")
Back to Journals » OncoTargets and Therapy » Volume 13
Downregulation of miR-7-5p Inhibits the Tumorigenesis of Esophagus Cancer via Targeting KLF4
Authors Shi W, Song J, Gao Z, Liu X, Wang W
Received 27 February 2020
Accepted for publication 1 July 2020
Published 24 September 2020 Volume 2020:13 Pages 9443—9453
DOI https://doi.org/10.2147/OTT.S251508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Woda Shi ,* Jianxiang Song ,* Zhengya Gao, Xingchen Liu, Wencai Wang
Department of Cardio-Thoracic Surgery, Yancheng Third People’s Hospital, Yancheng, Jiangsu 224000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wencai Wang Email [email protected]
Background: Esophageal cancer (EC) is one of the aggressive gastrointestinal malignancies. It has been reported that microRNAs (miRNAs) play key roles during the tumorigenesis of EC. To identify novel potential targets for EC, differential expressed miRNAs (DEG) between EC and adjacent normal tissues were analyzed with bioinformatics tool.
Methods: The differential expression of miRNAs between EC and adjacent normal tissues was analyzed. CCK-8 and Ki67 staining were used to detect the cell proliferation. Flow cytometry was performed to test the cell apoptosis. The correlation between miR-7-5p and KLF4 was detected by dual-luciferase report assay. Gene and protein expression in EC cells or in tissues were measured by qRT-PCR and Western blot, respectively. Cell migration and invasion were detected with transwell assay. Xenograft mice model was established to investigate the role of miR-7-5p in EC tumorigenesis in vivo.
Results: MiR-7-5p was found to be negatively correlated with the survival rate of patient with EC. In addition, downregulation of miR-7-5p significantly inhibited the growth and invasion of EC cells. Meanwhile, miR-7-5p directly targeted KLF4 in EC cells. Moreover, downregulation of miR-7-5p inhibited the tumorigenesis of EC via inactivating MAPK signaling pathway in vivo.
Conclusion: Downregulation of miR-7-5p notably suppressed the progression of EC via targeting KLF4. Thus, miR-7-5p might serve as a new target for the treatment of EC.
Keywords: esophageal cancer, miR-7-5p, KLF4, MAPK signaling
Introduction
Esophageal cancer (EC) has become one of the aggressive malignancies which can cause cancer-related death all over the world.1 Meanwhile, esophageal squamous cell carcinoma (ESCC) is the major histologic type that contains about 95% cases of EC, while the percentage of esophageal adenocarcinoma is still low.2 Nowadays, surgical operation is the major method for EC treatment, while about 70% of cases were regarded to be in the late stage.3 When tumor is in late stage, chemotherapy is the only method since surgery is invalid.4,5 Therefore, it is necessary to find new strategies for the patients with late-stage EC.
MicroRNAs (miRNAs) are a novel class of 21–24 nucleotides, noncoding small ribonucleic acids, which regulate gene expression by suppression of mRNA translation or degradation of mRNA.6,7 Therefore, miRNAs have been considered as important biological regulators for multiple diseases. Previous reports have indicated the importance of miRNAs in modulating cancer-related signaling pathways.8,9 Additionally, miRNAs may be related to types of malignant tumors and serve as key factors for tumorigenesis of many cancers.10 However, the function of miRNAs during the progression of EC remains to be understood. In this research, we used a systemic bioinformatics analysis to identify differentially expressed miRNAs that are essential for the tumorigenesis of EC. Meanwhile, Kruppel-like factor 4 (KLF4) is a key mediator in cancer cell growth.11 On the other hand, Mitogen-activated protein kinase (MAPK) signaling is known to be participated in the progression of cancer.12 Additionally, KLF4 is known to mediate MAPK signaling during the tumorigenesis.13 However, the correlation between KLF4 and MAPK signaling in EC remains unclear.
In the current study, we aimed to detect the differential expressed miRNAs, which is closely associated with the tumorigenesis of EC. We hope our research can supply a new idea for the development of novel therapeutic strategies against EC.
