Back to Journals » Lung Cancer: Targets and Therapy » Volume 9
Dramatic response to alectinib in a lung cancer patient with a novel VKORC1L1-ALK fusion and an acquired ALK T1151K mutation
Authors Zhu VW ![]() , Schrock AB, Bosemani T, Benn BS, Ali SM, Ou SI
, Schrock AB, Bosemani T, Benn BS, Ali SM, Ou SI ![]()
Received 7 September 2018
Accepted for publication 4 October 2018
Published 8 November 2018 Volume 2018:9 Pages 111—116
DOI https://doi.org/10.2147/LCTT.S186804
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Antonio Araujo
Viola W Zhu,1 Alexa B Schrock,2 Thangavijayan Bosemani,3 Bryan S Benn,4 Siraj M Ali,2 Sai-Hong Ignatius Ou1
1Chao Family Comprehensive Cancer Center, Division of Hematology/Oncology, Department of Medicine, University of California, Irvine School of Medicine, Orange, CA, USA; 2Clinical Development, Foundation Medicine, Inc., Cambridge, MA, USA; 3Department of Radiological Sciences, University of California, Irvine School of Medicine, Orange, CA, USA; 4Division of Pulmonary Diseases and Critical Care Medicine, Department of Medicine, University of California, Irvine School of Medicine, Orange, CA, USA
Abstract: ALK-rearranged lung cancer defines a distinctive molecular cohort of patients whose outcomes are significantly improved by the availability of ALK inhibitors. Thus, it is imperative for clinicians to screen appropriate patients for this driver mutation with a molecular testing platform capable of capturing all ALK fusions. Here, we report a novel VKORC1L1-ALK fusion and an ALK T1151K resistance mutation detected in a lung cancer patient who had been on crizotinib for over 8 years. Alectinib induced a dramatic response in this patient demonstrating its clinical activity against T1151K. This case illustrates the importance of performing repeat biopsy to explore mechanism(s) of resistance when patients experience disease progression on an ALK inhibitor. The approach has a direct therapeutic impact particularly when an ALK resistance mutation is identified.
Keywords: VKORC1L1, T1151, fusion, resistance, crizotinib, lorlatinib
Introduction
In the US, there are now four approved ALK inhibitors for patients with metastatic ALK-rearranged lung cancer and many more at various development stages. Because of significant clinical benefits these ALK inhibitors provide, it is imperative for clinicians to screen appropriate patients for this driver mutation with a molecular testing platform capable of capturing all ALK fusions. For patients who progress on an ALK inhibitor, performing repeat biopsy to look for mechanism(s) of resistance is highly recommended. Identifying an ALK resistance mutation may guide selection of another ALK inhibitor with known activity against the specific mutation. Here, we report a novel VKORC1L1-ALK fusion with an acquired ALK T1151K resistance mutation detected in a patient who had disease progression after being on crizotinib for over 8 years. He had a dramatic response to alectinib illustrated in the following narrative indicating that T1151K confers sensitivity to alectinib.
Case presentation
A 54-year-old Caucasian man, former smoker of <5 pack years, originally underwent resection for locally advanced adenocarcinoma of the lung in April 2006 followed by adjuvant chemoradiation with weekly carboplatin/paclitaxel for focally positive margin. He was found to have metastatic disease in his pelvis in November 2008 for which he received palliative radiation. In October 2009, a computed tomography (CT) scan performed for cough showed multiple bilateral subcentimeter lung nodules and a large left pleural effusion. However, the patient had elected not to receive any treatment until February 2010 when he presented to our cancer center to discuss participation in clinical trial. The molecular profiling performed at the Massachusetts General Hospital revealed an ALK rearrangement, so he was enrolled in the PROFILE 1001 study (NCT00585195) in March 2010. He had a sustained partial response (PR, –27% by RECIST 1.1) to crizotinib at 250 mg twice daily confirmed by a contrast CT scan in October 2012. He declined further scans afterwards, and so he was switched to commercial supply of crizotinib in February 2013.
