")
Back to Journals » International Journal of Nanomedicine » Volume 13
Doxorubicin and anti-VEGF siRNA co-delivery via nano-graphene oxide for enhanced cancer therapy in vitro and in vivo
Authors Sun Q, Wang X, Cui C, Li J, Wang Y
Received 18 January 2018
Accepted for publication 8 March 2018
Published 27 June 2018 Volume 2018:13 Pages 3713—3728
DOI https://doi.org/10.2147/IJN.S162939
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Qi Sun,1–3 Xiaoli Wang,1–3 Chunying Cui,1–3 Jing Li,1–3 Yifan Wang1–3
1Department of Pharmaceutics, School of Pharmaceutical Sciences, Capital Medical University, Beijing, China; 2Beijing Area Major Laboratory of Peptide and Small Molecular Drugs, Beijing, China; 3Engineering Research Center of Endogenous Prophylactic of Ministry of Education of China, Beijing, China
Background: Graphene oxide (GO) has attracted intensive interest in biological and medical fields in recent years due to its unique physical, chemical, and biological properties. In our previous work, we proved that GO could deliver small interfering RNA (siRNA) into cells and downregulate the expression of the desired gene.
Methods: This study investigated the potential of a modified GO nanocarrier for co-delivery of siRNA and doxorubicin (DOX) for enhanced cancer therapy. Fourier transform infrared spectroscopy, laser particle size analyzer, UV-visible spectroscopy, gel electrophoresis retardation, and in vitro release assay were studied.
Results: The results of real-time polymerase chain reaction revealed that the expression of vascular endothelial growth factor (VEGF) mRNA was decreased 46.84%±3.72% (mean ± SD). Enzyme-linked immunosorbent assay indicated that the expression of VEGF protein was downregulated to 52.86%±1.10% (mean ± SD) in vitro. In vivo tumor growth assay GO-poly-L-lysine hydrobromide/folic acid (GPF)/DOX/siRNA exhibited gene silencing and tumor inhibition (66.95%±2.35%, mean ± SD) compared with naked siRNA (1.62%±1.47%, mean ± SD) and DOX (33.63%±5.85%, mean ± SD). GPF/DOX/siRNA exhibited no testable cytotoxicity.
Conclusion: The results indicated that co-delivery of siRNA and DOX by GPF could be a promising application in tumor clinical therapy.
Keywords: graphene oxide, siRNA delivery, co-delivery carrier, cervical carcinoma
Introduction
Tumors have always been a major threat to human health. Nowadays, chemotherapeutic drugs are widely used to treat cancer; however, the use of chemotherapeutic drugs alone has several drawbacks: firstly, chemotherapy has low selectivity and low antitumor efficiency; secondly, frequently using chemotherapeutic drugs could lead to multidrug resistance and reduce the sensitivity of cells to drugs, which is not conducive to tumor suppression.1 Research has shown that the combination of drug treatment and gene therapy can effectively retain their respective advantages and greatly enhance the treatment of cancer.2,3
RNA interference therapy can transform double-stranded RNA into 20–25 base pair small interfering RNAs (siRNAs) and directs them to disrupt their targeted mRNAs.4–9 As a result, it can result in the downregulation of the targeting gene expression. However, cell membrane prevents the crossing of the naked siRNA because of its negative charge, water solubility, and high molecular weight.10–12 Thus, an efficient carrier to overcome the poor intracellular uptake and instability problems is required.
Co-delivery of siRNA and drug is a promising approach to improve therapeutic efficacy. Combination therapy has been widely used in the treatment of cancer in recent years because of the inherent difficulty of tumor therapy and its complex mechanism of action.13 Currently, a large number of studies have shown that the combination of chemotherapy and gene therapy improved the efficiency of cancer treatment, reduced the side effects of chemotherapeutic drugs, increased the sensitivity of tumor cells to drugs, and reduced the multidrug resistance problem.14–18 The prerequisite for using this combination method is to construct a carrier for co-delivery of drugs and genes. Various studies have reported that the co-delivery of siRNA and anticancer drug suppressed tumor growth greatly. Susa et al studied the MDR1 siRNA nanocarrier to treat osteosarcoma cell lines KHOS(R2) and U-2OS(R2), and the expression of MDR1 protein (P-glycoprotein [P-gp]), drug retention, and immunofluorescence were explored.14 Xiong and Lavasanifar constructed a micellar system from degradable poly(ethylene oxide)-block-poly(ε-caprolactone) block copolymers with functional groups on both blocks, and this system was used to improve the efficacy of doxorubicin (DOX) in multidrug resistant MDA-MB-435 human tumor models that P-gp overexpressed by the simultaneous intracellular delivery of DOX and siRNA against P-gp expression.15 Saraswathy and Gong discussed the synergistic effects of combinatorial anticancer drug and siRNA therapy in various cancer models employing multifunctional drug/siRNA co-delivery nanocarriers.16 Cao et al prepared a diblock copolymer, polyethylene glycol-polycaprolactone (PEI-PCL), for biodegradable co-delivery of DOX and BCL-2 siRNA.17 Dong et al prepared a self-assembled gene/chemotherapeutic drugs polymer consisting of two parts, PEI-HZ-DOX and PEI-PEG-folate. DOX was linked to PEI by pH-sensitive hydrazone and siRNA was loaded on the carrier by the action of electrostatic adsorption.18
Graphene oxide (GO) is the oxidative product of graphite.19,20 Previous studies have shown that functionalized GO has been extensively used in drug research as a nanocarrier.21–26 With good biocompatibility and biodegradability, GO had been used for siRNA delivery and showed remarkable success in gene knockdown. GO has a single atomic structure with a larger specific surface area, negative charge, and good water dispersion. It can be functionally modified through its epoxy, hydroxyl, and carboxyl groups. For instance, Wang et al developed a GO-based gene delivery system by covalently conjugating PEG and PEI to improve its stability and gene transfection efficiency. Polyethylene glycol conjugation along with π–π stacked pyrenemethylamine was performed to deliver siRNA into cancer cells. PEI-PEG bifunctionalized GO has been synthesized, and transfected into human hepatic cancer Bel-7402 cells with plasmid DNA (pDNA) to complete gene transfer.27,28 Octaarginine was used to modify GO to deliver pDNA.29 Folic acid (FA)-modified GO was used as a novel vector to increase gene delivery efficiency and successfully loaded the SI-Stas3 gene into cells.30
In our previous work, we proved that GO can deliver siRNA into cells and downregulate the expression of the desired gene. This study investigated the potential of a modified GO nanocarrier for co-delivery of siRNA and DOX for enhanced cancer therapy. A novel carrier, GO-poly-L-lysine hydrobromide/FA (GPF), was characterized by Fourier transform infrared (FTIR) spectroscopy, laser particle size analyzer, UV-visible (UV-vis) spectroscopy, and gel electrophoresis retardation. Real-time polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA), and in vivo tumor growth assay were performed to evaluate the transfection efficiency by GPF/DOX/vascular endothelial growth factor (VEGF)-siRNA.
Materials and methods
Materials
The GO sheet, poly-L-lysine hydrobromide (PLL, molecular weight =30,000–70,000 Da), and FA were purchased from Sigma-Aldrich (St Louis, MO, USA). Dialysis membranes (molecular weight cutoff [Mwco]: 8,000–14,000) were purchased from SpectrumLabs (Los Angeles, CA, USA). Lipo™2000 was purchased from Thermo Fisher Scientific (Waltham, MA, USA). HyClone Laboratories Inc (Logan, UT, USA) provided Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), and trypsin. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), penicillin, and streptomycin were purchased from Sigma-Aldrich. Bicinchoninic acid (BCA) protein kit was purchased from Pierce (Rockford, IL, USA). Human VEGF ELISA kit, mouse VEGF ELISA kit, High Capacity cDNA Reverse Transcription Kit, High Capacity RNA-to-cDNA Kit, and Trizol were obtained from Thermo Fisher Scientific. Matrigel was obtained from BD Biosciences (San Jose, CA, USA). VEGF-siRNA and fluorescein-labeled VEGF-siRNA (FAM-VEGF-siRNA, Cy3-VEGF-siRNA) were purchased from Gene-Pharma Co, Ltd (Shanghai, China). HeLa cell line was obtained from the Chinese Academy of Medical Sciences tumor cells bank.
Preparation of GPF/DOX/VEGF-siRNA
Preparation of GPF
GO-PLL was synthesized using a previously described method.31–33 In brief, firstly, dispersed GO (2 mg) was added into 4 mL of deionized (DI) water by means of an ultrasonic cleaner. The GO solution (0.5 mg/mL) was ultrasonicated again by using an ultrasonic probe for 2 hours (2 seconds interval model) to obtain nano-size GO. Ten milligrams of PLL, 4 mL of GO solution, and 4 mL of KOH solution (pH 9.0) was mixed, and then 2 mL KOH solution (pH 11.0) was used to adjust the pH value to 9.0. The reaction mixture was stirred at 70°C for 24 hours. GO-PLL was acquired by centrifugation (125 g, 20 minutes). DI water was used to wash the suspension three times. Finally, the GO-PLL was collected and freeze dried (Scheme 1, step I). According to the results of a previous study, the final selection ratio of GO and PLL is 1:5 (w/w).
 | Scheme 1 Scheme for GPF and GPF/DOX/VEGF-siRNA preparation. (I) KOH, 70°C, 24 hours, DI water. (II) Room temperature, 24 hours. (III) Stirred at room temperature, without light, 12 hours, DI water. (IV) Incubation at 37°C, 30 minutes, DEPC water. |
Dispersed GO-PLL powder (2 mg), FA (2 μg), and EDC·HCl (2 mg) were activated, respectively, for 15 minutes. The mixture was stirred at room temperature for 24 hours in acetate buffer (pH 4.7).34–38 After the reaction, the pH of the solution was adjusted to a value of 9.0 by NaOH and the product was collected, centrifuged (125 g, 15 minutes), and washed with DI water three times. Eventually, GPF was obtained and freeze dried. Dialysis (Mwco: 8,000–14,000) was used to purify GPF in DI water for 48 hours. It was found that 2 μg of FA was loaded to 2 mg of GO-PLL (Scheme 1, step II).
