxyl and amine groups. In this reaction, the amino group from the GSN monoclonal antibody forms an amide bond with the carboxyl group on the PLGA surface.36 Figure 3A showed that GSN-PLGA-PFH-DOX marked by DiI fluorescent dye emitted red fluorescence, Figure 3B showed FITC-labeled secondary antibody emitted green fluorescence (FITC-labeled secondary antibodies can bind to GSN monoclonal antibodies attached to the surface of NPs), and Figure 3C showed orange fluorescence after the two fluorescence channels were superimposed. This result shows that the GSN monoclonal antibody was successfully attached to the surface of NPs. Flow cytometry revealed that the connection rate of GSN monoclonal antibody and NPs was 98.31% (Figure 3D), which was higher than that in the control group (0.073%) (P<0.05) (Figure 3E). These results show that the GSN monoclonal antibody was successfully connected, laying the foundation for subsequent targeted experiments.
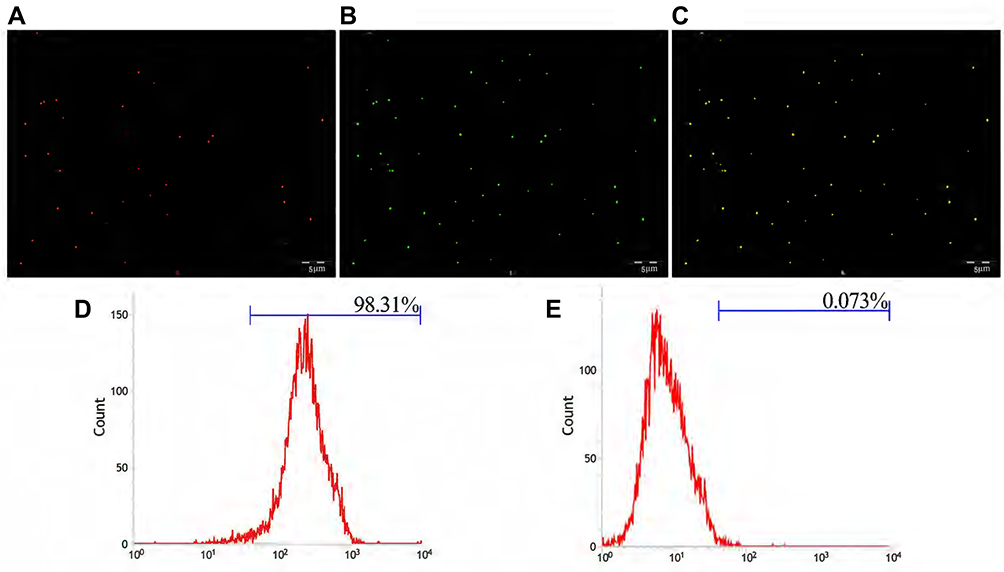 |
Figure 3 (A) The DiI-labelled NPs emitted red fluorescence. (B) FITC-labelled secondary antibody emitted green fluorescence after combination with NPs. (C) Both showed Orange fluorescence under the fusion channel. (D) Connection rate of the GSN monoclonal antibody on GSN-PLGA-PFH-DOX NPs. (E) Connection rate of the control group (Scale bar: 5 μm). |
In vitro Phase Transition and Drug Release Activated by LIFU
Ultrasound-mediated drug delivery NPs can be monitored by US imaging and induced to release therapeutic agents using external US radiation after they are detected in the target tissues by diagnostic US. Liquid PFH encapsulated in NPs changed to the gas phase under LIFU irradiation. Subsequently, larger microbubbles are formed and exploded, which leads drugs to be released from drug delivery systems. We selected adequate irradiation power and duration of LIFU to induce the phase transition of PFH incorporated in NPs. Using an LIFU transducer, the US energy was focused onto the NPs. DFY software was applied to evaluate the value of the average US signal intensity. GSN-PLGA-PFH-DOX NPs had the highest average B-mode and CEUS signal (Figure 4A–C) values at US parameters of 8 W/3 min, suggesting that a large number of PLGA NPs were converted to microbubbles. With the increase in irradiation duration, some PLGA microbubbles were destroyed and collapsed, decreasing the intensities of harmonic US images, which also laid the foundation for subsequent drug release therapy. In addition, LIFU irradiation is the focus, making the process of triggering the phase change both efficiently and safely. Therefore, a US irradiation parameter of 8 W/3 min was selected to activate the phase transition of PLGA NPs and release the loaded drug after bubble collapse. Following conversion of NPs into bubbles, tumour imaging signals could be collected, and the drug could be released after the collapse of the microbubbles. During the process of the LIFU-responsive phase transition, we observed the variations in PFH-loaded NPs every 1 min via OM. The size of the NPs increased approximately 200-fold from their original size (Figure 4D).
