Back to Journals » Drug Design, Development and Therapy » Volume 15
Drug Discovery and Development in Rare Diseases: Taking a Closer Look at the Tafamidis Story
Authors Burton A, Castaño A, Bruno M, Riley S, Schumacher J, Sultan MB, See Tai S, Judge DP, Patel JK, Kelly JW
Received 31 October 2020
Accepted for publication 20 January 2021
Published 18 March 2021 Volume 2021:15 Pages 1225—1243
DOI https://doi.org/10.2147/DDDT.S289772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Arianna Burton,1 Adam Castaño,1 Marianna Bruno,1 Steve Riley,2 Jennifer Schumacher,1 Marla B Sultan,3 Sandi See Tai,4 Daniel P Judge,5 Jignesh K Patel,6 Jeffery W Kelly7
1Medical Affairs, Pfizer Inc, New York, NY, USA; 2Clinical Pharmacology, Pfizer Inc, Groton, CT, USA; 3Global Product Development, Pfizer Inc, New York, NY, USA; 4Global Product Development, Pfizer Inc, Collegeville, PA, USA; 5Department of Medicine, Medical University of South Carolina, Charleston, SC, USA; 6Department of Cardiology, Cedars-Sinai Smidt Heart Institute, Los Angeles, CA, USA; 7Department of Chemistry, The Scripps Research Institute, La Jolla, CA, USA
Correspondence: Adam Castaño Email [email protected]
Abstract: Rare diseases are increasingly recognized as a global public health priority. Governments worldwide currently provide important incentives to stimulate the discovery and development of orphan drugs for the treatment of these conditions, but substantial scientific, clinical, and regulatory challenges remain. Tafamidis is a first-in-class, disease-modifying transthyretin (TTR) kinetic stabilizer that represents a major breakthrough in the treatment of transthyretin amyloidosis (ATTR amyloidosis). ATTR amyloidosis is a rare, progressive, and fatal systemic disorder caused by aggregation of misfolded TTR and extracellular deposition of amyloid fibrils in various tissues and organs, including the heart and nervous systems. In this review, we present the successful development of tafamidis spanning 3 decades, marked by meticulous laboratory research into disease mechanisms and natural history, and innovative clinical study design and implementation. These efforts established the safety and efficacy profile of tafamidis, leading to its regulatory approval, and enabled post-approval initiatives that further support patients with ATTR amyloidosis.
Keywords: amyloidosis, cardiomyopathy, heart failure, transthyretin, treatment, Vyndaqel/Vyndamax
Introduction
Rare diseases have an extensive impact on public health, despite their relatively low prevalence, and development of safe and effective treatments for rare disease is increasingly recognized as a global priority.1–3 Rare diseases are designated as conditions occurring in fewer than 200,000 people in the USA4 and 5 per 10,000 people in the EU.5 Development of new therapies for rare diseases poses unique challenges, particularly from a clinical and regulatory perspective. To help mitigate the inherent difficulties, governments worldwide have enacted legislation to stimulate research and investment for orphan drugs targeting rare diseases.6,7 Nonetheless, orphan drug discovery and development remains a daunting, risk-laden process, requiring substantial financial resources, incisive decision-making, and ingenuity to overcome obstacles related chiefly to minimal and, in some instances, lack of precedence in trial design and previously defined endpoints, small patient populations, and limited natural history of disease information.8 Despite improvements in rare disease awareness, knowledge of pathophysiology, and legislative support, 95% of rare diseases do not have an approved treatment available in the USA.9 Among the orphan drugs identified and investigated to date, only a small fraction successfully emerge from rigorous testing in preclinical and clinical studies to gain regulatory approval each year.
The introduction of tafamidis, a first-in-class transthyretin (TTR) kinetic stabilizer, marked a critical breakthrough in the pharmacologic treatment of transthyretin amyloidosis (ATTR amyloidosis) with cardiomyopathy (ATTR-CM) or polyneuropathy (ATTR-PN). This rare systemic disorder is characterized by the extracellular aggregation of native and/or variant TTR into several distinct structures, including amyloid fibrils, which deposit in various tissues and organs.10 The discovery and development of tafamidis for ATTR amyloidosis relied on a combination of meticulous laboratory investigation into the biological and chemical mechanisms underlying TTR aggregation, an innovative clinical development program, and determination and perseverance to support development of a treatment for patients with this rare disease.11
In this review, we highlight challenges associated with drug development in rare diseases and describe the translational success story of tafamidis. We identify research milestones that led to the development of the TTR kinetic stabilizer and its rational structure-based design. We summarize the essential findings of the pivotal tafamidis clinical trials that established the safety and efficacy profile of the drug and resulted in its regulatory approval for the treatment of ATTR-PN outside the US, and more recently, ATTR-CM in the US and globally. Finally, we present the ongoing work and challenges associated with rare disease amelioration in a population-driven health system.
Drug Development in Rare Diseases
Rare diseases such as ATTR amyloidosis often lead to premature death, have a debilitating impact on patients’ physical functioning, ability to work, and quality of life (QoL), and place a heavy emotional and financial burden on families.12,13 Historically, therapeutic advances in rare diseases have been impeded by a poor understanding of their pathophysiology and natural history, and the high cost and complexity of conducting clinical investigations for diseases that affect small populations.14
Advances
Legislative and Regulatory Incentives
Nearly 40 years ago, the US Congress passed the Orphan Drug Act to encourage drug/biologic development for the treatment of rare diseases.12 Subsequently, other governments worldwide similarly granted greater priority to rare disease investigation and the development of orphan drugs.15 Moreover, drug regulatory authorities in some countries have established designations (eg, fast-track and breakthrough therapy designations in the USA; Sakigake status in Japan) that allow for expedited development and review of select medications used for life-threatening diseases with an unmet medical need that demonstrate substantial benefit over existing therapies early in their clinical development. In the USA and elsewhere, tafamidis was granted orphan drug designation as well as fast-track and breakthrough therapy designations, and priority review, expediting the regulatory submission, review, and approval for this novel agent.
Obstacles
Despite the regulatory programs intended to incentivize and support new treatments for rare diseases, many challenges remain in both the preclinical and clinical phases of rare disease research.12
Limited Knowledge Base
Identification and characterization of appropriate in vitro and/or animal models for preclinical studies may be hindered by limited knowledge of disease pathophysiology and/or genetics. For clinical research, limited information and a poor understanding of the natural history of the disease makes selection of study endpoints difficult, and often impedes identification of the patient population best suited for assessment. These obstacles undoubtedly had an impact on the development of tafamidis, as the natural progression of ATTR amyloidosis and contributing environmental and genetic factors were not well understood when its clinical trial program was initiated.
Low Clinical Trial Enrollment
According to research conducted by the Tufts Center for the Study of Drug Development (CSDD), patient recruitment for rare disease clinical trials is more challenging than for non-rare disease clinical trials due to substantially higher rates of screening and randomization failure.16 Approximately 81% of screened patients were ineligible to enroll and 56% failed to be randomized in rare disease clinical trials, versus 57% and 36% of patients that fell into the categories in non-rare disease trials.
Several factors can hamper recruitment of patients for randomized controlled clinical trials of orphan drugs, including the small and widely dispersed patient populations affected, and disease heterogeneity. Patients with rare diseases frequently receive various misdiagnoses over several years before a correct diagnosis is determined, increasing their disease burden and rendering some patients ineligible for clinical studies because of advanced disease.17,18 Due to the low prevalence of rare diseases, a greater number of study sites are often needed globally to ensure adequate enrollment of even small numbers of patients, which may require additional support measures to reduce the burden of participation, eg, support for travel and/or lodging—all investments made without guarantee of success. Finally, there may be competing clinical trials for the same rare disease patients, limiting an already small number of patients eligible to enroll.
Establishing a Suitable Trial Design
Designing trials with clinically appropriate methodology, including diagnostic procedures, dose(s), safety/efficacy endpoints, and a statistical analysis plan, is also particularly challenging for rare diseases. Determining appropriate diagnostic criteria for study eligibility and endpoints/outcomes for measuring treatment response for rare disease research is often complicated by the absence of accepted guidelines, validated prognostic factors, or indicators of disease progression. Limited patient numbers often preclude conduct of dose-ranging studies to characterize dose–response relationships useful for dose selection in future studies. In addition, efficacy endpoints must be selected that are clinically meaningful and in compliance with regulatory requirements for product approval; validated efficacy endpoints are not usually available for rare diseases. These endpoints may require analysis using innovative statistical methodology to compensate for the inclusion of relatively small patient populations. Maurer et al used the novel Finkelstein–Schoenfeld statistical method to analyze data for the primary endpoint in the pivotal tafamidis clinical trial in ATTR-CM to enable a robust analysis with the study’s relatively small sample size (detailed below).19
Drug Development Cost
The economic burden associated with rare disease drug development is an additional possible deterrent, but few cost studies have been conducted. In one report, the clinical costs associated with developing orphan drugs were estimated to be half those associated with non-orphan drugs.20 Using publicly available drug cost data, Jayasundara et al estimated that the out-of-pocket and capitalized (10.5% per year over the clinical phase testing and regulatory approval period) clinical costs of each approved orphan drug were $166 million and $291 million in 2013 US dollars (or $202 and $354 million in 2020 US dollars), respectively.20 However, the investigators acknowledged that additional research is needed to address factors that may substantially increase these costs, such as longer development timelines and lower probability of success for orphan drugs. Based on the Tufts CSDD research, the overall clinical and regulatory duration (ie, investigational new drug application to approval decision) for rare diseases is approximately 4 years longer than that for all diseases.16 The prolonged timelines associated with orphan drug development can potentially increase costs.
