Back to Journals » International Journal of Nanomedicine » Volume 18
Doxorubicin-Loaded Platelet Decoys for Enhanced Chemoimmunotherapy Against Triple-Negative Breast Cancer in Mice Model
Authors Dong H ![]() , Gao M, Lu L, Gui R, Fu Y
, Gao M, Lu L, Gui R, Fu Y
Received 26 January 2023
Accepted for publication 18 June 2023
Published 30 June 2023 Volume 2023:18 Pages 3577—3593
DOI https://doi.org/10.2147/IJN.S403339
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Farooq A. Shiekh
Hang Dong, Meng Gao, Lu Lu, Rong Gui, Yunfeng Fu
Department of Blood Transfusion, the Third Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China
Correspondence: Yunfeng Fu, Department of Blood Transfusion, the Third Xiangya Hospital, Central South University, Tongzipo Road 138, Changsha, Hunan, 410013, People’s Republic of China, Email [email protected]
Introduction: Triple-negative breast cancer (TNBC) is a highly aggressive subtype with a poor prognosis. Current single-agent checkpoint therapy has limited effectiveness in TNBC patients. In this study, we developed doxorubicin-loaded platelet decoys (PD@Dox) for chemotherapy and induction of tumor immunogenic cell death (ICD). By combining PD-1 antibody, PD@Dox has the potential to enhance tumor therapy through chemoimmunotherapy in vivo.
Methods: Platelet decoys were prepared using 0.1% Triton X-100 and co-incubated with doxorubicin to obtain PD@Dox. Characterization of PDs and PD@Dox was performed using electron microscopy and flow cytometry. We evaluated the properties of PD@Dox to retain platelets through sodium dodecyl sulfate-polyacrylamide gel electrophoresis, flow cytometry, and thromboelastometry. In vitro experiments assessed drug-loading capacity, release kinetics, and the enhanced antitumor activity of PD@Dox. The mechanism of PD@Dox was investigated through cell viability assays, apoptosis assays, Western blot analysis, and immunofluorescence staining. In vivo studies were performed using a TNBC tumor-bearing mouse model to assess the anticancer effects.
Results: Electron microscopic observations confirmed that platelet decoys and PD@Dox exhibited a round shape similar to normal platelets. Platelet decoys demonstrated superior drug uptake and loading capacity compared to platelets. Importantly, PD@Dox retained the ability to recognize and bind tumor cells. The released doxorubicin induced ICD, resulting in the release of tumor antigens and damage-related molecular patterns that recruit dendritic cells and activate antitumor immunity. Notably, the combination of PD@Dox and immune checkpoint blockade therapy using PD-1 antibody achieved significant therapeutic efficacy by blocking tumor immune escape and promoting ICD-induced T cell activation.
Conclusion: Our results suggest that PD@Dox, in combination with immune checkpoint blockade therapy, holds promise as a potential strategy for TNBC treatment.
Keywords: drug delivery, doxorubicin, immunogenic cell death, immune checkpoint blockade therapy
Introduction
Triple-negative breast cancer (TNBC) is characterized by an earlier onset, higher recurrence rate, and poorer prognosis compared to other breast cancer subtypes.1,2 TNBC lacks the expression of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2, making targeted therapies less effective. Chemotherapy remains the primary treatment option for TNBC due to the absence of clear molecular targets and tumor heterogeneity.3 In recent years, immune checkpoint inhibitors (ICIs) such as anti-programmed cell death protein-1 (aPD-1), anti-programmed cell death protein-ligand 1, and anti-cytotoxic T lymphocyte antigen-4 monoclonal antibodies have shown promise in various cancer types, including melanoma, lung, and kidney cancers.4–6 However, breast cancer is considered “immunologically cold” with relatively low T cell infiltration, limiting the efficacy of single-agent checkpoint therapy in most breast cancer patients. Therefore, combination therapies involving ICIs and other immunological approaches have emerged as potential strategies to convert immunologically “cold” tumors into “hot” tumors, primed for immune response.7,8
Immunogenic cell death (ICD) is a specific type of apoptotic cell death characterized by the coordinated release of danger-associated molecular patterns.9 During ICD, dying tumor cells release molecules such as calreticulin (CRT), adenosine triphosphate (ATP), and high mobility group protein B1 (HMGB-1), which promote the maturation of dendritic cells (DCs) and activate innate T cells, ultimately leading to the generation of cytotoxic T lymphocytes.10 Several chemotherapeutic agents, including doxorubicin (Dox), oxaliplatin, cyclophosphamide, and paclitaxel, have been reported to induce ICD and enhance antitumor immunity.11 Dox, an anthracycline, is widely used in the treatment of various solid malignancies.12 However, its clinical application is limited by its short biological half-life and significant side effects, particularly on the heart and kidneys.13 Therefore, there is a need for an ideal drug delivery system to minimize systemic toxicity and improve treatment efficacy. Biologic drug carriers have emerged as a promising therapeutic platform for cancer, offering advantages such as increased intracellular uptake of drugs and reduced side effects compared to conventional formulations.14 Among the biologic drug carriers, platelets have garnered considerable attention due to their excellent biocompatibility, prolonged circulation time, and ease of production.15
Platelets, anucleated blood cells derived from megakaryocytes, are involved in various physiological processes such as hemostasis, vascular function maintenance, and tissue repair.16 Recent studies have explored the use of platelet membranes to camouflage nanoparticles, enabling them to evade the phagocytic system.17 This has led to the development of smart drug delivery systems with improved tumor-targeting capabilities, utilizing platelets as carrier vehicles.18 Moreover, natural platelets themselves can serve as drug carriers for tumor treatment. Wu et al have optimized the preservation conditions for doxorubicin-loaded platelets, making them more suitable for clinical applications.19 However, there are some limitations associated with using natural platelets as drug carriers, including increased risk of platelet aggregation, thrombosis, and limited drug loading capacity.20 In this study, we utilized platelet decoys based on previous research.21 Platelet decoys are stripped of their inherent functional capabilities such as activation and aggregation, while still retaining their cytoskeleton and functional cell surface receptors that enable binding interactions with other cells.
In this study, we developed an intelligent drug delivery system utilizing doxorubicin-loaded platelet decoys (PD@Dox) for the treatment of TNBC. This drug delivery system is designed to achieve simultaneous chemotherapy and immunotherapy by triggering ICD. Furthermore, it synergizes with aPD-1 to enhance the antitumor activity. We successfully prepared platelet decoys capable of delivering an optimal amount of Dox to the tumor site and enhancing drug uptake through the binding interaction between platelets and tumor cells. Importantly, our study demonstrated that platelet decoys did not activate platelets or induce thrombus formation within the coagulation system. Furthermore, the released doxorubicin from PD@Dox induced immune activation by triggering ICD, leading to the activation of antigen-presenting cells and subsequent recruitment of effector T-cells to the tumor. Ultimately, our findings highlight the potential of PD@Dox to enhance tumor therapy through the combination of chemotherapy and immunotherapy, achieved by integrating aPD-1 to achieve chemoimmunotherapy in vivo.
Materials and Methods
Materials
Prostaglandin E1 was ordered from AdooQ BioScience (Irvine, CA). Dox and Adenosine 5’-diphosphate (ADP) were purchased from Solarbio Science & Technology (Beijing, China). The Cell Counting Kit-8 cell counting kit was purchased from APE x BIO Technology (Houston, USA). Fetal bovine serum and RPMI-1640 were purchased from Procell Life Technology (Wuhuan, China). DAPI was produced by Servicebio Technology (China). BCA protein assay kit and Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel kit were purchased from Boster Biological Technology (Wuhan, China). Cell culture plates and Transwell chamber with 0.4-mum-pore polycarbonate-free filters were purchased from Corning Incorporated (New York, NY, USA). Anti-CD41-APC, anti-CD42b-FITC, anti-GPIIb/IIIa-FITC were purchased from BioLegend (BioLegend, San Diego, CA, USA). Anti-CD86-FITC and anti-CD80-PE were obtained from BD Biosciences (San Jose, CA). Did dye, Annexin V-FITC apoptosis detection kit and cell cycle detection kit were acquired from Beyotime Biotechnology (Shanghai, China). The antibodies including B cell lymphoma-2 (Bcl-2), Bcl-2 associated X protein (Bax), cleaved caspase-3 and cleaved caspase-9 proteins were from Abmart (Shanghai, China). The anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was from Cell Signaling Technology Inc (Danvers, MA, USA). The anti-CRT and anti-HMGB-1 were from Immunoway (TX, USA).
Cells and Animals
4T1 cells were obtained from the Cancer Research institute, Central South University in Hunan, China. DC2.4 cells and MDA-MB-231 cells were purchased from Procell Life Technology Co. The authenticity of the cells was ensured by performing STR analysis, which confirmed their identity. Additionally, the cells were tested and found to be free of mycoplasma contamination. All cells were incubated at 37°C in a 5% CO2 incubator.
For the animal experiments, six-week-old Balb/c mice were purchased from Hunan SJA Experimental Animal Co., Ltd (China). The research involving animals was conducted following the approved guidelines of the National Standard of the People’s Republic of China (GB/T35892-2018) titled “Laboratory Animals - Guidelines for Ethical Review of Animal Welfare.” The experimental protocols were also approved by the Central South University Standards for the Laboratory Animals Welfare and Ethical Review (Permit Number: CSU-2022-0071).