Materials and Methods
Cell Culture
Eca-109 and TE-9 cell lines were obtained from Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) with 10% FBS (Thermo Fischer Scientific), 1% penicillin and streptomycin (Thermo Fisher Scientific) at 37°C, 5% CO2.
Reagents
APTO-253 (KLF4 promoter, 5 μM) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Tissue Collection
In total, 10 pairs of EC tissues and adjacent normal tissues were collected from Yancheng Third People’s Hospital between September 2017 and August 2018. The clinical and pathological data of these patients were collected with their written informed consents. Each tissue sample was stored at −80°C until RNA extraction. The present study was approved by the Ethics Committee of Yancheng Third People’s Hospital.
Bioinformatics Analysis
The gene expression data EC and adjacent normal tissue (controls) were obtained from the dataset (GSE114110) and the Cancer Genome Atlas (TCGA). The survival curve was calculated based on the data from TCGA.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from EC cell lines or tissues using TRIzol reagent (TaKaRa, Tokyo, Japan). cDNA was synthesized using the reverse transcription kit (TaKaRa, Ver.3.0). Real-Time qPCRs were performed in triplicate under the following protocol: 2 mins at 94°C, followed by 35 cycles (30 s at 94°C and 45 s at 55°C). The primer for miR-7-5p, β-actin and U6 were obtained from GenePharma (Shanghai, China). MiR-7-5p: forward, 5ʹ- TGCGCTCAGCAAACATTTATTG-3ʹ and reverse 5ʹ- CCAGTGCAGGGTCCGAGGTATT-3ʹ. β-actin: forward, 5ʹ-AGCGAGCATCCCCCAAAGTT-3ʹ and reverse 5’-GGGCACGAAGGCTCATCATT-3ʹ. U6: forward, 5ʹ-CGCTTCGGCAGCACATATAC-3ʹ and reverse 5ʹ- AAATATGGAACGCTTCACGA-3ʹ. 2−ΔΔCt method was used to quantify the results. U6 or β-actin was used for normalization.
Cell Transfection
Eca-109 or TE-9 cells were transfected with miR-7-5p agonist, miR-7-5p antagonist or NC by Lipofectamine 2000 according to the previous reference.14 MiRNA agonist, antagonist and NC were purchased from GenePharma (Shanghai, China).
CCK-8 Assay
EC cells were seeded in 96-well plates (5×103 per well) overnight. Briefly, cells were treated with negative control (NC), miR-7-5p agonist or miR-7-5p antagonist for 0, 24, 48 and 72 h, respectively. Then, cells were treated with 10 μL CCK-8 reagents (Beyotime, Shanghai, China) and further incubated for 2 h at 37°C. Finally, the absorbance of EC cells was measured at 450 nm using a microplate reader.
Cell Apoptosis Analysis
EC cells were seeded in a 6-well plate (1×106/well). The residue was resuspended with 100 μL binding buffer after centrifuged at 1000 rpm/min for 5 min. Then, 5 μL Annexin V-FITC and propidium (PI) were added in the system for 15 min. The cell apoptotic rate was measured by flow cytometer (BD, Franklin Lake, NJ, USA) and the results were analyzed using the Fluorescence-activated Cell Sorting (FACS, BD, Franklin Lake, NJ, USA).
Immunofluorescence
EC cells were treated with NC, miR-7-5p agonist or miR-7-5p antagonist for 72 h. Next, cells were blocked at room temperature and then incubated with anti-Ki67 antibody (Abcam; 1:1000) at 4°C overnight. Then, cells were incubated with secondary antibody (IgG, Abcam; 1:5000) at 37°C for 1 h. The nuclei were stained with DAPI (Beyotime, Shanghai, China) for 5 min. Finally, cells were observed under a fluorescence microscope.