Systemic ATTR Amyloidosis: An Overview
ATTR amyloidosis is a rare, progressive, fatal disease caused by TTR misfolding, and aggregation and deposition of amyloid fibrils in the extracellular space of many tissues and organs, leading to end-organ degeneration. Strong genetic and pharmacologic evidence supports the amyloid hypothesis, ie, that TTR aggregation causes the death of post-mitotic tissue and is a critical triggering factor in the pathogenesis of ATTR amyloidosis–based degenerative diseases.21–23 The pathogenesis of ATTR amyloidosis is associated with the destabilization of the tetrameric TTR protein. Tetramer dissociation is the rate-limiting step in TTR aggregation, which also requires subsequent monomer misfolding for misassembly to produce soluble misfolded TTR aggregates and insoluble aggregates, including amyloid fibrils (Figure 1).
 |
Figure 1 The TTR amyloidogenic cascade. When destabilized by TTR pathogenic variants, the tetrameric protein TTR dissociates into monomers that misfold and misassemble into aggregates, including amyloid fibrils associated with transthyretin amyloidosis. Abbreviation: TTR, transthyretin. |
In patients with autosomal dominant hereditary (or variant) ATTR amyloidosis (ATTRv), TTR tetramer destabilization is caused by the incorporation of pathogenic variant subunits into TTR heterotetramers, otherwise composed of wild-type TTR subunits. More than 150 TTR gene variants have been linked to amyloidosis in ATTRv, with varying degrees of penetrance and different tissue tropism.24–26 Depending on the variant, ATTRv can present as a principal cardiomyopathy (ATTR-CM), a primary sensory, motor, and autonomic polyneuropathy (ATTR-PN), or a mixed phenotype that has cardiac and neurologic manifestations.27,28 In patients with wild-type ATTR amyloidosis (ATTRwt), aging-associated tetramer dissociation, misfolding, and aggregation of TTR occurs in the absence of a genetic variant.29 Other genetic and/or environmental contributors to ATTRwt remain poorly understood, as does the tissue tropism. ATTRwt typically affects the heart and autonomic nervous system, particularly in older males, increasing the risk of cardiomyopathy, diastolic dysfunction, arrhythmias, and heart failure, but may also cause damage in other tissues/organs.
Although ATTR amyloidosis has been considered a rare disease, growing evidence suggests that the disease (particularly ATTRwt) may be more common than previously recognized. The prevalence of ATTRv varies by region due to allele frequencies that fluctuate by geographic distribution. Based on a comprehensive literature review, Schmidt et al estimated that hereditary ATTR-PN has a global prevalence of 10,186 persons (range, 5526–38,468).30 In two recent cohort studies, the reported prevalence of the Val122Ile (p.Val142Ile) pathogenic variant, the most common in the USA, was approximately 3% among African Americans,31,32 but disease penetrance is still unknown.33 Importantly, this genetic variant was found to be significantly associated with heart failure in carriers of either African or Hispanic/Latino ancestry.32 Regions with endemic ATTRv have been reported in Japan, Portugal, Sweden, Cyprus, and Majorca, but the disease is now documented in 29 countries worldwide, including China, India, parts of Europe, and the USA.34
The prevalence of ATTRwt, the most common form of ATTR amyloidosis and an increasingly recognized cause of heart failure, has been difficult to establish. In a study of patients >60 years of age who had heart failure with preserved left ventricular ejection fraction and left ventricular wall thickness ≥12 mm, ATTRwt was identified by radionuclide scintigraphy in approximately 13% of screened patients.35 Based on prospectively assessed myocardial biopsies conducted in patients with heart failure with preserved ejection fraction, Hahn et al recently reported a prevalence of ATTR cardiac amyloidosis of 10%, with ATTRwt observed in ~7%.36 Recently, in a cohort of patients with heart failure and increased myocardial wall thickness (intraventricular septum >14 mm), approximately 20% of patients had ATTRwt when screened with radionuclide imaging.37 In autopsy studies, the reported frequency of ATTRwt deposits has ranged from 10% to 25% of elderly patients.38–41
In clinical practice, both hereditary and sporadic forms of ATTR amyloidosis are challenging to identify, primarily due to the wide range of possible symptoms that potentially involve multiple organs.42 ATTR-CM has been shown to be frequently misdiagnosed, or its diagnosis delayed until late in the disease course, because of low disease awareness/misperceptions of the disease and clinical features that overlap with more common etiologies of heart failure.17,43 The expectancy that ATTR-CM predominantly occurs in men may reduce clinicians’ suspicion of the disease in women. In a recent literature review,44 among over 4500 patients with ATTR-CM, 17% were women; the proportions of women with variant ATTR-CM (29%) and undefined disease (18%) were higher than that of women with wild-type ATTR-CM (9%). Importantly, in the referenced review, some cardiac structural/functional differences, including lower interventricular septal and posterior wall thickness and higher left ventricular ejection fractions, were observed in women versus men across all ATTR-CM subtypes. Left ventricular wall thickness >12 mm is a recommended threshold for ATTR-CM diagnosis in both sexes, suggesting that women may be underdiagnosed as a result of their smaller cardiac anatomy. Additional research is warranted to better elucidate these differences and inform the identification of these patients in clinical practice.
The natural progression of ATTR amyloidosis is also not well documented. Nearly a decade ago, Ruberg et al prospectively evaluated morbidity and mortality in 29 patients with wild-type and Val122lle (p.Val142Ile) variant ATTR-CM in the Transthyretin Amyloidosis Cardiac Study (TRACS).45 They found no differences between patients with ATTRwt and ATTRv in clinical characteristics, biomarker levels, or echocardiographic variables at baseline. However, after approximately 16 months, patients with ATTRwt had significantly lower rates of mortality (22% vs 73%) and cardiovascular hospitalization (28% vs 64%) than patients with ATTRv. In patients with ATTRwt and ATTRv, median survival from diagnosis was approximately 3.6 and 2.2 years, respectively. Similarly, in untreated patients with ATTRwt and ATTRv cardiomyopathy, median overall survival was recently estimated to be 3.6 and 2.5 years, respectively, after diagnosis.46,47
Until the introduction of tafamidis, treatment options available for ATTR amyloidosis were limited to symptomatic management or liver and/or heart transplantation. Because the liver produces more than 90% of TTR circulating in the bloodstream,48 liver transplantation is a disease-modifying treatment for ATTRv. Liver transplantation is effective at slowing disease progression and improving survival in patients with the inherited form of the disease, particularly when performed in those with early-onset Val30Met (p.Val50Met) hereditary ATTR-PN.49–52 However, in a subset of patients with ATTRv with both cardiac and neurologic manifestations, this surgical approach failed to prevent the progression of amyloid cardiomyopathy, potentially because circulating wild-type TTR continues to be deposited on existing variant TTR substrate, even in the absence of circulating variant TTR.53,54 Liver transplantation is also associated with a first-year mortality rate of approximately 10%, due to vascular fragility in patients with ATTR amyloidosis, and the risk of complications in transplant patients overall and substantial chronic immunosuppression-related morbidity.50,52,55
Patients with ATTR amyloidosis involving severe cardiomyopathy may undergo combined liver and heart transplantation or heart transplant alone.56–58 However, as with single-organ transplant, the benefits of liver/heart surgery can be offset by the risk of perioperative morbidity and mortality and the adverse effects of chronic immunosuppression.50–52 As a result, heart transplantation, with or without combined liver transplantation, is generally considered a viable option primarily for selected patients with advanced ATTR-CM.33,59 The limitations of organ transplantation and the scarcity of donor organs highlighted the need for development of pharmacologic therapies that block the amyloid cascade in patients with ATTR amyloidosis, and the need for early diagnosis and treatment to prevent advanced refractory disease.
Research Milestones in the Development of TTR Kinetic Stabilizers
Findings from a broad network of studies played a critical role in the development of small molecules such as tafamidis designed to halt the progression of ATTR amyloidosis (Figure 2).19,60–68 TTR (formerly called prealbumin) is a tetrameric protein that transports holo-retinol binding protein in the bloodstream and is a minor carrier of the thyroid hormone thyroxine (T4) in the blood.69–71 In humans, most thyroid hormone is transported by albumin or thyroid-binding globulin in the blood, which leaves most T4-binding sites on TTR unoccupied.72,73 Although TTR is the major T4 carrier in the brain, most of the TTR tetramer is unliganded in the brain due to the T4 to TTR tetramer stoichiometry. These observations were important to the development of kinetic-stabilizing agents capable of binding to TTR at the unoccupied T4 binding sites.
 |
Figure 2 Timeline of the major translational milestones in the development of the TTR kinetic stabilizer tafamidis. Superscript numbers refer to references. Abbreviations: ATTR-ACT, Tafamidis in Transthyretin Cardiomyopathy Clinical Trial; ATTR, amyloidosis, transthyretin amyloidosis; ATTR-CM, transthyretin amyloid cardiomyopathy; ATTR-PN, transthyretin amyloid polyneuropathy; qd, once daily; THAOS, Transthyretin Amyloidosis Outcomes Survey; TTR, transthyretin. |
In Portuguese families, in the early 1990s, Coelho et al observed that some carriers of the Val30Met (p.Val50Met) pathogenic variant either had no symptoms of ATTR-PN or had uncharacteristically mild symptoms.74 Gene sequencing studies revealed that these individuals were compound heterozygotes expressing the disease-causing Val30Met (p.Val50Met) variant on one allele and a Thr119Met (p.Thr139Met) variant on the second allele (instead of wild-type TTR), conferring protection against ATTR-PN.74 Incorporation of Thr119Met (p.Thr139Met) subunits into tetramers otherwise comprised of disease-associated subunits increased the kinetic stability of the tetramer, which slowed tetramer dissociation and suppressed disease.61,62,74,75 It was subsequently discovered that the dissociation barrier (ie, the energy required for the formation of misfolded monomers) increased proportionally with each Thr119Met (p.Thr139Met) subunit incorporated into a tetramer, demonstrating that increasing the kinetic barrier associated with misfolding can stabilize the native tetramer state and slow or prevent TTR aggregation, thus ameliorating ATTR amyloidosis.76
Based on a thorough investigation into the mechanisms underlying TTR aggregation, researchers were able to establish that dissociation of the native TTR tetramer was the slowest and first step of the aggregation cascade or process.61,62,76–82 The observed efficiency of TTR aggregation after TTR monomer misfolding also helped focus drug discovery on blocking TTR aggregation before rate-limiting tetramer dissociation.83 This milestone led to the hypothesis that small-molecule ligand stabilization could prevent TTR dissociation into monomers and thereby block all TTR misfolding and aggregation, including amyloid fibril formation. Thus, the concept of TTR small-molecule “kinetic stabilizers” emerged, ie, small molecules capable of selectively binding to native TTR tetramer over the dissociative transition state, slowing tetramer dissociation dramatically, to inhibit TTR aggregation. Importantly, the kinetic stabilizer strategy is conservative, in that its clinical success does not require an understanding of which aggregate structure(s) are driving the degenerative phenotypes, because kinetic stabilizer treatment reduces the concentration of all misfolded TTR structures and aggregates derived from newly synthesized TTR.