Preparation of GPF/DOX and GPF/DOX/siRNA
GPF (1 mg) was dispersed into 1 mL DI water, mixed with DOX of 1, 2, 3, 4, and 5 mg, respectively, and stirred for 12 hours without sunlight. The mixture was centrifuged (125 g, 15 minutes) at least three times until the supernate turned colorless. The supernate were collected, centrifuged, and measured at UV 480 nm absorbance.38 The standard curve of concentration versus absorbance was drawn, and the concentration of DOX was obtained (Scheme 1, step III). Then, GPF/DOX and VEGF-siRNA were prepared at the optimum N/P ratio. The samples were incubated and shaken gently at 37°C for 30 minutes (Scheme 1, step IV).
Characterization
The structure of GO, PLL, GO-PLL, and GPF was determined by FTIR spectra and UV-vis absorption spectra. UV-vis absorption was measured by a UV-vis spectrophotometer and the samples were dispersed in DI water. Zeta potentials, hydrodynamic sizes, and optical property were determined by a zeta potential analyzer.
Agarose gel retardation assay
The electrostatic adsorption of VEGF-siRNA onto GPF was analyzed quantitatively and qualitatively by agarose gel retardation assay. GPF/VEGF-siRNA and GO/VEGF-siRNA were prepared and tested at different w/w ratios (10:1, 15:1, 20:1, 25:1, 30:1, 35:1, 40:1). Before loading into the agarose gel, all samples were incubated and shaken gently at 37°C for 30 minutes. The process was stopped by adding 5 μL of loading buffer. The samples was electrophoresed in 1% (w/v) agarose gel containing ethidium bromide (2 mg/mL) at a constant voltage of 120 V for 20 minutes.39–41 The results were measured by a UV transilluminator.
Heparin and anti-RNase A degradation assay
Above all, 7.5 μL of naked VEGF-siRNA (0.075 nmol VEGF-siRNA) and 37.5 μL of GPF/VEGF-siRNA (0.075 nmol VEGF-siRNA) were precisely measured. The samples were co-incubated with an equal volume of RNase A solution (0.1 mg/mL) at 37°C for 30 minutes. Five drops of EDTA solution (5 mM) were added at the set time point to stop the degradation. Heparin sodium (0.8 mg/mL, 20 μL) was added at 37°C for 30 minutes incubation in order to replace siRNA from GPF, and the samples were subjected to agarose gel electrophoresis finally.42
Release profile
Dialysis assay was performed to evaluate the release of DOX from GPF and GO. PBS solution (pH 7.4 and pH 5.0) was selected as the release medium in order to investigate the release characteristics in different media. Here, 19.8 μg of DOX, GO/DOX, and GPF/DOX were separately dispensed into 500 μL of a different release medium. Dialysis bags (Mwco: 8,000–14,000) were placed in the flask (n=3) and shaken at 37°C (130 times/min). The buffer (5 mL) was collected and replaced with fresh buffer (5 mL) at different time points, respectively. The absorbance was recorded at 480 nm. The percentages of cumulative release were calculated according to the standard curve.43 All experiments were repeated three times.
Cellular uptake of GPF/DOX/VEGF-siRNA44
The relocation of VEGF-siRNA and DOX was recorded by confocal laser scanning microscopy. HeLa cells (2.5×105 cells/dish) were seeded in 35 mm glass bottomed dish and incubated with complete medium for 24 hours. When cells were attached onto the dishes, 1 mL of serum-free medium containing FAM-VEGF-siRNA (100 nM), DOX (100 nM), GPF/FAM-VEGF-siRNA (100 nM), GPF/DOX (100 nM), GPF/DOX/FAM-VEGF-siRNA (100 nM), and Lipo™2000/FAM-VEGF-siRNA (100 nM) were used to transfect cells for another 4 hours. After removing the medium and washing the cells three times with cold PBS solution (pH 7.4, 0.01 nM), 1 mL of Hoechst 33342 (4 μg/mL) was added into the dishes to stain the cell nuclei at 37°C for 20 minutes. Then, the liquid was discarded, and cells were washed with cold PBS solution. The intracellular location of VEGF-siRNA and DOX was observed by a laser scanning confocal microscope and analyzed using Leica CLSM software (Leica Microsystems, Wetzlar, Germany).
Cell proliferation inhibitory assay46
Anti-proliferation activity of GPF/DOX/VEGF-siRNA was evaluated by MTT assay. Briefly, HeLa cells were seeded in 96-well plates at a density of 4×103/well and cultured overnight for cell attachment. The cells were incubated with fresh medium containing 10, 40, 80, and 120 nM of GPF, naked VEGF-siRNA, DOX, GPF/NC, Lipo™2000/NC, GPF/VEGF-siRNA, GPF/DOX, GPF/DOX/VEGF-siRNA, and Lipo™2000/VEGF-siRNA (solution: 20 μL/well), respectively, at 37°C for 6 hours. The medium of each group was replaced by complete medium and incubated for another 44 hours. Then, 25 μL of MTT solution (5 mg/mL) was added into each well to measure the cell viability under 570 nm. All experiments were repeated three times.
Real-time PCR
Real-time PCR was used to evaluate the expression of VEGF mRNA in HeLa cells.47 HeLa cells (2×105 cells/well) were seeded in six-well dishes and transfected by GPF/VEGF-siRNA (100 nM), naked VEGF-siRNA (100 nM), GPF/NC (100 nM), GPF/DOX/NC (100 nM), GPF/DOX/VEGF-siRNA (100 nM), and Lipo™2000/VEGF-siRNA (100 nM) in the medium without FBS for 4 hours. The cells were cultured with complete medium for another 44 hours. The total cell RNA was extracted by Trizol reagent. The concentration of the RNA was recorded by Nanodrop-1000. High Capacity RNA-to-cDNA Kit was used to measure the RNA concentration. cDNA (80 ng) was subjected to a 7500 Real-time PCR System according to standard instructions. The mRNA expression of VEGF was evaluated against GAPDH mRNA. The Ct value was calculated using delta-delta Ct (2−ΔΔCt) method. All experiments were repeated three times.
ELISA
HeLa cells (3.0×105 cells/well) were seeded into six-well plates. After culturing overnight, the medium was replaced with naked VEGF-siRNA (100 nM), GPF/NC (100 nM), GPF/VEGF-siRNA (100 nM), GPF/DOX/NC (100 nM), GPF/DOX/VEGF-siRNA (100 nM), Lipo™2000/NC (100 nM), and Lipo™2000/VEGF-siRNA (100 nM) for 4 hours. The complete medium was used to culture for another 44 hours. The supernatant was collected and centrifuged (125 g, 4°C) for 10 minutes. The protein concentration was measured by BCA Protein Quantification Kit. Human VEGF ELISA kit was used to measure the amount of VEGF protein in accordance with the manufacturer’s instruction.48 A microplate reader was used to record the optical density (OD) values at 450 nm. All experiments were repeated three times.
Tumor growth inhibitory assay in vivo
Institute of Cancer Research (ICR) mice (6-week-old, 18–22 g) were obtained from the Animal Department of Capital Medical University. All animal studies were performed in accordance with the Health Guidelines of the Capital Medical University, and the Institutional Animal Ethics Committee of Capital Medical University approved the protocols and in vivo experiments. Tumor xenograft was made by inoculating 2.0×106 S180 cells into the mice for the experiments. The mice were inhabited in cages (five mice/cage) at constant temperatures (22°C–25°C) and suitable humidity (50%±2.0%). Tumor volume was measured by dial caliper and calculated as: volume (mm3) = length × width2/2. After the volume of the tumor reached 150 mm3, the mice were randomly divided into seven groups (n=10). The mice were treated with GPF/DOX/VEGF-siRNA (9.75 mg/kg), GPF/VEGF-siRNA (9.3 mg/kg), GPF/DOX (9.45 mg/kg), DOX (0.45 mg/kg), naked VEGF-siRNA (0.3 mg/kg), positive control (2.0 μmol/kg), and normal saline (NS) solution, respectively, once every other day via intravenous injection. The amount of GPF/DOX/VEGF-siRNA applied was calculated with VEGF-siRNA (0.3 mg/kg) and GPF (9.0 mg/kg). After 10 days, the mice were sacrificed to harvest, weigh, and photograph. Brain, liver, spleen, heart, and kidney were separately harvested in order to detect organ toxicity. Besides, mouse VEGF ELISA kit was used to detect the VEGF content in the collected serum of mice. The targeting effect of GPF/DOX/VEGF-siRNA was evaluated compared with the groups NS and GO-PLL/DOX/VEGF-siRNA. Tumor-bearing mice were randomly divided into four groups. The mice were treated with NS, DOX (clinical dosage), GPF/DOX/VEGF-siRNA, and GO-PLL/DOX/VEGF-siRNA (n=10).
Cytotoxicity assay45
MTT assay was used to evaluate the cytotoxicity of GPF against HeLa cells. HeLa cells were seeded into 96-well plates with complete medium and were cultured overnight for cell attachment. Then, 5, 10, 20, 40, 60, 80, 100, and 120 μg/mL of GPF in 20 μL of culture medium were added into each experimental well, while 20 μL of fresh medium was added into the control wells and cultured for another 48 hours. After incubation, 25 μL of MTT solution (5 mg/mL) was added into each well. The cells were incubated for another 4 hours. After removing the medium, 150 μL of DMSO was added into each well carefully. The 96-well plates were shaken for 10 minutes. Finally, the OD was recorded at 570 nm. All experiments were repeated three times. The cell viability was calculated as follows:
|
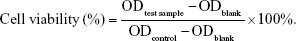
Statistics analysis
All experiments were performed at least three times. Quantitative data were expressed as mean ± SD. Paired two-sample Student’s t-test was used for statistical analysis. P-value <0.05 was considered to be of statistical significance and P-value <0.01 was considered very significant.
Results and discussion
Zeta potential and dispersion stability
Zeta potential is an important parameter and indicates the stability of the colloidal suspension. As shown in Figure 1, when FA is gradually added, the zeta potential of GPF is reduced from 34.38 to 22.42 mV. GO was shown to have a zeta potential of −45.65±0.70 mV (Figure 1A). For loading the negatively charged siRNA, the zeta potential of GO-PLL and GPF were 34.38±1.21 mV (Figure 1B) and 22.42±3.71 mV (Figure 1C). Because of the successful modification with PLL and FA, GPF was highly dispersed, and the nano-dispersion was stable and no aggregation was observed.