Transwell Assay
Transwell plates (24‑well, Corning, New York, NY, USA) were used for cell invasion and migration detection. For the cell migration assay, 2×105 EC cells were seeded into the upper chambers of the 24‑well plates in 200 μL of serum‑free RPMI 1640 medium supplemented with 0.2% bovine serum albumin. The lower chambers contained RPMI 1640 medium supplemented with 1% FBS. After 24 h of incubation at 37°C, the non‑migrating cells were gently removed from the upper side of each chamber with a cotton swab, while the cells that had migrated were fixed with 95% alcohol for 10 min and stained with 1% crystal violet (Sigma, Grand Island, NY, USA) for 5 min. Finally, cells were counted under an inverted light microscope (Olympus) at 400x magnification.
For the invasion assay, the upper chambers of the 24‑well plates were pretreated with 50 μL of Matrigel (12.5 mg/l, BD Biosciences, Franklin Lake, NJ, USA). Then, EC cells (1×106 cells/mL) in FBS-free medium were seeded into the upper chambers. The lower chambers contained RPMI 1640 medium supplemented with 1% FBS. The cells were incubated at 37°C for 24 h, and cells that had attached to the underside of the membrane were fixed and stained with 1% crystal violet solution. Finally, the number of invading cells was counted under a microscope at 400 x magnifications.
MiR-7-5p Target Genes Prediction
Potential target genes of miR-7-5p were predicted by using targetscan (http://www.targetscan.org/vert_71/) and miRDB (http://www.mirdb.org/). The data were selected for further analysis.
Luciferase Reporter Assay
Both wild-type and mutant constructs of KLF4 were cloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Fitchburg, WI, USA). EC cells were seeded in a 24-well plate and co-transfected with wild type or mutate type KLF4 3ʹUTR, NC or miR-7-5p agonist by Lipofectamine 2000. After 48 h of transfection, luciferase activity was measured using dual-luciferase reporter assay system (Promega).
Western Blot Detection
Total protein was isolated from cell lysates or tissues by using RIPA buffer and then quantified by BCA protein assay kit (Beyotime, Shanghai, China). Proteins were resolved on 10% SDS-PAGE and transferred onto PVDF membranes. After blocking, the membranes were incubated with primary antibodies at 4°C overnight. Then, the membranes were incubated with secondary anti-rabbit antibody (Abcam; 1:5000) at room temperature for 1 h. The primary antibodies were as follows: anti-p38 (Abcam, Cambridge, MA, USA; 1:1000), anti-ERK (Abcam; 1:1000), anti-JNK (Abcam; 1:1000), anti-KLF4 (Abcam; 1:1000) and anti-β-actin (Abcam; 1:1000). The density of the bands was measured using ImageJ software. Signals were detected with enhanced chemiluminescence using β-actin as the internal standard (Kodak).
In vivo Study
BALB/c nude mice (6–8 weeks old) were purchased from Vital River (Beijing, China). All in vivo experiments were performed in accordance with the National Institutes of Health guide for the care and use of laboratory animals, following a protocol approved by the Ethics Committees of Yancheng Third People’s Hospital.
The left flank of mice was administered with 1×107 Eca-109 cells by subcutaneous injection according to the previous reference.15 Mice were randomly stratified into three groups: Blank, antagonist control (NC) group and miR-7-5p antagonist group. Antagonist we used has been chemically modified, and it has been confirmed to be stably expressed in mice as previously described.16 The mice were intra-tumor treated with 50 nM miR-7-5p antagonist twice a week for 4 weeks when the tumor reached 150 mm3.17 The tumor volume was measured weekly as described.18 At the end of the experiments, body weight of each mouse was examined. Mice were sacrificed for the collection of tumor tissues. Then, tumor tissues were weighed.
Statistical Analysis
Each group was performed at least three independent experiments and all data were expressed as the mean ± standard deviation (SD). Differences were analyzed using a one-way analysis of variance (ANOVA) followed by Tukey’s test (more than 2 groups, Graphpad Prism7). P<0.05 was considered to indicate a statistically significant difference.