In April 2018, the patient developed dry cough while on crizotinib for which a contrast CT scan of the chest was performed that showed complete opacification of the left hemithorax (Figure 1). Restaging scans confirmed disease progression in both lungs, thoracic lymph nodes, pleura, and bone. He was immediately switched to alectinib at 600 mg twice daily while comprehensive genomic profiling (CGP) via Foundation Medicine, Inc. (Cambridge, MA, USA) was performed on a plasma-based liquid biopsy which subsequently identified a novel VKORC1L1-ALK fusion as well as an ALK T1151K resistance mutation. This CGP assay covers 62 genes to ×5,000 unique coverage and includes intron baiting for rearrangements of six genes (ALK, EGFR, FGFR3, PDGFR-α, ROS1, and RET). Alectinib was continued based on predicted efficacy of this ALK inhibitor against T1151K. A follow-up contrast CT scan of the chest 14 weeks later showed a dramatic response (–76% by RECIST 1.1) including resolution of a 2.9 cm left paravertebral mass at T1–2 level, decrease in size of the right pulmonary nodule from 1.8 to 1.1 cm, and significant improvement of the left lung aeration (Figure 1). Of note, prior to this follow-up scan, the patient underwent endobronchial cryosurgery for tissue extraction and spray cryotherapy of an endobronchial mass in order to prevent any significant pulmonary hemorrhage and for airway recanalization, but we did not obtain enough tissue for CGP. At the time of this manuscript preparation, the patient has been on alectinib for over 23 weeks with no reported symptoms. A written informed consent was obtained to have the case details published, and institutional approval was not required to publish this case report.
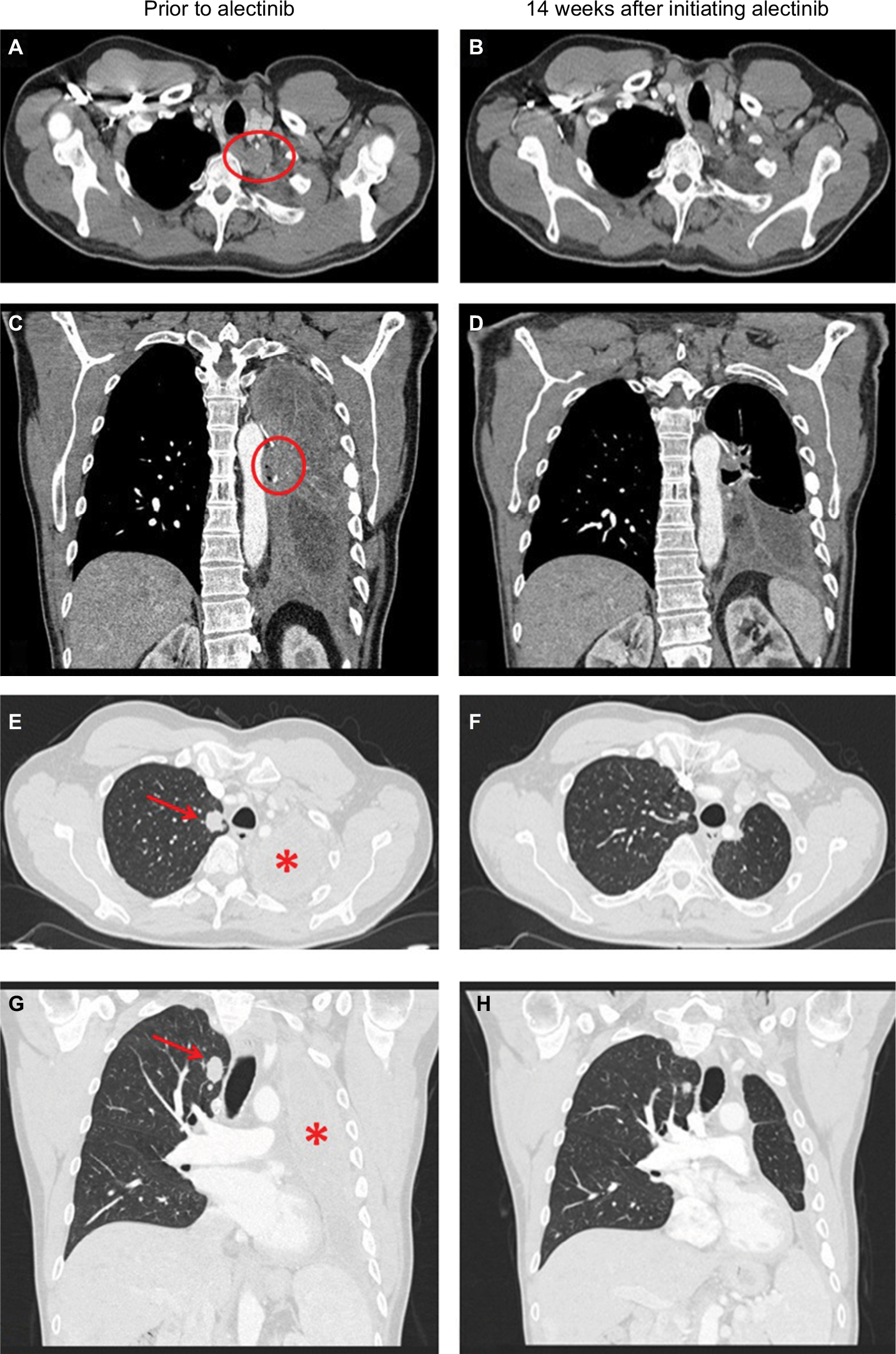 | Figure 1 Contrast CT scans of the chest showing a left paravertebral mass at T1–2 level (indicated by a red circle) prior to use of alectinib (A and C) and resolution of this mass 14 weeks after initiating alectinib (B and D). Contrast CT scans of the chest showing decrease in size of the right pulmonary nodule (indicated by a red arrow) and significant improvement of the left lung aeration (indicated by a red star) 14 weeks after initiating alectinib (F and H) as compared with prior to use of alectinib (E and G). Abbreviation: CT, computed tomography. |