Preparation of Doxorubicin-Loaded Platelet Decoys
Clinical-grade platelets were obtained from the Xiangya Third Hospital, collected from healthy donors in sodium citrate and suspended in 100% plasma. Mouse platelets were prepared according to published protocols.22 It is important to note that the study adhered to ethical guidelines and made efforts to minimize the use of animals whenever possible.23 Human platelets were utilized for the in vitro experiments, while mouse-derived platelets were specifically used for the animal experiments. Platelet decoys, prepared as previously described, were washed once with Tyrode buffer and obtained by centrifugation at 1000g for 10 minutes. Dox was added and incubated with shaking for 1 hour at room temperature. PD@Dox was washed with Tyrode buffer by centrifugation at 1000g for 10 min to remove excess Dox. The same treatments were applied to prepare nature platelets loaded Dox.
Characterization of Doxorubicin-Loaded Platelet Decoys
The morphology of the samples was observed under a transmission electron microscope (TEM, FEI Tecnai Spirit G2 BioTWIN, Hillsboro, OR) and a scanning electron microscopy (SEM, Hitachi, S-3400N, Japan). The size and granularity of the samples were analyzed using flow cytometry analysis (Cytek, Fremont, CA, USA). The protein concentration in platelets, platelet decoys, and PD@Dox was measured using a total protein assay kit (BCA method). SDS-PAGE was performed to detect the protein in platelet decoys and PD@Dox and confirm the retention of membrane proteins. Furthermore, the samples were stained anti-CD41-APC and anti-CD42b-FITC. After 20 minutes of incubation in the dark at room temperature, the samples were diluted and analyzed by flow cytometry.
Doxorubicin Load and Release by Platelet Decoys
Drug load was determined by lysing a 1 mL pellet of PD@Dox and nature platelets loaded with Dox (1×109). The mean fluorescence intensity of Dox in cells was measured by flow cytometry at different time points and Dox concentrations to assess drug retention. The maximum loading efficiency of Dox was calculated using fluorescence spectroscopy at 470/585 nm with the equation:
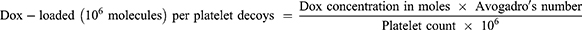
Avogadro’s number is a constant equal to approximately 6.022×1023 particles/mol.
For drug release, PD@Dox was incubated in phosphate-buffered saline (PBS) at pH 7.4, 6.5, or 5.5, and samples were collected at specific time intervals. To mimic the tumor microenvironment, PD@Dox was also incubated in the supernatant of 4T1 cell conditioned medium compared to PBS and RPMI-1640 medium. The release kinetics of Dox were evaluated by separating the supernatant from the pellet and quantifying Dox concentration using fluorescence spectroscopy at predetermined time points (12, 24, 48, and 72 hours).
Detection of Doxorubicin-Loaded Platelet Decoys Activated and Aggregated Normal Platelets
Platelets, platelet decoys, and PD@Dox (1×109) were incubated with 50 µM ADP for 30 min. FITC-conjugated anti-human PAC-1 (10 µg/mL) was added and incubated for 20 min. Flow cytometry was performed to analyze expression of PAC-1. The Platelet Function Analyzer (PFA-100) was used to assess platelet aggregation induced by platelet decoys and PD@Dox in vitro. Platelet decoys and PD@Dox were added to normal platelet samples (1:4 by volume), and platelet control groups were prepared using platelets in platelet-poor plasma. Platelet function analysis was performed after incubation with agonists (50 µM ADP) for 2 h at 37°C. Thromboelastometry (TEG) was utilized to evaluate platelet function. Samples were added to warmed TEG-sample cups with isotonic CaCl2 solution, and the TEG measurements were conducted according to the manufacturer’s instructions.
Interaction Between Doxorubicin-Loaded Platelet Decoys and 4T1 Cells in vitro
To investigate the interaction between platelet decoys and tumor cells, PD@Dox labeled with DiD dye was incubated with 4T1 cells for 4 hours. Cell adhesion and Dox uptake were assessed using flow cytometry. And then heparin (1µM) was added to the medium to determine the involvement of PAC-1 and P-Selectin. The effect of heparin on the Dox+DiD+ population was evaluated. The effect of platelet decoys on Dox delivery to breast tumor cells was evaluated by treating 4T1 cells separately with free Dox and PD@Dox for 0.5 and 2 hours. Fluorescence intensity was measured to compare the cellular uptake efficiency of Dox between the two groups.
Cytotoxicity of Doxorubicin-Loaded Platelet Decoys on 4T1 Cells in vitro
4T1 cells and MDA-MB-231 cells were seeded at a density of 3×103 cells/well in 96-well plates and allowed to attach overnight. The cells were then treated with different substances including PBS, platelet decoys, free Dox, and PD@Dox with a doxorubicin concentration of 2.5 μg/mL in all doxorubicin-related groups. After 24 hours of incubation, the cytotoxicity was assessed using a cell counting kit-8 assay. They were calculated as a percent of the control using the following formula: Cell viability (%) =
In vitro Apoptosis Assay and Cell Cycle
4T1 cells (5x105 cells/well) were treated with PBS, platelet decoys, free Dox, and PD@Dox for 24 hours ([Dox] =2.5 μg/mL for all Dox-related groups). Afterward, the cells were collected, washed, and suspended in PBS. Apoptosis was assessed using an Annexin V/PI apoptosis kit and flow cytometry. Cell cycle distribution was determined using a cell cycle assay kit and flow cytometry according to the manufacturer’s instructions.
In vitro Assays of Immunogenic Cell Death
4T1 cells (2x105 cells/well) were cultured in 12-well plates overnight. The cells were then treated with platelet decoys, free Dox, or PD@Dox for 24 hours ([Dox] =2.5μg/mL for all Dox-related groups), while the control group received PBS. After treatment, the culture medium was collected and assessed using an ATP assay kit to measure expression. The expression levels of CRT and HMGB-1 were analyzed by Western blotting, and surface CRT exposure was detected through immunofluorescence staining.
In vitro Immunogenic Cell Death-Induced Maturation of Dendritic Cells
A co-culture system was established using a 6-well transwell plate. In the upper wells, 4T1 cells were seeded and treated with platelet decoys, free Dox, or PD@Dox for 12 hours ([Dox] =2.5μg/mL for all Dox-related groups). Simultaneously, DC2.4 were seeded in the lower wells. After 24 hours of co-incubation, the DC2.4 were analyzed by flow cytometry after staining with anti-CD80-FITC and anti-CD86-PE antibodies.
Western Blot Analysis
4T1 cells (5x105 cells/well) were seeded in 6-well plates and treated with PBS, platelet decoys, free Dox, and PD@Dox for 24 hours ([Dox] =2.5μg/mL for all Dox-related groups). The cells were then collected and homogenized using RIPA, followed by incubation on ice for 30 minutes. The samples were sonicated, centrifuged at 14,000 rpm for 20 minutes at 4°C, and the supernatant was collected. The protein concentration was determined using a BCA assay kit. Western blot analysis was performed to detect the protein expressions of cleaved caspase-9, cleaved caspase-3, Bcl-2, Bax, CRT, and HMGB-1. GAPDH was used as the internal loading control, and the analysis was carried out following standard protocols.
In vivo Animal Experiments
A 4T1 tumor-bearing mice model was established by injecting 4T1 cells (5 x 106) suspended in PBS subcutaneously into the right flank of BALB/c mice. The mice were divided into five groups (n = 5): PBS, PD, free Dox, PD@Dox and PD@Dox+aPD-1. The mice in each group received intravenous injections of PBS, free Dox, and PD@Dox([Dox] = 1 mg/kg, for all Dox-related groups, 100 μL PBS for each mouse) via the tail vein on days 1, 5, 7 and 13. In the PD@Dox+aPD-1 group, each mouse received intratumoral administration of aPD-1 (0.2 mg/mL, 100 μL PBS for each mouse) on days 3, 7 and 11.
Tumor volumes and body weights of the mice were measured every two days for 14 days. Tumor volume (V) was calculated using the formula 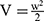 , where L and W represent the longest and shortest tumor diameters, respectively. Relative tumor volume and body weight were calculated based on the measurements on day 1. Serum concentrations of creatine kinase-MB (CK-MB), alanine transaminase (ALT), and blood urea nitrogen (BUN) were measured to evaluate myocardial injury, hepatic function, and renal function.
, where L and W represent the longest and shortest tumor diameters, respectively. Relative tumor volume and body weight were calculated based on the measurements on day 1. Serum concentrations of creatine kinase-MB (CK-MB), alanine transaminase (ALT), and blood urea nitrogen (BUN) were measured to evaluate myocardial injury, hepatic function, and renal function.
For the evaluation of PD@Dox distribution, DiD-labeled PD@Dox was administered intravenously when the tumor volume reached 150–200 mm3. The fluorescence emitted by the near-infrared fluorescent dye DiD was captured at different time points using a bioluminescence imaging system (IVIS Spectrum, USA). Ex vivo imaging of major organs and tumors was performed, and semiquantitative analysis of the fluorescence signals was conducted to assess the accumulation and biodistribution of PD@Dox.
At the end of the 14-day treatment period, the tumor-bearing mice were sacrificed. Vital organs (heart, liver, spleen, lung, and kidney) and tumor tissues were harvested and fixed in 4% paraformaldehyde for preparation of paraffin sections. H&E staining was performed on the vital organ sections to observe the histological changes caused by different treatments. To evaluate the anti-tumor effects, tumor tissues were stained with anti-Ki67 antibody detect cell proliferation. Immunohistochemical techniques were used to detect the protein expression of CRT, confirming the immunological effect induced by PD@Dox+aPD-1. Immunofluorescence staining was performed to assess the presence of CD8+ cells.
