")
Back to Journals » International Journal of Nanomedicine » Volume 19
Drug-Coformer Loaded-Mesoporous Silica Nanoparticles: A Review of the Preparation, Characterization, and Mechanism of Drug Release
Authors Budiman A, Wardhana YW, Ainurofiq A, Nugraha YP, Qaivani R, Lukmanul Hakim SNA, Aulifa DL
Received 10 November 2023
Accepted for publication 19 December 2023
Published 12 January 2024 Volume 2024:19 Pages 281—305
DOI https://doi.org/10.2147/IJN.S449159
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Misra
Arif Budiman,1 Yoga Windhu Wardhana,1 Ahmad Ainurofiq,2 Yuda Prasetya Nugraha,3 Ridhatul Qaivani,4 Siti Nazila Awaliyyah Lukmanul Hakim,4 Diah Lia Aulifa4
1Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Universitas Padjadjaran, Bandung, West Java 45363, Indonesia; 2Pharmaceutical Technology and Drug Delivery, Department of Pharmacy, Universitas Sebelas Maret, Surakarta, Central Java, 57126, Indonesia; 3School of Pharmacy, Bandung Institute of Technology, Bandung, West Java, 40132, Indonesia; 4Department of Pharmaceutical Analysis and Medicinal Chemistry, Faculty of Pharmacy, Universitas Padjadjaran, Bandung, West Java, 45363, Indonesia
Correspondence: Arif Budiman, Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Universitas Padjadjaran, Jl. Raya Bandung Sumedang Km. 21, Sumedang, West Java, 45363, Indonesia, Email [email protected]
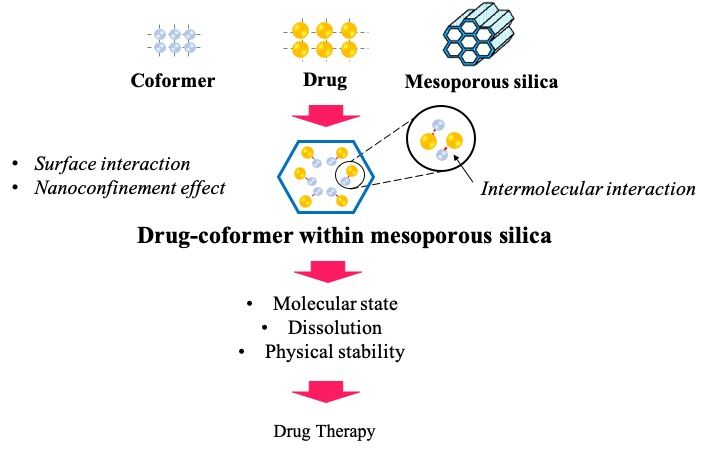
Abstract: Drug-coformer systems, such as coamorphous and cocrystal, are gaining recognition as highly effective strategies for enhancing the stability, solubility, and dissolution of drugs. These systems depend on the interactions between drug and coformer to prevent the conversion of amorphous drugs into the crystalline form and improve the solubility. Furthermore, mesoporous silica (MPS) is also a promising carrier commonly used for stabilization, leading to solubility improvement of poorly water-soluble drugs. The surface interaction of drug-MPS and the nanoconfinement effect prevent amorphous drugs from crystallizing. A novel method has been developed recently, which entails the loading of drug-coformer into MPS to improve the solubility, dissolution, and physical stability of the amorphous drug. This method uses the synergistic effects of drug-coformer interactions and the nanoconfinement effect within MPS. Several studies have reported successful incorporation of drug-coformer into MPS, indicating the potential for significant improvement in dissolution characteristics and physical stability of the drug. Therefore, this study aimed to discuss the preparation and characterization of drug-coformer within MPS, particularly the interaction in the nanoconfinement, as well as the impact on drug release and physical stability.
Keywords: mesoporous silica nanoparticles, coamorphous, cocrystal, hydrogen bond, dissolution, physical stability
Graphical Abstract:
Introduction
The aqueous solubility of drugs is important in pharmaceutical formulation, influencing bioavailability, particularly in the creation of oral dosage forms. Drugs are subjected to adsorption from the intestinal sites after dissolving in the gastrointestinal fluid.1–3 Over the last two decades, approximately 75% of new chemical entities (NCEs) in pharmaceutical study showed poor water solubility and have been successfully improved using traditional methods such as solubilizing, complexing, and salt formation. The limitations of these methods include their effectiveness in enhancing solubility, the permissible concentration of excipients, and the potential occurrence of different side effects associated with the excipients and co-solvents.4–6 Consequently, there is a need to explore innovative strategies to address the formulation of poorly water-soluble drugs.7
Amorphization is a promising strategy for improving the aqueous solubility of drugs. Generally, amorphous drugs have disordered structures and higher free energies compared to their crystalline counterparts.8,9 Due to this higher energy state, dissolution tends to occur more quickly, as their crystal structure is not disrupted. However, a pure amorphous API is thermodynamically unstable and easy to recrystallize during storage after being dispersed in dissolution medium. These properties contribute to the difficulty of using amorphous drugs alone in a solid formulation.10,11
In the past decade, mixing specific low molecular weight compounds (coformer) with poorly water-soluble drugs at the molecular level has been developed as an alternative method to stabilize the amorphous drug and improve dissolution profiles. This coamorphous system is characterized as a single-phase amorphous solid system composed of binary or multi-components.12,13 Based on the selection of coformers, coamorphous systems can be grouped into two categories, namely drug-excipient and drug-drug. In drug-excipient coamorphous systems, the excipients such as sugars, nicotinamide, amino acids, and carboxylic acid, are extensively used as coformers to improve solubility and physical stability. Meanwhile, in drug-drug coamorphous systems, two drug components can effectively stabilize each other in the amorphous state to achieve the desired physical stability and dissolution profiles.14 The use of two pharmacologically relevant drugs in these systems has potential benefits for synergistic effects. The formation of intermolecular interactions, such as hydrogen bonding as well as π–π between drug and the excipient plays a role in preventing conversion of the amorphous drug into the crystalline form.15,16
Cocrystal is a neutral crystalline material formed by combining two or more distinct molecular compounds at different stoichiometry, distinguished from solvates or simple salts.17 The crystal structure of cocrystal is different from the starting material (drug and coformer), leading to unique physicochemical properties that surpass pure materials. These properties make cocrystal a highly attractive system in the pharmaceuticals field, particularly for poorly water-soluble drugs.18 The formation of cocrystal with a suitable coformer in the pharmaceutical field has the potential to improve solubility by modifying the crystal structure, thereby improving dissolution, stability, and bioavailability of poorly water-soluble drugs.19
The pharmaceutical industry has developed an interest in the use of mesoporous silica (MPS) in amorphous drug delivery systems due to their stabilization capacity.20 MPS is characterized by small pores ranging from 2–50 nm, with large specific surface areas greater than 300 m2/g, and considerable extra surface-free energy. Consequently, drug adsorption on an MPS surface enables the system to develop into a lower free energy state, stabilizing the amorphous form.21 MPS also prevents the crystallization of the amorphous form by spatial confinement when the pore width is less than the critical crystal nuclei. The encapsulated drug can also rapidly release and generate supersaturated solutions due to the displacement of drug molecules from the silica surface by water.22–24 This unique property improves dissolution and generates supersaturation, significantly increasing the use of the delivery systems.25
Recently, a novel method combining coamorphization/cocrystallization and encapsulation, known as the loading of drug-coformer into MPS has been introduced to improve the solubility, dissolution, and physical stability of poorly water-soluble drugs. Several studies have reported the successful incorporation of drug-coformer into MPS. Skerupska et al stated that cocrystal-loaded MPS showed higher molecular mobility compared to those outside the pores. The interaction mode between drug and coformer within MPS has also been investigated.7,26,27 Bi et al reported that the molecular state of the cocrystal system changed into a coamorphous state after being loaded into MPS leading to variation in physical stability and dissolution characteristics.28 Therefore, this study aims to summarize and discuss the characterization of drug-coformer within MPS, specifically the mechanism of drug-coformer interaction affecting pharmaceutical properties such as solubility, dissolution, and physical stability of drugs. The journals of drug-coformer loaded MPS used to compile this review were obtained from the Scopus, Pubmed, and Google Scholar databases that were released during the last ten years.
Drug-Coformer System
Cocrystal
Cocrystal is a promising strategy for precisely adjusting solubility, dissolution, and bioavailability, without changing their molecular structure. These multicomponent solids contain two or more distinct molecular components in a single homogeneous crystalline phase, with well-defined stoichiometry, as shown in Figure 1.29–31 Cocrystal is formed by hydrogen-bonded assemblies between the neutral molecules and coformer, leading to the change in the solubility characteristics.32 In recent years, cocrystal has gained significant attention from the pharmaceutical industry. This leads to the development of a pharmaceutical cocrystal composed of an active pharmaceutical ingredient (API) or a small molecule as coformer commonly selected from substances appearing on the GRAS (generally regarded as safe) status.33,34
 |
Figure 1 Illustration of cocrystal system. Notes: Data from Bavishi and Borkhataria.19 |
The coformer is often selected based on its functional groups capable of forming hydrogen bonds with the drug molecules. The general guidelines have been developed to predict hydrogen bond interactions resulting in crystal formation.35,36 These guidelines are made based on the analysis of hydrogen bond interactions and the packing of molecular structures: (1) all acidic hydrogen atoms participate in hydrogen bonding within the crystal structure, (2) when there is an adequate supply of hydrogen bond donors, all good hydrogen bond acceptors participate in hydrogen bonding, and (3) intramolecular hydrogen bond in a six-membered ring form in preference to intermolecular hydrogen bond.37,38
Coformers used in the pharmaceutical cocrystal are generally hydrophilic molecules. Moreover, their mechanism in the solution consists of three main steps, namely (1) breaking intermolecular bonds of the drug-coformer, (2) breaking intermolecular bonds of each component in the solvent, and (3) forming intermolecular bonds between cocrystal molecules and the solvent molecules. The limitations of cocrystal in dissolving hydrophobic drug molecules in aqueous media include their solvation and inability to break away from the crystal lattice. The presence of hydrophilic coformer molecules can also decrease the solvation barrier of cocrystals of hydrophobic drugs, proportional to the coformer. Consequently, the aqueous solubility of the coformer is correlated with the solubility of cocrystal.39,40
In dissolution medium, particularly when the solvent is introduced, the supersaturation of cocrystal is reached as the components dissolved. As illustrated in Figure 2, the solubility of cocrystal and drug as well as cocrystal and coformer intersect at eutectic points, indicating a stability region of the solid phase either for each component or mixture.
 |
Figure 2 Triangular phase diagram that illustrates dissolution paths (arrows) leading to cocrystal stability regions (shaded areas). The highest supersaturation of cocrystal can be achieved by saturation from both drug and coformer, conditions associated with water contents below the eutectic points. Notes: Used with permission of Royal Society of Chemistry, from Good D, Miranda C, Rodríguez-Hornedo N. Dependence of cocrystal formation and thermodynamic stability on moisture sorption by amorphous polymer. CrystEngComm. 2011;13(4):1181–1189;41 permission conveyed through Copyright Clearance Center, Inc. |
Coamorphous
Conversion of crystalline drugs into the amorphous state could improve the solubility and dissolution profile of poorly water-soluble drugs. In recent years, some low molecular weight excipients, such as amino and organic acids, have been used for the development of the amorphous system, namely coamorphous. The combination of two drugs is also intended to form coamorphous to improve dissolution and generate combination therapy, including ezetimibe and simvastatin,42 as well as famotidine and ibuprofen.43 Consequently, coamorphous systems, based on the types of coformers, could be classified into drug-excipient and drug-drug.