Discussion
We believe this is the first report of VKORC1L1-ALK fusion detected in a lung cancer patient. Ever since the discovery of the transforming EML4-ALK fusion gene by Soda and Rikova et al in 2007, various ALK fusion partners have been identified including TFG, KIF5B, KCL1, PTPN3, STRN, HIP1, TPR, BIRC6, DCTN1, SQSTM1, SOCS5, CLIP4, CLTC, PRKAR1A, PPM1B, EIF2AK3, CRIM1, GCC2, DYSF, ITGAV, VIT, PLEKHA7, and CUX1.1–19 A list of chronologically published fusion partners in ALK-rearranged lung cancer with references is summarized in Table 1. The VKORC1L1 gene encodes an enzyme known as vitamin K 2,3-epoxide reductase, whose catalytic function is critical for the vitamin K cycle. The vitamin K-dependent proteins have various physiological roles that are not just restricted to coagulation and hemostasis. Although little is known of the functions of these proteins in direct tumorigenesis, they may contribute to remodeling of tumor microenvironment as vascular endothelial cell survival depends on vitamin K. In addition, the mRNA and protein expressions of VKORC1L1 in lung have been reported.20 The VKORC1L1 gene is located on chromosome 7q11.21 and only contains three exons with 531 nucleotides. The encoded protein product has 176 amino acids (aa) with four endoplasmic reticulum transmembrane domains. In this case, exon 1 of VKORC1L1 (aa 1–64) was fused with exons 20–29 (aa 1058–1620) of the ALK gene generating the final fusion protein which contains the full ALK tyrosine kinase domain (aa 1116–1329) (Figure 2). Interestingly, our patient’s tumor was found to harbor ALK rearrangement in 2010, but the specific fusion partner could not be identified by fluorescence in situ hybridization at the time.