Drug Design
Based on the kinetic stabilization observations, a strong rationale was constructed for the development of small-molecule kinetic stabilizers that bind to the largely unoccupied T4 binding pockets and stabilize the native TTR tetramer as potential pharmacotherapy to prevent the progression of ATTR amyloidogenesis.76 Structure-based design and biochemical assays were used to iteratively improve the potency of the kinetic stabilizers in several structurally distinct small-molecule families or chemotypes that inhibited TTR aggregation by kinetically stabilizing the tetrameric energetic ground state of TTR.62,76,84,85 The prioritization of candidate kinetic stabilizers was based on assessments of stabilizer performance under denaturing conditions,62 TTR aggregation inhibition as a function of kinetic stabilizer concentration under acidic conditions,76,78,79,84 and the degree of selective binding of the kinetic stabilizer to TTR in human plasma.86
Candidate TTR kinetic stabilizers from repurposing efforts included several nonsteroidal anti-inflammatory drugs (NSAIDs), such as flufenamic acid, diflunisal, meclofenamic acid, and mefenamic acid.68,85,87 However, NSAID activity was considered potentially detrimental in candidate TTR kinetic stabilizers because cyclooxygenase inhibition should be avoided in patients with ATTR-CM, who often have renal blood flow impairment.85 In addition, the risk of serious gastrointestinal, renal, and cardiac toxicity with chronic use of NSAIDs, particularly in aging populations with other medical conditions, weighed against their development for the treatment of ATTR amyloidosis.88
Medicinal chemistry and structure-activity data on novel kinetic stabilizer chemotypes focused attention on the benzoxazole carboxylic acids. The benzoxazole derivative 2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid, or tafamidis, was identified as an oral candidate capable of substantially slowing the rate-limiting step in the TTR amyloid cascade and was selected for clinical development.89 Tafamidis lacked NSAID activity, and exhibited an acceptable toxicity profile and no thyroid-related metabolic effects.89
Tafamidis Pharmacodynamic/Pharmacokinetic Studies
In nonclinical in vitro and ex vivo experiments, tafamidis bound with high selectivity and affinity, and negative cooperativity, to recombinant wild-type TTR and both wild-type and variant forms of TTR in human plasma.89,90 This compound was shown to dose-dependently kinetically stabilize TTR under denaturing and physiological conditions.85 Tafamidis exhibited comparable potency in inhibiting tetramer dissociation of wild-type native TTR and a broad range of TTR variants, including the two most common amyloidogenic variants, Val30Met (p.Val50Met) and Val122Ile (p.Val142Ile).89
Tafamidis was shown to stabilize TTR variants ex vivo using an immunoturbidometric assay in nonclinical and clinical studies.89 Evidence from nonclinical studies suggested that a tafamidis:TTR stoichiometry of 1–1.5:1 was sufficient to stabilize TTR and reduce TTR tetramer dissociation in human plasma.91 In two Phase 1, randomized, double-blind, ascending dose tolerance studies conducted in healthy volunteers receiving single oral doses of tafamidis (up to 120 mg or 480 mg), increasing plasma tafamidis:TTR stoichiometry was associated with increases in mean percent TTR stabilization.91 In an ex-vivo study, the immunoturbidometric assay was used to compare tetrameric TTR stability in plasma samples from patients with ATTR amyloidosis in the absence or presence of tafamidis.91 Tafamidis increased the stability of the native tetrameric structure of all but 1 of the 27 amyloidogenic, non-Val30Met (p.Val50Met) and non-Val122Ile (p.Val142Ile) TTR variants analyzed. Finally, a >73% reduction in TTR tetramer dissociation rate was observed in patients with ATTRwt treated with tafamidis 20 mg orally once daily versus untreated age-matched controls; in the tafamidis-treated patients, the tafamidis concentration in plasma was strongly correlated with the rate of tetramer dissociation.90
In addition to the ability to bind TTR tetramers with high affinity and selectivity to slow TTR tetramer dissociation, tafamidis needed to demonstrate appropriate pharmacokinetic properties prior to investigation in clinical trials. In clinical investigations conducted in healthy volunteers, tafamidis meglumine 20 mg administered orally, once daily, as soft gelatin capsules exhibited a favorable pharmacokinetic profile.91,92
The Tafamidis Clinical Trial Program
A brief review of the efficacy and safety profile of tafamidis demonstrated in Phase 2 and 3 clinical investigations is provided in this section, with additional information on the studies’ methodology and efficacy outcomes summarized in Table 1.
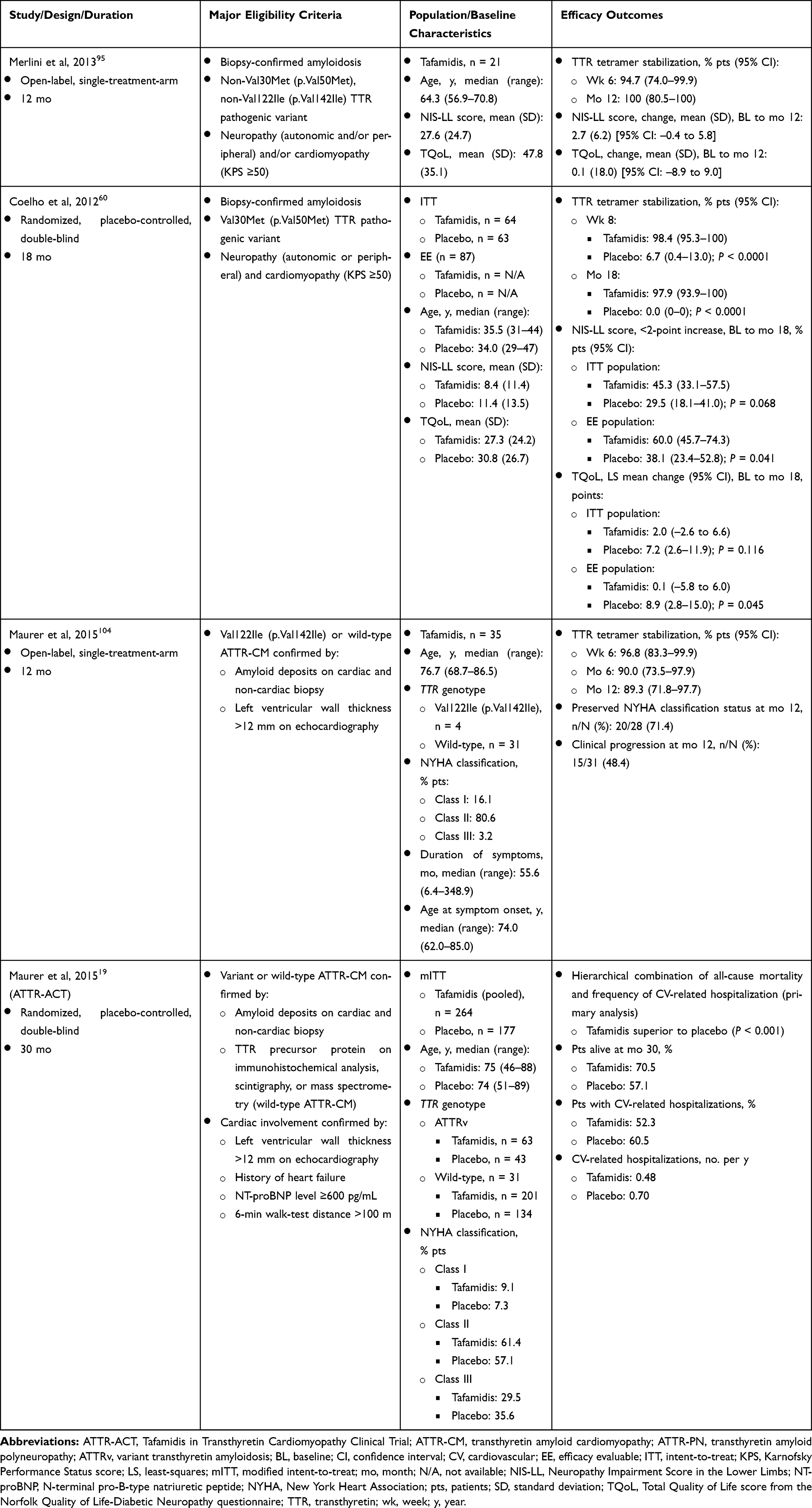 |
Table 1 Summary of Methods and Results from Phase 2/3 Clinical Studies of Tafamidis in Patients with ATTR-PN and ATTR-CM |
Transthyretin Amyloid Polyneuropathy
In 2011, in the EU, tafamidis became the first medication approved for the treatment of patients with ATTR-PN based on the clinical efficacy/safety profile demonstrated in polyneuropathy clinical trials.93 It is currently approved as treatment for this disease in more than 40 countries worldwide.94 A better understanding of the natural history of the polyneuropathy phenotype allowed testing in this population first, while knowledge of ATTR-CM was evolving. Moreover, testing of a 20-mg dose of tafamidis preceded testing of higher doses as more information was obtained on potential dose-dependent effects on TTR tetramer stabilization.