Statistical Analysis
All experiments were performed in triplicate, and the data were presented as mean ± standard deviation. Statistical analysis was conducted using GraphPad Prism 8 software. Student’s t-test was used for comparisons between two groups. For multiple comparisons involving more than two groups, Tukey’s post hoc tests and one-way analysis of variance (ANOVA) were employed. Statistical significance was indicated by* for P< 0.05, ** for P< 0.01, *** for P< 0.001, and **** for P< 0.0001.
Results
Preparation and Characterization of Doxorubicin-Loaded Platelet Decoys
In this study, platelet decoys were prepared by extracting natural platelets using 0.1% Triton X-100, which is a mild cell permeabilizer that removes most membranes, intracellular contents, and granules while preserving the cell shape and surface proteins.24 To obtain PD@Dox, Dox was co-incubated with platelet decoys. The characterization of platelet decoys and PD@Dox was examined using TEM and SEM. The images confirmed that both platelet decoys and PD@Dox displayed a round shape similar to normal platelets (Figure 1A). However, platelet decoys and PD@Dox experienced a reduction in intracellular granules, leading to a decrease in cytoplasmic electron density as observed in Figure 1B and C. In flow cytometry analysis, Forward scatter (FSC) represents the parameter for size, while side scatter(SSC) represents the parameter for granularity. Platelet decoys exhibited a 25.1% decrease in intracellular granules compared to natural platelets, whereas PD@Dox displayed slightly higher granularity than platelet decoys.The total protein content of platelet decoys and PD@Dox, measured using the BCA method, was found to be reduced by approximately 47% compared to pure platelets (Figure 1D). SDS-PAGE protein analysis (Figure 1E) indicated that the surface protein expression of platelet decoys and PD@Dox was similar to that of pure platelets. Flow cytometric analysis confirmed that platelet decoys and PD@Dox retained specific membrane adhesion receptors such as CD41 and CD42b, although their expression levels were reduced compared to normal platelets. CD41 expression was reduced by 6.4% and 22.5% in platelet decoys and PD@Dox, respectively, while CD42b expression was reduced by 60.0% and 55.5% (Figure 1F).
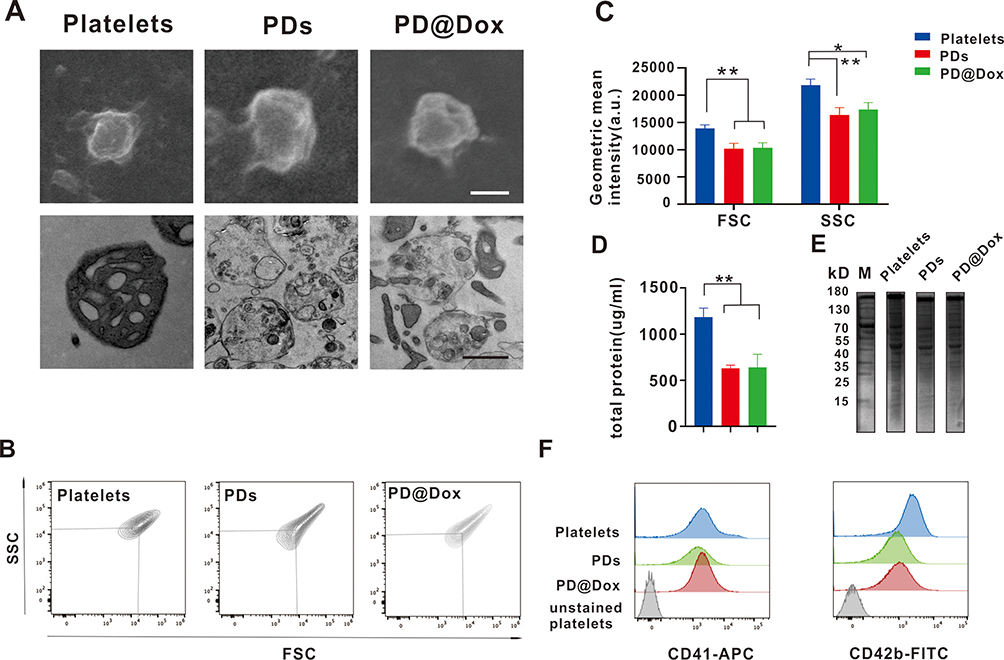 |
Figure 1 Characterization of Dox loaded platelet decoy. (A) SEM(top) and TEM(bottom) images showing the morphology and ultrastructure of platelets, PDs, and PD@Dox(Scale bar: 1 μm). (B) Flow cytometry density plots indicating FSC and SSC. (C) Geometric mean of FSC and SSC for platelets, PDs and PD@Dox. (D) Comparison of total protein concentration among platelets, PDs, and PD@Dox. (E) SDS-PAGE protein analysis with M, platelets, PDs, and PD@Dox. (F) Flow cytometry analysis of CD41 and CD42b expression. *P<0.05, **P<0.01. Abbreviations: SEM, scanning electron microscopy; TEM, transmission electron microscope; PDs, platelet decoys; PD@Dox, Dox loaded platelet decoys; FSC, forward scatter; SSC, side scatter; SDS-PAGE, Sodium dodecyl sulfate-polyacrylamide gel electrophoresis; M, marker. |
Doxorubicin Load and Release of Platelet Decoys
The uptake of Dox by platelet decoys exhibited a dose- and time-dependent pattern, with saturation observed at the highest dose after 6 hours. Notably, the PD group displayed higher fluorescence signals compared to the pure platelet group under the same conditions, indicating that platelet decoys can enhance the uptake efficiency of free Dox (Figure 2A and B). The quantity of Dox loaded per particle was measured to be 10.18×106±0.93 molecules from platelets, while in each platelet decoy, it was 16.18×106±0.66 molecules (Figure 2C). Moreover, the release kinetics of Dox from platelet decoys were significantly faster under acidic pH conditions compared to other pH conditions (Figure 2D). In cancer cell cultures, the release of Dox from platelet decoys exposed to 4T1 cell cultures for 72 hours was significantly higher (83.27±2.17%) compared to exposure to RPMI-1640 medium (75.62±0.47%) or PBS (75.20±0.78%) (Figure 2E).
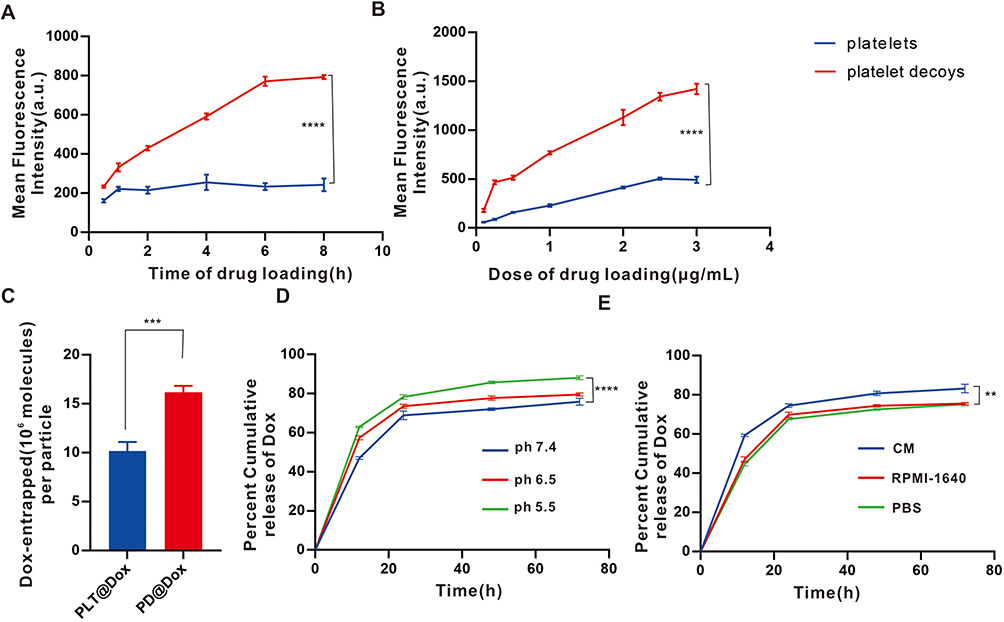 |
Figure 2 Loading and releasing of Dox. (A) Time-dependent uptake of Dox by platelets and decoys assayed through flow cytometry. (B) Dose-dependent uptake of Dox by platelets and decoys assayed through flow cytometry. (C) Loading efficiency of Dox by platelets and PDs determined by fluorescence spectrophotometry. (D) Comparison of the kinetics of Dox release at different pH levels (5.5, 6.5 and 7.4). (E) Comparison of the kinetics of Dox release in the presence of CM, RPMI-1640 and PBS. **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: Dox, doxorubicin; PDs, platelet decoys; CM, conditioned medium cultured with 4T1 cells; RPMI-1460, cell culture control medium, PBS, phosphate-buffered saline. |
Doxorubicin-Loaded Platelet Decoys Did Not Active and Aggregate Under Stimuli
The glycoprotein IIb/IIIa complex (clone PAC-1), expressed by activated platelets, plays a crucial role in fibrin binding and undergoes conformational changes upon activation by substances such as ADP. These conformational changes are essential for platelet aggregate.25 To assess whether the PAC-1 receptors undergo conformational changes on platelet decoys when stimulated by agonists, we exposed decoys and platelets to supraphysiological concentrations of ADP and quantified the binding of PAC-1 antibodies using flow cytometry. As shown in Figure 3A, PAC-1 expression on the surface of normal platelets significantly increased upon ADP stimulation. In contrast, the expression of PAC-1 remained unchanged on platelet decoys and PD@Dox. We further evaluated the aggregation capability of platelet decoys and PD@Dox using the Platelet Aggregation Meter. Our results demonstrated that platelet decoys and PD@Dox inhibited platelet aggregation induced by the supraphysiological concentrations of ADP, while more than 50% of intact platelets aggregated under the same conditions (Figure 3B and C). In vitro thromboelastography functional analysis was performed to assess hemostatic activity and the contribution of platelet decoys and PD@Dox to fibrin clot strength. The reaction time for platelet decoys and PD@Dox was shorter than that of cryopreserved Dox-loaded platelets and not significantly different from that of platelet-poor plasma (negative control) or fresh platelets (Figure 3D). Moreover, the maximum amplitude values of platelet decoys and PD@Dox, which directly reflect the functional contribution of platelets to fibrin clot strength, were significantly lower than those of normal platelets (Figure 3E). Thus, PD@Dox retained partial adhesion receptors on their surface but did not respond to physiological stimuli for aggregation.