To investigate drug-releasing behaviour under LIFU irradiation, four groups were formed, namely, GSN-PLGA-DOX (without LIFU radiation), GSN-PLGA-DOX+LIFU radiation (8 W, 3 min), GSN-PLGA-PFH-DOX (without LIFU radiation), and GSN-PLGA-PFH-DOX+LIFU radiation (8 W, 3 min). As displayed in Figure 4E, DOX was continuously released from the NPs without LIFU radiation, but burst release of the drug was observed in the NPs after LIFU exposure. For GSN-PLGA-DOX+LIFU, LIFU radiation triggered 10% more drug release at the 8 h time point with subsequent slow drug release from the NPs. The reason may be that the release of this part of the drug is caused by DOX that only adheres to the surface or is not tightly wrapped in NPs, and LIFU irradiation accelerates this process. Comparatively, only approximately 29% of the drug was released in the initial 8 h. The percentage of cumulative drug release increased up to 60% under LIFU triggering at the 8 h time point. In the GSN-PLGA-PFH-DOX group, drugs were released slowly from the NPs within 72 h, and no obvious burst drug-releasing profiles were observed because liquid PFH did not change to the gas phase without external stimulus. Although the mechanism of US-triggered drug release is not yet fully understood, it has been reported37 that the liquid-gas phase transition of PFH under US radiation causes the gaps between the shell polymer molecules to become larger, which may facilitate the escape of drug molecules from the bubbles.
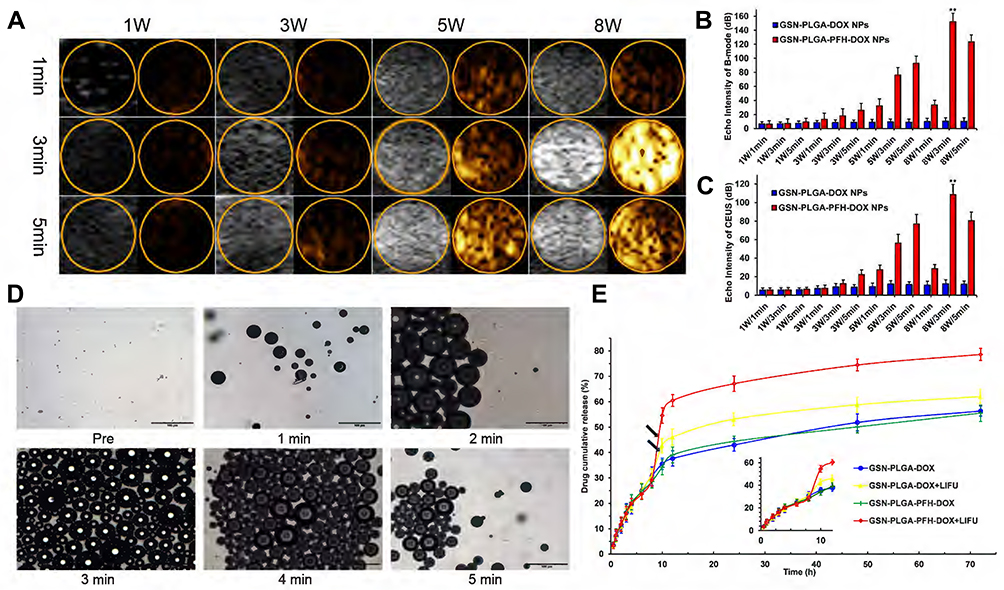 |
Figure 4 (A) Corresponding B-mode and CEUS images of the phase transition process of the GSN-PLGA-PFH-DOX NPs under LIFU irritation with different powers and times, as observed in vitro. Quantitative echo intensity of GSN-PLGA-PFH-DOX NPs and GSN-PLGA-DOX NPs in B-mode (B) and CEUS imaging (C) at different time points and powers under LIFU irradiation (**P<0.001 vs the other groups). (D) OM images of the LIFU-responsive phase transition process with a power of 8 W/5 min at different time points. (E) Drug-release profiles triggered by LIFU irritation (black arrow: ultrasound administration). |
In vitro Targeted Ability
The observation results by LSCM for the in vitro uptake experiment are shown in Figure 5A. More GSN-PLGA-PFH-DOX NPs were taken up by Hca-F cells, which resulted in strong cell fluorescence (1853.64±153.69 a.u.) (Figure 5B). When the target GSN on the surface of Hca-F cells was sealed, a markedly decreased amount of GSN-PLGA-PFH-DOX NPs was absorbed, which resulted in weak cell fluorescence (768.33±151.42 a.u.). Meanwhile, fewer PLGA-PFH-DOX NPs were unconnected to the GSN monoclonal antibody and were absorbed by Hca-F cells (701.63±181.53 a.u.).