Results
Differentially Expressed miRNAs in EC
To analyze the differentially expressed miRNAs in EC, bioinformatics analysis was used. As indicated in Figure 1A-D, differentially expressed miRNAs in GSE114110 data set (DS) and TCGA were presented using Volcano Plot. In addition, Volcano Plot of differentially expressed miRNAs depicting up- and downregulated miRNAs in EC, compared with the matched normal tissues. Among these differentially expressed miRNAs in GSE114110 DS and TCGA, 13 were commonly downregulated, whereas 35 were commonly upregulated (Figure 1E).
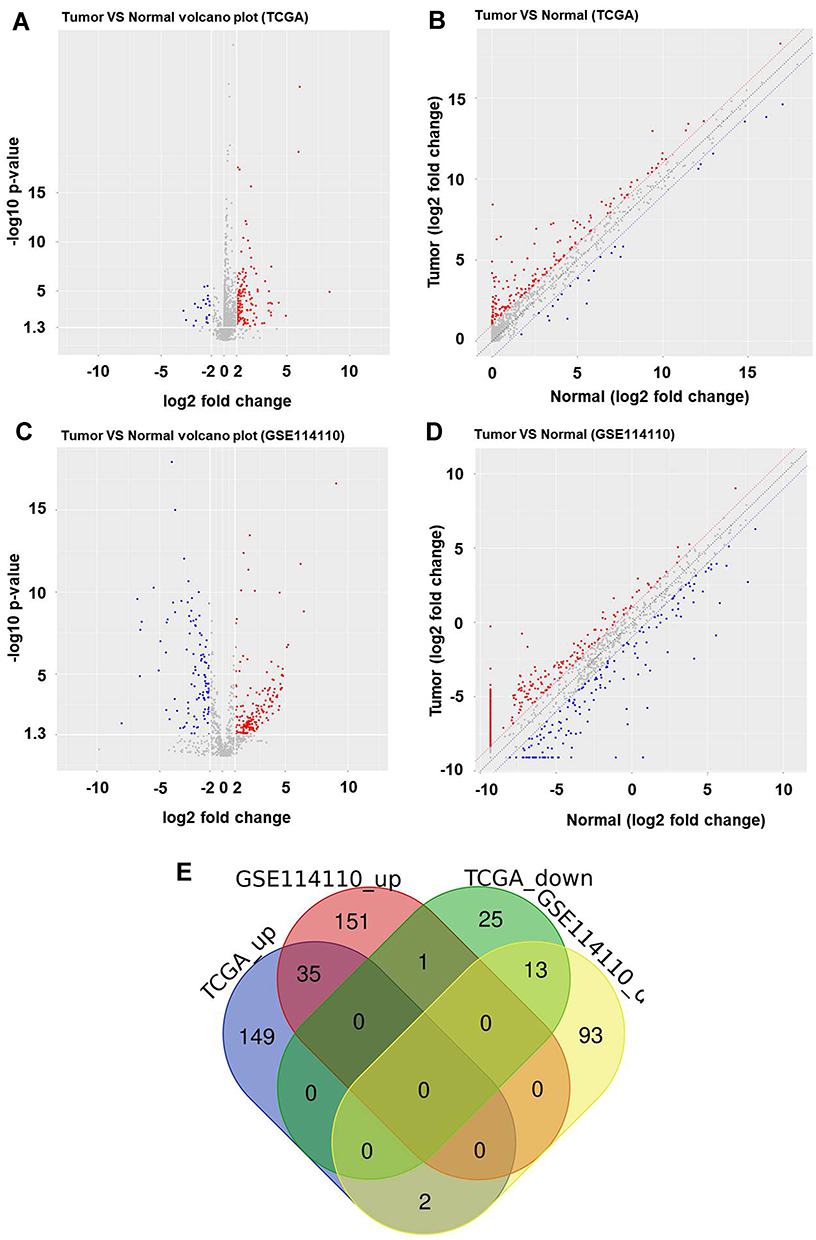 |
Figure 1 Differentially expressed miRNAs in EC. (A-D) The differentially expressed miRNAs in EC were presented in GSE114110 and TCGA using Volcano Plot. Red indicated a high expression level while blue indicated a low expression level. (E) Among these differentially expressed miRNAs in GSE114110 DS, 13 were downregulated, whereas 35 were upregulated. |
Moreover, among the overlap of the differentially expressed miRNAs, five most differentially expressed miRNAs were presented (Figure 2A). Among these five miRNAs, high expression of miR-7-5p was negatively correlated with survival rate of patients with EC (Figure 2B). Besides, the expression of miR-7-5p was significantly upregulated in EC tissues, compared with that in normal tissues (Figure 2C). These data suggested that miR-7-5p might play an important role during the tumorgenesis of EC. Thus, miR-7-5p was selected for investigation in the following experiments.