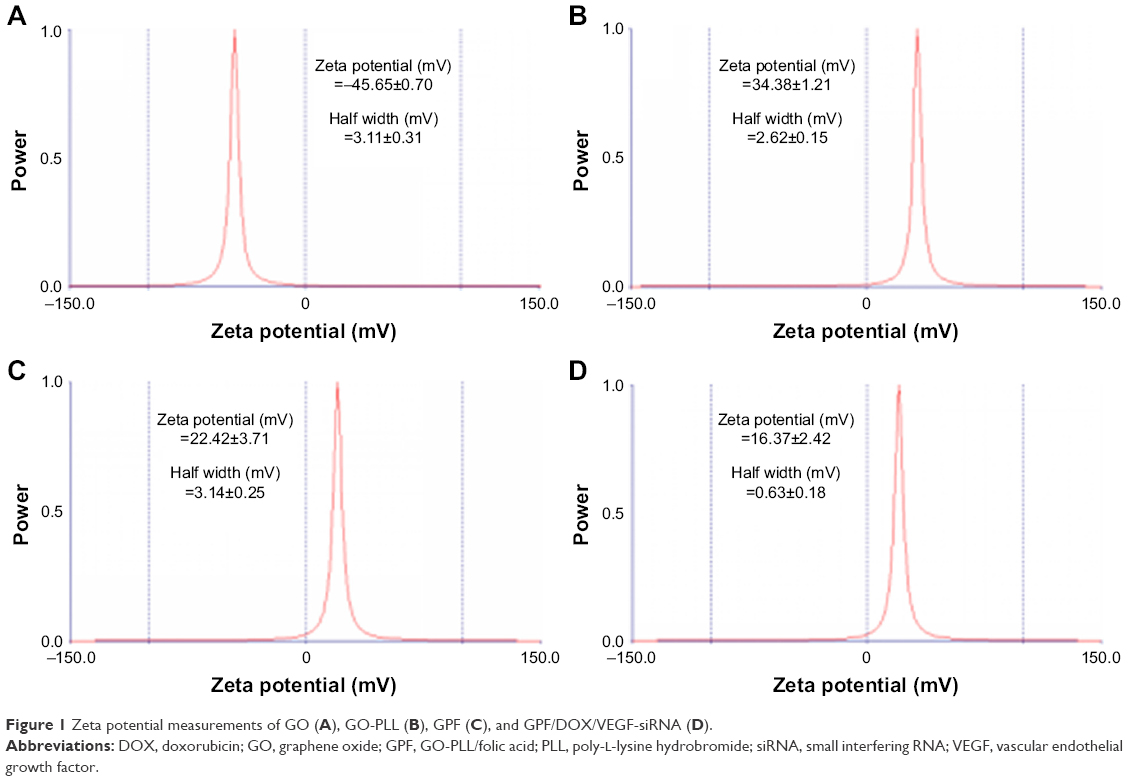 | Figure 1 Zeta potential measurements of GO (A), GO-PLL (B), GPF (C), and GPF/DOX/VEGF-siRNA (D). |
FTIR, UV-vis, transmission electron microscopy (TEM), zeta potential, and Tyndall effect
To confirm the successful preparation of GPF, the FTIR measurement was used (Figure 2). FTIR spectra showed a broad peak of CO2H groups at 3,200–3,500 cm−1. Comparing with the FTIR spectrum of GO (red) and PLL (purple), the spectrum of GO-PLL (green) showed a broad peak of CO2H groups at 3,200–3,500 cm−1, the peak of the carbonyl groups of the amide at ~1,680 cm−1, suggesting that PLL was covalently coupled with the GO successfully. Meanwhile, in the spectrum of GPF (blue), the strong absorption peaks of amide I (1,645 cm−1) and amide II (1,515 cm−1) were indicative of the existence of GPF.
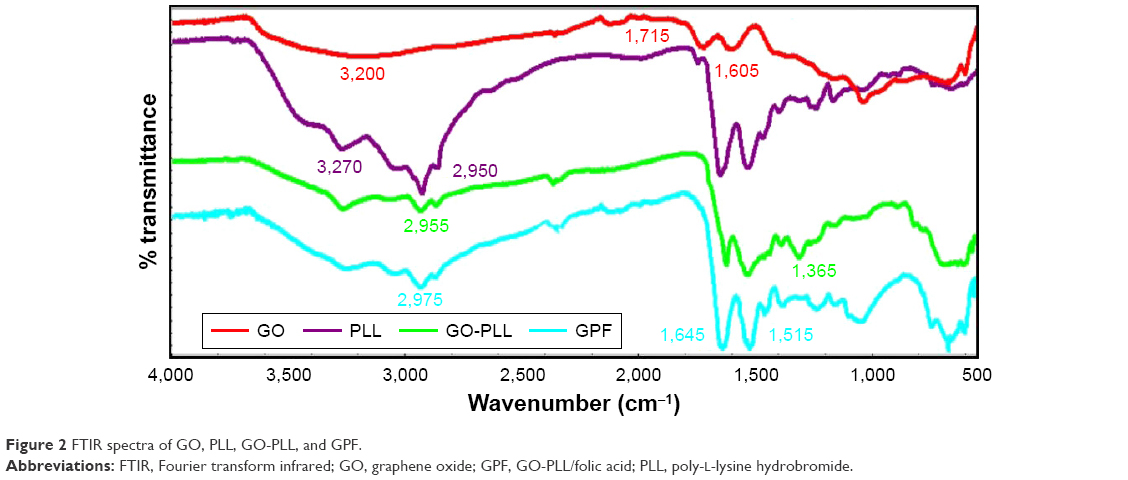 | Figure 2 FTIR spectra of GO, PLL, GO-PLL, and GPF. |
By means of UV-vis spectroscopy, the modified GO, PLL, GO-PLL, and GPF were further characterized (Figure S1). Because of π-π* transition of the aromatic ring, the absorption peak of GO (blue) was 243 nm. Compared to the absorption peak of PLL (red), GO-PLL (green) showed the characteristic peak at 207 nm after the ring-opening reaction. A red-shift was observed from 235 to 270 nm of shoulder peak of GO-PLL, which indicated the ring-opening reaction of GO and PLL. After the conjugation of FA, GPF (purple) was observed to have a shoulder peak at 275 nm.
By means of TEM, the morphology of GO, GPF, and GPF/DOX/VEGF-siRNA was studied as shown in Figure 3. The typical wrinkle sheet-like nanostructure with diameter sizes from 100 to 250 nm can be observed in Figure 3A. On the contrary, there were no obvious changes of nanoscale size in GPF (Figure 3B) and GPF/DOX/VEGF-siRNA (Figure 3C). The horizontal and vertical distances of GO were 210.37 and 3.366 nm, respectively. Compared to GO, the vertical distance of GPF increased to 17.247 nm due to the existence of PLL and FA, while the horizontal distance was 183.94 nm. Because of the loading of VEGF-siRNA and DOX, the horizontal and vertical distances of GPF/DOX/VEGF-siRNA changed to 21.343 and 158.61 nm, respectively.
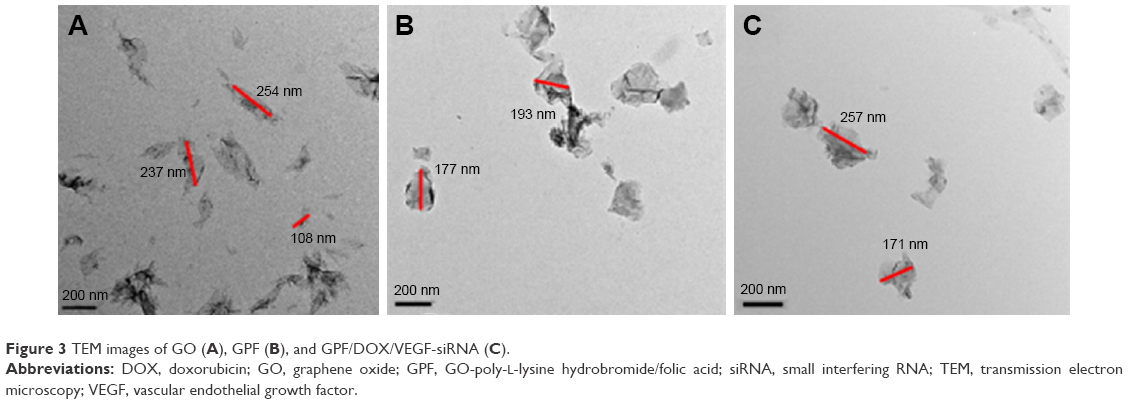 | Figure 3 TEM images of GO (A), GPF (B), and GPF/DOX/VEGF-siRNA (C). |
Cellular uptake of the nanocarriers depends on the size as well as the surface characteristics.49,50 To investigate the particle size of the carriers, the Tyndall phenomenon was observed and the results are shown in Figure 4. The results indicated that GO and GPF aqueous suspension reached the nano-level, which was observed by means of a single laser beam (λ=650 nm). As shown in Figure 4B, the precipitation of GO in PBS and DMEM was seen obviously which indicated that GO could be stable in water only after 48 hours while GPF could stay stable in water, PBS, and DMEM medium (Figure 4A).
 | Figure 4 The dispersibility and stability of GPF (A), GO (B), and the Tyndall effect of GO and GPF (C). |
UV determination
Table 1 shows that with the increase of the mass ratio of DOX onto GPF, the drug loading and entrapment efficiency of GPF gradually increased. In addition, with the increase of adsorbed DOX, it was found that both the positive conductance of the GPF surface and VEGF-siRNA loading were increased. In order to achieve the synergistic effect of chemical and genetic effects, the final ratio of GPF and DOX adsorption was 1:1, which was introduced in the following experiments.
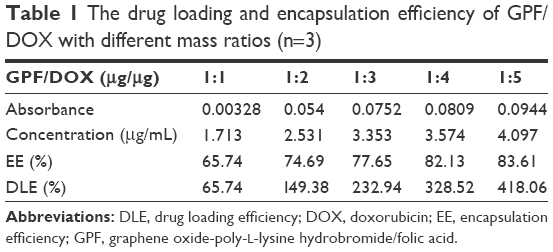 | Table 1 The drug loading and encapsulation efficiency of GPF/DOX with different mass ratios (n=3) |
Agarose gel retardation assay
The agarose gel retardation assay analyzed the electrostatic adsorption of VEGF-siRNA onto GPF as shown in Figure 5. GO/VEGF-siRNA and GPF/VEGF-siRNA at different w/w ratios (0:1, 10:1, 15:1, 20:1, 25:1, 30:1, 35:1, 40:1) were tested, respectively. VEGF-siRNA in each GPF/VEGF-siRNA group was retarded totally or partially, which indicated that VEGF-siRNA was absorbed onto GPF effectively. According to the phenomenon that the band of VEGF-siRNA disappeared at the w/w ratio of 25:1, we concluded that 0.1 mg of VEGF-siRNA could be loaded onto about 2.5 mg of GPF. On the basis of the gel retardation assay, GPF/VEGF-siRNA was prepared by the w/w ratio 25:1 (w/w). The results showed that GO was unable to retard the migration of VEGF-siRNA. Overall, it was of crucial significance to modify the surface of GO for its ability of loading VEGF-siRNA.