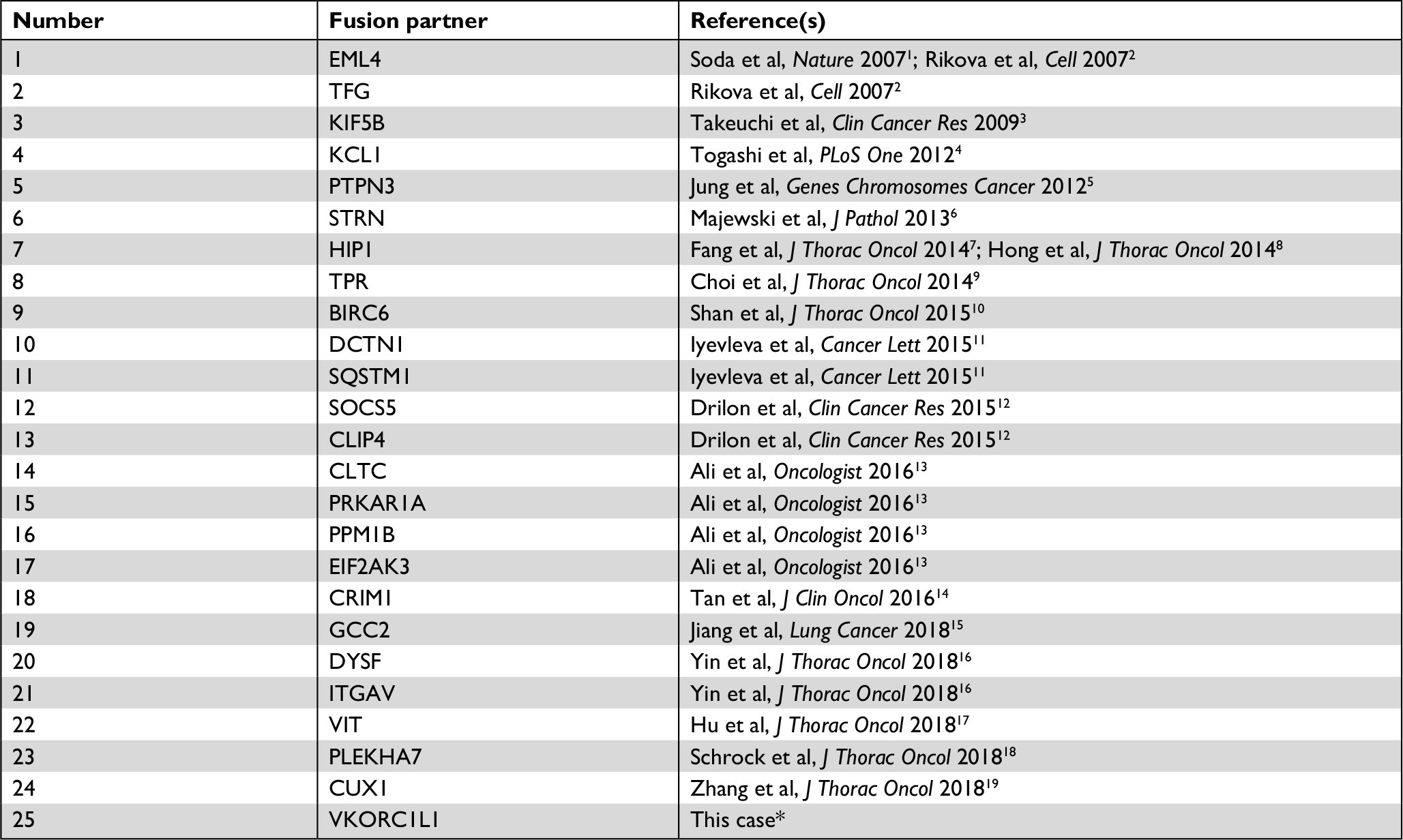 | Table 1 Published fusion partners in ALK-rearranged lung cancer Notes: *This fusion partner was detected in conjunction with an acquired ALK resistance mutation. |
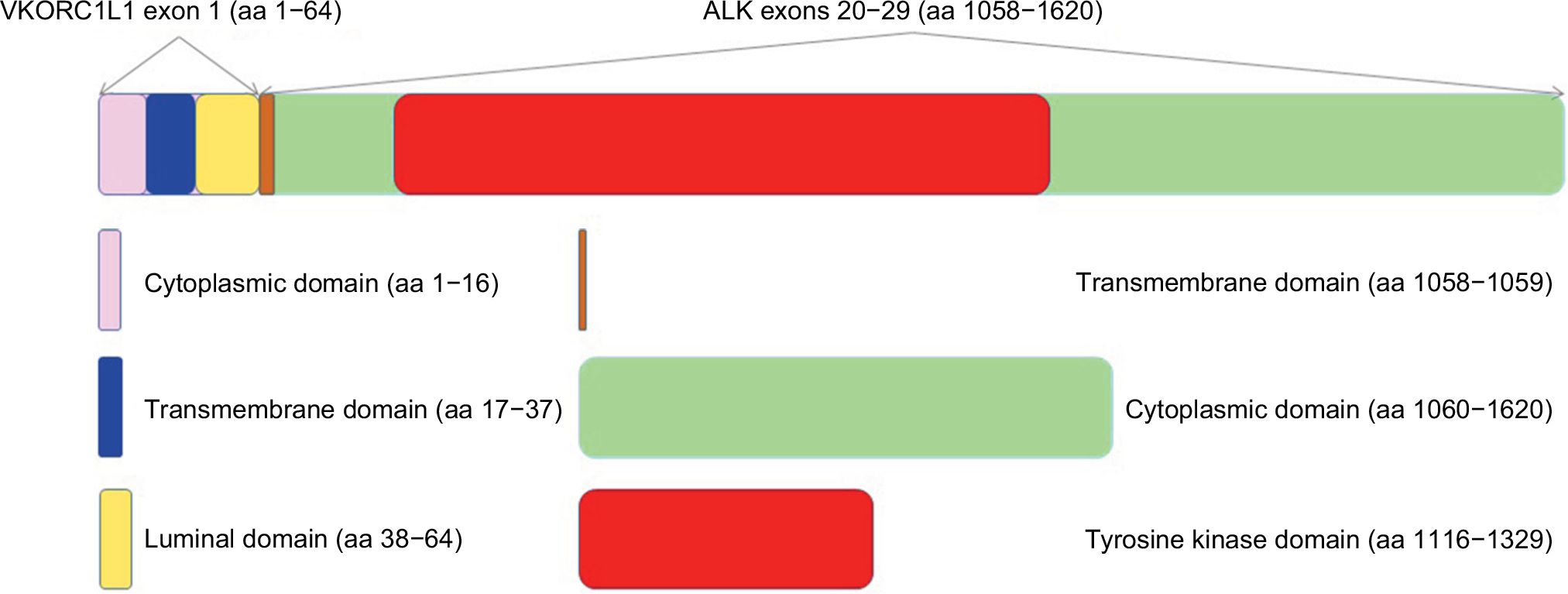 | Figure 2 Schema of the VKORC1L1-ALK fusion protein. Notes: The VKORC1L1 gene only contains three exons with 531 nucleotides. The encoded protein product has 176 aa with four endoplasmic reticulum transmembrane domains (aa 17–37, aa 92–112, aa 114–134, and aa 135–155). In this case, exon 1 of VKORC1L1 (aa 1–64) is fused with exons 20–29 (aa 1058–1620) of the ALK gene generating the final fusion protein which contains the full ALK tyrosine kinase domain (aa 1116–1329). Of note, the luminal domain of the VKORC1L1 protein itself contains aa 38–91, but the final VKORC1L1-ALK fusion protein only contains aa 38–64 which is highlighted in yellow. Similarly, the transmembrane domain of the ALK protein itself contains aa 1039–1059, but the final VKORC1L1-ALK fusion protein only contains aa 1058–1059 which is highlighted in brown. Abbreviation: aa, amino acid. |
The other significance of this report is the demonstration of the sensitivity of the ALK T1151K resistance mutation to alectinib in a clinical patient case. The identification of T1151K as a resistance mutation to crizotinib was reported in 2011 by Zhang et al through an accelerated mutagenesis screen.21 Our group was the first to report this resistance mutation in an ALK-rearranged lung cancer patient with prior exposure to crizotinib and ceritinib. Based on co-crystal structures of ceritinib/ALK, alectinib/ALK, and lorlatinib/ALK, we concluded that T1151K confers resistance to ceritinib which may be rescued by alectinib or lorlatinib. We also speculated that