Coamorphous systems commonly generate a spring-parachute dissolution profile often observed in amorphous systems, as represented in Figure 3.44–46 This profile entails a rapid dissolution that occurred in the coamorphous system at the beginning of the dissolution test, followed by nucleation and crystal growth. The “Spring” effect is obtained from the high-energy state of the coamorphous system, promoting dissolution of the drug when it dissolves, along with the coformer, leading to the supersaturated solution of the drug. Several factors such as dissolution media, the difference of free energy between the crystal phase and amorphous phase, and release rates and coformers can influence the supersaturated solution of the drug in a coamorphous system.44 Meanwhile, the “Parachute” effect delays or prevents the recrystallization of the dissolved drug and maintains a supersaturated level for a certain period due to the interaction between drug and coformers.45 Numerous studies have reported that strong interactions between drug and coformers could maintain supersaturation and prevent the recrystallization of the amorphous drug in the coamorphous system.47–49
 |
Figure 3 The spring and parachute concept to achieve high apparent solubility for poorly water-soluble drug. (1) The crystalline (stable) form has low solubility. (2) A metastable species (amorphous phase) shows peak solubility but quickly drops to the low solubility of the crystalline form. (3) Highly soluble drug forms are maintained for a long enough time in the metastable zone (coamorphous phase). Notes: Adapted from Progress in Crystal Growth and Characterization of Materials, Volume 62/Edition 3, Bavishi and Borkhataria, Spring and parachute: how cocrystals enhance solubility, pages 1-8, Copyright 2016, with permisison from Elsevier.19 |
Mesoporous Silica (MPS)
MPS is formed through a surfactant micelle templating method, using tetraethyl orthosilicate (TEOS) or 3-mercaptopropyl trimethoxysilane (MPTMS) as the silica precursor to produce the pore structures within the silica particle.50 The production of silica particles with different structures, particle sizes, pore sizes, and shapes can be generated by controlling various conditions such as surfactant type, temperature, pH, concentration, and ionic strength. Meanwhile, the most common architectures of MPS are hexagonally arranged cylindrical pores (hexagonal phase) and bicontinuous cylindrical porous networks (cubic phase).51
MPS has several advantages and uses various strategies to enhance oral delivery, in vivo uptake, and bioavailability of poorly water-soluble drugs, as illustrated in Figure 4. The ability of MPS to load a high concentration of drug in a molecular or an amorphous form and control drug release can significantly improve the bioavailability of poorly water-soluble drugs. Previous investigations have established that the characteristics of MPS, such as structure, surface, and pore size, can be engineered to precisely control drug release.52–55
 |
Figure 4 Schematic illustration of the relationship between drug loading, porous silica properties, drug dissolution, and oral absorption. Notes: Copyright © 2003 Taylor & Francis Ltd. Adapted from Capovilla G, Beccaria F, Montagnini A, et al. Short-term nonhormonal and nonsteroid treatment in West syndrome. Epilepsia. 2003;44(8): 1085–1088.51 |
Drug release from MPS depends on the wetting of the pores by the aqueous release media, either in dissolution medium or gastrointestinal fluids. The dissolution process relies on the size, connectivity, and length, as well as the existence of constrictions and “dead ends” in the structure. During this process, drug molecules are displaced by adsorption on the silica surface through interaction with the solvent. The interaction between dissolution medium and silica surface as well as the access to the porous architecture could be an important step for enhanced drug release.56,57 The interaction of drug-drug, drug-silica, drug-solvent, and solvent-silica is also essential in drug release from MPS.
Biocompatibility and Toxicity of MPS Nanoparticles
Since MPS are inorganic nanoparticles and are difficult to degrade in the body, understanding the toxicity mechanism is necessary in the drug formulation to ensure maximum activity with minimum or no toxicity. The routes of administration could affect the distribution and toxicity of MPS. A previous study reported that hypodermic and intramuscular injections could cross the biological barriers into the liver with a low absorption rate. Meanwhile, in oral administration, the MPS nanoparticles are absorbed in the intestinal tract and persist in the liver. The MPS nanoparticles are localized in the liver and spleen when administered by intravenous injection. After intramuscular and hypodermic injections, an inflammatory response was also observed around the injection sites. The MPS nanomaterials were mainly excreted through urine and feces.58 The particle shape, size, and composition of MPS nanoparticles also play critical roles in biodistribution and toxicity. An increase in the composition of MPS nanoparticles can decrease in vivo biodegradation, systematic absorption, and excretion, especially in liver distribution and urinary excretion. The renal damage during the period of urinary excretion, including vascular congestion, and renal tubular necrosis can be induced by MPS shape-dependency.59 The cytotoxicities of MPS nanoparticles are caused by mitochondrial dysfunction, membrane peroxidation, and DNA damage, leading to cell death. However, low concentrations of MPS nanoparticles are more biocompatible than their higher doses. The silanol groups on the MPS surface can cause hemolysis of mammalian red blood cells, thus their use for intravenous drug delivery is limited. In contrast, positively charged ammonium functionalization on the MPS surface prevents the toxicity of MPS nanoparticles due to their exclusion from endocytosis. Short rod-shaped MPS nanoparticles are easily trapped in the liver, while long rods of MPS nanoparticles are distributed in the spleen. PEG functionalization on the surface of MPS nanoparticles is predominant in the lungs and excreted in urine and feces. The clearance rate of short rod MPS nanoparticles is more rapid than long rod MPS nanoparticles by both routes of excretion. This indicated that the clearance rate of MPS nanoparticles is primarily dependent on their particle shape.60
Drug-Coformer Within MPS Systems
Previous studies have reported the loading drug and coformer into MPS as shown in Table 1.
 |
Table 1 The Studies of Drug-Coformer Loaded MPS |
Preparation Drug-Coformer Loading into MPS
The loading of drugs into MPS can be accomplished through various methods. Based on the previous studies, the loading drug and coformer into MPS are divided into two, namely solvent-free and solvent-based methods, as shown in Figure 5.
 |
Figure 5 The different methods used to load drug-coformer into MPS. Notes: Adapted from Trzeciak K, Chotera-ouda A, Bak-sypien II, Potrzebowski MJ. Mesoporous silica particles as drug delivery systems—the state of the art in loading methods and the recent progress in analytical techniques for monitoring these processes. Pharmaceutics. 2021;13(7):950.67 Creative Commons Attribution License. |
Solvent-Based Methods
The solvent-based methods for loading drug-coformer into MPS are challenging. This is because the procedure requires multiple stages using large amounts of solvents, which are often difficult to control in terms of filling factor. The removal of the solvent to the acceptable levels specified in the guidelines of the International Conference on Harmonization (ICH) Q3 (R5) is necessary, as some residual solvents commonly used may remain in MPS despite its disposal.68 To address these issues, less toxic and safer solvents, such as ethanol and supercritical, or near-critical, CO2 can be used as an alternative in the drug loading process or for human pharmaceutical applications.69–71 The selection of a solvent has a significant impact on the loading process into MPS. Moreover, the highest API solubility in the solvent is not necessarily the best candidate for achieving a high filling factor.