In a Phase 2, open-label, single-treatment-arm study, 21 patients with ATTRv due to non-Val30Met (p.Val50Met) pathogenic variants (excluding Val122Ile [p.Val142Ile]) received tafamidis 20 mg once daily for 12 months.95 TTR was stabilized in 95% of 19 patients with evaluable data after 6 weeks of treatment, and 100% of 17 patients with non-missing data after 12 months. Mean changes in the health-related QoL from baseline to months 6 and 12 were negligible, and overall nutritional status was maintained over the 12-month treatment period, with some variation between months 6 and 12.
In the pivotal double-blind trial (ClinicalTrials.gov identifier: NCT00409175), 128 patients with early-stage Val30Met (p.Val50Met) disease were randomized to receive tafamidis 20 mg or placebo for up to 18 months.60 A significant difference between tafamidis and placebo was not achieved for the coprimary efficacy endpoints in the primary analysis of the intent-to-treat population (Figure 3A, 3B). Based on these results, tafamidis was denied regulatory approval in the USA. However, in this study, patients receiving tafamidis had a significant reduction in neurologic deterioration at both months 12 and 18 compared with patients receiving placebo (Figure 3C). At month 18, tafamidis-treated patients had 52% less neurologic deterioration than patients who received placebo. The latter clinical benefits were sufficient to warrant approval of the drug by regulatory authorities in the EU, Japan, and other countries. The incidences of serious adverse events and adverse events leading to discontinuation from the study were similar in the tafamidis and placebo groups over this time period (Table 2A). It is worth noting that the selection of endpoints for this study, which was among the first randomized controlled studies conducted in a population with this rare disease, was based on the limited natural history information available at the time, including estimates of disease progression and clinically meaningful treatment effects. The outcomes from this study and evolving understanding of the disease substantially informed the design of other ATTR amyloidosis treatment trials.
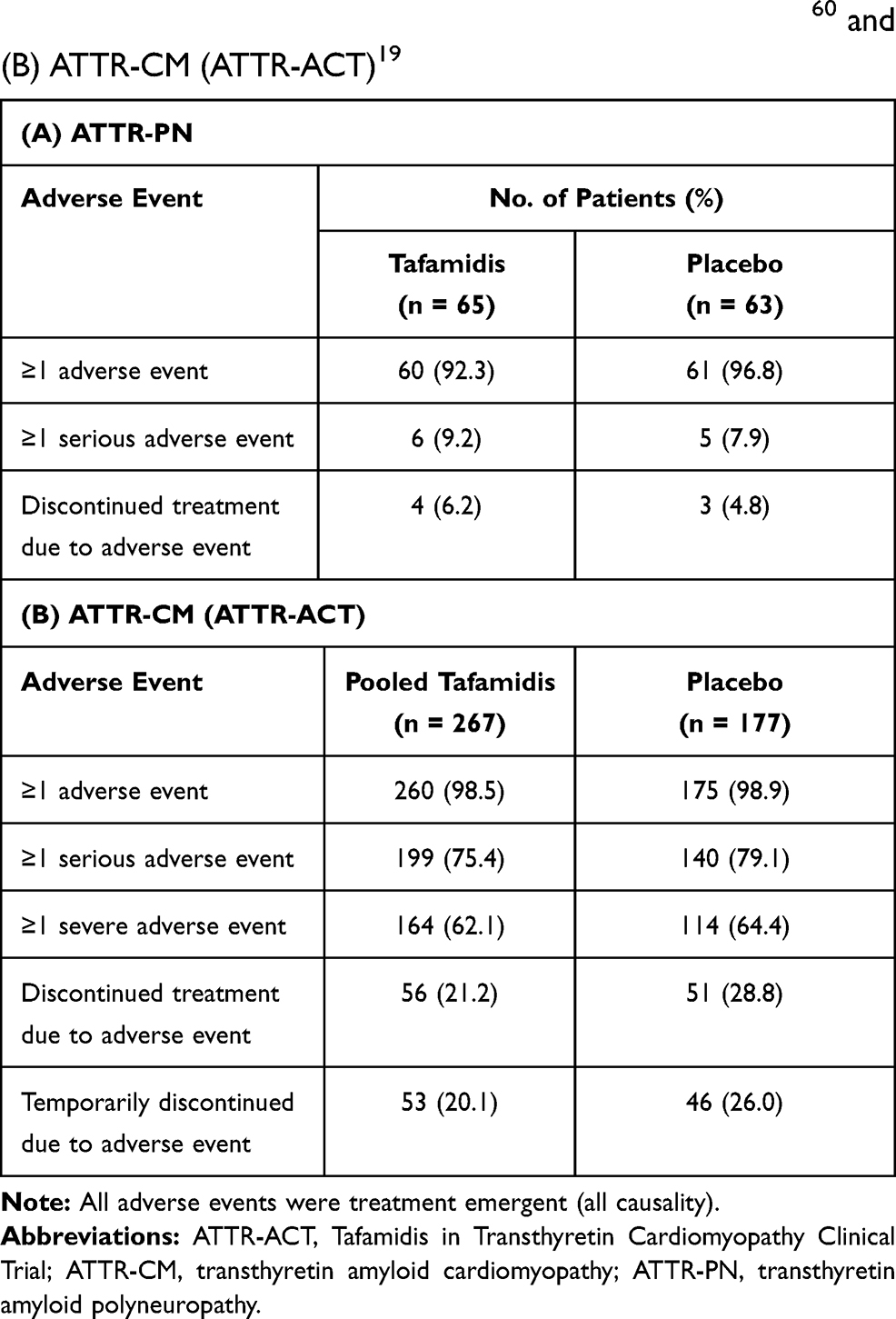 |
Table 2 Adverse Events Reported by Treatment Group in Pivotal Randomized Controlled Trials of Tafamidis in (A) ATTR-PN60 and (B) ATTR-CM (ATTR-ACT)19 |
 |
Figure 3 (A) Proportion of NIS-LL respondersa at month 18 in the ITTb and EE populations. (B) LS mean (±SE) change from baseline to month 18 in the TQoL score from the Norfolk QoL-DN in the ITTb and EE populations. (C) LS mean (±SE) changes from baseline at months 6, 12, and 18 in the NIS-LL in the ITT population.90,97 aResponse = increase of <2 in the NIS-LL overall score. bProportion of NIS-LL responders and LS mean change from baseline in TQoL score at month 18 in the ITT population were coprimary endpoints of the study. Abbreviations: EE, efficacy evaluable; ITT, intent-to-treat; LS, least-squares; NIS-LL, Neuropathy Impairment Score in the Lower Limbs; Norfolk QoL-DN, Norfolk Quality of Life-Diabetic Neuropathy questionnaire; SE, standard error; TQoL, Total Quality of Life score from the Norfolk QoL-DN. |
Patients who continued to receive tafamidis in an open-label extension (ClinicalTrials.gov identifier: NCT00791492) had stable rates of change in scores measuring neurologic function and QoL over 30 months.96 Patients switching from placebo to tafamidis showed significant reductions in neurologic and QoL deterioration in the extension trial, but treatment benefits were not as great as in patients who initiated tafamidis earlier. Adverse event rates were similar in both groups, and no patients died during the extension study or had adverse events that were life-threatening or led to study discontinuation.
Evidence from open-label extension studies providing data from up to 8.5 years of treatment (ClinicalTrials.gov identifiers: NCT00409175, NCT00791492, NCT00630864, and NCT00925002) has further supported the sustained beneficial effects of tafamidis in delaying disease progression and demonstrates tafamidis tolerability in ATTR-PN.98–100 Additionally, evaluation of mortality in patients receiving long-term tafamidis therapy in prospective interventional trials showed approximately 75% and 85% of patients with ATTR-PN caused by non-Val30Met (p.Val50Met) and Val30Met (p.Val50Met) pathogenic variants were alive at 8 and 9 years, respectively, after receiving their first tafamidis dose.99 None of the 11 deaths reported in the latter analysis were related to tafamidis treatment; four of the deaths occurred following liver transplantation.
When comparing survival among patients with Val30Met (p.Val50Met) ATTR-PN treated with tafamidis or undergoing liver transplantation to untreated controls in an observational multihospital cohort study, Coelho et al found that both tafamidis and surgery provided survival benefits compared with natural disease progression.101 Tafamidis was associated with a significant reduction in mortality risk of 63% versus liver transplantation in patients with early-onset disease. In a recent study comparing the effects of tafamidis and liver transplantation in patients with hereditary ATTR-PN, patients receiving treatment with tafamidis had significantly longer overall survival compared with those undergoing surgery.102
Transthyretin Amyloid Cardiomyopathy
Tafamidis is the first pharmacotherapy approved to treat patients with wild-type or hereditary ATTR-CM, with approval in the USA and Japan received in 2019, and the EU in 2020. Currently available for the treatment of ATTR-CM in nearly 50 countries worldwide, tafamidis is the only pharmacotherapy shown to reduce all-cause mortality and cardiovascular-related hospitalizations in patients with ATTR-CM.103
Evidence of its safety and efficacy in ATTR-CM was provided in Phase 2 and 3 clinical trials. In a post hoc analysis of findings from the aforementioned Phase 2, open-label ATTR-PN study,95 Damy et al assessed the efficacy and safety of tafamidis 20 mg daily, administered for 12 months, on cardiac structure and function in 21 patients with non-Val30Met (p.Val50Met) and non-Val122Ile (p.Val142Ile) disease.104 At baseline, a majority of patients presented with signs/symptoms of cardiac involvement, including New York Heart Association (NYHA) class II heart failure (n = 11), conduction abnormalities (n = 13), N-terminal pro-B-type natriuretic peptide (NT-proBNP) >300 pg/mL (n = 14), and/or interventricular septal thickness >12 mm (n = 17); mean left ventricular ejection fraction was 60.3% in this study population. During the 12-month tafamidis treatment period, patients had no clinically meaningful deterioration in conduction, left ventricular thickness, or left ventricular systolic or diastolic function, and NT-proBNP levels were stable. Moreover, echocardiographic and electrocardiographic findings showed no clinically relevant changes with tafamidis over time. The cardiac findings from this Phase 2 tafamidis trial in patients with ATTR-PN provided critical information and momentum for the development of additional ATTR-CM studies.