brigatinib may not be able to completely overcome the T1151K/M resistance mutations according to an in vitro assay as well as the strong interaction of brigatinib with the P-loop of the ALK protein which may be interrupted by the T1151 mutation leading to drug resistance.22 The patient described in our previous case report has been on lorlatinib for over two years with a PR (–32% by RECIST 1.1). The dramatic response seen in this case would require longer follow-up to determine if the response is durable. Interestingly, as alectinib only has weak interaction with the P-loop of the ALK protein while lorlatinib has moderate interaction, alectinib may be more potent than lorlatinib to rescue patients with tumors harboring the ALK T1151K resistance mutation, which could explain a greater response seen in this patient treated with alectinib.
One limitation of this case report is that from a molecular standpoint it remains unclear how the ALK protein is activated by its fusion partner VKORC1L1 as this protein product does not appear to contain a dimerization domain similar to EML4. It would be ideal to demonstrate positive ALK immunohistochemistry or the final VKORC1L1-ALK fusion protein and phospho-ALK by Western blot for instance. However, our patient declined tissue biopsy and his therapeutic bronchoscopy did not yield enough tissue to perform such laboratory assays. It is fortunate that liquid biopsy revealed this novel ALK fusion and an acquired ALK resistance mutation. More importantly, the dramatic response seen in follow-up scans highly suggests that his disease process remains dependent on the ALK signaling pathway.