The Adsorption Method
The adsorption method is a simple method for loading drug and coformer into the pores of MPS. Subsequently, MPS is dispersed in the concentrated drug-coformer solution, allowing drug molecules to be adsorbed on the pore walls. Drug-coformer loaded MPS are separated from the solution by centrifugation or filtration, followed by drying of particles to remove the residual solvent.72 This method is suitable for thermally sensitive substances, as the loading process does not require high temperatures. However, it is inefficient in some cases due to the need for a high concentration of drug in the solutions to achieve effective drug loading. When the concentration is excessively high, drug molecules block the mesopores of MPS, potentially reducing the available surface area for drug loading.73 Bi et al, and Ali et al have successfully used the adsorption method to incorporate the IBU-NA and VAL-VAN into MPS.28,61
The Solvent Evaporation Method
The solvent evaporation method is often used to incorporate drugs into MPS through the combination of adsorption and rapid solvent evaporation.74 During this process, silica is dispersed in the volatile organic solution, namely ethanol and dichloromethane, containing the drug, followed by drying through fast solvent evaporation using a rotary evaporator75 to obtain drug-loaded MPS.52,76 This method allows sufficient time for drug molecules to rearrange and aggregate inside MPS. The solvent evaporation process may affect the physical state of the drug, its localization, and the rate of drug release.74 A previous study by Budiman et al reported the loading of drug-coformer into MPS using the solvent evaporation method to successfully load RTN and SAC into MPS.62 Ohita et al also reported cocrystal formation of caffeine-oxalic acid within MPS.63
Diffusion-Supported Loading (DiSupLo)
Diffusion-supported loading (DiSupLo) is a novel and efficient method for loading drug-coformer into the pores of MPS. In this method, physical mixture of drug-coformer and MPS, with the desired proportion is transferred to an opened weighing vessel as a solid matter. This is followed by placing the mixture in a closed vessel containing ethanol for 3 h at room temperature. The layer thickness of physical mixture of drug-coformer should be small and experimentally optimized. Finally, the ethanol is thermally removed to obtain drug-loaded samples. The DiSupLo is environmentally friendly and economical due to its fast process requiring a minimum amount of solvent. Furthermore, it is the simplest method for loading drug-MPS into MPS pores without special equipment and specific experimental conditions such as high temperature, stirring, high pressure, and grinding.66 This method allows for a high drug-coformer loading degree and incorporates the whole drug-coformer with an appropriate weight ratio of the starting drug-coformer/MPS mixture. Trzeciak et al have successfully incorporated ibuprofen and flurbiprofen into MCM-41 using the DiSupLo method.66 Meanwhile, a cocrystal of BA-4-FBA was formed within MPS after being prepared by the DiSupLo method.65
Solvent-Free Methods
Solvent-free methods can achieve a high degree of drug loading without consuming significant time. Moreover, the concentration of drug in MPS is easily predictable, as it is directly influenced by the ratio between API and MPS. This method is an environmentally friendly method and does not require an organic solvent for drug loading.67
The Melt Method
The melt method is carried out by heating a physical mixture of cocrystal and MPS above the melting point of the cocrystal.25,77 This method is efficient and significantly reduces the time for incorporating drug-coformer (cocrystal) into MPS. However, its limitations include suitability for thermally stable drug and coformer characterized by a low viscosity after melting. The high molten viscosity of drug-coformer can reduce the penetration of drug-coformer into MPS and block mesopores.23,78 Skerupska et al reported a series of studies, where drug-coformers (benzoic acid-fluorinated benzoic acid, naproxen-picolinamide, and ibuprofen-nicotinamide) were incorporated into MPS using the melt method. The result showed that the interaction mode between drug and coformer was formed within MPS after being evaluated using two-dimensional solid-state NMR spectroscopy.7,26,27 Salas-Zuniga also stated that the PZQ-GLU cocrystal was formed after incorporation into MPS using the melt method.64
Co-Milling
Co-milling is a common method of producing sub-micrometric particles and facilitating solid-state amorphization.79,80 Previous studies have reported that milling using a planetary ball mill is an effective method for incorporating organic compounds such as benzoic acid and 4-fluoro benzoic acid into the pores of MPS.81 The results suggest that the guest-loading process is highly controllable, with a maximum loading capacity of over 50% at a 1:1 ratio. This shows the potential of the co-milling method as an alternative to incorporating drug-coformer into MPS. Moreover, this method is simple and time-efficient, making it applicable on an industrial scale. Treciak et al also reported the incorporation of BA and 4-FBA into MPS using a co-milling method. Solid-state NMR studies clarified that the interaction mode between the BA and 4-FBA was similar to cocrystal, indicating the formation of BA −4-FBA within MPS.65
Characterization of Drug-Coformer Loaded MPS
Nitrogen Adsorption/Desorption Analysis
Physical gas sorption is a well-established method used to characterize the textural properties of solid surfaces and changes after various actions. This method can precisely determine the amount of gas adsorbed on the surface material measuring the porous properties and structure. The isotherm obtained from the adsorption measurements can provide information on the pore volume (PV), surface area (SA), and pore size distribution (PSD).82–84 Regarding MPS, the changes in the porous structure and drug-loaded MPS are estimated based on the course of nitrogen adsorption-desorption isotherms of type IV at 77K and a wide range of relative pressures (p/p0).67 Skorupska et al reported the nitrogen adsorption/desorption analysis to assess the loading of NPX: PA into the pores of SBA-15. According to BET calculations, the SA, PV, and mean pore diameter of SBA-15 are 747 m2g−1, 1.21 cm3g−1, and 100 Å, respectively. A significant decrease in specific surface area of 24% and 39% was observed for samples with NPX:PA cocrystal/MPS ratios equal to 1:1 and 1:2 (weight-to-weight). The SA of NPX:PA cocrystal/MPS was also found to be 177.9 m2g−1 and 287.9 m2g−1, respectively. The analysis of MPS volumes, relative pressures, and pore diameter indicated the successful encapsulation of the NPX:PA cocrystal into MPS. After short-term ethanol diffusion, MPS diameter for the NPX:PA/SBA-15 was slightly smaller compared to NPX:PA/SBA-15. This phenomenon occurred due to the high affinity of the polar solvents in the pores of MPS nanoparticles.27
Scanning Electron Microscopy (SEM)
SEM serves as a powerful tool for investigating various nanostructured materials, including MPS. This tool can provide detailed structural characterization on the nanometer scale such as external diameter, surface area, morphology, and pore architecture. Furthermore, it is one of the most popular microscopic tools for characterization of nanomaterials. SEM creates images of the sample including API within MPS by scanning the surface with an electron beam, visualizing, and providing a 3D view.67 Salas-Zuniga et al (2022) used SEM analysis to examine the morphology of SBA-15, PZQ-GLU cocrystal, and PZQ-GLU/SBA-15 50:50 w/w.64 The micrographs of SBA-15 particles showed a uniform rod-like morphology, as described by Zhang et al,85 while the PZQ–GLU cocrystal indicated a smooth surface composed of micrometer-sized crystals. PZQ-GLU/SBA-15 50:50 w/w showed rod-like morphology and there were no visible crystalline particles at the surface. This indicated that cocrystal of PZQ-GLU was loaded inside the nanopores of SBA–15.86 Ali et al also reported SEM images of VAL-VAN coamorphous loaded MPS, indicating a rougher surface morphology.61 The smooth appearance of VAL-VAN coamorphous loaded MPS was due to its partial entry into the pore volume, leading to a uniform texture of the particles.87
Powder X-ray Diffraction (PXRD)
Powder X-ray diffraction (PXRD) has been used to analyze the degree of crystallinity of drug-coformer within MPS. In a previous study by Ali et al (2019) the X-ray diffraction results were reported for VAL-VAN within MPS. The X-ray diffractogram showed distinct crystalline peaks for VAL between 20° and 40°, indicating its crystalline nature.61 However, these peaks disappeared in X-ray patterns of VAL-VAN coamorphous and VAL-VAN/MPS, indicating that co-amorphous forms were retained in the pores of MPS.88–90 Salas-Zuniga et al (2022) also reported the PXRD results of PZQ within SBA-15 and PZQ-GLU within SBA-15. As illustrated in Figure 5, the PXRD pattern of SBA-15 showed a wide halo typical of amorphous MPS, while the PZQ-GLU cocrystal showed diffraction peaks of crystalline materials. When SBA-15 and PZQ-GLU were combined at a 50:50 weight-to-weight ratio, broad halo and low-intensity diffraction peaks, resembling a cocrystalline solid, were observed, as presented in Figure 6. Diffractograms in composites with a decreased cocrystal concentration showed an amorphous phase-like appearance.64 Based on these results, PXRD measurement can be used to determine the maximum amount of drug-coformer (cocrystal) within MPS, which is attributed to the presence of diffraction peaks typical of cocrystal.
 |
Figure 6 PXRD patterns of PZQ-GLU cocrystal loaded SBA-15 with various weight ratios. Notes: Adapted with permission from Salas-Zúñiga R, Mondragón-Vásquez K, Alcalá-Alcalá S, et al. Nanoconfinement of a pharmaceutical cocrystal with praziquantel in mesoporous silica: the influence of the solid form on dissolution enhancement. Mol Pharm. 2022;19(2):414–431.64 [41]. Copyright 2022 American Chemical Society. |
Differential Scanning Calorimetry (DSC) Measurement
DSC is used to characterize the physical state, location of drug, and coformer within MPS by observing their glass transition temperature of melting point. Observations have shown that when drug and coformer are confined in the pores of MPS, the melting point of cocrystal and/or Tg of coamorphous system would be lower compared to the crystalline state. Salas-Zuniga et al conducted a DSC measurement of PZQ-GLU-loaded SBA-15 at a 50:50 w/w ratio. In this study, the crystalline solid of PZQ and PZQ-GLU showed a single melting transition at 138.8 °C and 123.0 °C, respectively. However, composite SBA-15/PZQ did not show an endothermic peak for crystalline PZQ, which was indicative of the amorphous state of the confined drug. Regarding PZQ-GLU loaded SBA-15 at 50:50 w/w ratio, two broad Tg were observed at lower temperatures (Tonset = 63.0 °C and Tonset = 117.2 °C) compared to the bulk cocrystal.64 Confined nanocrystals in MPS were reported to show a broadening of the melting peak and depression of the melting temperature.91–93 The decrease in the melting temperature due to nanocrystallization of the drug has been explained quantitatively by the Gibbs−Thomson equation.94,95 A previous study also reported that the Tg of the drug was commonly reduced by confinement in a porous structure of MPS due to an intrinsic size effect. This phenomenon indicated that the mobility of drug within MPS was higher than outside MPS.95–98
Budiman et al conducted an analysis of the MDSC curve of RTN-SAC 1:1 coamorphous within TMPS, as shown in Figure 7. The result showed that the heat capacity changes (ΔCp) of the glass transition event or RTN-SAC coamorphous decreased with a reduction in the RTN-SAC concentration. At a low weight ratio of RTN-SAC 1:1 coamorphous/TMPS = 3:7, the glass transition was not detected in the MDSC curve, indicating that all of the RTN and SAC were successfully incorporated into TMPS. Furthermore, the glass transition event for the RTN-SAC 1:1 coamorphous/TMPS with a higher RTN-SAC weight ratio was observed. This indicated that the RTN amorphous or SAC was unloaded in the TMPS. Based on the results, the Tg of RTN-SAC 1:1 coamorphous/TMPS = 5:5 and 7:3 were different from coamorphous. This phenomenon occurred because the remaining compositions of RTN and SAC unloaded into the TMPS were lower than 1:1, resulting in a reduced Tg at a higher RTN-SAC coamorphous weight ratio.62
 |
Figure 7 Reversing heat flow MDSC curves of RTN crystal, RTN-SAC 1:1 coamorphous, RTN-SAC 1:1 coamorphous/TMPS = 7:3, 5:5, and 3:7. Notes: Adapted from International Journal of Pharmaceutics, Volume 600, Budiman A, Higashi K, Ueda K, Moribe K, Effect of drug-coformer interactions on drug dissolution from a coamorphous in mesoporous silica, pages 120492. Copyright 2021, with permisison from Elsevier.62 |