Cytotoxicity Assay, Cell Apoptosis and Half Inhibition Concentration
In our study, the drug loading of GSN-PLGA-PFH-DOX was 6.85±0.34%%; therefore, the amount of DOX in 300 μg/mL NPs was equivalent to that in the free DOX group (20 μg/mL). To investigate the cytotoxicity and cell apoptosis efficiency of NPs, various groups of agents were incubated with Hca-F cells for 24 h. As shown in Figure 5C and D, compared with the control group, the survival rates of Hca-F cells in the GSN-PLGA-PFH and GSN-PLGA-PFH+LIFU groups were not significantly decreased, and the cell apoptosis rates were not significantly increased, which indicated that the cytotoxicity of nondrug-loaded PLGA NPs to cells was very low. The cytotoxicity levels of the PLGA-PFH-DOX NP and GSN-PLGA-PFH-DOX NP groups were lower than that of the free DOX group. Comparatively, the cell viability rates of groups treated with PLGA-PFH-DOX NPs, GSN-PLGA-PFH-DOX NPs and free DOX were significantly decreased after LIFU irradiation. Meanwhile, the cell apoptosis rates of these three groups were significantly increased. The reason may be that the mechanical and cavitation effects of LIFU irradiation increase the permeability of the cell membrane, which increases the amount of nanoparticles or drugs entering the cells, thereby increasing the therapeutic effect. In addition, before the application of combined LIFU irradiation, the cytotoxicity of GSN-PLGA-PFH-DOX NPs was less than that of free DOX, indicating that the DOX contained in GSN-PLGA-PFH-DOX NPs was relatively stable without significant burst release. After combined LIFU irradiation, the cytotoxicity of GSN-PLGA-PFH-DOX NPs was significantly higher than that of free DOX (P<0.05), indicating that targeted delivery of drugs was increased under the targeted effect of GSN monoclonal antibody. Meanwhile, the blasting effect of the NPs and the release of DOX after combined LIFU irradiation further enhanced the cytotoxicity of NPs. The results of the half inhibition concentration experiment are shown in Figure 5E. When the concentration of GSN-PLGA-PFH-DOX NPs was 0.1 mg·mL−1, the inhibition rate of Hca-F cells was 29.11±2.14%, and when the concentration of NPs was 0.25 mg·mL−1, the cell inhibition rate increased to 65.03±2.35%. The IC50 of NPs calculated by GraphPad Prism 9 software (GraphPad Software Inc., San Diego, CA, USA) was 0.17 mg·mL−1.