 |
Figure 3 PD@Dox did not active and aggregate under stimuli. (A) Flow cytometry histogram showing the expression of PAC-1 on platelets, PDs, and PD@Dox upon ADP stimulation. (B) Histogram of the maximum platelet aggregation rates in the presence of platelets, PDs, and PD@Dox under ADP stimulation. (C) Histogram of the average platelet aggregation rates in the presence of platelets, PDs, and PD@Dox under ADP stimulation. (D) Histogram of the reaction time in the presence of platelets, PDs, and PD@Dox under Ca2+ stimulation. (E) Histogram of maximum amplitude in the presence of platelets, PDs, and PD@Dox under Ca2+ stimulation. *P<0.05, **P<0.01. Abbreviations: ns, non-significance; PPP, platelet-poor plasma; ADP, Adenosine 5’-diphosphate; Dox, doxorubicin; PDs, platelet decoys; PD@Dox, Dox loaded platelet decoys. |
Doxorubicin-Loaded Platelet Decoys Enhanced Cell Adhesion and Integration into Tumor Tissue
Previous studies have extensively investigated the intricate interaction between tumor cells and platelets. This interaction is mediated through molecules such as PAC-1 and P-Selectin, which can be blocked by heparin treatment.26–28 In our study, PD@Dox, labeled with DiD, a lipophilic fluorescent dye, underwent a 4h incubation with 4T1 cells and flow cytometry was used to assess cell adhesion and Dox uptake. As shown in Figure 4A, the population of 4T1 cells displaying both Dox and DiD fluorescence (Dox+DiD+) amounted to 42.67%. However, when the attachment of platelets to 4T1 cells was blocked via heparin intervention, the Dox+DiD+ population dwindled to 20.87%. These findings indicate that platelet decoys can adhere to tumor cell surfaces, and this adhesion is diminished in the presence of heparin, confirming the involvement of PAC-1 and P-Selectin in the specific adhesion between tumor cells and platelet decoys. To assess the effective delivery of Dox by platelet decoys to breast tumor cells in vitro, 4T1 cells were treated separately with free Dox and PD@Dox for 0.5 hours and 2 hours. The fluorescence intensity of the PD@Dox-treated group was significantly higher than that of the free Dox-treated group (Figure 4B), suggesting that platelets and tumor cells, which have an adhesive relationship, contribute to the enhanced cellular uptake efficiency of Dox.
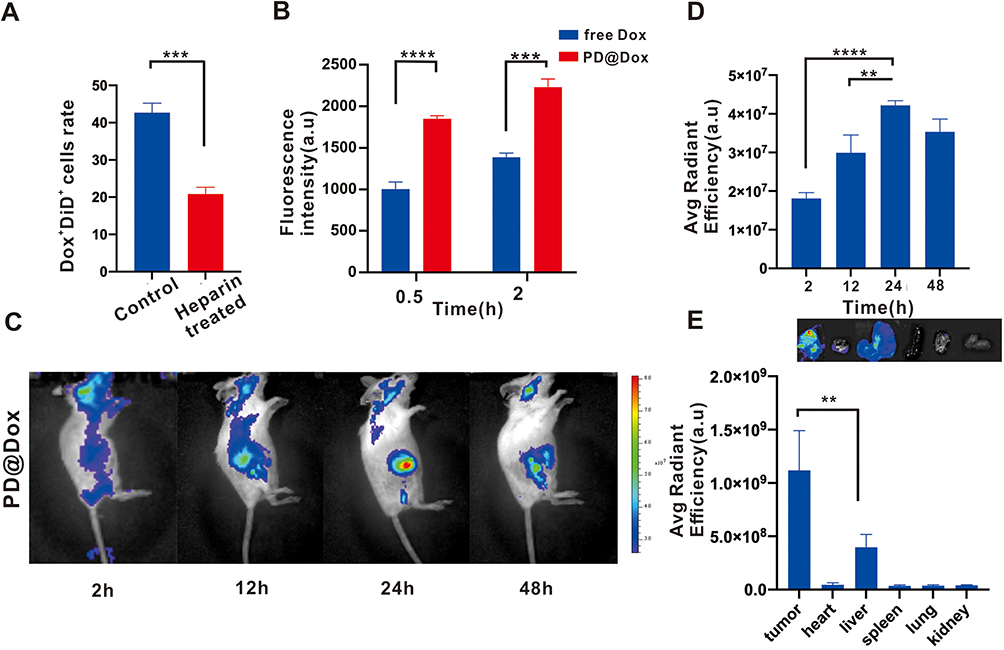 |
Figure 4 PD@Dox enhanced cell adhesion and integration into tumor tissue. (A) Flow cytometry histogram of the percentage of 4T1 cells positive (Dox+DiD+) when incubated with PD@Dox, with or without heparin treatment. (B) Flow cytometry measurements of 4T1 cells after 0.5 and 2 hours of incubation with PD@Dox and free Dox. (C) Representative images displaying the fluorescence distribution of DiD-labeled PD@Dox. (D) Fluorescence intensities of tumors at various time intervals postinjection. (E) Ex vivo fluorescence imaging and intensities of major organs 24 hours after injection. **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: Dox, doxorubicin; PD@Dox, Dox loaded platelet decoys. |
To investigate the localization of PD@Dox in tumor tissue, we labeled PD@Dox with DiD and evaluated the fluorescence intensity at the tumor sites. Figure 4C and D showed that the fluorescence intensity of DiD reach its peak 24 hours after injection, indicating successful accumulation of PD@Dox at the tumor site. Additionally, the fluorescence intensities of major organs 24 hours post-injection demonstrated that PD@Dox predominantly accumulated in tumor tissues, while minimizing potential side effects on other organs (Figure 4E). These findings indicate that PD@Dox effectively targets and integrates into tumor tissue, offering a targeted and localized drug delivery approach.
Anti-Tumor Effect of Doxorubicin-Loaded Platelet Decoys in vitro
To evaluate the cytotoxicity of Dox on cell survival, we treated 4T1 and MDA-MB-231 with platelet decoys, free Dox, and PD@Dox for 24 hours, and measured cell viability using the CCK8 assay. As shown in Figure 5A, we found that the cytotoxic effect of Dox and PD@Dox on breast cells was significantly enhanced when treated with pure platelet decoys in breast cell lines. The PD@Dox group exhibited a stronger anti-tumor effect than the free Dox group at the same Dox dose. In addition, we investigated the anti-tumor mechanism of PD@Dox. The number of apoptotic cells (Annexin V-FITC+/PI+ and Annexin V-FITC+/PI−) in 4T1 cells with platelet decoys, Dox, and PD@Dox for 24 hours. The percentages of apoptotic cells were 25.54±5.74% and 40.90±10.92% for Dox and PD@Dox, respectively, whereas it was 6.58±3.60% for platelet decoys (Figure 5B and C). Flow cytometry analysis revealed cell cycle arrest at the G2/M phase in PD@Dox-treated 4T1 cells, indicating a delay in cell cycle progression (Figure 5D and E).