Fourier Transform Infrared (FT-IR) Spectroscopy
FT-IR spectroscopy is a universal study tool used to examine the chemical status of MPS surfaces. In recent years, FT-IR spectroscopy has become an indispensable tool for pharmaceutical analysis. Understanding the position of the absorption bands of Si–O bond serves as the basis for analyzing MPS using FT-IR spectroscopy.99–101 According to previous investigations, the changes in the appearance of functional groups on the FT-IR spectra of MPS and drug indicates the interactions occurring between drug and MPS.102 The reduction in the intensity of drug functional group bands loaded into MPS can be attributed to a strong molecular interaction between drug and MPS.103 In binary systems loaded into MPS, it is often challenging to investigate the interaction between drug and coformer. Consequently, FT-IR spectra are initially investigated to acquire insight into a probable molecular interaction between drug and coformer as comparative data for drug-coformer interaction within MPS. Bi et al used FT-IR spectroscopy as the first investigation to clarify the interaction between IBU and NA within MPS, as illustrated in Figure 8. In cocrystal system, the N-H stretching peaks of NA shifted from 3364 cm−1 to 3401 cm−1 and from 3160 cm−1 to 3182 cm−1, while the C=O stretching peak of IBU adjusted from 1721 cm−1 to 1707 cm−1. These results indicated the formation of hydrogen bonds between the carboxyl group of IBU and the acylamino group of NA. The cocrystal system showed a π-π interaction between IBU and NA molecule when the two peaks were at 1506 cm−1 (IBU) and 1485 cm−1 (NA), reflecting the skeleton vibrations of the aromatic rings, merged into a single one (1516 cm−1). The IBU-NA-loaded MPS showed the same spectrum properties as cocrystal system. The similarity between the IR spectra of The IBU-NA loaded MPS and cocrystal system yields an indirect confirmation of the existence of IBU-NA interaction in MPS.28
 |
Figure 8 FTIR spectra for each sample. Notes: Reprinted from Journal of Pharmaceutical Sciences, Volume 106/Edition 10, Bi Y, Xiao D, Ren S, Bi S, Wang J, Li F, The binary system of ibuprofen-nicotinamide under nanoscale confinement: from cocrystal to coamorphous state, pages 3150–3155,28 Copyright 2017, with permisison from Elsevier. |
FTIR spectroscopy was used to investigate the molecular interactions of VAL and VAN within MPS. The spectra obtained confirmed the formation of a co-amorphous system, followed by its subsequent loading into MPS. The resulting spectrum corroborated the co-amorphous formation system and its incorporation into MPS, with VAN showing an O-H peak of VAN above 3100 cm−1.104 Meanwhile, the observed absence of VAN in the VAL-VAN mixture suggested the possibility of intermolecular interactions occurring in the coamorphous system. The shift of the C=O peak of VAL from 1728 cm−1 to 1715 cm−1 was also attributed to hydrogen bonding interactions, which can immobilize the O-atom leading to a retarded C=O stretching. As the majority of VAN and VAL peaks disappeared, only the Si-O-Si peak at 1099 cm−1 was observed, suggesting the confinement of VAL-VAN coamorphous in the pores of MPS. Additionally, the presence of diverse Si-OH groups was included in the interaction within the coamorphous system, resulting in the disappearance of the VAN and VAL peaks.61
Raman Spectroscopy
Raman spectroscopy is a method used for identifying the molecular composition from the vibration frequencies of molecular bonds.105–108 This method has been used to evaluate small Raman shifts, allowing for the direct differentiation of intermolecular bonds in addition to intramolecular bonds.109,110 The deformations of the Raman peak in intramolecular bonds show the adsorption of molecular crystals on the surfaces,111,112 playing an important role in cocrystallization of porous materials. Therefore, Raman spectroscopy could be a potential method of analyzing pharmaceutical cocrystal in MPS with high resolution in frequency and spatial regions. Ohta et al reported the Raman spectra of caffeine-oxalic acid cocrystal with that of SBA-16, and compared to caffeine, (b) oxalic acid, and (c) caffeine-oxalic acid cocrystal without SBA-16. The spectrum of the intramolecular bond in cocrystal above 400 cm–1 is similar to caffeine but the peak frequencies of the two spectra are slightly different. This is because the intermolecular bond of caffeine and oxalic acid modified the electron distribution of the intramolecular bond in caffeine. Subsequently, each vibrational mode is slightly modified when the molecules are trapped in SBA-16. This phenomenon occurs because the intermolecular interactions from SBA-16 modify the electron distribution of the intra- and inter-molecules. The peak frequencies at 32, 56, and 83 cm–1 are slightly shifted when cocrystals are synthesized in SBA-16, indicating a dominant formation on the surface of SBA-16 without aggregation.63
The Raman spectra are examined in the range from 530 to 670 cm−1 to provide a more detailed description of the formation mechanism of cocrystal in SBA-16. In caffeine crystals and caffeine-oxalic acid cocrystal, two peaks originating from intramolecular bond of δ(CNC) and δ(OCN) in a caffeine molecule are observed in this region.113–115 These peak's alterations in frequency and bandwidth are observed when caffeine crystals and cocrystal are added to SBA-16. The Raman spectra of cocrystal with SBA-16 are shown in Figure 9 along with the curve fitted using the four spectra (pink) and SBA-16 (sand). For each spectrum, the molar ratios of cocrystal (caffeine/oxalic acid) are (a) 27/13, (b) 54/27, (c) 80/40, and (d) 107/54 μmol, respectively. By comparing the pink and black curves for all spectra, the cocrystal with SBA-16 can be broken down into its parts. These include cocrystal without SBA-16, caffeine with and without SBA-16, and the background of SBA-16. This analysis offers valuable insights into the cocrystallization of caffeine and oxalic acid within SBA-16, as shown by the peak intensities of each component at different molar quantities.63
 |
Figure 9 Raman spectra of cocrystal with MPS (black line) and the curve (pink line) fitted by cocrystal without MPS (red line), caffeine crystal with and without MPS (blue and green lines), and background of MPS (sand line). Notes: Adapted from Analytical Sciences, Volume 33/Edition 1, Ohta R, Ueno Y, Ajito K, Raman spectroscopy of pharmaceutical cocrystals in nanosized pores of mesoporous silica, pages 47–52, Copyright 2017, with permisison from Springer Nature.63 |
The formation of caffeine-oxalic acid cocrystal in SBA-16 is explained by the relative peak intensities of each component at different molar quantities, as shown in Figure 10(a). This procedure can be divided into two categories. In the first phase (A), where the amount of caffeine is smaller than 67 μmol, the peak intensity of caffeine with SBA-16 linearly increases, while the cocrystal remains constant. This observation indicates that caffeine and oxalic acid are trapped on the surface of SBA-16, forming monolayers without, as schematically shown in Figure 10(b). In the second phase (B) presented in Figure 10(c), the peak intensity of caffeine with SBA-16 saturates at roughly 67 μmol, indicating that caffeine and oxalic acid have completely occupied the surface of SBA-16. After saturation occurs, the peak intensity of cocrystal without SBA-16 increases in tandem with caffeine without SBA-16. As illustrated in Figure 10(d), cocrystal is produced on the monolayer caffeine and oxalic acid, stacking by layers without aggregation. The growth rate proceeds slowly enough to regulate the thickness for a few-nanometer-scale cocrystal due to the huge surface area of SBA-16. Additionally, by selecting the proper molar ratios of caffeine and oxalic acid, it is possible to create a bi-layer and tri-layer cocrystal. In this situation, the use of 134 and 201 μmol would yield bi-layer and tri-layer cocrystal, respectively.63
 |
Figure 10 (a) Relative peak intensities of cocrystal without MPS and caffeine with and without MPS at various molar ratios. The molar amount dependence is categorized into situations A and B. Schematic illustration of cocrystal caffeine and oxalic acid on the surface of MPS (b) in the situation A, (c) at the boundary of A and B, and (d) in the situation B. Notes: Adapted from Analytical Sciences, Volume 33/Edition 1, Ohta R, Ueno Y, Ajito K, Raman spectroscopy of pharmaceutical cocrystals in nanosized pores of mesoporous silica, pages 47–52, Copyright 2017, with permisison from Springer Nature.63 |
Solid-State NMR (SS NMR)
Solid-state (SS) NMR is a valuable tool used for investigating the molecular and physicochemical properties of API, providing the broadest possible spectrum of structural information on condensed matter.116 Furthermore, SS NMR can be applied to any solid physical state either crystalline or amorphous, and materials of different complexity, from pure APIs and excipients to amorphous solid dispersions.117 The development of fast magic angle spinning (MAS)118–121 and single-pulse experiment (SPE), makes the SS NMR the main method to study mesoporous materials, particularly drugs confined in MPS, drug-coformer interaction within MPS, and API-MPS interactions.122,123 The cross-polarization/magic angle spinning experiments have been conducted by Bi et al using a double-tuned cross-polarization/magic angle spinning probe to justify the formation of coamorphous systems of IBU-NA loaded MPS. The spectra of the NCA system show a 0.4–0.5 ppm downfield shift of the methyl resonances of IBU and a 0.6 ppm upfield shift of the carboxyl resonance when compared to the spectrum of physical mixture of IBU and NA-loaded MPS. These changes in chemical shifts are attributed to the molecular interactions between IBU and NA within MPS. Theoretically, the very weak molecular interaction of drug-coformer in a coamorphous system offers a unique opportunity to distinguish the coamorpous system from the physical mixture of each amorphous component.28 Skorupska et al reported the 1H MAS and 19F MAS spectra of BA/FBA within MCM41 recorded with the spinning rate of 28 kHz at ambient temperature. The differences in spectra between BA/FBA cocrystal and BA/FBA loaded MCM41 were observed, with significantly better resolution in the proton spectra for BA/FBA loaded MCM41, particularly in the fluorine data. This indicated that cocrystal-loaded MPS was in a fast exchange regime due to rapid molecular motion.7
Skorupska et al also used an SPE 13C MAS measurement to investigate NPX:PA cocrystal loaded MCM-41 to confirm the location of drug-coformer within MPS. The 13C CP/MAS NMR was conducted to show the spectrum of physical mixture of NPX:PA/MCM-41, as presented in Figure 11a. Specifically, the 13C CP/MAS spectrum observed in the NPX:PA/MCM-41 after thermal treatment was only the NPX component. When conducting the SPE 13C MAS measurement with a short relaxation delay, which was suitable for mobile components, mainly PA resonances were monitored in the sample of NPX:PA/MCM-41 after thermal treatment. Based on these results, the molecular dynamics between NPX and PA were significantly different, consisting of rigid NPX and mobile PA, which were observed and unobserved in the cross-polarization, respectively. This showed that the PA was located inside the pore, while NPX was on the silica surface of MPS.27
 |
Figure 11 13C MAS NMR of each sample.27 Notes: Adapted with permission from Skorupska E, Jeziorna A, Potrzebowski MJ. Thermal solvent-free method of loading of pharmaceutical cocrystals into the pores of silica particles: a Case of naproxen/picolinamide cocrystal. J Phys Chem C. 2016;120(24):13169–13180. Copyright 2016 American Chemical Society. |
A 1H−19F HOESY MAS measurement was conducted to further analyze the interactions between BA and FBA within MPS. This method was found suitable in a system with fast molecular motion. Figure 12 showed a HOESY spectrum with clear correlation peaks suggesting the proximity between proton and fluorine spins. This indicated that the cross-peaks reflected the π- π-stacking interactions more possibly rather than contacts inside the isolated heterodimer.27
 |
Figure 12 1H−19F HOESY MAS NMR spectra of BA/FBA loaded MCM-41 = 1:1 w/w with a spinning rate of 12 kHz. Notes: Adapted with permission from Skorupska E, Jeziorna A, Potrzebowski MJ. Thermal solvent-free method of loading of pharmaceutical cocrystals into the pores of silica particles: a Case of naproxen/picolinamide cocrystal. J Phys Chem C. 2016;120(24):13169–13180.27 Copyright 2016 American Chemical Society. |