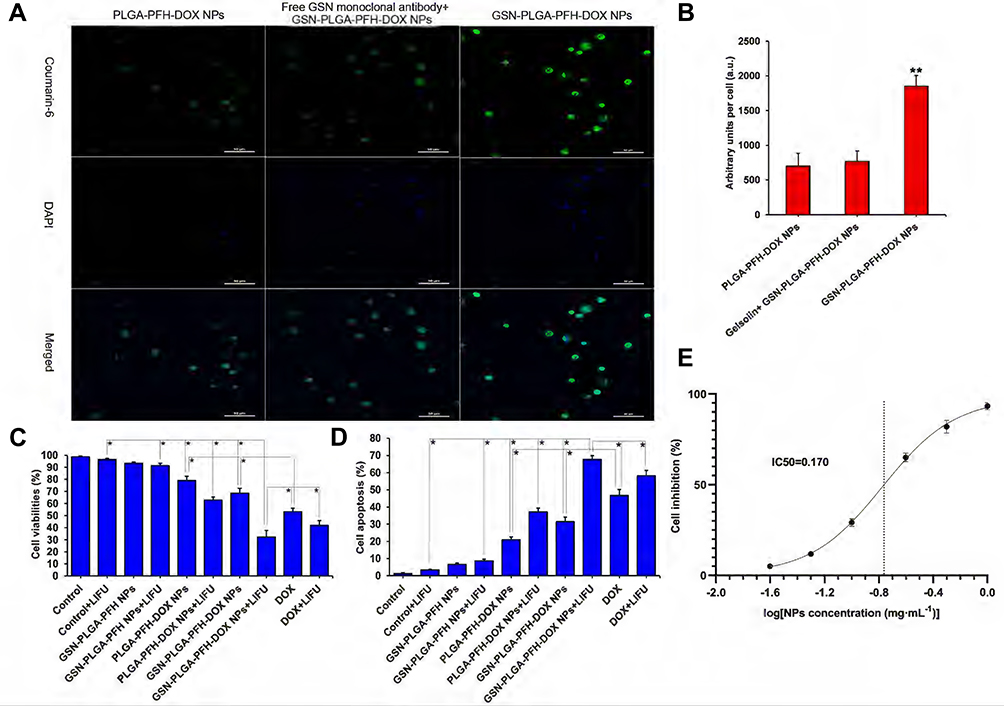 |
Figure 5 (A) Cell uptake observed by LSCM (Coumarin-6: green, DAPI: blue) (Scale bar=50 μm). (B) Histogram of the quantitative study of the cellular uptake efficiencies of different groups (**P<0.001 vs the other two groups). (C) Cell viability rates of various therapeutic agents, *P<0.05. (D) Cell apoptosis rate of various therapeutic agents, *P<0.05. (E) Fitting curve of half inhibition concentration of NPs. |
Conclusion
In summary, we successfully developed gelsolin-targeted and doxorubicin-loaded phase transition nanoparticles (GSN-PLGA-PFH-DOX NPs) as a targeted drug carrier and an efficient UCA for both the imaging and therapy of tumours and metastatic lymph nodes. The GSN-PLGA-PFH-DOX NPs exhibited efficient targeted capability to tumour tissues in vitro and correspondingly enhanced US imaging. Over 72 h of incubation, DOX was slowly and steadily released after the initial burst release. Furthermore, US stimulation can accelerate drug release on demand. LIFU stimulation, causing a controllable release of encapsulated drug from the GSN-PLGA-PFH-DOX NPs, may allow cytotoxic drugs to accumulate at higher levels in tumours, which can maximally kill Hca-F cells with fewer side effects and prevent tumour recurrence and metastasis. The combination of targeted imaging and anticancer drug transportation based on GSN-PLGA-PFH-DOX NPs as the contrast agent for ultrasonography could be attractive in the future development of nanomedicine and noninvasive medicine for simultaneous accurate diagnosis, drug delivery and therapy for cancers or other diseases.
Acknowledgment
The authors would like to thank Ms. Lan Hao for her assistance with the laser scanning confocal microscopy. This work was financially supported by the Young Talents Fund of Lianyungang First People’s Hospital (NO. QN1808, QN202118) and the Key Project of Bengbu Medical College (No. 2020Byzd344).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yi S, Zheng J, Lv P, et al. Controlled drug release from cyclodextrin-gated mesoporous silica nanoparticles based on switchable host-guest interactions. Bioconjug Chem. 2018;29(9):2884–2891. doi:10.1021/acs.bioconjchem.8b00416
2. Cheng YJ, Zhang AQ, Hu JJ, He F, Zeng X, Zhang XZ. Multifunctional peptide-amphiphile end-capped mesoporous silica nanoparticles for tumor targeting drug delivery. ACS Appl Mater Interfaces. 2017;9(3):2093–2103. doi:10.1021/acsami.6b12647
3. Shen Y, Li M, Liu T, et al. A dual-functional HER2 aptamer-conjugated, pH-activated mesoporous silica nanocarrier-based drug delivery system provides in vitro synergistic cytotoxicity in HER2-positive breast cancer cells. Int J Nanomedicine. 2019;31(14):4029–4044. doi:10.2147/IJN.S201688
4. Luo W, Wen G, Yang L, et al. Dual-targeted and pH-sensitive doxorubicin prodrug-microbubble complex with ultrasound for tumor treatment. Theranostics. 2017;7(2):452–465. doi:10.7150/thno.16677
5. Keum DH, Mun JH, Hwang BW, et al. Smart microbubble eluting theranostic stent for noninvasive ultrasound imaging and prevention of restenosis. Small. 2017;13(10):1602925. doi:10.1002/smll.201602925
6. Chertok B, Langer R, Anderson DG. Spatial control of gene expression by nanocarriers using heparin masking and ultrasound-targeted microbubble destruction. ACS Nano. 2016;10(8):7267–7278. doi:10.1021/acsnano.6b01199