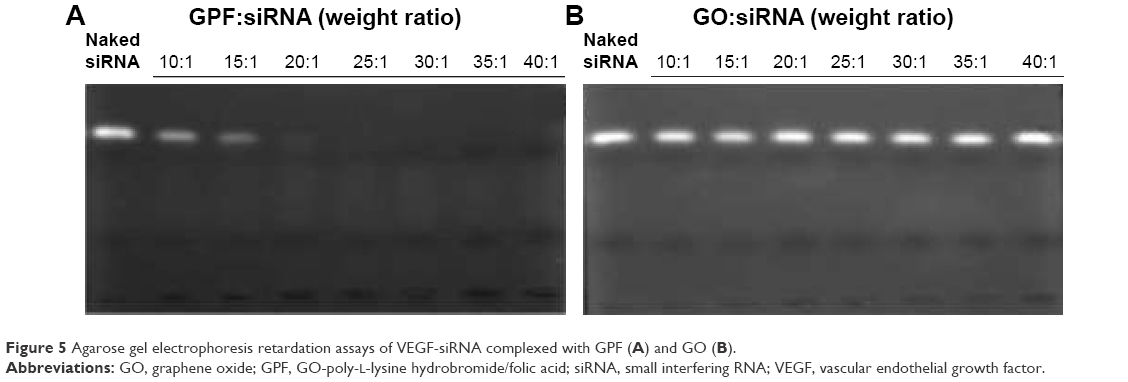 | Figure 5 Agarose gel electrophoresis retardation assays of VEGF-siRNA complexed with GPF (A) and GO (B). |
Release experiments
Figure 6 shows the cumulative release profile of DOX, GO/DOX, and GPF/DOX at pH 7.4 (physiological) and pH 5.0 (tumor mimicking) buffer medium. It was observed that GO (gray) and GPF (orange) have pH sensitivity and sustained release behavior, and there was a significant difference compared to DOX (blue) alone. At 2 hours, the releases of DOX, GO/DOX, and GPF/DOX at pH 7.4 buffer solution were 17.49%, 2.90%, and 4.99%, respectively (Figure 6). The cumulative releases of DOX, GO/DOX, and GPF/DOX at pH 5.0 buffer solution were 20.16%, 4.32%, and 5.88%, respectively (Figure 6). The cumulative releases of GO/DOX and GPF/DOX at pH 7.4 buffer were only 39.36% and 59.61%, respectively, while the DOX group was completely released in the two buffers within 216 hours. The cumulative releases of GO/DOX and GPF/DOX at pH 5.0 buffer were 48.45% and 77.47%, respectively. Figure 6 shows that after a burst release of 11.19%±3.67% a constant release of DOX under pH 7.4 condition is monitored within 216 hours, while after a burst release of 14.05%±1.84% a constant release of DOX was observed under pH 5.0 condition. The cumulative release of DOX within 216 hours at pH 7.4 and pH 5.0 was 58.66%±3.20% and 77.47%±1.46%, respectively. The results indicate that the release of DOX under pH 5.0 was much slower. This indicated that DOX was adsorbed onto GO by the π–π bond interaction which induced the sustained release of DOX, while the modification of PLL and FA resulted in the adsorption of DOX by GPF. It was proved that, compared to the tumor environment (pH 5.0), the amount of GPF/DOX release reduced significantly.
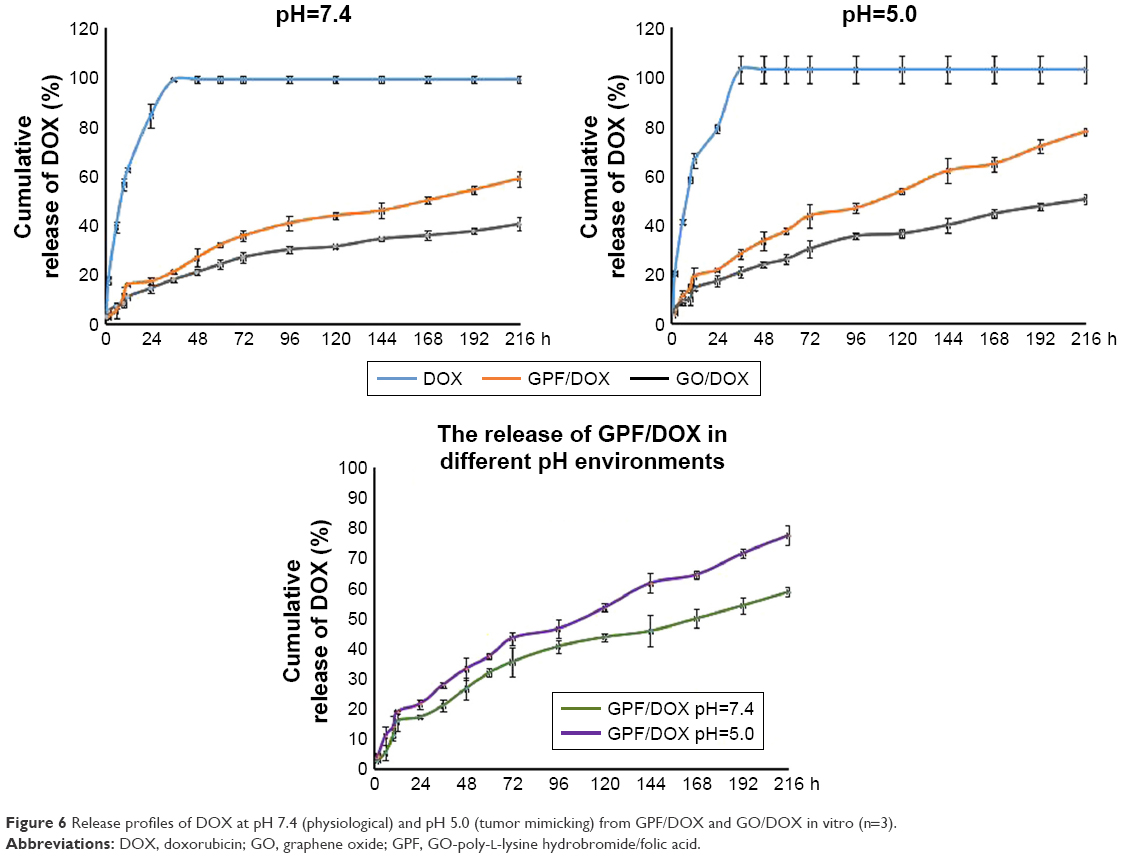 | Figure 6 Release profiles of DOX at pH 7.4 (physiological) and pH 5.0 (tumor mimicking) from GPF/DOX and GO/DOX in vitro (n=3). |
Degradation of GPF/VEGF-siRNA with heparin and anti-RNase A
With the strong negative charge, heparin can be used to replace siRNA from GPF. GPF/VEGF-siRNA and RNase A were loaded and co-incubated for different time points (Figure 7). The competitive replacement of siRNA by heparin can be used to evaluate the siRNA protective effect of the vector. As shown in Figure 7, the free VEGF-siRNA was completely degraded by RNase A within 5 minutes, while it was difficult to degrade GPF/VEGF-siRNA on the material until 6 hours by RNase A (Figure 7). The results indicated that the prepared GPF had a good protective effect and stabilizing function for VEGF-siRNA.
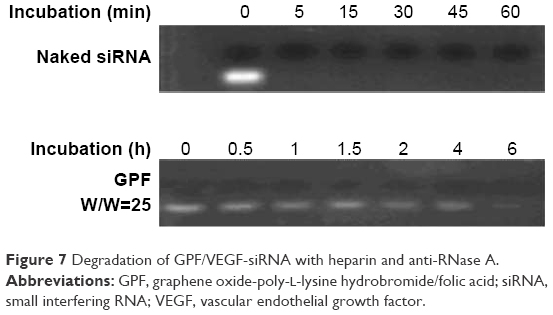 | Figure 7 Degradation of GPF/VEGF-siRNA with heparin and anti-RNase A. |
Cellular uptake of GPF/DOX/VEGF-siRNA
The cellular uptake of GO and GPF/VEGF-siRNA was studied by a laser scanning confocal microscope. The images of HeLa cells treated with blank medium, naked FAM-VEGF-siRNA, DOX, Lipo™2000/FAM-VEGF-siRNA, GPF/DOX, GPF/FAM-VEGF-siRNA, and GPF/DOX/FAM-VEGF-siRNA were captured and are shown in Figure 8. Clear fluorescent signal was observed in the cytoplasm in the DOX group (Figure 8C), GPF/FAM-VEGF-siRNA group (Figure 8D), GPF/DOX group (Figure 8E), and GPF/DOX/FAM-VEGF-siRNA group (Figure 8G). As a negative control, green fluorescence could not be seen in Figure 8B, which indicated that naked FAM-VEGF-siRNA could not be internalized by cells. Green fluorescent signal (VEGF-siRNA) was observed entering the cytoplasm, and red fluorescent signal (DOX) accessed the nucleus in GPF/DOX/FAM-VEGF-siRNA group, compared to GPF/siRNA or GPF/DOX alone. As a positive control, fluorescent signal was also observed in Figure 8F, weaker than that in GPF/DOX/FAM-VEGF-siRNA group. The experiment demonstrated that co-delivery of siRNA and DOX was distributed in the uptake of siRNA in the cytoplasm and DOX in nucleus, which resulted in an enhanced accumulation of siRNA and drug and the suppression of the desired gene.