Previous studies have conducted SS NMR to confirm the interaction of RTN-SAC within TMPS. 13C PST/MAS NMR (υ = 15 kHz) was performed to detect both mobile and rigid components. For RTN amorphous/TMPS, the relative peak intensity of thiazole groups peaks at 179 ppm increased in the 13C PST/MAS NMR spectrum compared to the 13C CP/MAS NMR spectrum, as presented in Figure 13. The difference in spectra between the 13C PST/MAS NMR and 13C CP/MAS NMR was detected at 179 ppm in the sample of RTN amorphous/TMPS. The enhanced peak in the PST/MAS NMR spectrum suggested that the thiazole groups in RTN were highly mobile in the RTN amorphous/TMPS. In contrast, the difference in spectra between the 13C PST/MAS NMR and 13C CP/MAS NMR was not significant in the RTN-SAC 1:1 coamorphous/TMPS. This indicated that the mobility of the thiazole groups in RTN within TMPS was suppressed through hydrogen bond interaction of RTN-SAC, as observed in the RTN-SAC coamorphous.62
 |
Figure 13 13C CP/MAS NMR spectra (υ = 15 kHz) and 13C PST/ MAS NMR spectra (υ = 15 kHz) of RTN amorphous/TMPS and RTN-SAC 1:1 coamorphous/TMPS. Notes: Adapted from International Journal of Pharmaceutics, Volume 600, Budiman A, Higashi K, Ueda K, Moribe K, Effect of drug-coformer interactions on drug dissolution from a coamorphous in mesoporous silica, pages 120492. Copyright 2021, with permisison from Elsevier.62 |
Evaluation of Dissolution Performance
The dissolution study was conducted to evaluate the behavior of drugs in the drug-coformer loaded MPS after dispersion in dissolution medium. Understanding this dissolution behavior is important in solid formulation development and regulatory assessment. Bi et al conducted the in vitro drug release experiments of IBU-NA/MPS under non-sink conditions. The results showed that IBU-NA cocrystal and IBU crystal reached their equilibrium solubility state after 150 min. In contrast, the IBU amorphous/MPS and IBU-NA/MPS showed rapid release, which was achieved within 5 min. This phenomenon occurred due to the nanosized effect of drugs induced by the confinement of MPS leading to monomolecular dispersion of drugs in the dissolution medium. Specifically, IBU-NA/MPS showed higher equilibrium solubility compared to that of IBU/MPS. The high solubility of IBU in IBU-NA/MPS was attributed to the hydrotropic effect of NA and the confinement effect of MPS. Moreover, the addition of an equal amount of NA into IBU/MPS in pure water can achieve the same solubility with IBU-NA/MPS, confirming the hydrotropic effect of NA.28
A previous study showed a different result as presented in Figure 14 compared to the report of Bi et al.62 In the RTN/MPS system, the rapid release occurred at the beginning of the dissolution test, suggesting good dispersibility in the dissolution medium. Meanwhile, in the RTN-SAC coamorphous/MPS, dissolution of RTN was initially slow compared to the RTN amorphous/MPS at the beginning of the dissolution test. The high concentration of RTN was maintained to be longer in the presence of SAC than in amorphous/MPS. A similar result was also observed in the physical mixture (PM) of the RTN amorphous/TMPS + SAC crystal, indicating that SAC crystal did not affect the dissolution of RTN amorphous/MPS. This result suggested that the incorporation of coformer into mesopores extended the maintenance time of drug supersaturation through hydrogen bond interaction between drug and coformer within MPS.62
 |
Figure 14 Dissolution profiles of each sample in 100 mM phosphate buffer (pH 6.8) at 37 ◦C. (n = 3, mean ± S. D). Notes: Adapted from International Journal of Pharmaceutics, Volume 600, Budiman A, Higashi K, Ueda K, Moribe K, Effect of drug-coformer interactions on drug dissolution from a coamorphous in mesoporous silica, pages 120492. Copyright 2021, with permisison from Elsevier.62 |
Trzeciak et al and Salas-Zuniga et al reported no significant difference in the dissolution profile of drugs between drug/MPS and drug-coformer/MPS systems. Although the rapid supersaturation of drugs occurs at the beginning of the dissolution test in the confined solids in MPS, the concentration of RTN gradually decreases due to RTN recrystallization in the dissolution medium even in the presence of coformer. This phenomenon occurs because the water contacting to drug or coformer can break their interaction after dispersion into water. The dissolution profile in the drug-coformer/MPS system could be similar to drug/MPS, despite the drug-coformer interaction being observed in the solid state.64,66
Physical Stability
In the drug-coformer-loaded MPS system, a physical stability test was conducted to evaluate the impact of drug-coformer interaction within MPS in the stabilization of the amorphous drug. The physical stability of VAL-VAN loaded MPS was monitored by evaluating their dissolution profiles upon storage. However, the dissolution profile of VAL in VAL-VAN loaded MPS did not significantly change over 8 weeks compared to the observation made on day 0. VAL-VAN coamorphous showed a significant decrease in the rate of dissolution (~35%) after the storage period due to the recrystallization, in comparison with day 0 (~94%). Physical stability improvement of VAL-VAN coamorphous after loaded MPS was attributed to the nanoconfinement effect, preventing the recrystallization and maintaining a stable dissolution profile of the amorphous drug.61,124 Physical stability of drug-coformer loaded MPS was also monitored from PXRD measurements. After 45 days of storage at 60 oC, some small crystalline peaks were observed on the PXRD pattern of drug amorphous/MPS, while the drug-coformer/MPS system maintained the halo patterns. This indicated that the presence of coformer within MPS can further improve physical stability of the amorphous drug.28 To confirm the impact of drug-coformer interaction within MPS on physical stability of the amorphous drug, the dispersed solid sample in dissolution medium of drug amorphous/MPS and coamarphous/MPS system were collected through centrifugation, leading to the preparation of freeze-dried samples. The PXRD patterns showed that some crystalline peaks of the drug were observed for the sample of drug amorphous/MPS due to recrystallization occurring during the dissolution test. In contrast, the sample of RTN-SAC 1:1 amorphous/TMPS showed a small RTN peak, indicating that drug-coformer interaction within MPS could improve physical stability of the amorphous drug after dispersion in water.62
Author Perspective
Molecular State of the Drug-Coformer Within MPS
The molecular state of the drug-coformer within MPS is discussed in Figure 15. In the pores of MPS, the molecular mobility of each component can be used to confirm the localization of drug and coformer. The cocrystal of BA-FBA within MPS showed high molecular mobility compared to the cocrystal without MPS, suggesting its location in the center of MPS due to an intrinsic effect. Meanwhile, in the case of the cocrystal of NPX-PA, NPX mobility was different from PA. NPX was observed in the cross-polarization (low mobility), while PA was not found in the cross-polarization (high mobility). This suggested that PA was located in the center of MPS, while NPX was on the silica surface. According to Chen et al, the two Tgs (Tg low and Tg high) were observed in the nifedipine confined in the porous media, commonly found in the small molecules loaded with the porous nanoconfinement. The change in Tg was affected by the interaction between the surface of MPS and the molecules, as well as the size of the confinement. The Tg of the amorphous drug within MPS was lower compared to the amorphous counterpart without MPS, indicating its high molecular mobility due to an intrinsic size effect. In contrast, the surface interaction between the pore wall and molecules led to a lower molecular mobility and a higher Tg compared to the amorphous drug located outside MPS. According to a previous study, the mobility of RTN was suppressed after loaded MPS, which was different from the commonly observed small molecules. The strong surface interactions between RTN and MPS could restrict the mobility of adsorbed RTN, forming a monolayer on the mesoporous surface.98,125,126 Strong interactions between the C=O of RTN and the Si-OH of MPS through hydrogen bonding have been reported.21 Consequently, when the RTN-SAC was incorporated into MPS, the RTN should be located in the surface or walls of MPS due to the surface interaction between RTN and MPS.
 |
Figure 15 Schematic illustration of drug-coformer/MPS located (a) in the center of MPS, and (b) on the surface of MPS. |
Drug Release Mechanism
The speculated mechanism of drug dissolution from drug-coformer-loaded MPS is summarized in Figure 16. The high supersaturation of drug within MPS occurred due to monomolecular dispersion in dissolution medium through the interaction of water molecules with the drug. This made the drug rapidly dissolve at the beginning of the dissolution test. Moreover, drug-coformer interaction and the nanoconfinement effect of MPS contributed to the improvement of solubility and stability from poorly water-soluble drugs. Based on the previous studies, the mechanism of drugs in the drug-coformer-loaded MPS system was divided into three parts, as illustrated in Figure 16. These included (1) the presence of coformer can increase the solubility of the amorphous drug due to the hydrotropic effect from coformer after dispersion in water. The hydrophobic interaction between drug with the hydrophobic part of the coformer was formed, leading to aggregation activity above its minimum hydrotrope concentration (MHC).127 (2) The presence of the coformer could suppress the water contact with the drug in MPS. This was because the hydrophilic part of the drug was occupied through hydrogen bonding with the coformer, leading to a slower dissolution rate compared to drug amorphous/MPS. The remaining hydrogen bond between the drug and coformer in the solution could inhibit drug crystallization and maintain supersaturation. Therefore, the formation of drug-cofomer interaction in the mesopore contributed to the maintenance of drug supersaturation for extended periods. (3) The presence of the coformer did not affect the dissolution behavior of the drug, as the remaining molecular interaction between the drug and coformer in the solution was not observed. Although the drug-coformer interaction was observed in the solid state, the dissolution profile of the drug in the drug-coformer/MPS system could be similar to drug/MPS.
 |
Figure 16 Schematic illustration of drug dissolution from drug-coformer/MPS. |
The Impact of This System on the Drug Therapy
The monomolecularly dispersed drug within MPS can then rapidly dissolve into the dissolution medium which plays a critical role in the absorption improvement of the poorly water-soluble drug. Previous studies reported that poorly water-soluble drugs’ absorption was enhanced when API was in a supersaturated state, which was commonly observed in the drug-loaded MPS. The addition of coformer in the MPS could increase the solubility or maintain the supersaturated solution of amorphous drugs in the aqueous compartment of the body, such as interstitial space resulting in the bioavailability improvement of amorphous drugs. Based on the Fick law of diffusion, the mechanism of drug absorption commonly occurs by passive diffusion, in which the drugs will move from a higher concentration to a lower concentration of drugs until equilibrium is reached. Thus, the solubility of the drug in the aqueous compartment of the body could significantly affect the amount of drug absorption via passive diffusion. The drug absorption of the drug’s crystal was lower compared to that of its amorphous state because of its poor water solubility.
Meanwhile, in the system of drug-coformer loaded MPS, the amount of drug absorption would be higher compared to drug-loaded MPS because the presence of coformer within MPS could improve the solubility of drugs and maintain the supersaturated solution in the aqueous compartment of the body through the intermolecular interaction between drug and coformer in the MPS. As a result, it was assumed that the absorption of poorly water-soluble drugs in the drug-coformer/MPS formulation was mainly passive diffusion where only molecularly dissolved API was absorbed by the intestinal epithelium. Furthermore, the permeability of the drug could only be increased by high concentrations of dispersed API. Therefore, the presence of coformer in drug-loaded MPS systems could enhance the concentration of molecularly dissolved API leading to the improvement of drug absorption. Improving the bioavailability of poorly water-soluble drugs could enhance their pharmacokinetics, efficacy, and safety.