The patient will undergo routine CT scans to monitor treatment response as well as surveillance brain magnetic resonance imaging as it is a common site of disease progression for patients with ALK-rearranged lung cancer. He does not have any brain metastasis at present. Should his disease progress again, we plan to explore resistance mechanism(s) by repeat biopsy. As patients are exposed to sequential ALK inhibitors, they are more likely to develop compound ALK resistance mutations. This phenomenon has been elegantly illustrated by Yoda et al in their approach to define lorlatinib-resistant ALK mutations.23 To prevent highly refractory compound mutations from emerging, one might argue for the upfront use of the most potent ALK inhibitor and designing next-generation ALK inhibitors that can overcome these resistance mutations.
Conclusion
ALK-rearranged lung cancer defines a distinctive molecular cohort of patients whose outcomes are significantly improved by the availability of ALK inhibitors. Thus, screening for this driver mutation by a comprehensive molecular testing platform is absolutely necessary in routine practice. This is the first case report of a novel VKORC1L1-ALK fusion detected in a lung cancer patient. The patient had been on crizotinib for over 8 years until emergence of an ALK T1151K resistance mutation that was successfully treated with alectinib, demonstrating for the first time the activity of alectinib against T1151K in the clinic.
Disclosure
VWZ has received honoraria from Roche-Foundation Medicine, Roche/Genentech, Takeda, and Biocept, and consulting fees from TP Therapeutics. ABS and SMA are employees of Foundation Medicine, Inc., a wholly owned subsidiary of Roche. SIO has received honoraria from Pfizer, Roche-Foundation Medicine, Roche/Genentech, and Takeda, and has stock ownership in TP Therapeutics. The other authors report no conflicts of interest in this work.