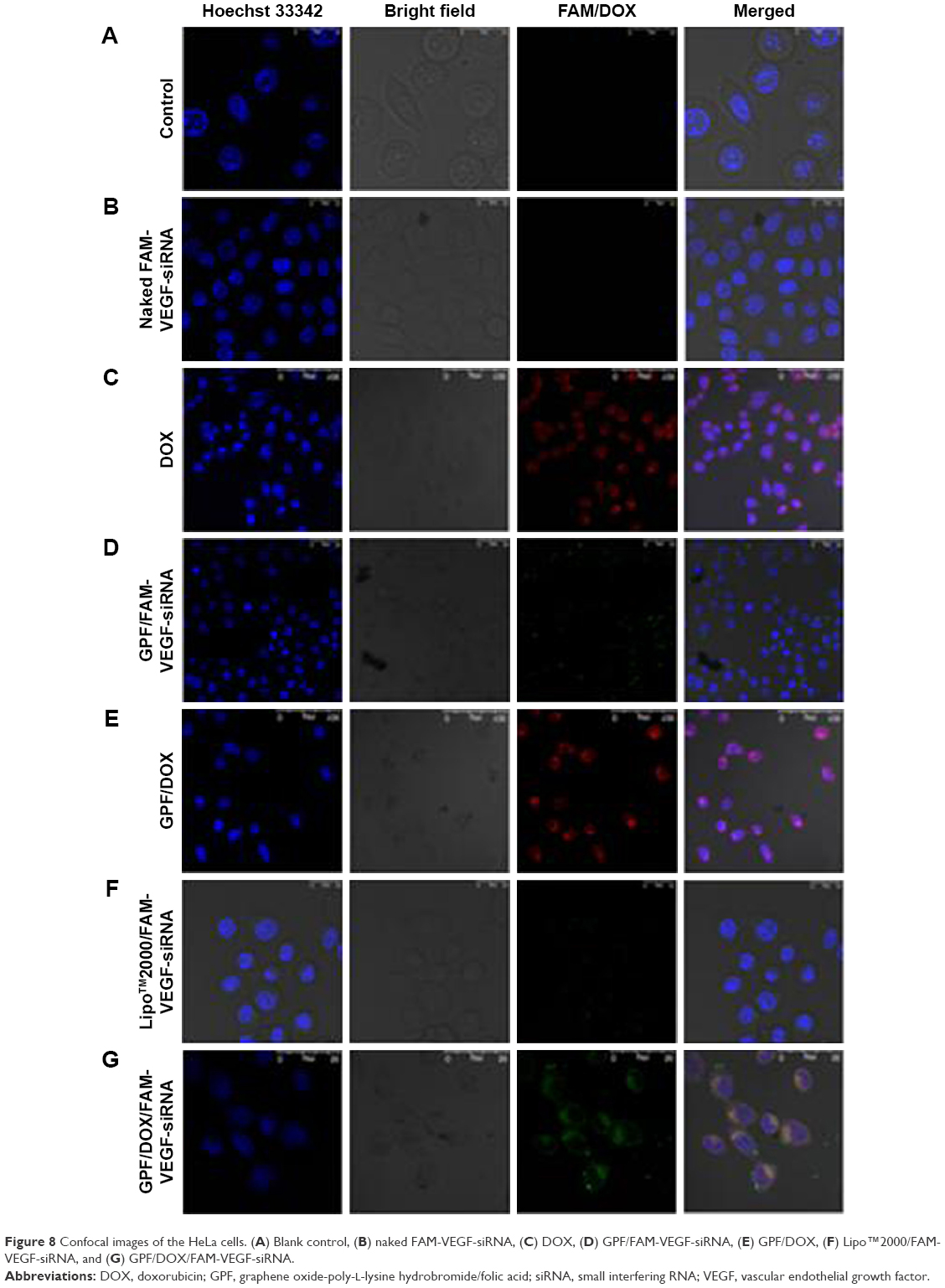 | Figure 8 Confocal images of the HeLa cells. (A) Blank control, (B) naked FAM-VEGF-siRNA, (C) DOX, (D) GPF/FAM-VEGF-siRNA, (E) GPF/DOX, (F) Lipo™2000/FAM-VEGF-siRNA, and (G) GPF/DOX/FAM-VEGF-siRNA. |
Cell proliferation inhibitory activity
Tumor growth can be efficiently inhibited by downregulating the expression of the VEGF gene.51,52 Following the previous study, the proliferation inhibitory activity of GPF/DOX/VEGF-siRNA was studied. The results are shown in Figure 9. GPF, naked VEGF-siRNA, GPF/NC, and Lipo™2000/NC served as negative controls, and Lipo™2000/VEGF-siRNA served as positive control. Compared to the blank control, naked VEGF-siRNA had no anti-proliferation effect (P>0.05), and the cell viability of GPF, GPF/NC, and Lipo™2000/NC still remained >95% at the maximum concentration (120 nM). However, results of the GPF/VEGF-siRNA group, DOX group, GPF/DOX group, GPF/DOX/NC group, and GPF/DOX/VEGF-siRNA group were decreased significantly (P<0.05), and the cell viabilities were reduced to 43.2%±0.6%, 46.8%±0.8%, 40.7%±0.3%, 41.8%±0.4%, and 29.0%±0.2%, respectively, at 120 nM of siRNA concentration (P<0.01). Through variance analysis (Excel-Visual Basic for Applications function), the results showed that there were no significant differences between the GPF group, VEGF-siRNA group, GPF/NC group, and Lipo™2000/NC, and the P-values were 0.20, 0.32, 0.26, and 0.07, respectively. Meanwhile, the P-values of DOX group, GPF/DOX group, GPF/DOX/NC group, GPF/VEGF-siRNA, GPF/DOX/VEGF-siRNA, and Lipo™2000/VEGF-siRNA were 5.68e-46, 7.10e-74, 4.97e-63, 2.83e-84, 5.27e-89, and 3.96e-52, which indicated that there were very significant differences between these groups. With increasing concentration of GPF/DOX/VEGF-siRNA, the inhibition efficiency improved significantly. Compared with the naked siRNA group, the co-carrier group demonstrated a stronger antitumor proliferation effect, which indicated that GPF was a potential co-delivery carrier of siRNA and drug for tumor therapy.
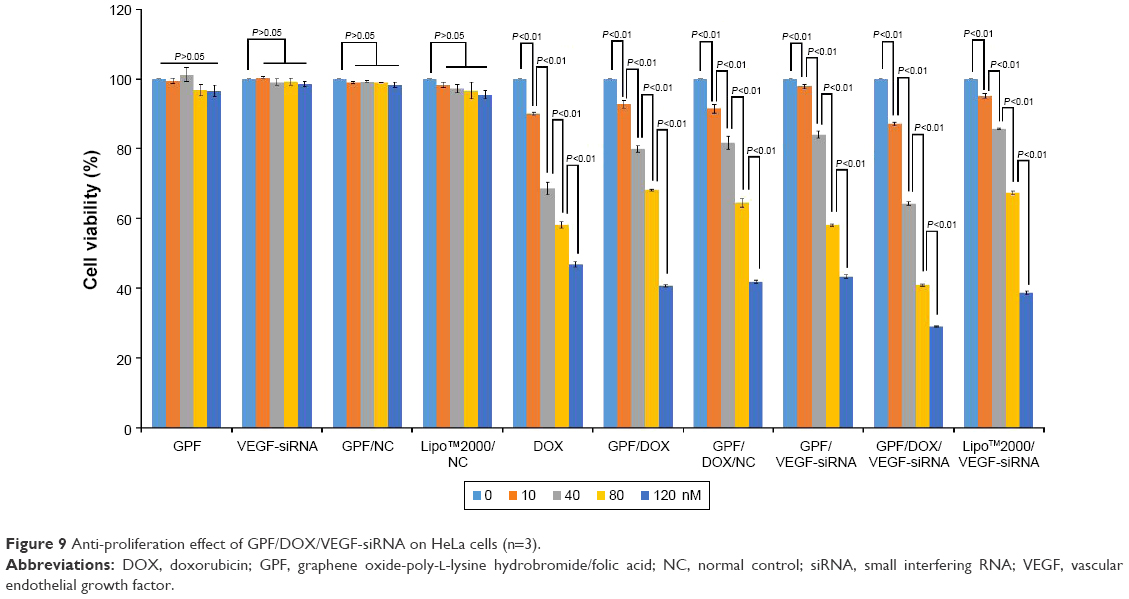 | Figure 9 Anti-proliferation effect of GPF/DOX/VEGF-siRNA on HeLa cells (n=3). |
Gene silencing efficiency of GPF/DOX/VEGF-siRNA on mRNA level
The expression of VEGF mRNA was evaluated by real-time PCR in HeLa cells, and the results are shown in Figure 10. Compared to the blank control, naked VEGF-siRNA and GPF/NC served as negative control and showed no significant gene silencing effects (P>0.05). As shown in Figure 10, the results indicated that the expression of VEGF mRNA in GPF/VEGF-siRNA group (0.5435±0.017) was close to the positive control group (0.5262±0.015) (P>0.05), but significantly decreased compared with the negative control group (0.9683±0.020) (P<0.01). GPF/DOX/VEGF-siRNA group showed no obvious difference from GPF/VEGF-siRNA at the mRNA level. Therefore, GPF could be applied as a useful carrier to deliver VEGF-siRNA and silence the specified gene expression.
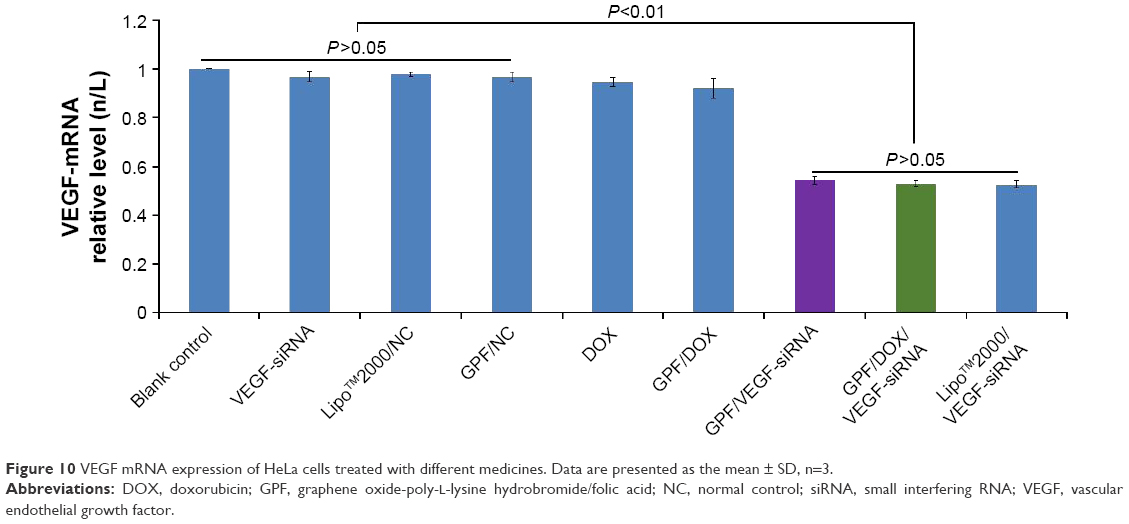 | Figure 10 VEGF mRNA expression of HeLa cells treated with different medicines. Data are presented as the mean ± SD, n=3. |
Gene silencing efficiency of GPF/DOX/VEGF-siRNA on protein level
The protein expression of VEGF was measured by ELISA to estimate the gene silencing efficiency of GPF/DOX/VEGF-siRNA. The result of ELISA is shown in Figure 11. There was no suppression effect on the expression of VEGF protein in naked VEGF-siRNA, GPF/NC, and Lipo™2000/NC groups. Compared with the Lipo™2000/VEGF-siRNA group (56.79%), GPF/VEGF-siRNA and GPF/DOX/VEGF-siRNA showed no significant difference, respectively (54.98% and 53.16%, P>0.05). Meanwhile, the gene silencing effect of the GPF/DOX/VEGF-siRNA group was not significantly different from that of GPF/VEGF-siRNA. It indicated that GPF could downregulate the expression of VEGF protein while the chemotherapeutic drug loaded.