Conclusions
In conclusion, this study contributes to elucidating mechanisms of drug-coformer within MPS and its dissolution and drug distribution in an aqueous environment. The drug-coformer within MPS system emerged as an appealing alternative for improving the limitations associated with the utilization of binary systems, such as drug loaded MPS and co-amorphous/cocrystal systems. The presence of coformer within MPS has the potential to improve their pharmaceutical properties, including solubility, dissolution profile, and physical stability. However, there were challenges in the preparation of the drug-coformer within MPS system, especially in choosing an appropriate coformer and a preparation method. Moreover, it would be difficult to characterize the mechanism of drug-coformer within MPS including the interaction mechanism due to their complex systems. Therefore, these could be challenges for researchers when developing an amorphous formulation of the drug-coformer within MPS system. Furthermore, this study provided fundamental insights into the drug-coformer within MPS system, which is significant when formulating strategies for improving the pharmaceutical properties of poorly water-soluble drugs.
Acknowledgments
We would like to thank Universitas Padjadjaran for supporting this work and APC.
Funding
This research was funded by Universitas Padjadjaran (Riset Kolaborasi Indonesia (RKI)) to A.B. (No. 2213/UN6.3.1/TU.00/2023, 11 Mei 2023). (CC-BY-NC).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Budiman A, Nurfadilah N, Muchtaridi M, Sriwidodo S, Aulifa DL, Rusdin A. The impact of water-soluble chitosan on the inhibition of crystal nucleation of alpha-mangostin from. Polymers. 2022;14(20):4370. doi:10.3390/polym14204370
2. Budiman A, Citraloka ZG, Muchtaridi M, Sriwidodo S, Aulifa DL, Rusdin A. Inhibition of crystal nucleation and growth in aqueous drug solutions: impact of different polymers on the supersaturation profiles of amorphous drugs—the case of alpha-mangostin. Pharmaceutics. 2022;14(11):2386. doi:10.3390/pharmaceutics14112386
3. Budiman A, Aulifa DL. Characterization of drugs with good glass formers in loaded-mesoporous silica and its theoretical value relevance with mesopores surface and pore-filling capacity. Pharmaceuticals. 2022;15(1):1–16. doi:10.3390/ph15010093
4. Al-Kassas R, Bansal M, Shaw J. Nanosizing techniques for improving bioavailability of drugs. J Control Release. 2017;260(28):202–212. doi:10.1016/j.jconrel.2017.06.003
5. Johnson JLH, He Y, Yalkowsky SH. Prediction of precipitation-induced phlebitis: a statistical validation of an in vitro model. J Pharm Sci. 2003;92(8):1574–1581. doi:10.1002/jps.10396
6. Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J Pharm Sci. 1996;85(10):1017–1025. doi:10.1021/js950534b
7. Skorupska E, Paluch P, Jeziorna A, Potrzebowski MJ. NMR study of BA/FBA cocrystal confined within mesoporous silica nanoparticles employing thermal solid phase transformation. J Phys Chem C. 2015;119(16):8652–8661. doi:10.1021/jp5123008
8. Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev. 2001;48(1):27–42. doi:10.1016/S0169-409X(01)00098-9
9. Hancock BC, Parks M. What is the true solubility advantage of amorphous pharmaceuticals? Pharm Res. 2000;17:397–403. doi:10.1023/A:1007516718048
10. Azad M, Moreno J, Davé R. Stable and fast-dissolving amorphous drug composites preparation via impregnation of Neusilin® UFL2. J Pharm Sci. 2018;107(1):170–182. doi:10.1016/j.xphs.2017.10.007
11. Yamamoto K, Kojima T, Karashima M, Ikeda Y. Physicochemical evaluation and developability assessment of co-amorphouses of low soluble drugs and comparison to the co-crystals. Chem Pharm Bull. 2016;64(12):1739–1746. doi:10.1248/cpb.c16-00604
12. Moinuddin SM, Ruan S, Huang Y, et al. Facile formation of co-amorphous atenolol and hydrochlorothiazide mixtures via cryogenic-milling: enhanced physical stability, dissolution and pharmacokinetic profile. Int J Pharm. 2017;532(1):393–400. doi:10.1016/j.ijpharm.2017.09.020
13. Newman A, Reutzel-Edens SM, Zografi G, et al. Coamorphous active pharmaceutical ingredient-small molecule mixtures: considerations in the choice of coformers for enhancing dissolution and oral bioavailability. J Pharm Sci. 2018;107:5–17. doi:10.1016/j.xphs.2017.09.024
14. Löbmann K, Grohganz H, Laitinen R, Strachan C, Rades T. Amino acids as co-amorphous stabilizers for poorly water soluble drugs - Part 1: preparation, stability and dissolution enhancement. Eur J Pharm Biopharm. 2013;85(3 PART B):873–881. doi:10.1016/j.ejpb.2013.03.014
15. Korhonen O, Pajula K, Laitinen R. Rational excipient selection for co-amorphous formulations. Expert Opin Drug Deliv. 2017;14(4):551–569. doi:10.1080/17425247.2016.1198770
16. Shi Q, Moinuddin SM, Cai T. Advances in coamorphous drug delivery systems. Acta Pharm Sin B. 2019;9(1):19–35. doi:10.1016/j.apsb.2018.08.002
17. Aitipamula S, Banerjee R, Bansal AK, et al. Polymorphs, salts, and cocrystals: what’s in a name? Cryst Growth Des. 2012;5(12):2147–2152. doi:10.1021/cg3002948
18. Duggirala NK, Perry ML, Almarsson Ö, Zaworotko MJ. Pharmaceutical cocrystals: along the path to improved medicines. Chem Commun. 2016;52(4):640–655. doi:10.1039/c5cc08216a
19. Bavishi DD, Borkhataria CH. Spring and parachute: how cocrystals enhance solubility. Prog Cryst Growth Charact Mater. 2016;62(3):1–8. doi:10.1016/j.pcrysgrow.2016.07.001
20. Pallavi P, Harini K, Alshehri S, et al. From synthetic route of silica nanoparticles to theranostic applications. Processes. 2022;10(12):2595. doi:10.3390/pr10122595
21. Dening TJ, Zemlyanov D, Taylor LS. Application of an adsorption isotherm to explain incomplete drug release from ordered mesoporous silica materials under supersaturating conditions. J Control Release. 2019;307(March):186–199. doi:10.1016/j.jconrel.2019.06.028
22. Qian KENK, Bogner RH. Spontaneous crystalline-to-amorphous phase transformation of organic or medicinal compounds in the presence of porous media, part 1 : thermodynamics of spontaneous amorphization. J Pharm Sci. 2011;100(7):2801–2815. doi:10.1002/jps
23. Mellaerts R, Jammaer JAG. Physical state of poorly water soluble therapeutic molecules loaded into SBA-15 ordered mesoporous silica carriers : a case study with itraconazole and ibuprofen. Langmuir. 2008;24(16):8651–8659. doi:10.1021/la801161g
24. Van Speybroeck M, Mellaerts R, Mols R, et al. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur J Pharm Sci. 2010;41(5):623–630. doi:10.1016/j.ejps.2010.09.002
25. Mccarthy CA, Ahern RJ, Dontireddy R, Ryan KB, Crean AM. Mesoporous silica formulation strategies for drug dissolution enhancement: a review. Expert Opin Drug Deliv. 2016;13(1):93–108. doi:10.1517/17425247.2016.1100165
26. Skorupska E, Kaźmierski S, Potrzebowski MJ. Solid state NMR characterization of ibuprofen:nicotinamide cocrystals and new idea for controlling release of drugs embedded into mesoporous silica particles. Mol Pharm. 2017;14(5):1800–1810. doi:10.1021/acs.molpharmaceut.7b00092
27. Skorupska E, Jeziorna A, Potrzebowski MJ. Thermal solvent-free method of loading of pharmaceutical cocrystals into the pores of silica particles: a Case of naproxen/picolinamide cocrystal. J Phys Chem C. 2016;120(24):13169–13180. doi:10.1021/acs.jpcc.6b05302
28. Bi Y, Xiao D, Ren S, Bi S, Wang J, Li F. The binary system of ibuprofen-nicotinamide under nanoscale confinement: from cocrystal to coamorphous state. J Pharm Sci. 2017;106(10):3150–3155. doi:10.1016/j.xphs.2017.06.005
29. Basavoju S, Boström D, Velaga SP. Indomethacin-saccharin cocrystal: design, synthesis and preliminary pharmaceutical characterization. Pharm Res. 2008;25(3):530–541. doi:10.1007/s11095-007-9394-1
30. Gao Y, Zu H, Zhang J. Enhanced dissolution and stability of Adefovir dipivoxil by cocrystal formation. J Pharm Pharmacol. 2011;63(4):483–490. doi:10.1111/j.2042-7158.2010.01246.x
31. Karki S, Friščić T, Fábián L, Laity PR, Day GM, Jones W. Improving mechanical properties of crystalline solids by cocrystal formation: new compressible forms of paracetamol. Adv Mater. 2009;21(38–39):3905–3909. doi:10.1002/adma.200900533
32. Good DJ, Rodríguez-Hornedo N. Solubility advantage of pharmaceutical cocrystals. Cryst Growth Des. 2009;9(5):2252–2264. doi:10.1021/cg801039j
33. Wouters J, Rome S, Quéré L. Monographs of Most Frequent Co-Crystal Formers; 2011.
34. Blagden N, Colesb SJ, Berry DJ. Pharmaceutical co-crystals—are we there yet? Cryst Eng Comm. 2014;16(26):5753–5761. doi:10.1039/C4CE00127C
35. Etter MC. Hydrogen bonds as design elements in organic chemistry. J Phys Chem. 1991;95(12):4601–4610. doi:10.1021/j100165a007
36. Kuminek G, Cao F, da Rocha ABDO, Cardoso SG, Rodríguez-Hornedo N. Cocrystals to facilitate delivery of poorly soluble compounds beyond-rule-of-5. Adv Drug Deliv Rev. 2016;101:143–166. doi:10.1016/j.addr.2016.04.022
37. Etter MC. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc Chem Res. 1990;23(4):120–126. doi:10.1021/ar00172a005
38. Donohue J. The hydrogen bond in organic crystals. J Phys Chem. 1952;56:502–510. doi:10.1021/j150496a023
39. Alhalaweh A, Roy L, Rodríguez-Hornedo N, Velaga SP. pH-dependent solubility of indomethacin-saccharin and carbamazepine- saccharin cocrystals in aqueous media. Mol Pharm. 2012;9(9):2605–2612. doi:10.1021/mp300189b
40. Roy L, Lipert MP, Rodríguez-Hornedo N. Co-crystal solubility and thermodynamic stability. Pharm Salts Co-Crystals. 2012;2012;247–279.