In a Phase 2, open-label trial conducted in 35 patients with early-stage wild-type ATTR-CM (ClinicalTrials.gov identifier: NCT00694161), tafamidis 20 mg daily was administered for 12 months to evaluate its safety and efficacy in altering disease progression.105,106 This was the first clinical assessment of tafamidis in patients with ATTR amyloidosis nearly exclusively of the cardiac phenotype. Patients had a median disease duration of 56 months, and 97% had mild-to-moderate heart failure (NYHA class I–II). During the treatment period, 89% of patients achieved TTR stabilization and 71% had preserved NYHA functional status. After 12 months, 10% had discontinued tafamidis prematurely, 6% died, 23% were hospitalized due to cardiovascular events, and 48% demonstrated clinical progression. Tafamidis was generally well tolerated, with no significant increase in NT-proBNP or clinically relevant changes in echocardiographic findings observed.
To further evaluate the effect of tafamidis on survival in earlier-stage ATTR-CM, Sultan et al compared survival data from tafamidis-treated patients in the above-mentioned open-label trial105 with those from untreated patients with ATTR-CM in the TRACS population,45 excluding patients with NYHA class ≥III to ensure patients had comparable baseline disease severity.106 In this post hoc analysis, patients with either wild-type or Val122Ile (p.Val142lle) genotypes treated with tafamidis had a significantly longer time to death than untreated patients. These encouraging findings suggested the need for further exploration of tafamidis in a larger controlled clinical study of patients with ATTR-CM.
In the pivotal, randomized controlled Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT; ClinicalTrials.gov identifier: NCT01994889), Maurer et al compared the safety and efficacy of tafamidis (20 mg and 80 mg daily) versus placebo after 30 months of treatment in 441 patients with wild-type and variant ATTR-CM.19 Investigation of the effects of the tafamidis 80 mg dose in this study was supported by two salient findings from previous studies conducted in healthy volunteers: first, the 80 mg dose was shown to provide near maximal TTR stabilization;91 and second, a supratherapeutic dose of tafamidis 400 mg was shown to be well tolerated.107 The 20 mg and 80 mg tafamidis dose groups were pooled and compared with placebo for analysis of primary and secondary efficacy endpoints. This pooling of the active treatment groups was conducted in agreement with US and EU regulatory authorities to address the small number of patients with this rare disease.
The primary analysis from ATTR-ACT utilized the Finkelstein–Schoenfeld statistical method, which demonstrated that tafamidis treatment was superior to placebo in reducing the hierarchical combination of all-cause mortality and frequency of cardiovascular-related hospitalization over 30 months (Figure 4). This statistical method was selected because it increases the power and sensitivity of analyses of smaller patient populations and prioritizes assessment of mortality while also addressing morbidity.108
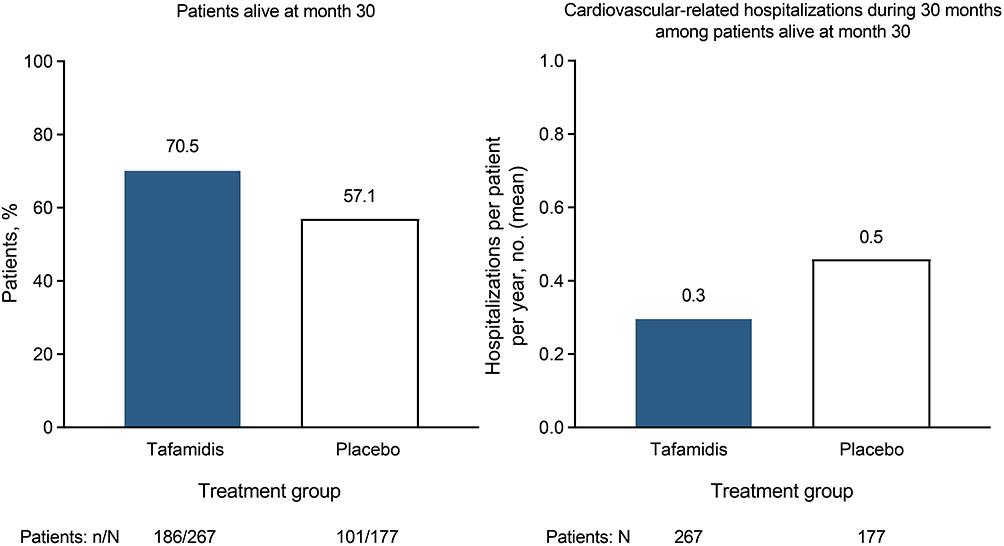 |
Figure 4 All-cause mortality and frequency of cardiovascular-related hospitalizations (primary efficacy analysis) in patients with ATTR-CM treated with tafamidis versus placebo in ATTR-ACT.19 The combined primary endpoint was hierarchically assessed using the Finkelstein–Schoenfeld method. P < 0.001. Abbreviations: ATTR-ACT, Tafamidis in Transthyretin Cardiomyopathy Clinical Trial; ATTR-CM, transthyretin amyloid cardiomyopathy. |
In addition, treatment with tafamidis reduced the risk of all-cause mortality and cardiovascular-related hospitalization by 30% and 32%, respectively, versus placebo.19 The number needed to treat with tafamidis to prevent one death was seven and a half over the 2.5-year treatment period, and the number needed to treat to prevent one hospitalization per year was four. These benefits were consistent across the prespecified subgroups analyzed in ATTR-ACT (ie, 80 mg vs 20 mg dose; ATTRv vs ATTRwt; and NYHA class I or II vs class III), except the frequency of cardiovascular-related hospitalizations was higher with tafamidis than placebo in patients with NYHA class III. The investigators speculated that the higher hospitalization rate in the latter subgroup of tafamidis-treated patients was a consequence of their longer survival during a period of more severe disease. Thus the greatest benefits were observed when tafamidis was administered earlier in the course of the disease. Overall, patients in the tafamidis group also demonstrated significant reductions in the decline of functional capacity, health status, and QoL compared with those in the placebo group. Similar incidence and types of adverse events were reported across the pooled tafamidis and placebo groups in the study (Table 2B).
In ATTR-ACT, median overall survival was not reached in either the placebo or pooled tafamidis treatment groups; 57.1% and 70.5% of patients in these groups, respectively, survived at 30 months. Therefore, a survival extrapolation analysis was conducted to estimate patients’ median overall survival beyond the study’s 30-month duration.109 According to this extrapolation analysis, conducted based on National Institute for Health and Care Excellence technical support guidelines, the estimated median overall survival was 17.5 months longer with tafamidis compared with placebo.109
ATTR-ACT was not designed to assess the relative efficacy of each tafamidis dose. However, the efficacy of the individual tafamidis 20 mg and 80 mg doses was recently assessed over a longer treatment period in a combined analysis of 30-month data from ATTR-ACT19 and interim data from the ongoing, open-label, long-term extension of ATTR-ACT110 (ClinicalTrials.gov identifiers: NCT01994889; NCT02791230). In ATTR-ACT, both tafamidis doses significantly reduced the combination of all-cause mortality and cardiovascular-related hospitalizations over 30 months versus placebo.19 After a median of 51 months’ follow-up, patients initially treated with tafamidis 80 mg had a significantly reduced risk of all-cause mortality compared with those initiated on the 20-mg dose.110
Development of a Single Capsule Tafamidis Formulation
After its regulatory approval for ATTR-PN in 2011, tafamidis was available as 20-mg tafamidis meglumine capsules. In ATTR-ACT, patients received tafamidis in doses of 20 mg (tafamidis meglumine 1 × 20 mg and 3 × placebo) or 80 mg (tafamidis meglumine 4 × 20 mg).19 Because the 80-mg tafamidis dose was under investigation (and subsequently approved in several countries) for the treatment of ATTR-CM, researchers sought to develop a single tafamidis meglumine 80-mg capsule for patient convenience. However, technical limitations (ie, concentration-dependent gelling in aqueous media) precluded the development of this formulation. Other single, solid, oral dosage formulations were therefore evaluated as more convenient options to the tafamidis meglumine 80-mg dose.
The tafamidis 61-mg free acid capsule formulation was selected on the basis of pharmacokinetic/safety results from several single-dose tafamidis studies, and simulations conducted to select the appropriate dose. This formulation is currently approved for the treatment of ATTR-CM in the EU, USA, and in many other countries worldwide. The tafamidis meglumine salt and free acid formulations have the same bioactive moiety (tafamidis) and biologic effects. In an open-label, multiple-dose, Phase 1 study, tafamidis 61-mg free acid capsules were shown to be bioequivalent (as measured by area under the concentration–time profile and maximum observed concentration) to tafamidis meglumine 80-mg (4 × 20-mg) capsules after 7 days of repeated oral dosing under fasted conditions in healthy volunteers.111 Both formulations also demonstrated an acceptable safety/tolerability profile in this population, with no serious adverse events or deaths reported. Importantly, as described above, in the ongoing open-label, long-term extension study of ATTR-ACT (ClinicalTrials.gov identifier: NCT02791230), all patients were successfully transitioned from tafamidis 80 mg or 20 mg (after a median of 39 months) to the new, single-capsule tafamidis free acid 61 mg formulation.110
Real-World Evidence
Many reports on ATTR amyloidosis have been published from studies conducted in a small number of centers within a single country because of the rare nature of the disease.112–120 Rare disease registries allow for the investigation of disease states and treatments in a real-world setting over longer periods of follow-up in generally larger and more heterogeneous global populations.121 The Transthyretin Amyloidosis Outcomes Survey (THAOS; ClinicalTrials.gov identifier: NCT00628745), established in 2007, is the largest ongoing, global, longitudinal observational survey of patients with ATTR amyloidosis, including both inherited and wild-type disease, and asymptomatic patients with TTR mutations. The primary objective of this survey is to better understand and characterize the natural history of ATTR amyloidosis by studying a large and heterogeneous patient population. THAOS is currently the largest global database on ATTR amyloidosis, with findings that have reinforced the genetic and phenotypic heterogeneity of the disease worldwide.22,122–124
THAOS also provides data for real-world analyses of the safety and effectiveness of treatment for ATTR amyloidosis. As part of a comprehensive safety summary of tafamidis in ATTR-PN, safety was evaluated in 661 patients in THAOS who were treated with tafamidis 20 mg/day (data cut: 3 January 2017).125 Data suggested that tafamidis was generally safe and well tolerated; after exposure to tafamidis for a mean of 28 months, 38% and 15% of patients experienced treatment-emergent adverse events and treatment-emergent serious adverse events, respectively. In an effectiveness analysis of data from THAOS (data cut: 6 January 2015), Mundayat et al compared outcomes in 252 tafamidis-treated patients with a predominantly neurological phenotype (93% with Val30Met [p.Val50Met]) with those in 167 matched untreated patients over a 2-year period.126 Tafamidis-treated patients had significantly lower rates of deterioration in neurologic function and in worsening of QoL than untreated patients, suggesting a slower rate of disease progression. Seven deaths occurred in the untreated group, and no deaths occurred in the tafamidis-treated group. The real-world evidence from THAOS is consistent with findings from tafamidis clinical trials,60,96 supporting the overall safety and effectiveness of this first-in-class kinetic stabilizer in ATTR-PN.