References
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | ||
Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. | ||
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. | ||
Togashi Y, Soda M, Sakata S, et al. KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One. 2012;7(2):e31323. | ||
Jung Y, Kim P, Jung Y, et al. Discovery of ALK-PTPN3 gene fusion from human non-small cell lung carcinoma cell line using next generation RNA sequencing. Genes Chromosomes Cancer. 2012;51(6):590–597. | ||
Majewski IJ, Mittempergher L, Davidson NM, et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. J Pathol. 2013;230(3):270–276. | ||
Fang DD, Zhang B, Gu Q, et al. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. J Thorac Oncol. 2014;9(3):285–294. | ||
Hong M, Kim RN, Song JY, et al. HIP1-ALK, a novel fusion protein identified in lung adenocarcinoma. J Thorac Oncol. 2014;9(3):419–422. | ||
Choi YL, Lira ME, Hong M, et al. A novel fusion of TPR and ALK in lung adenocarcinoma. J Thorac Oncol. 2014;9(4):563–566. | ||
Shan L, Jiang P, Xu F, et al. BIRC6-ALK, a novel fusion gene in ALK break-apart FISH-negative lung adenocarcinoma, responds to crizotinib. J Thorac Oncol. 2015;10(6):e37–e39. | ||
Iyevleva AG, Raskin GA, Tiurin VI, et al. Novel ALK fusion partners in lung cancer. Cancer Lett. 2015;362(1):116–121. | ||
Drilon A, Wang L, Arcila ME, et al. Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in “driver-negative” lung adenocarcinomas. Clin Cancer Res. 2015;21:3631–3639. | ||
Ali SM, Hensing T, Schrock AB, et al. Comprehensive genomic profiling identifies a subset of crizotinib-responsive ALK-rearranged non-small cell lung cancer not detected by fluorescence in situ hybridization. Oncologist. 2016;21(6):762–770. | ||
Tan DS, Kim DW, Thomas M, et al. Genetic landscape of ALK+ non-small cell lung cancer (NSCLC) patients (pts) and response to ceritinib in ASCEND-1. J Clin Oncol. 2016;34(suppl; abstr 9064). | ||
Jiang J, Wu X, Tong X, et al. GCC2-ALK as a targetable fusion in lung adenocarcinoma and its enduring clinical responses to ALK inhibitors. Lung Cancer. 2018;115:5–11. | ||
Yin J, Zhang Y, Zhang Y, Peng F, Lu Y. Reporting on two novel fusions, DYSF-ALK and ITGAV-ALK, coexisting in one patient with adenocarcinoma of lung, sensitive to crizotinib. J Thorac Oncol. 2018;13(3):e43–e45. | ||
Hu S, Li Q, Peng W, et al. VIT-ALK, a novel alectinib-sensitive fusion gene in lung adenocarcinoma. J Thorac Oncol. 2018;13(5):e72–e74. | ||
Schrock AB, Zhu VW, Hsieh WS, et al. Receptor tyrosine kinase fusions and BRAF kinase fusions are rare but actionable resistance mechanisms to EGFR tyrosine kinase inhibitors. J Thorac Oncol. 2018;13(9):1312–1323. | ||
Zhang M, Wang Q, Ding Y, et al. CUX1-ALK, a Novel ALK Rearrangement That Responds to Crizotinib in Non-Small Cell Lung Cancer. J Thorac Oncol. Epub 2018 Aug 7. | ||
Oldenburg J, Watzka M, Bevans CG. VKORC1 and VKORC1L1: Why do Vertebrates Have Two Vitamin K 2,3-Epoxide Reductases? Nutrients. 2015;7(8):6250–6280. | ||
Zhang S, Wang F, Keats J, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78(6):999–1005. | ||
Zhu VW, Cui JJ, Fernandez-Rocha M, Schrock AB, Ali SM, Ou SI. Identification of a novel T1151K ALK mutation in a patient with ALK-rearranged NSCLC with prior exposure to crizotinib and ceritinib. Lung Cancer. 2017;110:32–34. | ||
Yoda S, Lin JJ, Lawrence MS, et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018;8(6):714–729. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.