41. Good D, Miranda C, Rodríguez-Hornedo N. Dependence of cocrystal formation and thermodynamic stability on moisture sorption by amorphous polymer. CrystEngComm. 2011;13(4):1181–1189. doi:10.1039/c0ce00592d
42. Childs SL, Rodríguez-Hornedo N, Reddy LS, et al. Screening strategies based on solubility and solution composition generate pharmaceutically acceptable cocrystals of carbamazepine. CrystEngComm. 2008;10(7):856–864. doi:10.1039/b715396a
43. Chadha R, Saini A, Arora P, Jain DS, Dasgupta A, Guru Row TN. Multicomponent solids of lamotrigine with some selected coformers and their characterization by thermoanalytical, spectroscopic and X-ray diffraction methods. CrystEngComm. 2011;13(20):6271–6284. doi:10.1039/c1ce05458a
44. Laitinen R, Löbmann K, Grohganz H, Priemel P, Strachan CJ, Rades T. Supersaturating drug delivery systems: the potential of co-amorphous drug formulations. Int J Pharm. 2017;532(1):1–12. doi:10.1016/j.ijpharm.2017.08.123
45. Sun DD, Wen H, Taylor LS. Non-sink dissolution conditions for predicting product quality and in vivo performance of supersaturating drug delivery systems. J Pharm Sci. 2016;105(9):2477–2488. doi:10.1016/j.xphs.2016.03.024
46. Taylor LS, Zhang GGZ. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv Drug Deliv Rev. 2016;101:22–142. doi:10.1016/j.addr.2016.03.006
47. Wang R, Han J, Jiang A, et al. Involvement of metabolism-permeability in enhancing the oral bioavailability of curcumin in excipient-free solid dispersions co-formed with piperine. Int J Pharm. 2019;561(20):9–18. doi:10.1016/j.ijpharm.2019.02.027
48. Wei Y, Zhou S, Hao T, et al. Further enhanced dissolution and oral bioavailability of docetaxel by coamorphization with a natural P-gp inhibitor myricetin. Eur J Pharm Sci. 2019;129:21–30. doi:10.1016/j.ejps.2018.12.016
49. Ojarinta R, Heikkinen AT, Sievänen E, Laitinen R. Dissolution behavior of co-amorphous amino acid-indomethacin mixtures: the ability of amino acids to stabilize the supersaturated state of indomethacin. Eur J Pharm Biopharm. 2017;112(85):85–95. doi:10.1016/j.ejpb.2016.11.023
50. Kresge CT, Roth WJ. The discovery of mesoporous molecular sieves from the twenty year perspective. Chem Soc Rev. 2013;42(9):3663–3670. doi:10.1039/c3cs60016e
51. Bremmell KE, Prestidge CA. Enhancing oral bioavailability of poorly soluble drugs with mesoporous silica based systems: opportunities and challenges. Drug Dev Ind Pharm. 2019;45(3):349–358. doi:10.1080/03639045.2018.1542709
52. Maleki A, Kettiger H, Schoubben A, Rosenholm JM, Ambrogi V, Hamidi M. Mesoporous silica materials: from physico-chemical properties to enhanced dissolution of poorly water-soluble drugs. J Control Release. 2017;262:329–347. doi:10.1016/j.jconrel.2017.07.047
53. Diab R, Canilho N, Pavel IA, Haffner FB, Girardon M, Pasc A. Silica-based systems for oral delivery of drugs, macromolecules and cells. Adv Colloid Interface Sci. 2017;249:346–362. doi:10.1016/j.cis.2017.04.005
54. Simovic S, Ghouchi-Eskandar N, Moom Sinn A, et al. Silica materials in drug delivery applications. Curr Drug Discov Technol. 2011;8(3):250–268. doi:10.2174/157016311796799026
55. Xu B, Li S, Shi R, et al. Multifunctional mesoporous silica nanoparticles for biomedical applications. Signal Transduct Target Ther. 2023;8(1):435. doi:10.1038/s41392-023-01654-7
56. McCarthy CA, Ahern RJ, Devine KJ, Crean AM. Role of drug adsorption onto the silica surface in drug release from mesoporous silica systems. Mol Pharm. 2018;15(1):141–149. doi:10.1021/acs.molpharmaceut.7b00778
57. Martín A, Morales V, Ortiz-Bustos J, et al.. Modelling the adsorption and controlled release of drugs from the pure and amino surface-functionalized mesoporous silica hosts. Microporous Mesoporous Mater. 2018;262:23–24. doi:10.1016/j.micromeso.2017.11.009
58. Fu C, Liu T, Li L, Liu H, Chen D, Tang F. The absorption, distribution, excretion and toxicity of mesoporous silica nanoparticles in mice following different exposure routes. Biomaterials. 2013;34(10):2565–2575. doi:10.1016/j.biomaterials.2012.12.043
59. Li L, Liu T, Fu C, et al. Biodistribution, excretion, and toxicity of mesoporous silica nanoparticles after oral administration depend on their shape. Nanomed Nanotech Biol Med. 2015;11(8):1915–1924. doi:10.1016/j.nano.2015.07.004
60. Huang X, Li L, Liu T, et al. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano. 2011;5(7):5390–5399. doi:10.1021/nn200365a
61. Hashim Ali K, Mohsin Ansari M, Ali Shah F, et al. Enhanced dissolution of valsartan-vanillin binary co-amorphous system loaded in mesoporous silica particles. J Microencapsul. 2019;36(1):10–20. doi:10.1080/02652048.2019.1579265
62. Budiman A, Higashi K, Ueda K, Moribe K. Effect of drug-coformer interactions on drug dissolution from a coamorphous in mesoporous silica. Int J Pharm. 2021;600(March):120492. doi:10.1016/j.ijpharm.2021.120492
63. Ohta R, Ueno Y, Ajito K. Raman spectroscopy of pharmaceutical cocrystals in nanosized pores of mesoporous silica. Anal Sci. 2017;33(1):47–52. doi:10.2116/analsci.33.47
64. Salas-Zúñiga R, Mondragón-Vásquez K, Alcalá-Alcalá S, et al. Nanoconfinement of a pharmaceutical cocrystal with praziquantel in mesoporous silica: the influence of the solid form on dissolution enhancement. Mol Pharm. 2022;19(2):414–431. doi:10.1021/acs.molpharmaceut.1c00606
65. Trzeciak K, Wielgus E, Kaźmierski S, et al.Unexpected factors affecting the kinetics of guest molecule release from investigation of binary chemical systems trapped in a single void of mesoporous silica particles. Eur J Chem Phys Phys Chem. 2022;24(7):25.
66. Trzeciak K, Kaźmierski S, Wielgus E, Potrzebowski MJ. DiSupLo - new extremely easy and efficient method for loading of active pharmaceutical ingredients into the pores of MCM-41 mesoporous silica particles. Microporous Mesoporous Mater. 2020;308:110506. doi:10.1016/j.micromeso.2020.110506
67. Trzeciak K, Chotera‐ouda A, Bak‐sypien II, Potrzebowski MJ. Mesoporous silica particles as drug delivery systems—the state of the art in loading methods and the recent progress in analytical techniques for monitoring these processes. Pharmaceutics. 2021;13(7):950. doi:10.3390/pharmaceutics13070950
68. European Medicines Agency. ICH guideline Q3C (R6) on impurities: guideline for residual solvents. Int Conf Harmon Tech Requir Regist Pharm Hum Use. 2019;31(August):1.
69. Eren ZS, Tunçer S, Gezer G, Yildirim LT, Banerjee S, Yilmaz A. Improved solubility of celecoxib by inclusion in SBA-15 mesoporous silica: drug loading in different solvents and release. Microporous Mesoporous Mater. 2016;235:211–223. doi:10.1016/j.micromeso.2016.08.014
70. Hillerström A, van Stam J, Andersson M. Ibuprofen loading into mesostructured silica using liquid carbon dioxide as a solvent. Green Chem. 2009;11:662–667. doi:10.1039/b821281c
71. Belhadj-Ahmed F, Badens E, Llewellyn P, Denoyel R, Charbit G. Impregnation of vitamin E acetate on silica mesoporous phases using supercritical carbon dioxide. J Supercrit Fluids. 2009;51:278–286. doi:10.1016/j.supflu.2009.07.012
72. Lehto VP, Riikonen J. Drug Loading and Characterization of Porous Silicon Materials. Woodhead Publishing Limited; 2014. doi:10.1533/9780857097156.3.337
73. Brás AR, Fonseca IM, Dionísio M, Schönhals A, Affouard F, Correia NT. Influence of nanoscale confinement on the molecular mobility of ibuprofen. J Phys Chem C. 2014;118(25):13857–13868. doi:10.1021/jp500630m
74. Wang Y, Zhao Q, Han N, et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine Nanotechnology, Biol Med. 2015;11(2):313–327. doi:10.1016/j.nano.2014.09.014
75. Abd-Elbary A, El Nabarawi MA, Hassen DH, Taha AA. Inclusion and characterization of ketoprofen into different mesoporous silica nanoparticles using three loading methods. Int J Pharm Pharm Sci. 2014;6(9):183–191.
76. He Y, Liang S, Long M, Xu H. Mesoporous silica nanoparticles as potential carriers for enhanced drug solubility of paclitaxel. Mater Sci Eng C. 2017;78:12–17. doi:10.1016/j.msec.2017.04.049
77. Uejo F, Limwikrant W, Moribe K, Yamamoto K. Dissolution improvement of fenofibrate by melting inclusion in mesoporous silica. Asian J Pharm Sci. 2013;8(6):329–335. doi:10.1016/j.ajps.2013.11.001
78. Niu X, Wan L, Hou Z, et al. Mesoporous carbon as a novel drug carrier of fenofibrate for enhancement of the dissolution and oral bioavailability. Int J Pharm. 2013;452(1–2):382–389. doi:10.1016/j.ijpharm.2013.05.016
79. Hampsey JE, De Castro CL, McCaughey B, Wang D, Mitchell BS, Lu Y. Preparation of micrometer- to sub-micrometer-sized nanostructured silica particles using high-energy ball milling. J Am Ceram Soc. 2004;87(7):1280–1286. doi:10.1111/j.1151-2916.2004.tb07723.x
80. Willart JF, Descamps M. ChemInform abstract: solid state amorphization of pharmaceuticals. ChemInform. 2009;40(3):905–920. doi:10.1002/chin.200903277
81. Trzeciak K, Kaźmierski S, Drużbicki K, Potrzebowski MJ. Mapping of guest localization in mesoporous silica particles by solid-state NMR and Ab initio modeling: new insights into benzoic acid and p-fluorobenzoic acid embedded in MCM-41 via ball milling. J Phys Chem C. 2021;125(18):10096–10109. doi:10.1021/acs.jpcc.1c01675
82. Sing KS. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl Chem. 1985;57(4):603–619. doi:10.1351/pac198557040603
83. Thommes M, Kaneko K, Neimark AV, et al.. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC technical report). Pure Appl Chem. 2015;87(9–10):1051–1069. doi:10.1515/pac-2014-1117
84. Rouquerol J, Avnir D, Everett DH, et al.. Guidelines for the characterization of porous solids. Stud Surf Sci Catal. 1994;87:1–9.