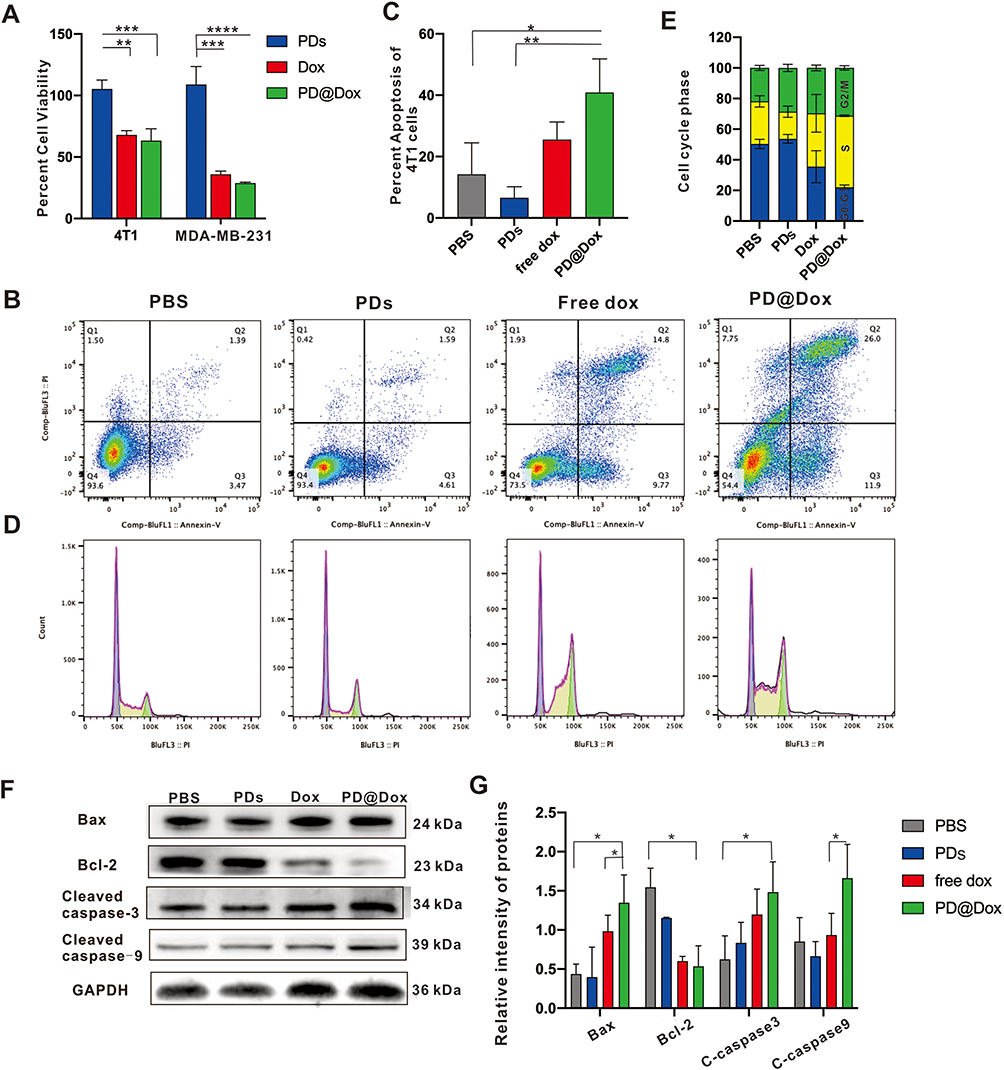 |
Figure 5 PD@Dox enhanced the antitumor effect of Dox in 4T1 cells. (A) Assessment of cell viability of 4T1 and MDA-MB-231 cells treated with PDs, free Dox, and PD@Dox at 24 hours. (B) FCM was used to investigate the apoptosis of 4T1 cells following different treatments. (C) Graph showing the comparative apoptosis levels of 4T1 cells following different treatments. (D) FCM was used to assess the cell cycle state of 4T1 cells following different treatments. (E) Graph showing the percentage of cells in G0/G1, S, and G2/M phases following different treatments. (F) Western blot was used to analysis protein expression of apoptosis-related genes following different treatments. (G) Graph showing the quantification of the bands in (F). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: Dox, doxorubicin; PDs, platelet decoys; PD@Dox, Dox loaded platelet decoys. |
To further investigate the apoptosis mechanism in 4T1 cells, we examined the protein levels of Bcl-2, Bax, cleaved caspase-3, cleaved caspase-9, and GAPDH using Western blot analysis. Compared to the control group, PD@Dox treatment significantly upregulated the levels of Bax, cleaved caspase-3, and cleaved caspase-9 proteins, while downregulating the level of Bcl-2 proteins (Figure 5F and G). These findings suggest that the combination of platelet decoys and Dox enhances cytotoxic effects in vitro.
Immunogenic Cell Death Effect of Doxorubicin-Loaded Platelet Decoys in vitro
Recent advances have highlighted the potential of Dox as an inducer of ICD by promoting the exposure of CRT on the tumor cell surface and the release of HMGB-1 and ATP from tumor cells.29,30 We aimed to investigate whether PD@Dox could enhance the ICD effect, 4T1 cells were subjected to different treatments and the hallmarks of ICD, including release of ATP, surface CRT, and HMGB-1 expression, were assessed. As shown in Figure 6A, PD@Dox group exhibited significantly higher ATP release compared to the free drug groups, likely due to the enhanced cellular uptake of Dox facilitated by platelet decoys (P < 0.05). Western blot analysis results also revealed upregulated expression of CRT and HMGB-1 in the PD@Dox group compared to the PBS and PD treatment groups (Figure 6B and C). Additionally, we examined CRT expression on the surface of 4T1 cells after the same treatments using immunofluorescence staining (Figure 6D). Free Dox induced some extent of CRT expression on the cell surface compared to the PBS group, while PD@Dox treatment led to a significant increase in CRT expression.
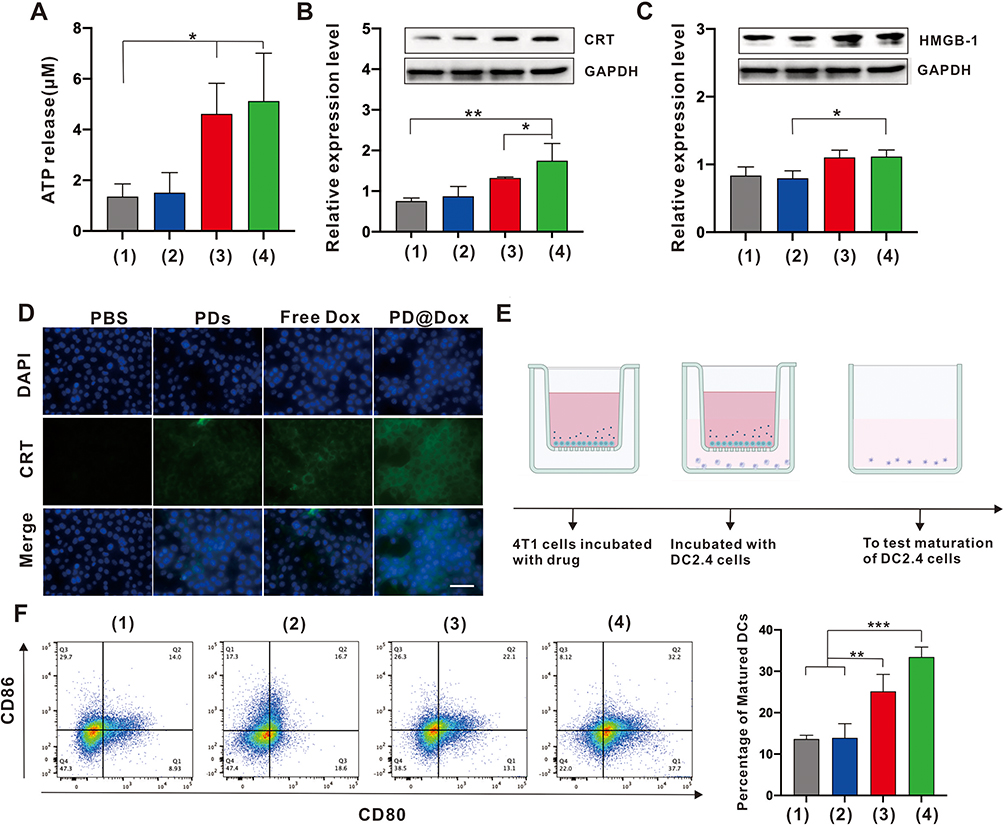 |
Figure 6 ICD effect of PD@Dox in vitro. (A) Histogram plot of extracellular ATP production by 4T1 cells after different treatments for 24 h. (B and C) Western blot and corresponding quantifications analysis of CRT and HMGB-1 expression on 4T1 cells following different treatments. (D) Immunofluorescence detection of CRT expressed on the surface of 4T1 cells following different treatments (scale bar = 50 μm). (E) Schematic illustration of 4T1 cells and DC2.4 cells coculture system. (F) Flow cytometric analysis of DCs maturation after 24 h coculture with ICD cancer cells generated through different treatments. In (A-C and F), (1) PBS; (2) PDs; (3) free Dox; (4)PD@Dox. *P<0.05, **P<0.01, ***P<0.001. Abbreviations: ICD, immunogenic cell death; Dox, doxorubicin; PDs, platelet decoys; PD@Dox, Dox loaded platelet decoys; CRT, calreticulin; ATP, adenosine triphosphate; HMGB-1, high mobility group protein B1; DCs, dendritic cells. |
Antigen presentation to T cells is typically mediated by DCs. Immature DCs efficiently capture and process ICD-associated markers or antigens, allowing them to mature and activate T lymphocytes, including CD8 cytotoxic T lymphocytes, which can then eliminate tumor cells. To further confirm the maturation of DCs induced by PD@Dox-mediated ICD, PD@Dox-treated 4T1 cells were cocultured with DC2.4 cells using a transwell system in vitro. After 4T1 cells were cultured in the upper wells with different agents for 6 hours, DC2.4 cells were seeded in the bottom wells and cocultured with the upper 4T1 cells for an additional 24 hours (Figure 6E). The DCs were collected and analyzed by flow cytometry for the expressions of mature DCs biomarkers (CD86+/CD80+). PD@Dox treatment significantly increased DCs maturation compared to the control group and the single-mode treatment groups (Figure 6F).