7. Burns PN, Wilson SR, Simpson DH. Pulse inversion imaging of liver blood flow: improved method for characterizing focal masses with microbubble contrast. Invest Radiol. 2000;35(1):58–71. doi:10.1097/00004424-200001000-00007
8. Karimi M, Zangabad PS, Mehdizadeh F, et al. Nanocaged platforms: modification, drug delivery and nanotoxicity. Opening synthetic cages to release the tiger. Nanoscale. 2017;9(4):1356–1392. doi:10.1039/c6nr07315h
9. Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–2663. doi:10.1021/acs.chemrev.5b00346
10. Xue L, Maihle NJ, Yu X, Tang SC, Liu HY. Synergistic targeting HER2 and EGFR with bivalent aptamer-siRNA chimera efficiently inhibits HER2-positive tumor growth. Mol Pharm. 2018;15(11):4801–4813. doi:10.1021/acs.molpharmaceut.8b00388
11. Cao Y, Liu F, Chen Y, et al. Drug release from core-shell PVA/silk fibroin nanoparticles fabricated by one-step electrospraying. Sci Rep. 2017;7(1):11913. doi:10.1038/s41598-017-12351-1
12. Xu H, Zhang D, Li J. Antibacterial nanoparticles with universal adhesion function based on dopamine and eugenol. J Bioresources Bioproducts. 2019;4(3):177–182.
13. Huang H, Mao L, Li Z, et al. Multifunctional polypyrrole-silver coated layered double hydroxides embedded into a biodegradable polymer matrix for enhanced antibacterial and gas barrier properties. J Bioresources Bioproducts. 2019;4(4):231–241.
14. Sanchez-Moreno P, Ortega-Vinuesa JL, Peula-Garcia JM, Marchal JA, Boulaiz H. Smart drug-delivery systems for cancer nanotherapy. Curr Drug Targets. 2018;19(4):339–359. doi:10.2174/1389450117666160527142544
15. Shahbaz SK, Foroughi F, Soltaninezhad E, Jamialahmadi T, Penson PE, Sahebkar A. Application of PLGA nano/microparticle delivery systems for immunomodulation and prevention of allotransplant rejection. Expert Opin Drug Deliv. 2020;17(6):767–780. doi:10.1080/17425247.2020.1748006
16. Zhang Y, Chang YQ, Han L, et al. Aptamer-anchored di-polymer shell-capped mesoporous carbon as a drug carrier for bi-trigger targeted drug delivery. J Mater Chem B. 2017;5(33):6882–6889. doi:10.1039/C7TB01528C
17. Chen Z, Sun M, Luo F, Xu K, Lin Z, Zhang L. Stimulus-response click chemistry based aptamer-functionalized mesoporous silica nanoparticles for fluorescence detection of thrombin. Talanta. 2018;178:563–568. doi:10.1016/j.talanta.2017.09.043
18. Zhang Y, Jiang S, Zhang D, Bai X, Hecht SM, Chen S. DNA-affibody nanoparticles for inhibiting breast cancer cells overexpressing HER2. Chem Commun (Camb). 2017;53(3):573–576. doi:10.1039/C6CC08495H
19. Shao F, Zhang L, Jiao L, et al. Enzyme-free immunosorbent assay of prostate specific antigen amplified by releasing pH indicator molecules entrapped in mesoporous silica nanoparticles. Anal Chem. 2018;90(14):8673–8679. doi:10.1021/acs.analchem.8b02019
20. Xie Y, Bagby TR, Cohen MS, Forrest ML. Drug delivery to the lymphatic system: importance in future cancer diagnosis and therapies. Expert Opin Drug Deliv. 2009;6(8):785–792. doi:10.1517/17425240903085128
21. Wu J, Meng JY, Du Y, et al. RACK1 promotes the proliferation, migration and invasion capacity of mouse hepatocellular carcinoma cell line in vitro probably by PI3K/Rac1 signaling pathway. Biomed Pharmacother. 2013;67(5):313–319. doi:10.1016/j.biopha.2013.01.011
22. Serrels B, Sandilands E, Serrels A, et al. A complex between FAK, RACK1, and PDE4D5 controls spreading initiation and cancer cell polarity. Curr Biol. 2010;20(12):1086–1092. doi:10.1016/j.cub.2010.04.042