85. Zhang Y, Wang J, Bai X, Jiang T, Zhang Q, Wang S. Mesoporous silica nanoparticles for increasing the oral bioavailability and permeation of poorly water soluble drugs. Mol Pharm. 2012;9(3):505–513. doi:10.1021/mp200287c
86. Sayed E, Karavasili C, Ruparelia K, et al. Electrosprayed mesoporous particles for improved aqueous solubility of a poorly water soluble anticancer agent: in vitro and ex vivo evaluation. J Control Release. 2018;278:142–155. doi:10.1016/j.jconrel.2018.03.031
87. Giunchedi P, Juliano C, Gavini E, Cossu M, Sorrenti M. Formulation and in vivo evaluation of chlorhexidine buccal tablets prepared using drug-loaded chitosan microspheres. Eur J Pharm Biopharm. 2002;53(2):233–239. doi:10.1016/S0939-6411(01)00237-5
88. Laitinen R, Löbmann K, Grohganz H, Strachan C, Rades T. Amino acids as Co-amorphous excipients for simvastatin and glibenclamide: physical properties and stability. Mol Pharm. 2014;11(7):2381–2389. doi:10.1021/mp500107s
89. Yamamura S, Gotoh H, Sakamoto Y, Momose Y. Physicochemical properties of amorphous precipitates of cimetidine-indomethacin binary system. Eur J Pharm Biopharm. 2000;49(3):259–265. doi:10.1016/S0939-6411(00)00060-6
90. Wang J, Chang R, Zhao Y, et al. Coamorphous loratadine-citric acid system with enhanced physical stability and bioavailability. AAPS Pharm Sci Tech. 2017;18(7):2541–2550. doi:10.1208/s12249-017-0734-0
91. Nartowski KP, Tedder J, Braun DE, Fábián L, Khimyak YZ. Building solids inside nano-space: from confined amorphous through confined solvate to confined “metastable” polymorph. Phys Chem Chem Phys. 2015;17(38):24761–24773. doi:10.1039/c5cp03880d
92. Nartowski KP, Malhotra D, Hawarden LE, Fábián L, Khimyak YZ. Nanocrystallization of rare tolbutamide form V in mesoporous MCM-41 Silica. Mol Pharm. 2018;15:4926–4932. doi:10.1021/acs.molpharmaceut.8b00575s
93. Hamilton BD, Ha JM, Hillmyer MA, Ward MD. Manipulating crystal growth and polymorphism by confinement in nanoscale crystallization chambers. Acc Chem Res. 2012;45(3):414–423. doi:10.1021/ar200147v
94. Dwyer LM, Michaelis VK, O’Mahony M, Griffin RG, Myerson AS. Confined crystallization of fenofibrate in nanoporous silica. CrystEngComm. 2015;17(41):7922–7929. doi:10.1039/c5ce01148e
95. Cheng S, McKenna GB. Nanoconfinement effects on the glass transition and crystallization behaviors of nifedipine. Mol Pharm. 2019;16(2):856–866. doi:10.1021/acs.molpharmaceut.8b01172
96. Koh YP, Simon SL. Trimerization of monocyanate ester in nanopores. J Phys Chem B. 2010;114(23):7727–7734. doi:10.1021/jp912235c
97. Li W, Quan P, Zhang Y, et al. Influence of drug physicochemical properties on absorption of water insoluble drug nanosuspensions. Int J Pharm. 2014;460(1–2):13–23. doi:10.1016/j.ijpharm.2013.10.038
98. Li Q, Simon SL. Curing of bisphenol M dicyanate ester under nanoscale constraint. Macromolecules. 2008;41(4):1310–1317. doi:10.1021/ma702144b
99. Wu H, Xiao Y, Guo Y, Miao S, Chen Q, Chen Z. Functionalization of SBA-15 mesoporous materials with 2-acetylthiophene for adsorption of Cr(III) ions. Microporous Mesoporous Mater. 2020;292(May 2019):109754. doi:10.1016/j.micromeso.2019.109754
100. Melnyk IV, Nazarchuk GI, Václavíková M, Zub YL. IR spectroscopy study of SBA-15 silicas functionalized with the ethylthiocarbamidepropyl groups and their interactions with Ag (I) and Hg (II) ions. Appl Nanosci. 2019;9:683–694. doi:10.1007/s13204-018-0761-5
101. Tomozawa M, Hong JW, Ryu SR. Infrared (IR) investigation of the structural changes of silica glasses with fictive temperature. J Non Cryst Solids. 2005;351(12–13):1054–1060. doi:10.1016/j.jnoncrysol.2005.01.017
102. Acharya M, Mishra S, Sahoo RN, Mallick S. Infrared spectroscopy for analysis of co-processed ibuprofen and magnesium trisilicate at milling and freeze drying. Acta Chim Slov. 2017;64(1):45–54. doi:10.17344/acsi.2016.2772
103. Kinnari P, Mäkilä E, Heikkilä T, Salonen J, Hirvonen J, Santos HA. Comparison of mesoporous silicon and non-ordered mesoporous silica materials as drug carriers for itraconazole. Int J Pharm. 2011;414(1–2):148–156. doi:10.1016/j.ijpharm.2011.05.021
104. Peng H, Xiong H, Li J, et al. Vanillin cross-linked chitosan microspheres for controlled release of resveratrol. Food Chem. 2010;121(1):23–28. doi:10.1016/j.foodchem.2009.11.085
105. Kong K, Kendall C, Stone N, Notingher I. Raman spectroscopy for medical diagnostics - from in-vitro biofluid assays to in-vivo cancer detection. Adv Drug Deliv Rev. 2015;89:121–134. doi:10.1016/j.addr.2015.03.009
106. Huang N, Short M, Zhao J, et al.. Full range characterization of the Raman spectra of organs in a murine model. Opt Express. 2011;19(23):22892–22909. doi:10.1364/OE.19.022892
107. Li S, Chen G, Zhang Y, et al.. Identification and characterization of colorectal cancer using Raman spectroscopy and feature selection techniques. Opt Express. 2014;22(21):25895–25908. doi:10.1364/OE.22.025895
108. Ajito K, Han C, Torimitsu K. Detection of glutamate in optically trapped single nerve terminals by Raman spectroscopy. Anal Chem. 2004;76(9):2506–2510. doi:10.1021/ac049969m
109. Moser C, Havermeyer F. Ultra-narrow-band tunable laserline notch filter. Appl Phys B Lasers Opt. 2009;95(3):597–601. doi:10.1007/s00340-009-3447-6
110. Parrott EPJ, Zeitler JA. Terahertz time-domain and low-frequency Raman spectroscopy of organic materials. Appl Spectrosc. 2015;69(1):1–25. doi:10.1366/14-07707
111. Hédoux A, Decroix AA, Guinet Y, Paccou L, Derollez P, Descamps M. Low- and high-frequency Raman investigations on caffeine: polymorphism, disorder and phase transformation. J Phys Chem B. 2011;115(19):5746–5753. doi:10.1021/jp112074w
112. Allesø M, Velaga S, Alhalaweh A, et al. Near-infrared spectroscopy for cocrystal screening. A comparative study with Raman spectroscopy. Anal Chem. 2008;80(20):7755–7764. doi:10.1021/ac8011329
113. Edwards HGM, Munshi T, Anstis M. Raman spectroscopic characterisations and analytical discrimination between caffeine and demethylated analogues of pharmaceutical relevance. Spectrochim Acta - Part A Mol Biomol Spectrosc. 2005;61(7):1453–1459. doi:10.1016/j.saa.2004.10.022
114. Pavel I, Szeghalmi A, Moigno D, Cîntă S, Kiefer W. Theoretical and pH dependent surface enhanced Raman spectroscopy study on caffeine. Biopolym Orig Res Biomol. 2003;72(1):25–37.
115. Kang J, Gu H, Zhong L, Hu Y, Liu F. The pH dependent Raman spectroscopic study of caffeine. Spectrochimica acta part A. Mol Biomol Spectrosc. 2011;78(2):757–762. doi:10.1016/j.saa.2010.11.055
116. Merisko-Liversidge EM, Liversidge GG. Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol Pathol. 2008;36(1):43–48. doi:10.1177/0192623307310946
117. Geppi M, Mollica G, Borsacchi S, Veracini CA. Solid-state NMR studies of pharmaceutical systems. Appl Spectrosc Rev. 2008;43(3):202–302. doi:10.1080/05704920801944338
118. Trébosc J, Wiench JW, Huh S, Lin VSY, Pruski M. Solid-state MMR study of MCM-41-type mesoporous silica nanoparticles. J Am Chem Soc. 2005;127(9):3057–3068. doi:10.1021/ja043567e
119. Trebosc J, Wiench JW, Huh S, Lin VSY, Pruski M. Studies of organically functionalized mesoporous silicas using heteronuclear solid-state correlation NMR spectroscopy under fast magic angle spinning. J Am Chem Soc. 2005;127(20):7587–7593. doi:10.1021/ja0509127
120. Wiench JW, Avadhut YS, Maity N, et al.. Characterization of covalent linkages in organically functionalized MCM-41 mesoporous materials by solid-state NMR and theoretical calculations. J Phys Chem B. 2007;111(15):3877–3885. doi:10.1021/jp067417x
121. Mao K, Kobayashi T, Wiench JW, et al.. Conformations of silica-bound (pentafluorophenyl) propyl groups determined by solid-state NMR spectroscopy and theoretical calculations. J Am Chem Soc. 2010;132(35):12452–12457. doi:10.1021/ja105007b
122. Baccile N. Application of advanced solid-state nmr techniques to the characterization of nanomaterials: a focus on interfaces and structure; 2010.
123. Shenderovich IG, Limbach HH. Solid state NMR for nonexperts: an overview of simple but general practical methods. Solids. 2021;2(2):139–154. doi:10.3390/solids2020009
124. Qian KK, Bogner RH. Application of mesoporous silicon dioxide and silicate in oral amorphous drug delivery systems. J Pharm Sci. 2012;101(7):2271–2280. doi:10.1002/jps
125. Chieng N, Aaltonen J, Saville D, Rades T. Physical characterization and stability of amorphous indomethacin and ranitidine hydrochloride binary systems prepared by mechanical activation. Eur J Pharm Biopharm. 2009;71(1):47–54. doi:10.1016/j.ejpb.2008.06.022
126. Kissi EO, Ruggiero MT, Hempel NJ, et al. Characterising glass transition temperatures and glass dynamics in mesoporous silica-based amorphous drugs. Phys Chem Chem Phys. 2019;21(35):19686–19694. doi:10.1039/c9cp01764j
127. Paul R, Chattaraj KG, Paul S. Role of hydrotropes in sparingly soluble drug solubilization: insight from a molecular dynamics simulation and experimental perspectives. Langmuir. 2021;37(16):4745–4762. doi:10.1021/acs.langmuir.1c00169
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.