Doxorubicin-Loaded Platelet Decoys-Mediated Combination Tumor Therapy in vivo
Numerous studies have indicated that combining PD-1/PD-L1 checkpoint blockade with other therapies significantly enhances antitumor efficacy and improves treatment outcomes compared to individual therapies alone.31,32 Thus, we aimed to investigate the therapeutic efficiency of PD@Dox in vivo by combining chemotherapy and immunotherapy. Female BALB/c mice bearing 4T1 tumor were subjected to different treatments as depicted in Figure 7A. The body weights of the tumor-bearing mice after different treatments did not exhibit significant variation, except for the free Dox group, which displayed significant weight loss during treatment, likely due to Dox-induced toxic effects (Figure 7B). Treatment with PD@Dox showed significant tumor inhibition, as shown in Figure 7C and D. Moreover, the combination of PD@Dox and aPD-1 demonstrated the most pronounced tumor suppression after treatment. As shown in Figure 7E, Ki67 staining of tumor slices was performed to assess tumor cell proliferation after different treatments. The PD@Dox+aPD-1 group exhibited the highest proportion of necrotic and apoptotic tumor cells among all groups, confirming the superior therapeutic effect of combining PD@Dox with aPD-1. To evaluate whether the significant tumor inhibition resulted from an immune effect induced by ICD, immunohistochemical staining was performed to examine the ICD marker, CRT, expression in tumors following various treatments. Remarkably, both Dox and PD@Dox treatments increased CRT exposure compared to PBS and PD treatment.
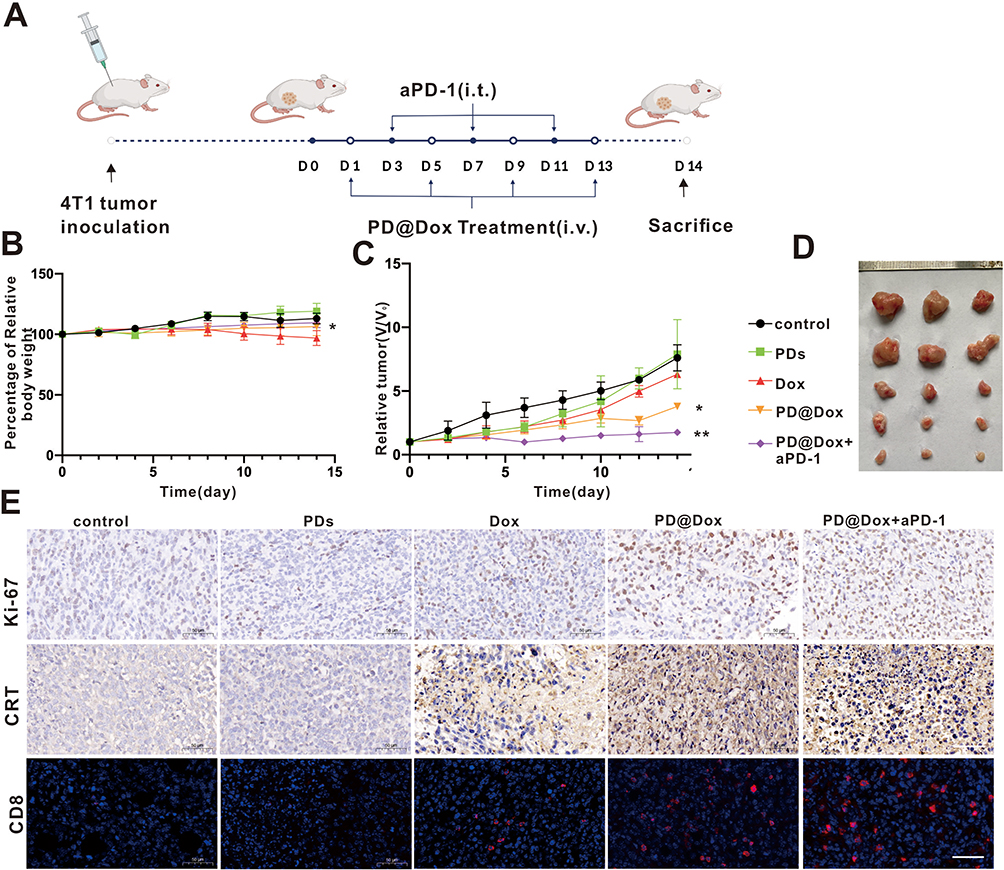 |
Figure 7 PD@Dox mediated combination tumor therapy in vivo. (A) Therapeutic schedule of PD@Dox+aPD-1 in 4T1 tumor-bearing mice. (B and C) The Relative body weights and relative tumor volume changes of tumor-bearing mice after different treatment processes. (D) Representative tumor photographs of tumor-bearing mice after different treatments. (E) Ki67 and CRT expression, as well as intratumoral infiltration of CD8+ T cells of tumor-bearing mice as after different treatment processes, respectively (scale bar: 50 μm). *P<0.05, **P<0.01. Abbreviations: i.v, intravenous injection; i.t, intratumoral injection; Dox, doxorubicin; PDs, platelet decoys; PD@Dox, Dox loaded platelet decoys; CRT, calreticulin. |
Furthermore, the PD@Dox+aPD-1 treatment induced a more significant expression of CRT than the other groups, indicating that the combination of PD@Dox and aPD-1 could generate a higher level of ICD, which may activate DCs and promote tumor antigen presentation to enhance the antitumor immune response subsequently. Additionally, the distribution of activated T cell phenotypes in tumors was directly assessed by immunofluorescent staining. Among all groups, the PD@Dox+aPD-1 treatment group exhibited the most extensive distribution of CD8+ T cells in tumor slices, followed by the PD@Dox and free Dox groups. These findings suggest that PD@Dox could induce dense apoptosis of tumor cells, trigger ICD, significantly promote infiltration of CD8+ T cells into tumors, stimulate a robust antitumor immune response, and demonstrate potent antitumor activity through synergistic immune checkpoint blockade therapy.
Safety Evaluation of Doxorubicin-Loaded Platelet Decoys
The severe side effects associated with Dox, such as cardiotoxicity, nephrotoxicity, hepatotoxicity, and short circulatory life, limit its use in chemotherapy.33 In this study, blood serum was collected from tumor-bearing mice after treatment, and the levels of CK-MB, ALT, and BUN were measured for safety evaluation. Figure 8A demonstrates that the level of CK-MB in the free Dox group was significantly increased compared to the control groups (P<0.0001). In contrast, the expression of CK-MB in the PD@Dox and PD@Dox+a-PD1 groups showed no significant change compared to the PBS group, indicating the safety of the Dox-loaded platelet decoys. Additionally, no significant differences in ALT and BUN levels were observed among all groups (Figure 8B and C).
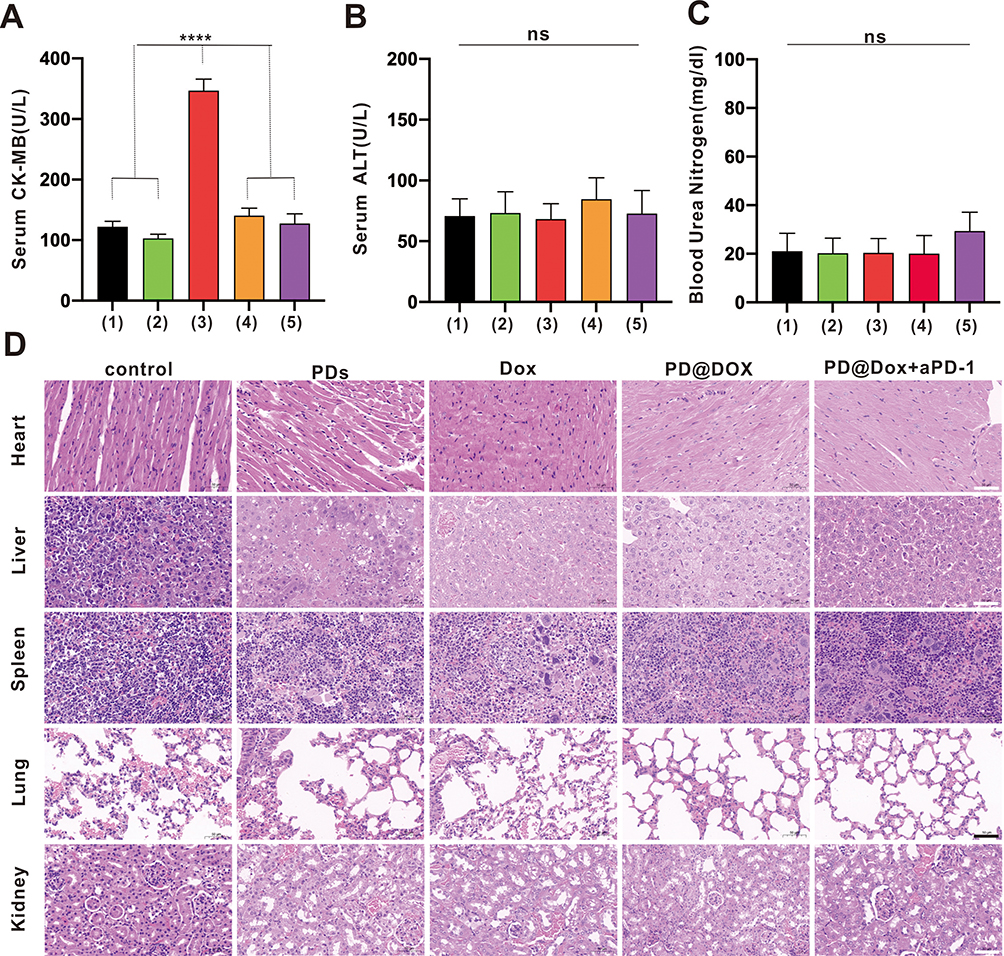 |
Figure 8 Safety evaluation of PD@Dox. (A-C) CK-MB, ALT and BUN were monitored on day 14 after treatment. (1) control; (2) PDs; (3) free Dox; (4)PD@Dox; (5)PD@Dox+aPD-1. (D) Hematoxylin-eosin staining of heart, liver, lung, spleen and kidney tissues obtained from animals in the different treatment groups (scale bar: 50 μm). ****P<0.0001. Abbreviations: ns, non-significance; CK-MB, creatine kinase-MB; ALT, alanine transaminase; BUN, blood urea nitrogen; Dox, doxorubicin; PDs, platelet decoys; PD@Dox, Dox loaded platelet decoys; CRT, calreticulin. |
Pathological examination of major organs, including the heart, liver, spleen, lung, and kidney, collected from the mice after different treatments, further confirmed the safety of PD@Dox. Figure 8D illustrates that there were no apparent signs of damage or inflammation infiltration in the major organs, except for the free Dox group, which exhibited some disarrangement and degeneration of the myocardium. These findings suggest that PD@Dox is non-toxic and exhibits good biocompatibility.