23. Chan MW, Arora PD, Bozavikov P, McCulloch CA. FAK, PIP5KIgamma and gelsolin cooperatively mediate force-induced expression of alpha-smooth muscle actin. J Cell Sci. 2009;122(Pt 5):2769–2781. doi:10.1242/jcs.044008
24. Qazi AS, Sun M, Huang Y, Wei Y, Tang J. Subcellular proteomics: determination of specific location and expression levels of lymphatic metastasis associated proteins in hepatocellular carcinoma by subcellular fractionation. Biomed Pharmacother. 2011;65(6):407–416. doi:10.1016/j.biopha.2011.04.028
25. Shieh DB, Godleski J, Herndon JE, et al. Cell motility as a prognostic factor in Stage I nonsmall cell lung carcinoma: the role of gelsolin expression. Cancer. 1999;85(1):47–57. doi:10.1002/(SICI)1097-0142(19990101)85:1<47::AID-CNCR7>3.0.CO;2-L
26. Silacci P, Mazzolai L, Gauci C, Stergiopulos N, Yin HL, Hayoz D. Gelsolin superfamily proteins: key regulators of cellular functions. Cell Mol Life Sci. 2004;61(19–20):2614–2623. doi:10.1007/s00018-004-4225-6
27. Van Den Abbeele A, De Corte V, Van Impe K, et al. Downregulation of gelsolin family proteins counteracts cancer cell invasion in vitro. Cancer Lett. 2007;255(1):57–70. doi:10.1016/j.canlet.2007.03.023
28. Zhou Y, Wang Z, Chen Y, et al. Microbubbles from gas-generating perfluorohexane nanoemulsions for targeted temperature-sensitive ultrasonography and synergistic HIFU ablation of tumors. Adv Mater. 2013;25(30):4123–4130. doi:10.1002/adma.201301655
29. Wang R, Zhou Y, Zhang P, et al. Phase-transitional Fe3O4/perfluorohexane microspheres for magnetic droplet vaporization. Theranostics. 2017;7(4):846–854. doi:10.7150/thno.17251
30. Rapoport N, Gao Z, Kennedy A. Multifunctional nanoparticles for combining ultrasonic tumor imaging and targeted chemotherapy. J Natl Cancer Inst. 2007;99(14):1095–1106. doi:10.1093/jnci/djm043
31. Wilson K, Homan K, Emelianov S. Biomedical photoacoustics beyond thermal expansion using triggered nanodroplet vaporization for contrast-enhanced imaging. Nat Commun. 2012;3:618. doi:10.1038/ncomms1627
32. Zhao YZ, Zhang M, Wong HL, et al. Prevent diabetic cardiomyopathy in diabetic rats by combined therapy of aFGF-loaded nanoparticles and ultrasound-targeted microbubble destruction technique. J Control Release. 2016;223:11–21. doi:10.1016/j.jconrel.2015.12.030
33. Chen S, Grayburn PA. Ultrasound-targeted microbubble destruction for cardiac gene delivery. Methods Mol Biol. 2017;1521:205–218.
34. Song Y, Huang Z, Xu J, et al. Multimodal SPION-CREKA peptide based agents for molecular imaging of microthrombus in a rat myocardial ischemia-reperfusion model. Biomaterials. 2014;35(9):2961–2970. doi:10.1016/j.biomaterials.2013.12.038
35. Peña-Bahamonde J, Nguyen HN, Fanourakis SK, Rodrigues DF. Recent advances in graphene-based biosensor technology with applications in life sciences. J Nanobiotechnology. 2018;16(1):75. doi:10.1186/s12951-018-0400-z
36. Liu J, Pu H, Liu S, Kan J, Jin C. Synthesis, characterization, bioactivity and potential application of phenolic acid grafted chitosan: a review. Carbohydr Polym. 2017;174:999–1017. doi:10.1016/j.carbpol.2017.07.014
37. Rapoport NY, Kennedy AM, Shea JE, Scaife CL, Nam KH. Controlled and targeted tumor chemotherapy by ultrasound-activated nanoemulsions/microbubbles. J Control Release. 2009;138(3):268–276. doi:10.1016/j.jconrel.2009.05.026
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.