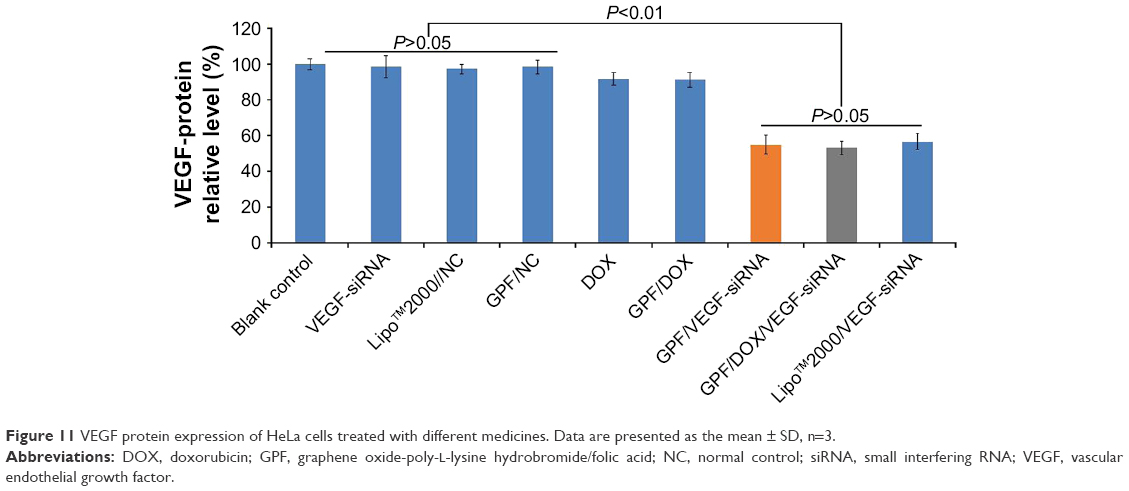 | Figure 11 VEGF protein expression of HeLa cells treated with different medicines. Data are presented as the mean ± SD, n=3. |
Tumor growth inhibitory activity in vivo
All of the in vivo experiments were approved by the Institutional Animal Ethics Committee of Capital Medical University. The effects of GPF/DOX/VEGF-siRNA on tumor growth were evaluated with S180 mice treated with GPF/DOX/VEGF-siRNA, naked VEGF-siRNA, DOX, GPF/DOX, and GPF/VEGF-siRNA. The results are shown in Figures 12–14. ICR mice were injected with NS and DOX (clinical dosage), respectively, and served as the blank control and positive control.
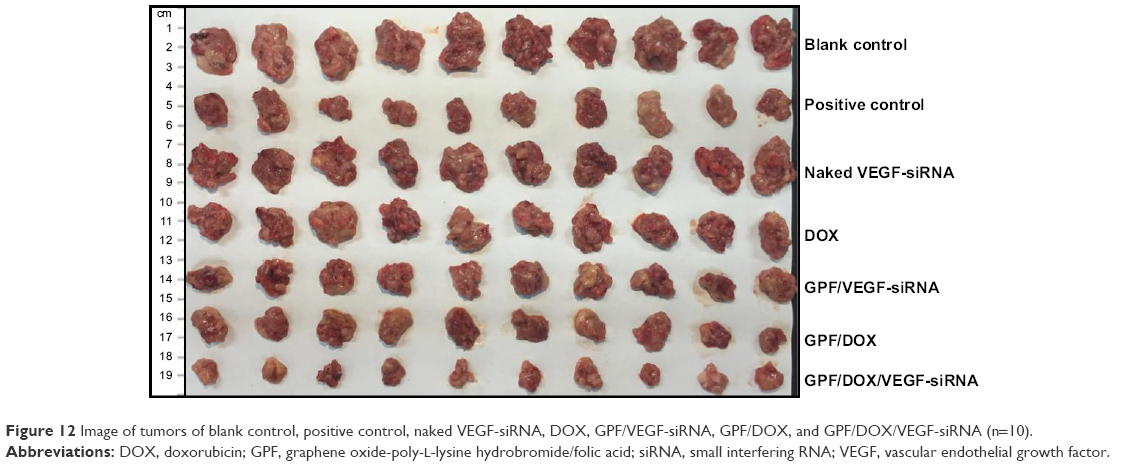 | Figure 12 Image of tumors of blank control, positive control, naked VEGF-siRNA, DOX, GPF/VEGF-siRNA, GPF/DOX, and GPF/DOX/VEGF-siRNA (n=10). |
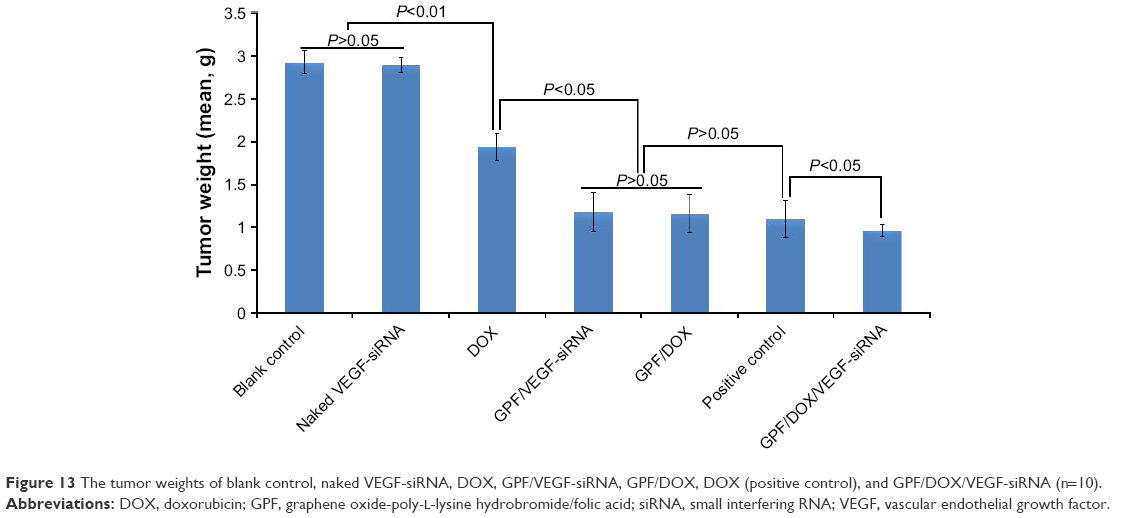 | Figure 13 The tumor weights of blank control, naked VEGF-siRNA, DOX, GPF/VEGF-siRNA, GPF/DOX, DOX (positive control), and GPF/DOX/VEGF-siRNA (n=10). |
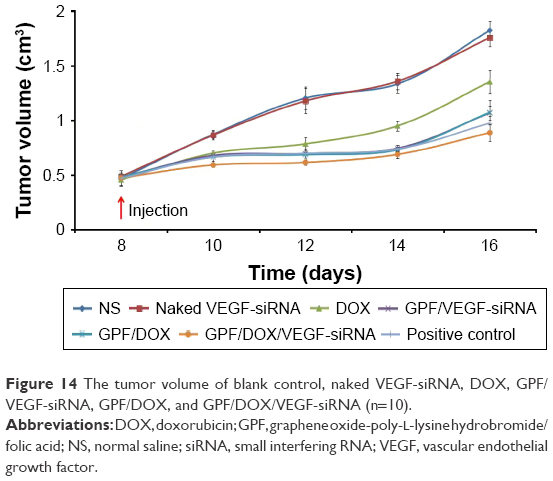 | Figure 14 The tumor volume of blank control, naked VEGF-siRNA, DOX, GPF/VEGF-siRNA, GPF/DOX, and GPF/DOX/VEGF-siRNA (n=10). |
The mean tumor weight of each group is shown in Figure 13. There was no significant difference between the naked VEGF-siRNA group and NS group (P>0.05), but the difference was much higher than that of controlled DOX, GPF/VEGF-siRNA, GPF/DOX, and GPF/DOX/VEGF-siRNA-treated groups (P<0.01). Moreover, the GPF/DOX/VEGF-siRNA-treated group had a significantly better effect compared with the groups of VEGF-siRNA and DOX.
The tumor inhibitory rate of GPF/DOX/VEGF-siRNA group (66.95%, P<0.05) demonstrated a significant difference compared with other groups (Figure S2). The results of GPF/DOX group (60.16%) and GPF/VEGF-siRNA (59.71%) group were much higher than that of the naked VEGF-siRNA group (1.25%). The effect of the positive group (62.54%) showed no significant difference with the groups of GPF/DOX and GPF/VEGF-siRNA (P>0.05).