As evidence from real-world studies accrues, data will also become available on the use of tafamidis in ATTR amyloidosis with cardiac manifestations. Among the first examples is a recent retrospective observational study of patients with ATTR-CM treated with tafamidis at the Oregon Health & Science University.127 In this real-world population, clinical outcomes, including mortality rates, were found to be comparable to those previously reported in ATTR-ACT.19
Discussion
Knowledge of ATTR amyloidosis has advanced over the past several decades, but unanswered questions about its pathophysiology, etiology, natural history, and prevalence have presented unique challenges and risks when considering drug development and clinical trial design. Although discovery of the complex underlying mechanisms of ATTR amyloidosis has involved decades of detailed investigation, it is far from complete. However, researchers’ identification of TTR tetramer dissociation as the rate-limiting step in ATTR amyloidogenesis proved to be a critical breakthrough, leading to the hypothesis that a small-molecule kinetic stabilizer could be developed to slow tetramer dissociation, stop the aggregation of newly biosynthesized TTR, and halt disease progression. This mechanistic laboratory research culminated in the discovery of tafamidis, the TTR kinetic stabilizer rationally and precisely designed to arrest the amyloid cascade in patients with TTR amyloidosis. As summarized in this review, an extensive series of laboratory and clinical research programs and regulatory milestones subsequently paved the way for the clinical success of this novel treatment (Figure 2; Table 1).
Important insights have been gained from the pioneering work involved in the discovery and development of tafamidis, including the need to adopt a tenacious and thoughtful approach to the clinical trial program that keeps pace with research findings on the disease itself. Knowledge garnered from facing and overcoming obstacles informed not only tafamidis trial design and conduct but also helped provide a roadmap for future experimental agents in rare diseases. For example, the original protocol of the ATTR-ACT study was amended to include radionuclide scintigraphy for eligibility assessment based on evolving diagnostic recommendations. This non-invasive scanning technique subsequently became more commonly used as its high sensitivity and specificity in diagnosing ATTR-CM was confirmed in both clinical research and practice settings. In addition, collaboration with patient advocacy groups and genetic testing companies were important to support and ensure recruitment of sufficient numbers of participants with ATTR amyloidosis in tafamidis clinical trials, which will continue to strengthen research initiatives for subsequent generations of amyloidosis therapies.
Clinicians and health systems have typically focused on conditions affecting large populations, allowing rare diseases to remain unrecognized until they have progressed to late stages. To help facilitate the early, rapid, and accurate identification of ATTR-CM, the Amyloidosis Research Consortium published diagnostic recommendations in 2019, including “red flag” signs and symptoms and a detailed diagnostic algorithm for patients with suspected disease.63 The advantages of noninvasive myocardial scintigraphy in the diagnosis of ATTR-CM—high specificity and sensitivity for ATTR-CM, imaging simplicity, ease of access, and lower cost versus endomyocardial biopsy—support broader use of this technique, which is expected to improve diagnosis, and potentially lead to earlier diagnosis of the disease.63 Moreover, work is ongoing on artificial intelligence and machine learning models using electronic health records and imaging technologies capable of identifying patients at risk of having ATTR-CM.128 These models may eventually be used to increase the index of suspicion of ATTR-CM to support prompt diagnosis of the disease in the clinical practice setting. Finally, educational initiatives and research/decision support tools are in development for systematic adoption by health systems to help ensure that patients with rare diseases such as ATTR-CM are not overlooked.
Despite newly available diagnostic algorithms, imaging modalities, technological innovations, and population health initiatives, there remains a need for greater awareness of ATTR amyloidosis among clinicians and patients, and more informed screening to identify at-risk patients and ensure earlier identification and use of a disease-modifying treatment. The availability of pharmacologic treatments such as tafamidis will encourage clinicians to consider ATTR amyloidosis in their differential diagnoses when patients present with possible manifestations of amyloid disease, improving the formerly very poor prognosis associated with this rare disease.
Abbreviations
ATTR-ACT, Tafamidis in Transthyretin Cardiomyopathy Clinical Trial; ATTR amyloidosis, transthyretin amyloidosis; ATTR-CM, transthyretin amyloid cardiomyopathy; ATTR-PN, transthyretin amyloid polyneuropathy; ATTRv, variant transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis; BL, baseline; CI, confidence interval; CV, cardiovascular; EE, efficacy evaluable; ITT, intent-to-treat; KPS, Karnofsky Performance Status score; LS, least-squares; mITT, modified intent-to-treat; mo, month; N/A, not available; NIS-LL, Neuropathy Impairment Score in the Lower Limbs; Norfolk QoL-DN, Norfolk Quality of Life-Diabetic Neuropathy questionnaire; NSAID, nonsteroidal anti-inflammatory drugs; NT-proBNP, N-terminal pro-B-type natriuretic peptide; NYHA, New York Heart Association; pts, patients; QoL, quality of life; SD, standard deviation; SE, standard error; T4, thyroxine; THAOS, Transthyretin Amyloidosis Outcomes Survey; TQoL, Total Quality of Life score from the Norfolk Quality of Life-Diabetic Neuropathy questionnaire; TRACS, Transthyretin Amyloidosis Cardiac Study; TTR, transthyretin; wk, week; y, year.
Disclosure
Medical writing support for this article was provided by Donna McGuire of Engage Scientific Solutions and was funded by Pfizer. AC, MB, SR, JS, MBS, and SST are full-time employees of Pfizer and may hold Pfizer stock and/or stock options. AB is a paid consultant to Pfizer in connection with the development of this manuscript. DPJ has received consultancy fees from Alnylam, Blade Therapeutics, GSK, and Pfizer and research grants from Array Biopharma, Eidos, and Pfizer. JWK discovered tafamidis at the Scripps Research Institute; is a shareholder of FoldRx Pharmaceuticals, acquired by Pfizer Inc in October 2010 (the companies that developed tafamidis into a drug); and receives royalty payments from tafamidis patents and sales. JP has received grant/research support from Akcea, Alnylam, Alexion, Eidos, and Pfizer; served as a consultant for Akcea, Alnylam, Cytokinetics, and Pfizer; and served on the speaker’s bureau for Akcea and Alnylam. The authors report no other conflicts of interest related to this work.
References
1. de Vrueh R, Baekelandt E. Rare diseases. In: Kaplan W, Wirtz V, Mantel-Teeuwisse A, Stolk P, Duthey B, Laing R, editors. Priority Medicines for Europe and the World 2013 Update. Geneva, Switzerland: World Health Organization; 2013:148–151.
2. Valdez R, Ouyang L, Bolen J. Public health and rare diseases: oxymoron no more. Prev Chronic Dis. 2016;13:E05. doi:10.5888/pcd13.150491
3. Dharssi S, Wong-Rieger D, Harold M, Terry S. Review of 11 national policies for rare diseases in the context of key patient needs. Orphanet J Rare Dis. 2017;12(1):63. doi:10.1186/s13023-017-0618-0
4. National Institute of Health [Internet]. Public law 97–414 97th congress. 1983. Available from: https://history.nih.gov/research/downloads/PL97-414.pdf.
5. The European Parliament and the Council of the European Union [Internet]. Regulation (EC) N°141/2000 of the European parliament and of the council of 16 December 1999 on orphan medicinal products. 2000. Available from: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2000:018:0001:0005:EN:PDF.
6. US Food & Drug Administration [Internet]. Designating an orphan product: drugs and biological products. 2020. Available from: https://www.fda.gov/industry/developing-products-rare-diseases-conditions/designating-orphan-product-drugs-and-biological-products.
7. Mariz S, Reese JH, Westermark K, et al. Worldwide collaboration for orphan drug designation. Nat Rev Drug Discov. 2016;15(6):440–441. doi:10.1038/nrd.2016.80
8. Mulberg AE, Bucci-Rechtweg C, Giuliano J, et al. Regulatory strategies for rare diseases under current global regulatory statutes: a discussion with stakeholders. Orphanet J Rare Dis. 2019;14(1):36. doi:10.1186/s13023-019-1017-5
9. Global Genes [Internet]. Rare diseases: facts and statistics. 2019. Available from: https://globalgenes.org/rare-facts/.
10. Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215–219. doi:10.1080/13506129.2018.1549825
11. Maurer MS, Mann DL. The tafamidis drug development program: a translational triumph. JACC Basic Transl Sci. 2018;3(6):871–873. doi:10.1016/j.jacbts.2018.10.001
12. Institute of Medicine (US) Committee on Accelerating Rare Diseases Research and Orphan Product Development. Rare Diseases and Orphan Products: Accelerating Research and Development. Washington, DC: National Academies Press (US); 2010.