Discussion
In this study, we successfully prepared PD@Dox using a detergent commonly used for blood transfusions. These PD@Dox showed potential advantages over most platelet-related drug delivery systems that are primarily modified from natural platelets. Previous evidence has demonstrated that natural platelets can be recruited and activated by tumor cells to form tumor microemboli, which can promote metastasis.34,35 Li et al have designed tumor-targeted drug delivery platelets that eliminate the adverse effects of free Dox and effectively kill lymphoma cells.18 Similarly, Xu et al proposed a strategy utilizing platelets with photo-controlled release properties for highly targeted and photodynamic therapy for glioblastoma.36 However, therapeutic nanosystems based on platelets used for cancer treatment carry a risk of thrombosis, which is associated with poor prognosis and is a significant cause of death.37,38 Our electron microscopic results demonstrated that the normal morphology of platelets was preserved in PD@Dox. Although some key receptors on the surface of platelets (eg, CD41 and CD42b) were expressed at a lower level, they retained their ability to bind physiological ligands. Adhesion experiments showed that PD@Dox adhered to the surface of tumor cells, and this binding was weakened by heparin-blocked the related receptors. However, platelet decoys and PD@Dox failed to activate when stimulated with high concentrations of ADP, indicating an inactive conformation of cell surface GPIIb/IIIa. Similarly, the aggregation capability of PD@Dox with other platelets was diminished in response to ADP induction under the platelet aggregometer assay. Moreover, in the TEG experiments, PD@Dox and PD did not contribute to fibrin clot strength regardless of ADP stimulation. These findings are consistent with a study conducted by Papa et al.21
The PD@Dox demonstrated higher encapsulation efficiency and drug load compared to native platelets due to the loss of intracellular granules. Interestingly, our findings suggest that PD@Dox is readily released in the acidic environment that mimics the tumor microenvironment. This is particularly significant because free Dox is associated with severe off-target side effects and toxicity when distributed systemically.39,40 In contrast, PD@Dox can selectively target tumor cells through surface receptors and release the drug in a sustainable and pH-responsive manner. This approach reduces systemic adverse effects and enhances the efficacy of anticancer drugs. In a study by Uslu et al, exosomes isolated from ADP-stimulated platelets loaded with doxorubicin were shown to induce apoptosis in MDA-MB-231 cells. It was observed that platelets have a high affinity for binding to MDA-MB-231 cells.41 Additionally, we evaluated changes in serum CK-MB, ALT, and BUN levels in the free Dox group compared to other groups and observed that the platelet decoys alleviated the degree of cardiotoxicity induced by Dox in mice. Consequently, PD@Dox enables targeted drug delivery to tumor sites, reducing the required dosage of cytotoxic drugs for chemotherapy.
Chemotherapy not only exerts direct cytotoxic effects but also induces ICD in cancer cells, which can modulate the immunosuppressive tumor microenvironment and enhance response to immune checkpoint blockade. To investigate the ICD effect induced by PD@Dox-mediated chemotherapy, we examined characteristic markers of ICD in 4T1 cells, including ATP release, surface expression of CRT and HMGB-1, and DC maturation using a transwell system. Several strategies have been proposed to combine drug delivery systems loaded with Dox to selectively target cancer cells, trigger ICD, and initiate immune responses against DCs or macrophages.42–44 However, controlling the sequential release of drugs to improve efficacy and reduce toxicity remains a challenge. In our study, we utilized the prolonged circulation and selective targeting of platelet decoys to enhance intratumoral delivery of Dox, induce ICD, and promote antitumor immunity.
While modern immunotherapy, particularly ICIs, has shown breakthroughs in the treatment of hematologic and solid tumors, monotherapy with ICIs has yielded disappointing results in TNBC.45 Clinical trials evaluating anti-PD-1/PD-L1 monotherapy in breast cancer have reported objective response rates (ORRs) ranging from 5% to 24%. For example, a Phase I clinical trial investigating single-agent atezolizumab in metastatic TNBC showed an ORR of only 10%.46 Subsequently, a Phase 1b trial in unselected metastatic TNBC patients receiving chemotherapy reported a disappointing ORR of 5.3%.7 Combination strategies using ICIs and other chemotherapeutic agents have been explored due to their synergistic effects. Inspired by the anticancer therapy mediated by PD@Dox and the observed in vitro ICD effects, we investigated the therapeutic efficiency of PD@Dox in a xenograft 4T1 tumor model by combining it with aPD-1. Our data demonstrated significant tumor volume reduction in the groups treated with Dox, PD@Dox, and aPD-1, with the PD@Dox+aPD-1 group showing the most pronounced effect. Histopathological analysis further confirmed the improved prognosis of the PD@Dox+aPD-1 combination group compared to other groups.
Although our study has provided valuable insights into the potential of platelet decoys as a drug delivery system, there are several limitations that should be acknowledged. Firstly, it is important to note that platelet decoys, being non-living entities, may have a shorter circulation time compared to live platelets or cold-stored platelets. To address this limitation, future research should focus on engineering strategies to enhance the longevity of platelet decoys in circulation, thus optimizing their effectiveness as a drug delivery platform. Additionally, it is worth noting that dose-escalation studies were not conducted in this study to evaluate the safety profile of administering a large quantity of platelet decoys. Such studies are necessary to assess potential effects on liver and spleen function, as well as other adverse effects. Addressing these limitations will be instrumental in advancing the clinical application of platelet decoys for targeted therapeutic interventions.
Conclusion
Our study highlights PD@Dox as an innovative drug delivery system with significant potential for treating triple-negative breast cancer. By exploiting its unique characteristics, PD@Dox offers new avenues for improving patient outcomes and overcoming the challenges associated with this aggressive subtype. Future research should focus on optimizing therapeutic efficacy, addressing remaining challenges, and advancing PD@Dox towards clinical translation. This advancement requires comprehensive evaluation of safety, scalability, and feasibility for clinical implementation. PD@Dox holds great promise in transforming TNBC therapy.
Acknowledgments
The authors thank Dr.Xinmin Nie provided with valuable guidance and Dr.Jian Li for providing experimental support.