Tumor tissues from GPF/DOX/VEGF-siRNA groups were much smaller than that of the controlled one, naked VEGF-siRNA groups, DOX, GPF/DOX, and GPF/VEGF-siRNA as shown in Figure 14. The antitumor effect of the co-administered group was significantly better than that of the control group and the single-loaded gene group and drug group, which indicated that co-delivery of VEGF-siRNA and DOX had a synergistic effect.53
The expression of VEGF protein in vivo is shown in Figure 15. ELISA was used to evaluate the relation between the tumor inhibitory activity and the suppressed VEGF expression. VEGF expression levels of NS, naked VEGF-siRNA, GPF/VEGF-siRNA, GPF/DOX, and GPF/DOX/VEGF-siRNA groups were analyzed. The serum concentration of VEGF in the GPF/DOX/VEGF-siRNA group (87.92±7.80 pg/mL) was the lowest significantly compared with that of NS groups (141.90±9.43 pg/mL), naked VEGF-siRNA groups (140.31±8.60 pg/mL), GPF/VEGF-siRNA (87.24±4.79 pg/mL), and GPF/DOX (140.70±5.49 pg/mL) (P<0.01), which indicated that GPF increased the gene silencing effect while loading the chemotherapeutic drug.
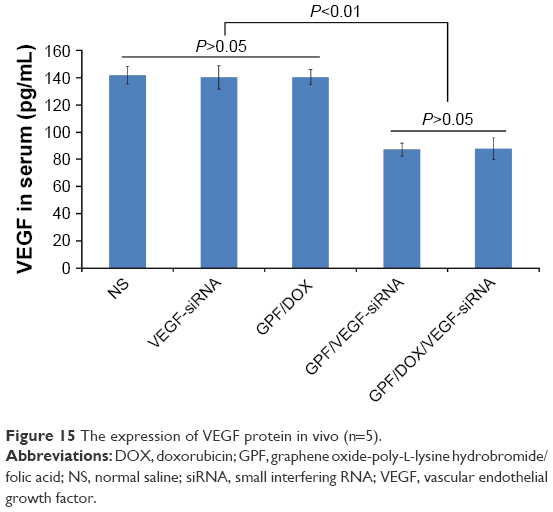 | Figure 15 The expression of VEGF protein in vivo (n=5). |
The effect of the targeting of GPF/DOX/VEGF-siRNA is shown in Figure S3. Mice injected with NS served as blank control and those injected with DOX (clinical dosage) served as positive control. Tumor tissues from GPF/DOX/VEGF-siRNA groups (0.9457±0.0738 g) and GO-PLL/DOX/VEGF-siRNA groups (1.9391±0.1586 g) were much smaller than that of the blank control groups (2.9731±0.1343 g). There was a significant difference between GPF/DOX/VEGF-siRNA groups and GO-PLL/DOX/VEGF-siRNA groups (P<0.01); whereas, there was no obvious difference between GPF/DOX/VEGF-siRNA groups and DOX groups. Compared with no targeting vectors, GPF/DOX/VEGF-siRNA had a perfect targeting effect.
Cytotoxicity assay
One of the main disadvantages of gene carriers is cytotoxicity. MTT assay was applied to evaluate the cytotoxicity of GPF against HeLa cells. The results are illustrated in Figure S4. Even when the concentration of GPF reached 120 μg/mL, the viability of cells treated with GPF remained >92%. Therefore, these results indicated that GPF could be a genetic drug carrier with no testable cytotoxicity.
Conclusion
In this study, nonviral vector GPF was developed. The physicochemical properties and bioactivity of the delivery system, including zeta potential, cell uptake, siRNA loading capacity, cytotoxicity, antiproliferative effects in vitro and in vivo, and gene silencing efficiency on mRNA and protein levels, were studied. GPF/DOX/VEGF-siRNA can not only play the antitumor role of chemotherapeutic drug DOX effectively, but can also effectively regulate the expression of VEGF. The results showed that GPF/DOX/siRNA exhibited gene silencing and tumor inhibition significantly compared with naked siRNA alone or DOX alone in vitro and in vivo. GPF/DOX/siRNA exhibited no testable cytotoxicity. Compared with the previous studies, it is a very familiar method to deliver chemotherapy drugs (DOX) into tumor cells.55 However, multidrug resistance is an important factor which should be regarded, and the toxicity of DOX also cannot be ignored. Functionalized GO as a nonviral carrier for siRNA delivery has been explored in previous research.54 The result of gene therapy is remarkable, but it also has some limitations, such as transfection efficiency, instability etc. In this study, modified GO had the ability to complete co-delivery of DOX and VEGF-siRNA into HeLa cells. What is more, better antitumor effect had been observed obviously in the results in vitro and in vivo. The results demonstrated that synergistic action may benefit the antitumor effect of drugs and transfection efficiency by co-delivery of DOX and anti-VEGF siRNA. In conclusion, co-delivery of siRNA and DOX by GPF could be a promising application in tumor clinical therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation (81502688), Beijing Natural Science Foundation Program and Scientific Research Key Program of Beijing Municipal Commission of Education (KM201810025019), and a basic-clinical key research grant (16JL72, 17JL67) from Capital Medical University, the Importation and Development of High-Caliber Talents Project of Beijing Municipal Institutions (2013–2015). The authors gratefully acknowledge the support from Beijing Area Major Laboratory of Peptide and Small Molecular Drugs, Engineering Research Center of Endogenous Prophylactic of Ministry of Education of China, and Beijing Laboratory of Biomedical Materials.
Disclosure
The authors report no conflicts of interest in this work.
References
Liu Z, Chen K, Davis C, et al. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008;68(16):6652–6660. | ||
Liu CW, Lin WJ. Systemic co-delivery of doxorubicin and siRNA using nanoparticles conjugated with EGFR-specific targeting peptide to enhance chemotherapy in ovarian tumor bearing mice. J Nanopart Res. 2013;15(10):1–14. | ||
Huang HY, Kuo WT, Chou MJ, Huang YY. Co-delivery of anti-vascular endothelial growth factor siRNA and doxorubicin by multifunctional polymeric micelle for tumor growth suppression. J Biomed Mater Res A. 2011;97(3):330–338. | ||
Li JH, Chia M, Shi W, et al. Tumor-targeted gene therapy for nasopharyngeal carcinoma. Cancer Res. 2002;62(1):171–178. | ||
Takeda T, Inaba H, Yamazaki M, et al. Tumor-specific gene therapy for undifferentiated thyroid carcinoma utilizing the telomerase reverse transcriptase promoter. J Clin Endocrinol Metab. 2003;88(8):3531–3538. | ||
Verma IM, Weitzman MD. Gene therapy: twenty-first century medicine. Annu Rev Biochem. 2005;74(74):711–738. | ||
Pardridge WM. Intravenous, non-viral RNAi gene therapy of brain cancer. Expert Opin Biol Ther. 2004;4(7):1103–1113. | ||
Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF, Wu TC. Prospects of RNA interference therapy for cancer. Gene Ther. 2006;13(6):464–477. | ||
Bazanperegrino M, Seymour LW, Harris AL. Gene therapy targeting to tumor endothelium. Cancer Gene Ther. 2007;14(2):117–127. | ||
Wang Y, Li Z, Han Y, Liang LH, Ji A. Nanoparticle-based delivery system for application of siRNA in vivo. Curr Drug Metab. 2010;11(2):182–196. | ||
Hutvágner G, Zamore PD. RNAi: nature abhors a double-strand. Curr Opin Genet Dev. 2002;12(2):225–232. | ||
Aliabadi HM, Landry B, Sun C, Tang T, Uludağ H. Supramolecular assemblies in functional siRNA delivery: where do we stand? Biomaterials. 2012;33(8):2546–2569. | ||
Zhang CG, Zhu WJ, Liu Y, et al. Novel polymer micelle mediated co-delivery of doxorubicin and P-glycoprotein siRNA for reversal of multidrug resistance and synergistic tumor therapy. Sci Rep. 2016;6:23859. | ||
Susa M, Iyer AK, Ryu K, et al. Inhibition of ABCB1 (MDR1) expression by an siRNA nanoparticulate delivery system to overcome drug resistance in osteosarcoma. PLoS One. 2010;5(5):10764. | ||
Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5(6):5202–5213. | ||
Saraswathy M, Gong S. Recent developments in the co-delivery of siRNA and small molecule anticancer drugs for cancer treatment. Mater Today. 2014;17(6):298–306. | ||
Cao N, Cheng D, Zou S, Ai H, Gao J, Shuai X. The synergistic effect of hierarchical assemblies of siRNA and chemotherapeutic drugs co-delivered into hepatic cancer cells. Biomaterials. 2011;32(8):2222–2232. | ||
Dong DW, Xiang B, Gao W, Yang ZZ, Li JQ, Qi XR. pH-responsive complexes using prefunctionalized polymers for synchronous delivery of doxorubicin and siRNA to cancer cells. Biomaterials. 2013;34(20):4849–4859. | ||
Chen D, Feng H, Li J. Graphene oxide: preparation, functionalization, and electrochemical applications. Chem Rev. 2012;112(11):6027–6053. | ||