13. Stewart M, Shaffer S, Murphy B, et al. Characterizing the high disease burden of transthyretin amyloidosis for patients and caregivers. Neurol Ther. 2018;7(2):349–364. doi:10.1007/s40120-018-0106-z
14. US Food and Drug Administration [Internet]. Rare diseases: common issues in drug development. Guidance for industry. 2019. Available from: https://www.fda.gov/media/119757/download.
15. Orphanet [Internet]. About orphan drugs. 2020. Available from: https://www.orpha.net/consor/cgi-bin/Education_AboutOrphanDrugs.php?lng=EN.
16. Tufts Center for the Study of Drug Development. Growth in Rare Disease R&D is Challenging Development Strategy and Execution. Vol. 21. Boston, MA: Tufts University; 2019.
17. Witteles RM, Bokhari S, Damy T, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 2019;7(8):709–716. doi:10.1016/j.jchf.2019.04.010
18. Gainotti S, Mascalzoni D, Bros-Facer V, et al. Meeting patients’ right to the correct diagnosis: ongoing international initiatives on undiagnosed rare diseases and ethical and social issues. Int J Environ Res Public Health. 2018;15(10):2072. doi:10.3390/ijerph15102072
19. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
20. Jayasundara K, Hollis A, Krahn M, Mamdani M, Hoch JS, Grootendorst P. Estimating the clinical cost of drug development for orphan versus non-orphan drugs. Orphanet J Rare Dis. 2019;14(1):12. doi:10.1186/s13023-018-0990-4
21. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8(1):31. doi:10.1186/1750-1172-8-31
22. Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161–172. doi:10.1016/j.jacc.2016.03.596
23. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641–2654. doi:10.1016/S0140-6736(15)01274-X
24. Waddington-Cruz M, Schmidt H, Botteman MF, et al. Epidemiological and clinical characteristics of symptomatic hereditary transthyretin amyloid polyneuropathy: a global case series. Orphanet J Rare Dis. 2019;14(1):34. doi:10.1186/s13023-019-1000-1
25. Rowczenio D, Quarta CC, Fontana M, et al. Analysis of the TTR gene in the investigation of amyloidosis: a 25-year single UK center experience. Hum Mutat. 2019;40(1):90–96. doi:10.1002/humu.23669
26. Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10(3):160–184. doi:10.3109/13506120308998998
27. Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451–2466. doi:10.1016/j.jacc.2015.09.075
28. Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520–528. doi:10.1093/eurheartj/ehs123
29. Brunjes DL, Castano A, Clemons A, Rubin J, Maurer MS. Transthyretin cardiac amyloidosis in older Americans. J Card Fail. 2016;22(12):996–1003. doi:10.1016/j.cardfail.2016.10.008
30. Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829–837. doi:10.1002/mus.26034
31. Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22(3):171–174. doi:10.3109/13506129.2015.1051219
32. Damrauer SM, Chaudhary K, Cho JH, et al. Association of the V122I hereditary transthyretin amyloidosis genetic variant with heart failure among individuals of African or Hispanic/Latino ancestry. JAMA. 2019;322(22):2191–2202. doi:10.1001/jama.2019.17935
33. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(22):2872–2891. doi:10.1016/j.jacc.2019.04.003
34. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404. doi:10.1038/s41582-019-0210-4
35. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–2594. doi:10.1093/eurheartj/ehv338
36. Hahn VS, Yanek LR, Vaishnav J, et al. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail. 2020;8(9):712–724. doi:10.1016/j.jchf.2020.04.007
37. Lindmark K, Pilebro B, Sundström T, Lindqvist P. Prevalence of wild type transtyrethin cardiac amyloidosis in a heart failure clinic. ESC Heart Fail. 2020. doi:10.1002/ehf2.13110
38. Shiozaki T, Sato N, Hayashi T, Kobayashi K, Asamura H. Wild-type ATTR amyloidosis may be associated with unexpected death among the elderly. Leg Med (Tokyo). 2019;41:101634. doi:10.1016/j.legalmed.2019.101634
39. Tanskanen M, Peuralinna T, Polvikoski T, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232–239. doi:10.1080/07853890701842988
40. Pomerance A. Senile cardiac amyloidosis. Br Heart J. 1965;27(5):711–718. doi:10.1136/hrt.27.5.711
41. Cornwell GG
42. Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020;21(1):198. doi:10.1186/s12875-020-01252-4
43. Connors LH, Sam F, Skinner M, et al. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation. 2016;133(3):282–290. doi:10.1161/CIRCULATIONAHA.115.018852
44. Bruno M, Castaño A, Burton A, Grodin JL. Transthyretin amyloid cardiomyopathy in women: frequency, characteristics, and diagnostic challenges. Heart Fail Rev. 2021;26(1):35–45. doi:10.1007/s10741-020-10010-8
45. Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222–228.e221. doi:10.1016/j.ahj.2012.04.015
46. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799–2806. doi:10.1093/eurheartj/ehx589
47. Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68(10):1014–1020. doi:10.1016/j.jacc.2016.06.033
48. Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40(3):242–246. doi:10.1111/j.1399-0004.1991.tb03085.x
49. de Carvalho M, Conceição I, Bentes C, Luís ML. Long-term quantitative evaluation of liver transplantation in familial amyloid polyneuropathy (Portuguese V30M). Amyloid. 2002;9(2):126–133. doi:10.3109/13506120208995245
50. Herlenius G, Wilczek HE, Larsson M, Ericzon BG. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial amyloidotic polyneuropathy world transplant registry. Transplantation. 2004;77(1):64–71. doi:10.1097/01.TP.0000092307.98347.CB
51. Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78(9):637–643. doi:10.1212/WNL.0b013e318248df18
52. Okamoto S, Wixner J, Obayashi K, et al. Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients’ survival. Liver Transpl. 2009;15(10):1229–1235. doi:10.1002/lt.21817
53. Stangou AJ, Hawkins PN, Heaton ND, et al. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation. 1998;66(2):229–233. doi:10.1097/00007890-199807270-00016
54. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277–282. doi:10.1080/13506120701614032
55. Takei Y, Ikeda S, Ikegami T, Hashikura Y, Miyagawa S, Ando Y. Ten years of experience with liver transplantation for familial amyloid polyneuropathy in Japan: outcomes of living donor liver transplantations. Intern Med. 2005;44(11):1151–1156. doi:10.2169/internalmedicine.44.1151
56. Dubrey SW, Burke MM, Hawkins PN, Banner NR. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant. 2004;23(10):1142–1153. doi:10.1016/j.healun.2003.08.027
57. Ruygrok PN, Gane EJ, McCall JL, Chen XZ, Haydock DA, Munn SR. Combined heart and liver transplantation for familial amyloidosis. Intern Med J. 2001;31(1):66–67. doi:10.1046/j.1445-5994.2001.00008.x
58. Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112(13):2047–2060. doi:10.1161/CIRCULATIONAHA.104.489187
59. Sousa M, Monohan G, Rajagopalan N, Grigorian A, Guglin M. Heart transplantation in cardiac amyloidosis. Heart Fail Rev. 2017;22(3):317–327. doi:10.1007/s10741-017-9601-z
60. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. doi:10.1212/WNL.0b013e3182661eb1
61. Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293(5539):2459–2462. doi:10.1126/science.1062245
62. Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299(5607):713–716. doi:10.1126/science.1079589
63. Maurer MS, Bokhari S, Damy T, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12(9):e006075. doi:10.1161/CIRCHEARTFAILURE.119.006075
64. McCutchen SL, Colon W, Kelly JW. Transthyretin mutation Leu-55-Pro significantly alters tetramer stability and increases amyloidogenicity. Biochemistry. 1993;32(45):12119–12127. doi:10.1021/bi00096a024
65. Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci U S A. 1996;93(26):15051–15056. doi:10.1073/pnas.93.26.15051
66. Peterson SA, Klabunde T, Lashuel HA, Purkey H, Sacchettini JC, Kelly JW. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc Natl Acad Sci U S A. 1998;95(22):12956–12960. doi:10.1073/pnas.95.22.12956
67. Razavi H, Powers ET, Purkey HE, et al. Design, synthesis, and evaluation of oxazole transthyretin amyloidogenesis inhibitors. Bioorg Med Chem Lett. 2005;15(4):1075–1078. doi:10.1016/j.bmcl.2004.12.022
68. Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–249. doi:10.1080/13506120600960882
69. Kopelman M, Cogan U, Mokady S, Shinitzky M. The interaction between retinol-binding proteins and prealbumins studied by fluorescence polarization. Biochim Biophys Acta. 1976;439(2):449–460. doi:10.1016/0005-2795(76)90082-9
70. Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268(5213):1039–1041. doi:10.1126/science.7754382
71. Monaco HL. The transthyretin-retinol-binding protein complex. Biochim Biophys Acta. 2000;1482(1–2):65–72. doi:10.1016/S0167-4838(00)00140-0
72. Bartalena L, Robbins J. Variations in thyroid hormone transport proteins and their clinical implications. Thyroid. 1992;2(3):237–245. doi:10.1089/thy.1992.2.237
73. Bartalena L, Robbins J. Thyroid hormone transport proteins. Clin Lab Med. 1993;13(3):583–598. doi:10.1016/S0272-2712(18)30427-X
74. Coelho T, Chorão R, Sousa A, Alves I, Torres MF, Saraiva MJM. Compound heterozygotes of transthyretin Met30 and transthyretin Met119 are protected from the devastating effects of familial amyloid polyneuropathy [abstract]. Neuromuscul Disord. 1996;6(Suppl 1):S20. doi:10.1016/0960-8966(96)88826-2
75. Longo Alves I, Hays MT, Saraiva MJ. Comparative stability and clearance of [Met30]transthyretin and [Met119]transthyretin. Eur J Biochem. 1997;249(3):662–668. doi:10.1111/j.1432-1033.1997.00662.x
76. Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38(12):911–921. doi:10.1021/ar020073i
77. Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31(36):8654–8660. doi:10.1021/bi00151a036
78. Lai Z, Colon W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35(20):6470–6482. doi:10.1021/bi952501g
79. Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A. 2002;99:16427–16432. doi:10.1073/pnas.202495199
80. Schneider F, Hammarstrom P, Kelly JW. Transthyretin slowly exchanges subunits under physiological conditions: a convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 2001;10(8):1606–1613. doi:10.1110/ps.8901
81. Wiseman RL, Green NS, Kelly JW. Kinetic stabilization of an oligomeric protein under physiological conditions demonstrated by a lack of subunit exchange: implications for transthyretin amyloidosis. Biochemistry. 2005;44(25):9265–9274. doi:10.1021/bi050352o
82. Wiseman RL, Powers ET, Kelly JW. Partitioning conformational intermediates between competing refolding and aggregation pathways: insights into transthyretin amyloid disease. Biochemistry. 2005;44(50):16612–16623. doi:10.1021/bi0511484
83. Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43(23):7365–7381. doi:10.1021/bi049621l
84. Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol. 2010;20(1):54–62. doi:10.1016/j.sbi.2009.12.009
85. Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421(2–3):185–203. doi:10.1016/j.jmb.2011.12.060
86. Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci U S A. 2001;98(10):5566–5571. doi:10.1073/pnas.091431798
87. Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84(5):545–552. doi:10.1038/labinvest.3700059
88. Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60(4):765–774. doi:10.1111/j.1532-5415.2011.03868.x
89. Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–9634. doi:10.1073/pnas.1121005109
90. Rappley I, Monteiro C, Novais M, et al. Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry. 2014;53(12):1993–2006. doi:10.1021/bi500171j
91. Coelho T, Merlini G, Bulawa CE, et al. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther. 2016;5(1):1–25. doi:10.1007/s40120-016-0040-x
92. Schmidt HH-J. Tafamidis for the treatment of transthyretin-associated familial amyloid polyneuropathy. Expert Opin Orphan Drugs. 2013;1:837–845. doi:10.1517/21678707.2013.841576
93. European Medicines Agency Committee for Medicinal Products for Human Use [Internet]. Assessment report: Vyndaqel (tafamidis meglumine). 2011. Available from: https://www.ema.europa.eu/en/documents/assessment-report/vyndaqel-epar-public-assessment-report_en.pdf.