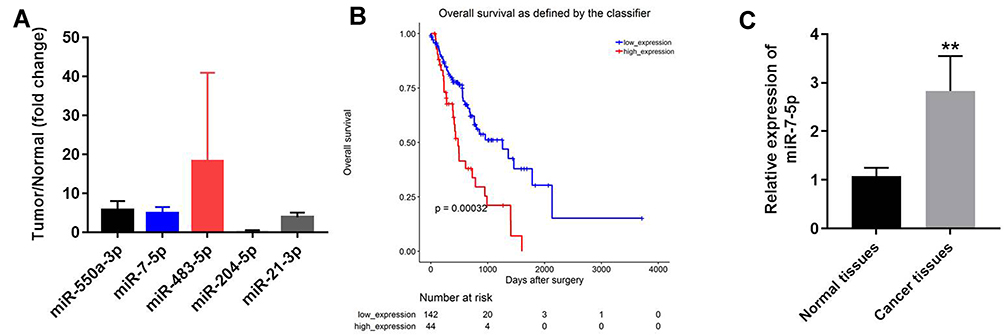 |
Figure 2 MiR-7-5p was selected for further investigation in the following experiments. (A) Among the overlap of differentially expressed miRNAs, five most significant differentially expressed miRNAs were presented. The fold changes (cancer tissues/adjacent normal tissues) were acquired from GSE and TCGA, respectively. The mean value of fold changes from these two datasets was calculated. (B) TCGA was performed to confirm the correlation between miR-7-5p and survive rate of EC. (C) The expression of miR-7-5p in tumor or in adjacent normal tissues was detected by qRT-PCR. **P < 0.01 compared to normal tissues. |
MiR-7-5p Antagonist Greatly Suppressed the Growth of EC Cells
To verify the efficiency of cell transfection, qRT-PCR was performed. As demonstrated in Figure 3A, the expression of miR-7-5p in EC cells was notably upregulated by miR-7-5p agonist, while it was downregulated in the presence of miR-7-5p antagonist. The result demonstrated that miR-7-5p was stably transfected into EC cells. Moreover, OD value of EC cells (Eca-109 and TE-9) was notably inhibited by miR-7-5p antagonist but increased by miR-7-5p agonist (Figure 3B and C). In summary, downregulation of miR-7-5p significantly inhibited the proliferation of EC cells.