Kim F, Cote LJ, Huang J. Graphene oxide: surface activity and two-dimensional assembly. Adv Mater. 2010;22(17):1954–1958. | ||
Liu G, Shen H, Mao J, et al. Transferrin modified graphene oxide for glioma-targeted drug delivery: in vitro and in vivo evaluations. ACS Appl Mater Interfaces. 2013;5(15):6909–6914. | ||
Liu Z, Robinson JT, Sun X, Dai H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008;130(33):10876–10877. | ||
Wang H, Gu W, Xiao N, Ye L, Xu Q. Chlorotoxin-conjugated graphene oxide for targeted delivery of an anticancer drug. Int J Nanomedicine. 2014;9(2):1433–1442. | ||
Kavitha T, Abdi SIH, Park SY. pH-sensitive nanocargo based on smart polymer functionalized graphene oxide for site-specific drug delivery. Phys Chem Chem Phys. 2013;15(14):5176–5185. | ||
Wen H, Dong C, Dong H, et al. Engineered redox-responsive PEG detachment mechanism in PEGylated nano-graphene oxide for intracellular drug delivery. Small. 2012;8(5):760–769. | ||
Xu H, Fan M, Elhissi AM, et al. PEGylated graphene oxide for tumor-targeted delivery of paclitaxel. Nanomedicine. 2015;10(8):1247–1262. | ||
Feng L, Yang X, Shi X, et al. Polyethylene glycol and polyethylenimine dual-functionalized nano-graphene oxide for photothermally enhanced gene delivery. Small. 2013;9(11):1989–1997. | ||
Feng L, Zhang S, Liu Z. Graphene based gene transfection. Nanoscale. 2011;3(3):1252–1257. | ||
Imani R, Emami SH, Faghihi S. Synthesis and characterization of an octaarginine functionalized graphene oxide nano-carrier for gene delivery applications. Phys Chem Chem Phys. 2015;17(9):6328–6339. | ||
Wang C. Multi-functionalized graphene oxide complex as a plasmid delivery system for targeting hepatocellular carcinoma therapy. RSC Adv. 2016;6(27):22461–22468. | ||
Shan C, Yang H, Han D, Zhang Q, Ivaska A, Niu L. Water-soluble graphene covalently functionalized by biocompatible poly-L-lysine. Langmuir. 2009;25(20):12030–12033. | ||
Wu C, He Q, Zhu A, et al. Synergistic anticancer activity of photo- and chemoresponsive nanoformulation based on polylysine-functionalized graphene. ACS Appl Mater Interfaces. 2014;6(23):21615–21623. | ||
Sun J, Chao J, Huang J, et al. Uniform small graphene oxide as an efficient cellular nanocarrier for immunostimulatory CpG oligonucleotides. ACS Appl Mater Interfaces. 2014;6(10):7926–7932. | ||
Mansouri S, Cuie Y, Winnik F, et al. Characterization of folate-chitosan-DNA nanoparticles for gene therapy. Biomaterials. 2006;27(9):2060–2065. | ||
Peng S, Wang Y, Wang Y, et al. Cu-RGDFRGDS: exploring the mechanism and high efficacy of the nanoparticle in antithrombotic therapy. Int J Nanomedicine. 2015;10:2925–2938. | ||
Qi L, Guo Y, Luan J, Zhang D, Zhao Z, Luan Y. Folate-modified bexarotene-loaded bovine serum albumin nanoparticles as a promising tumor-targeting delivery system. J Mater Chem B. 2014;2(47):8361–8371. | ||
Mansouri S, Cuie Y, Winnik F, et al. Characterization of folate-chitosan-DNA nanoparticles for gene therapy. Biomaterials. 2006;27(9):2060–2065. | ||
Li W, Shi J, Zhang C, et al. Co-delivery of thioredoxin 1 shRNA and doxorubicin by folate-targeted gemini surfactant-based cationic liposomes to sensitize hepatocellular carcinoma cells. J Mater Chem B. 2014;2(30):4901–4910. | ||
Yang F, Huang W, Li Y, et al. Anti-tumor effects in mice induced by survivin-targeted siRNA delivered through polysaccharide nanoparticles. Biomaterials. 2013;34(22):5689–5699. | ||
Cui C, Wang Y, Zhao W, et al. RGDS covalently surfaced nanodiamond as a tumor targeting carrier of VEGF-siRNA: synthesis, characterization and bioassay. J Mater Chem B. 2015;3(48):9260–9268. | ||
Xie Y, Qiao H, Su Z, Chen M, Ping Q, Sun M. PEGylated carboxy methyl chitosan/calcium phosphate hybrid anionic nanoparticles mediated hTERT siRNA delivery for anticancer therapy. Biomaterials. 2014;35(27):7978–7991. | ||
Cao Y, Shi LL, Cao QR, Yang M, Cui JH. In-vitro characterization and oral bioavailability of organic solvent-free solid dispersions containing telmisartan. Iran J Pharm Res. 2016;15(2):385–394. | ||
Yang H, Bremner DH, Tao L, Li H, Hu J, Zhu L. Carboxymethyl chitosan-mediated synthesis of hyaluronic acid-targeted graphene oxide for cancer drug delivery. Carbohydr Polym. 2016;135:72–78. | ||
Yu J, Xin X, Zheng M, et al. Fabrication and characterization of nuclear localization signal-conjugated glycol chitosan micelles for improving the nuclear delivery of doxorubicin. Int J Nanomedicine. 2012;7:5079–5090. | ||
Mohyeldin SM, Mehanna MM, Elgindy NA. The relevancy of controlled nanocrystallization on rifampicin characteristics and cytotoxicity. Int J Nanomedicine. 2016;11:2209–2222. | ||
Arun C, Swaleha Z, Saba T, et al. Fungus-mediated biological synthesis of gold nanoparticles: potential in detection of liver cancer. Int J Nanomedicine. 2011;6:2305–2319. | ||
Yang P, Jia T, Zhang Y, et al. MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int J Nanomedicine. 2012;7:5957–5967. | ||
Cheng HY, Lai LJ, Fu-Hsiang K. Rapid and sensitive detection of rare cancer cells by the coupling of immunomagnetic nanoparticle separation with ELISA analysis. Int J Nanomedicine. 2012;7:2967–2973. | ||
Ding M, He X, Wang Z, et al. Cellular uptake of polyurethane nanocarriers mediated by gemini quaternary ammonium. Biomaterials. 2011;32(35):9515–9524. | ||
Sara S, Solmaz MD, Ahmad YK. Effect of the surface modification, size, and shape on cellular uptake of nanoparticles. Cell Biol Int. 2015;39(8):881–890. | ||
Wang W, Fang C, Wang X, et al. Modifying mesoporous silica nano-particles to avoid the metabolic deactivation of 6-mercaptopurine and methotrexate in combinatorial chemotherapy. Nanoscale. 2013;5(14):6249–6253. | ||
Maver U, Godec A, Bele M, et al. Novel hybrid silica xerogels for stabilization and controlled release of drug. Int J Pharm. 2007;330(1–2):164–174. | ||
Al Faraj A, Shaik AS, Ratemi E, Halwani R. Combination of drug-conjugated SWCNT nanocarriers for efficient therapy of cancer stem cells in a breast cancer animal model. J Control Release. 2016;225:240–251. | ||
Ren L, Zhang Y, Cui C, Bi Y, Ge X. Functionalized graphene oxide for anti-VEGF siRNA delivery: preparation, characterization and evaluation in vitro and in vivo. RSC Adv. 2017;7(33):20553–20566. | ||
Zhang Q, Chi H, Tang M, et al. Mixed surfactant modified graphene oxide nanocarriers for DOX delivery to cisplatin-resistant human ovarian carcinoma cells. RSC Adv. 2016;6(90):87258–87269. |
Supplementary materials
Nucleotide sequence
The sequences of mus-vascular endothelial growth factor (VEGF)-small interfering RNA (siRNA): 5′-CGAGGCAGCUUGAGUUAAATT-3′ (sense); 5′-UUUAACUCAAGCUGCCUCGTT-3′ (antisense).
The sequences of homo-VEGF-siRNA: 5′-GCGCAAGAAAUCCCGGUAUTT-3′ (sense); 5′-AUACCGGGAUUUCUUGCGCTT-3′ (antisense).
The sequences of negative control: 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense); 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense).
The scramble sequence of siRNA served as normal control.
Primer pairs for VEGF (forward sequence: 5′-ATCGAGACCCTGGTGGACA-3′; reverse sequence: 5′-CCGCCTCGGCTTGTCACA-3′).
Primer pairs for GAPDH (forward sequence: 5′-CAAATTCCATGGCACCGTCA-3′; reverse sequence: 5′-GGAGTGGGTGTCGCTGTTGA-3′).
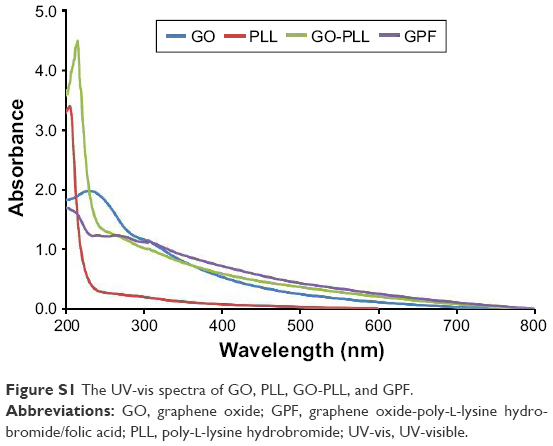 | Figure S1 The UV-vis spectra of GO, PLL, GO-PLL, and GPF. |
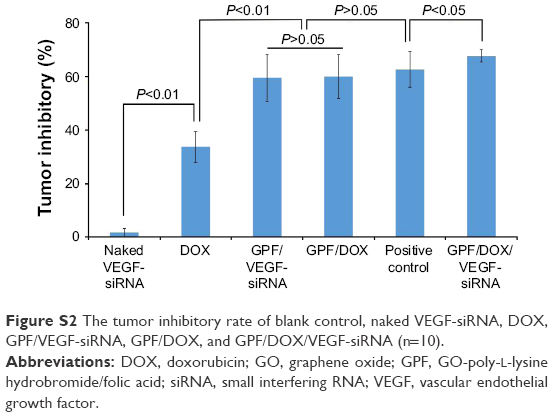 | Figure S2 The tumor inhibitory rate of blank control, naked VEGF-siRNA, DOX, GPF/VEGF-siRNA, GPF/DOX, and GPF/DOX/VEGF-siRNA (n=10). |
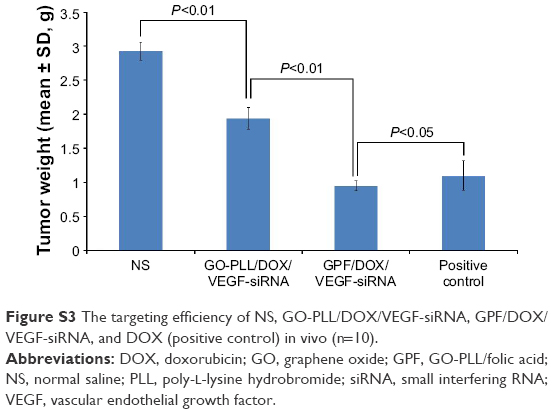 | Figure S3 The targeting efficiency of NS, GO-PLL/DOX/VEGF-siRNA, GPF/DOX/VEGF-siRNA, and DOX (positive control) in vivo (n=10). |
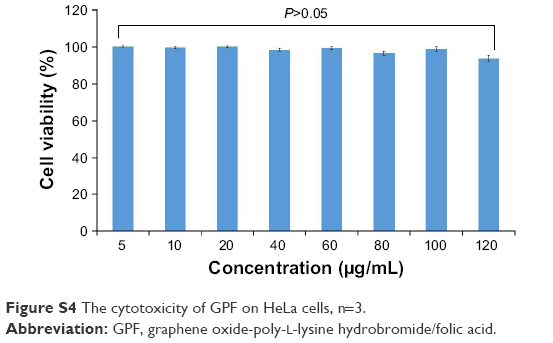 | Figure S4 The cytotoxicity of GPF on HeLa cells, n=3. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.