94. Lamb YN, Deeks ED. Tafamidis: a review in transthyretin amyloidosis with polyneuropathy. Drugs. 2019;79(8):863–874. doi:10.1007/s40265-019-01129-6
95. Merlini G, Planté-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6(6):1011–1020. doi:10.1007/s12265-013-9512-x
96. Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–2814. doi:10.1007/s00415-013-7051-7
97. European Medicines Agency [Internet]. Tafamidis summary of product characteristics 2011. Available from: https://www.ema.europa.eu/en/documents/product-information/vyndaqel-epar-product-information_en.pdf. Accessed February 4, 2021.
98. Barroso FA, Judge DP, Ebede B, et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017;24(3):194–204. doi:10.1080/13506129.2017.1357545
99. Merlini G, Coelho T, Waddington Cruz M, Li H, Stewart M, Ebede B. Evaluation of mortality during long-term treatment with tafamidis for transthyretin amyloidosis with polyneuropathy: clinical trial results up to 8.5 years. Neurol Ther. 2020;9(1):105–115. doi:10.1007/s40120-020-00180-w
100. Waddington Cruz M, Amass L, Keohane D, Schwartz J, Li H, Gundapaneni B. Early intervention with tafamidis provides long-term (5.5-year) delay of neurologic progression in transthyretin hereditary amyloid polyneuropathy. Amyloid. 2016;23(3):178–183. doi:10.1080/13506129.2016.1207163
101. Coelho T, Inês M, Conceição I, Soares M, de Carvalho M, Costa J. Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018;91(21):e1999–e2009. doi:10.1212/WNL.0000000000006543
102. Socie P, Benmalek AB, Lallemand AL, et al. 1170 Tafamidis versus liver transplantation as first-line therapy for hereditary transthyretin amyloidosis [abstract]. Eur Heart J. 2019;40(Suppl1):
103. US Food and Drug Administration [Internet]. Highlights of prescribing information: VYNDAQEL and VYNDAMAX. 2019. Available from: https://www.fda.gov/media/126283/download.
104. Damy T, Judge DP, Kristen AV, Berthet K, Li H, Aarts J. Cardiac findings and events observed in an open-label clinical trial of tafamidis in patients with non-Val30Met and non-Val122Ile hereditary transthyretin amyloidosis. J Cardiovasc Transl Res. 2015;8(2):117–127. doi:10.1007/s12265-015-9613-9
105. Maurer MS, Grogan DR, Judge DP, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail. 2015;8(3):519–526. doi:10.1161/CIRCHEARTFAILURE.113.000890
106. Sultan MB, Gundapaneni B, Schumacher J, Schwartz JH. Treatment with tafamidis slows disease progression in early-stage transthyretin cardiomyopathy. Clin Med Insights Cardiol. 2017;11:1179546817730322. doi:10.1177/1179546817730322
107. Klamerus KJ, Watsky E, Moller R, Wang R, Riley S. The effect of tafamidis on the QTc interval in healthy subjects. Br J Clin Pharmacol. 2015;79(6):918–925. doi:10.1111/bcp.12561
108. Finkelstein DM, Schoenfeld DA. Combining mortality and longitudinal measures in clinical trials. Stat Med. 1999;18(11):1341–1354. doi:10.1002/(SICI)1097-0258(19990615)18:11<1341::AID-SIM129>3.0.CO;2-7
109. Li B, Alvir J, Stewart M. Extrapolation of survival benefits in patients with transthyretin amyloid cardiomyopathy receiving tafamidis: analysis of the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial. Cardiol Ther. 2020;9(2):535–540. doi:10.1007/s40119-020-00179-2
110. Damy T, Garcia-Pavia P, Hanna M, et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail. 2020. doi:10.1002/ejhf.2027
111. Lockwood PA, Le VH, O’Gorman MT, et al. The bioequivalence of tafamidis 61-mg free acid capsules and tafamidis meglumine 4 × 20-mg capsules in healthy volunteers. Clin Pharmacol Drug Dev. 2020;9(7):849–854. doi:10.1002/cpdd.789
112. Yamashita T, Ueda M, Misumi Y, et al. Genetic and clinical characteristics of hereditary transthyretin amyloidosis in endemic and non-endemic areas: experience from a single-referral center in Japan. J Neurol. 2018;265(1):134–140. doi:10.1007/s00415-017-8640-7
113. Cappelli F, Frusconi S, Bergesio F, et al. The Val142Ile transthyretin cardiac amyloidosis: not only an Afro-American pathogenic variant? A single-centre Italian experience. J Cardiovasc Med. 2016;17(2):122–125. doi:10.2459/JCM.0000000000000290
114. Martens B, De Pauw M, De Bleecker JL. Single-centre experience on transthyretin familial amyloid polyneuropathy: case series and literature review. Acta Neurol Belg. 2018;118(2):179–185. doi:10.1007/s13760-018-0906-z
115. Chao HC, Liao YC, Liu YT, et al. Clinical and genetic profiles of hereditary transthyretin amyloidosis in Taiwan. Ann Clin Transl Neurol. 2019;6(5):913–922. doi:10.1002/acn3.778
116. Samuelsson K, Radovic A, Press R, et al. Screening for Fabry disease and hereditary ATTR amyloidosis in idiopathic small-fiber and mixed neuropathy. Muscle Nerve. 2019;59(3):354–357. doi:10.1002/mus.26348
117. Aus Dem Siepen F, Hein S, Prestel S, et al. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol. 2019;108(12):1324–1330. doi:10.1007/s00392-019-01467-1
118. Andreou S, Panayiotou E, Michailidou K, et al. Epidemiology of ATTRV30M neuropathy in Cyprus and the modifier effect of complement C1q on the age of disease onset. Amyloid. 2018;25(4):220–226. doi:10.1080/13506129.2018.1534731
119. Carr AS, Pelayo-Negro AL, Evans MR, et al. A study of the neuropathy associated with transthyretin amyloidosis (ATTR) in the UK. J Neurol Neurosurg Psychiatry. 2016;87(6):620–627. doi:10.1136/jnnp-2015-310907
120. Alves-Ferreira M, Coelho T, Santos D, et al. A trans-acting factor may modify age at onset in familial amyloid polyneuropathy ATTRV30M in Portugal. Mol Neurobiol. 2018;55(5):3676–3683. doi:10.1007/s12035-017-0593-4
121. Gagne JJ, Thompson L, O’Keefe K, Kesselheim AS. Innovative research methods for studying treatments for rare diseases: methodological review. BMJ. 2014;349:g6802. doi:10.1136/bmj.g6802
122. Damy T, Kristen AV, Suhr OB, et al. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J. 2019. doi:10.1093/eurheartj/ehz173
123. Coelho T, Maurer MS, Suhr OB. THAOS - the Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76. doi:10.1185/03007995.2012.754348
124. Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis - report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS One. 2017;12(4):e0173086. doi:10.1371/journal.pone.0173086
125. Huber P, Flynn A, Sultan MB, et al. A comprehensive safety profile of tafamidis in patients with transthyretin amyloid polyneuropathy. Amyloid. 2019;26(4):203–209. doi:10.1080/13506129.2019.1643714
126. Mundayat R, Stewart M, Alvir J, et al. Positive effectiveness of tafamidis in delaying disease progression in transthyretin familial amyloid polyneuropathy up to 2 years: an analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther. 2018;7(1):87–101. doi:10.1007/s40120-018-0097-9
127. Chandrashekar P, Dale Z, Rashdan L, et al. Functional class, biomarker stability, and clinical outcomes of patients with transthyretin cardiac amyloidosis treated with tafamidis. Circulation. 2020;142:A17077.
128. Huda A, Castano A, Niyogi A, et al. A machine learning model of medical claims data for identification of wild-type transthyretin amyloid cardiomyopathy. Nat Commun. In press 2021.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.