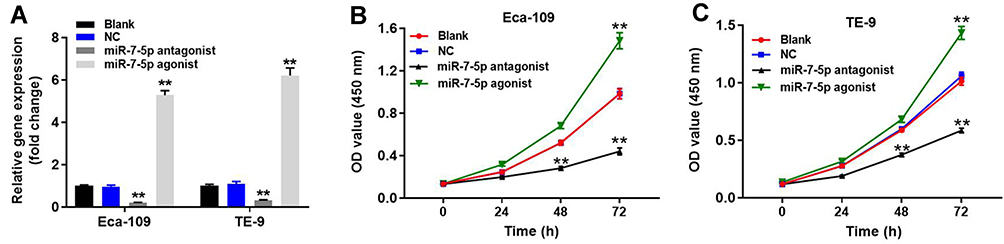 |
Figure 3 Downregulation of miR-7-5p significantly inhibited the cell proliferation of EC. (A) Eca-109 or TE-9 cells were transfected with NC, miR-7-5p antagonist or miR-7-5p agonist for 24 h. Then, cell transfection efficiency was verified by qRT-PCR. After incubation of 0, 24, 48 or 72 h, OD value of (B) Eca-109 or (C) TE-9 cells was detected by CCK-8 assay. **P < 0.01 compared to control. |
MiR-7-5p Antagonist Notably Induced the Apoptosis of EC Cells
For the purpose of detecting the effect of miR-7-5p on apoptosis of EC cells, flow cytometry was used. As demonstrated in Figure 4A and B, miR-7-5p antagonist obviously induced the apoptosis of Eca-109 cells. Consistently, the apoptosis rate of TE-9 cells was notably increased by the downregulation of miR-7-5p (Figure 4C and D). Since Eca-109 cells were more sensitive to miR-7-5p antagonist compared with TE-9 cells, Eca-109 cells were used in the following experiments. Altogether, miR-7-5p antagonist notably induced the apoptosis of EC cells.
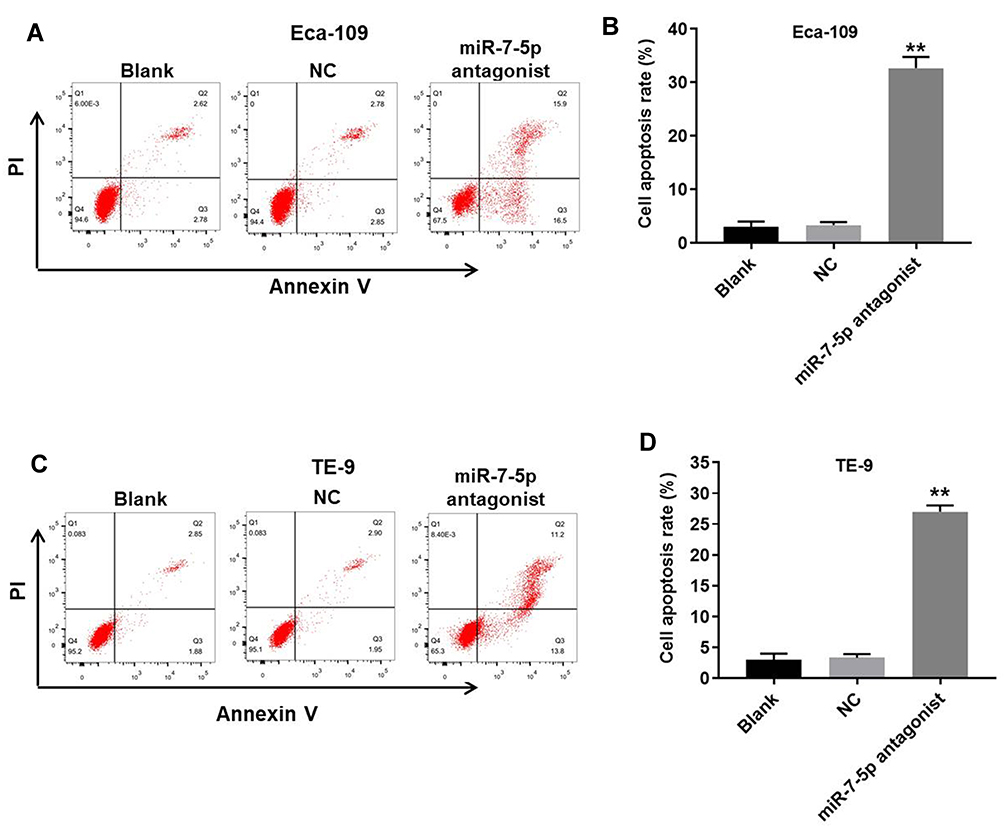 |
Figure 4 MiR-7-5p inhibition notably induced the apoptosis of EC cells. (A, B) The rate of apoptotic Eca-109 cells was detected by FACS after double staining with Annexin V and PI. X-axis: the level of Annexin-V FITC fluorescence; Y-axis: the PI fluorescence. (C, D) The rate of apoptotic TE-9 cells was detected by FACS after double staining with Annexin V and PI. X-axis: the level of Annexin-V FITC fluorescence; Y-axis: the PI fluorescence. **P < 0.01 compared to control. |
MiR-7-5p Antagonist Significantly Suppressed the Migration and Invasion of EC Cells