Funding
This work was supported by the Natural Science Foundation of Hunan Province (Grant No.2022JJ30885), the Independent Innovation Projects of Postgraduates of Central South University (grant no. 2021zzts0404) and Hunan Provincial Innovation Foundation for Postgraduate China Hunan Provincial Science and Technology Department (Grant No. CX20210376).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019;9(2):176–198. doi:10.1158/2159-8290.CD-18-1177
2. Gazinska P, Grigoriadis A, Brown JP, et al. Comparison of basal-like triple-negative breast cancer defined by morphology, immunohistochemistry and transcriptional profiles. Mod Pathol. 2013;26(7):955–966. doi:10.1038/modpathol.2012.244
3. Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–1281. doi:10.1200/JCO.2007.14.4147
4. Viscardi G, Tralongo AC, Massari F, et al. Comparative assessment of early mortality risk upon immune checkpoint inhibitors alone or in combination with other agents across solid malignancies: a systematic review and meta-analysis. Eur J Cancer. 2022;177:175–185. doi:10.1016/j.ejca.2022.09.031
5. Srivastava S, Mohanty A, Nam A, Singhal S, Salgia R. Chemokines and NSCLC: emerging role in prognosis, heterogeneity, and therapeutics. Semin Cancer Biol. 2022;86(Pt 2):233–246. doi:10.1016/j.semcancer.2022.06.010
6. Flynn M, Pickering L, Larkin J, Turajlic S. Immune-checkpoint inhibitors in melanoma and kidney cancer: from sequencing to rational selection. Ther Adv Med Oncol. 2018;10:1758835918777427. doi:10.1177/1758835918777427
7. Schmid P, Rugo HS, Adams S, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet Oncol. 2020;21(1):44–59. doi:10.1016/S1470-2045(19)30689-8
8. Adams S, Diéras V, Barrios CH, et al. Patient-reported outcomes from the Phase III IMpassion130 trial of atezolizumab plus nab-paclitaxel in metastatic triple-negative breast cancer. Ann Oncol. 2020;31(5):582–589. doi:10.1016/j.annonc.2020.02.003
9. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875. doi:10.1038/nrc3380
10. Dosset M, Joseph ELM, Rivera Vargas T, Apetoh L. Modulation of Determinant Factors to Improve Therapeutic Combinations with Immune Checkpoint Inhibitors. Cells. 2020;9(7):1727. doi:10.3390/cells9071727
11. Jiang M, Zeng J, Zhao L, et al. Chemotherapeutic drug-induced immunogenic cell death for nanomedicine-based cancer chemo-immunotherapy. Nanoscale. 2021;13(41):17218–17235. doi:10.1039/d1nr05512g
12. Singh M, Nicol AT, DelPozzo J, et al. Demystifying the Relationship Between Metformin, AMPK, and Doxorubicin Cardiotoxicity. Front Cardiovasc Med. 2022;9:839644. doi:10.3389/fcvm.2022.839644
13. Bagdasaryan AA, Chubarev VN, Smolyarchuk EA, et al. Pharmacogenetics of drug metabolism: the role of gene polymorphism in the regulation of doxorubicin safety and efficacy. Cancers. 2022;14(21):5436. doi:10.3390/cancers14215436
14. Ke WR, Chang RYK, Chan HK. Engineering the right formulation for enhanced drug delivery. Adv Drug Deliv Rev. 2022;191:114561. doi:10.1016/j.addr.2022.114561
15. Kong L, Yang C, Zhang Z. Organism-generated biological vesicles in situ: an emerging drug delivery strategy. Adv Sci. 2022;e2204178. doi:10.1002/advs.202204178
16. Vadasz B, Chen P, Yougbaré I, et al. Platelets and platelet alloantigens: lessons from human patients and animal models of fetal and neonatal alloimmune thrombocytopenia. Genes Dis. 2015;2(2):173–185. doi:10.1016/j.gendis.2015.02.003
17. Deng C, Zhao X, Chen Y, et al. Engineered platelet microparticle-membrane camouflaged nanoparticles for targeting the golgi apparatus of synovial fibroblasts to attenuate rheumatoid arthritis. ACS Nano. 2022;16(11):18430–18447. doi:10.1021/acsnano.2c06584
18. Li QR, Xu HZ, Xiao RC, et al. Platelets are highly efficient and efficacious carriers for tumor-targeted nano-drug delivery. Drug Deliv. 2022;29(1):937–949. doi:10.1080/10717544.2022.2053762
19. Wu YW, Huang CC, Changou CA, Lu LS, Goubran H, Burnouf T. Clinical-grade cryopreserved doxorubicin-loaded platelets: role of cancer cells and platelet extracellular vesicles activation loop. J Biomed Sci. 2020;27(1):45. doi:10.1186/s12929-020-00633-2
20. Pandey S, Shaikh F, Gupta A, Tripathi P, Yadav JS. A recent update: solid lipid nanoparticles for effective drug delivery. Adv Pharm Bull. 2022;12(1):17–33. doi:10.34172/apb.2022.007
21. Papa AL, Jiang A, Korin N, et al. Platelet decoys inhibit thrombosis and prevent metastatic tumor formation in preclinical models. Sci Transl Med. 2019;11(479):eaau5898. doi:10.1126/scitranslmed.aau5898
22. Hu Q, Sun W, Wang J, et al. Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat Biomed Eng. 2018;2(11):831–840. doi:10.1038/s41551-018-0310-2
23. du Sert N P, Ahluwalia A, Alam S, et al. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020;18(7):e3000411. doi:10.1371/journal.pbio.3000411
24. Lau N, Haeberle AL, O’Keeffe BJ, et al. SopF, a phosphoinositide binding effector, promotes the stability of the nascent Salmonella-containing vacuole. PLoS Pathog. 2019;15(7):e1007959. doi:10.1371/journal.ppat.1007959
25. Bivard A, Lin L, Parsonsb MW. Review of stroke thrombolytics. J Stroke. 2013;15(2):90–98. doi:10.5853/jos.2013.15.2.90
26. Schwarz S, Gockel LM, Naggi A, et al. Glycosaminoglycans as Tools to Decipher the Platelet Tumor Cell Interaction: a Focus on P-Selectin. Molecules. 2020;25(5):1039. doi:10.3390/molecules25051039
27. Tesfamariam B. Involvement of platelets in tumor cell metastasis. Pharmacol Ther. 2016;157:112–119. doi:10.1016/j.pharmthera.2015.11.005
28. Fabricius HÅ, Starzonek S, Lange T. The role of platelet cell surface P-selectin for the direct platelet-tumor cell contact during metastasis formation in human tumors. Front Oncol. 2021;11:642761. doi:10.3389/fonc.2021.642761
29. Cheng Y, Wang C, Wang H, et al. Combination of an autophagy inhibitor with immunoadjuvants and an anti-PD-L1 antibody in multifunctional nanoparticles for enhanced breast cancer immunotherapy. BMC Med. 2022;20(1):411. doi:10.1186/s12916-022-02614-8
30. D C, Z Q, J M, et al. Tumors and their microenvironment dual-targeting chemotherapy with local immune adjuvant therapy for effective antitumor immunity against breast cancer. Adv Sci. 2019;6(6). doi:10.1002/advs.201801868
31. Qiu F, Becker KW, Knight FC, et al. Poly(propylacrylic acid)-peptide nanoplexes as a platform for enhancing the immunogenicity of neoantigen cancer vaccines. Biomaterials. 2018;182:82–91. doi:10.1016/j.biomaterials.2018.07.052
32. Shevtsov M, Sato H, Multhoff G, Shibata A. Novel approaches to improve the efficacy of immuno-radiotherapy. Front Oncol. 2019;9:156. doi:10.3389/fonc.2019.00156
33. Farag MR, Moselhy AAA, El-Mleeh A, et al. Quercetin alleviates the immunotoxic impact mediated by oxidative stress and inflammation induced by doxorubicin exposure in rats. Antioxidants. 2021;10(12):1906. doi:10.3390/antiox10121906
34. Xu XR, Zhang D, Oswald BE, et al. Platelets are versatile cells: new discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci. 2016;53(6):409–430. doi:10.1080/10408363.2016.1200008
35. Wang L, Wang X, Guo E, Mao X, Miao S. Emerging roles of platelets in cancer biology and their potential as therapeutic targets. Front Oncol. 2022;12:939089. doi:10.3389/fonc.2022.939089
36. Xu HZ, Li TF, Ma Y, et al. Targeted photodynamic therapy of glioblastoma mediated by platelets with photo-controlled release property. Biomaterials. 2022;290:121833. doi:10.1016/j.biomaterials.2022.121833
37. Koizume S, Miyagi Y. Tissue factor in cancer-associated thromboembolism: possible mechanisms and clinical applications. Br J Cancer. 2022;127(12):2099–2107. doi:10.1038/s41416-022-01968-3
38. Campia U, Moslehi JJ, Amiri-Kordestani L, et al. Cardio-oncology: vascular and metabolic perspectives: a scientific statement from the American heart association. Circulation. 2019;139(13):e579–e602. doi:10.1161/CIR.0000000000000641
39. Rao NV, Rho JG, Um W, et al. Hyaluronic Acid Nanoparticles as Nanomedicine for Treatment of Inflammatory Diseases. Pharmaceutics. 2020;12(10):931. doi:10.3390/pharmaceutics12100931
40. Thapa R, Wilson GD. The Importance of CD44 as a Stem Cell Biomarker and Therapeutic Target in Cancer. Stem Cells Int. 2016;2016:2087204. doi:10.1155/2016/2087204
41. Uslu D, Abas BI, Demirbolat GM, Cevik O. Effect of platelet exosomes loaded with doxorubicin as a targeted therapy on triple-negative breast cancer cells. Mol Divers. 2022. doi:10.1007/s11030-022-10591-6
42. Zhan M, Qiu J, Fan Y, et al. Phosphorous dendron micelles as a nanomedicine platform for cooperative tumor chemoimmunotherapy via synergistic modulation of immune cells. Adv Mater. 2023;35(3):e2208277. doi:10.1002/adma.202208277
43. Jeong SD, Jung BK, Lee D, et al. Enhanced Immunogenic Cell Death by Apoptosis/Ferroptosis Hybrid Pathway Potentiates PD-L1 Blockade Cancer Immunotherapy. ACS Biomater Sci Eng. 2022;8(12):5188–5198. doi:10.1021/acsbiomaterials.2c00950
44. Synergistic combination of targeted nano-nuclear-reactors and anti-PD-L1 nanobodies evokes persistent T cell immune activation for cancer immunotherapy - PubMed. Available from: https://pubmed.ncbi.nlm.nih.gov/36496381/.
45. Rizzo A, Cusmai A, Massafra R, et al. Pathological complete response to neoadjuvant chemoimmunotherapy for early triple-negative breast cancer: an updated meta-analysis. Cells. 2022;11(12):1857. doi:10.3390/cells11121857
46. Emens LA, Cruz C, Eder JP, et al. Long-term clinical outcomes and biomarker analyses of atezolizumab therapy for patients with metastatic triple-negative breast cancer: a phase 1 study. JAMA Oncol. 2019;5(1):74–82. doi:10.1001/jamaoncol.2018.4224
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Targeted Nanobubbles of PD-L1 mAb Combined with Doxorubicin as a Synergistic Tumor Repressor in Hepatocarcinoma
Chen Y, Luo X, Liu Y, Zou Y, Yang S, Liu C, Zhao Y
International Journal of Nanomedicine 2022, 17:3989-4008
Published Date: 7 September 2022