Transwell assay was performed to test the migration and invasion of EC cells. As indicated in Figure 5A-D, migration and invasion of EC cells were notably inhibited in the presence of miR-7-5p antagonist but promoted by miR-7-5p agonist. In addition, miR-7-5p agonist notably promoted the proliferation of EC cells (Figure 5E and F). In contrast, miR-7-5p antagonist exhibited an inhibitory effect on EC cell proliferation (Figure 5E and F). Taken together, miR-7-5p antagonist notably inhibited the migration and invasion of EC cells.
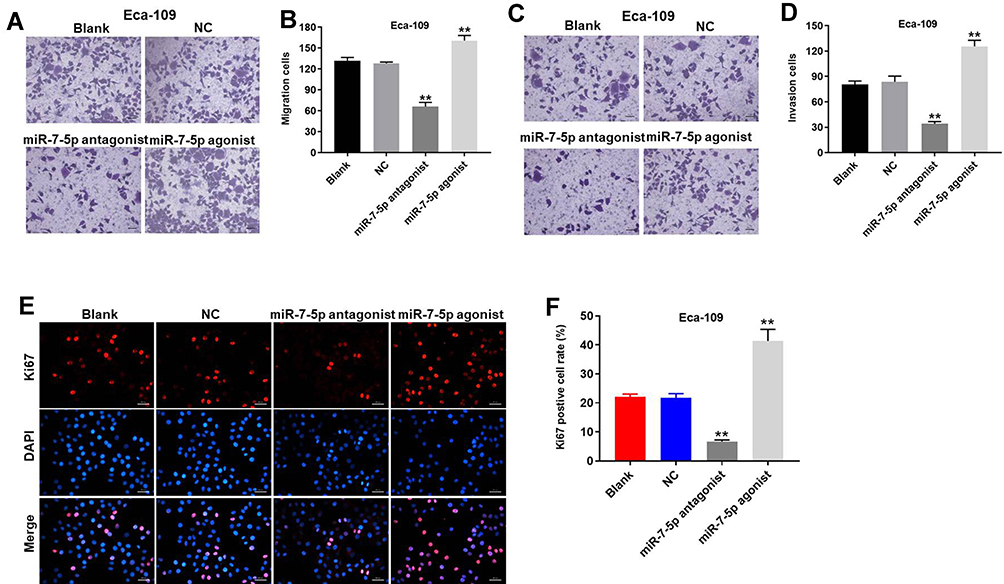 |
Figure 5 Downregulation of miR-7-5p significantly inhibited the migration and invasion of EC cells. (A, B) The migration of Eca-109 cells was tested using transwell migration assay; ×400 magnification. (C, D) The invasion of Eca-109 cells was tested using transwell invasion assay; ×400 magnification. (E) The proliferation of Eca-109 cells was detected by Ki67 staining. Then, (F) Ki67 positive cell rate was calculated. **P < 0.01 compared to control. |
KLF4 Was the Direct Target of miR-7-5p in EC Cells
To investigate the target gene of miR-7-5p, targetscan, miRDB and dual-luciferase report assay were used. As demonstrated in Figure 6A and B, KLF4 was found to be the direct target of miR-7-5p. In addition, the data of dual-luciferase indicated that the co-transfection of the wild-type KLF4 vector (WT-KLF4) with miR-7-5p agonist significantly reduced luciferase activities compared with mutant KLF4 vector (MT-KLF4) (Figure 6C). To sum up, KLF4 was the direct target of miR-7-5p in EC cells.
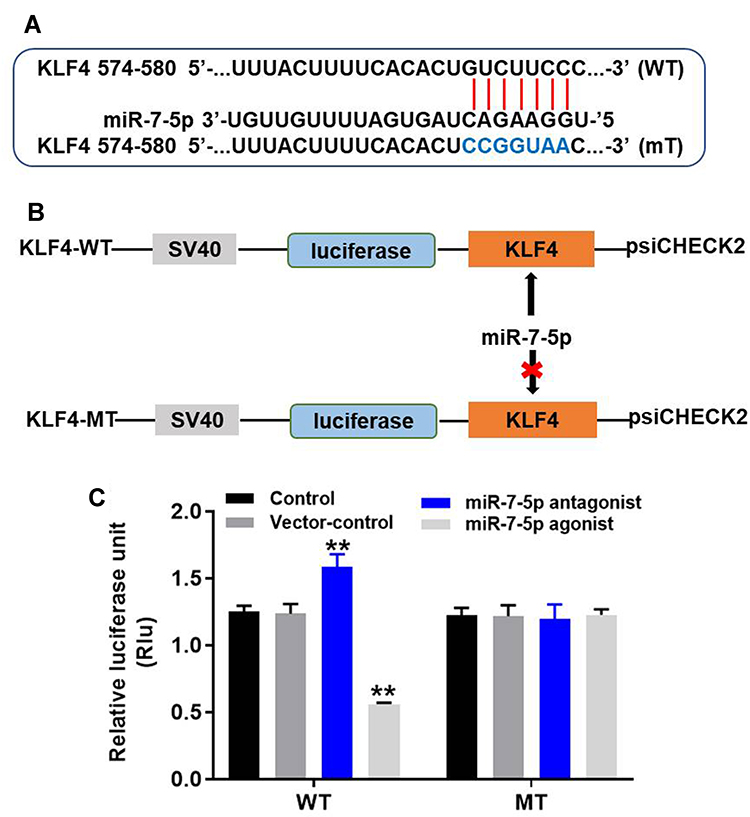 |
Figure 6 KLF4 was the direct target of miR-7-5p. (A, B) Gene structure of KLF4 at the position of 574–580 indicated the predicted target site of miR-7-5p in its 3ʹUTR. (C) The luciferase activity was measured in Eca-109 cells following co-transfecting with WT/MT KLF4 3′-UTR plasmid and miR-7-5p with the dual-luciferase reporter assay. **P < 0.01 compared to control. |
MiR-7-5p Agonist Promoted the Tumorigenesis of EC via Activation of MAPK Signaling
To explore the mechanism by which miR-7-5p mediated the tumorigenesis of EC, Western blot was performed. As demonstrated in Figure 7A and B, the expression of KLF4 in EC cells was significantly upregulated by miR-7-5p antagonist or APTO-253 (KLF4 promoter). However, overexpression of miR-7-5p notably decreased the level of KLF4 in EC cells. Besides, APTO-253 partially rescued the inhibitory effect of miR-7-5p agonist on KLF4 expression. In contrast, miR-7-5p agonist-induced activation of p-p38 and p-ERK in EC cells were reversed by APTO-253 (Figure 7A, 7 and D). Meanwhile, miR-7-5p antagonist or APTO-253 alone significantly inhibited the expression of these two proteins (Figure 7A, C and D). However, miR-7-5p agonist, antagonist or KLF4 had very limited effect on the expression of p-JNK in EC cells (Figure 7E). Besides, miR-7-5p agonist increased the cell proliferation of EC, which was significantly reversed by APTO-253 (Figure 7F). Altogether, miR-7-5p promoted the progression of EC via the mediation of KLF4/MAPK axis.
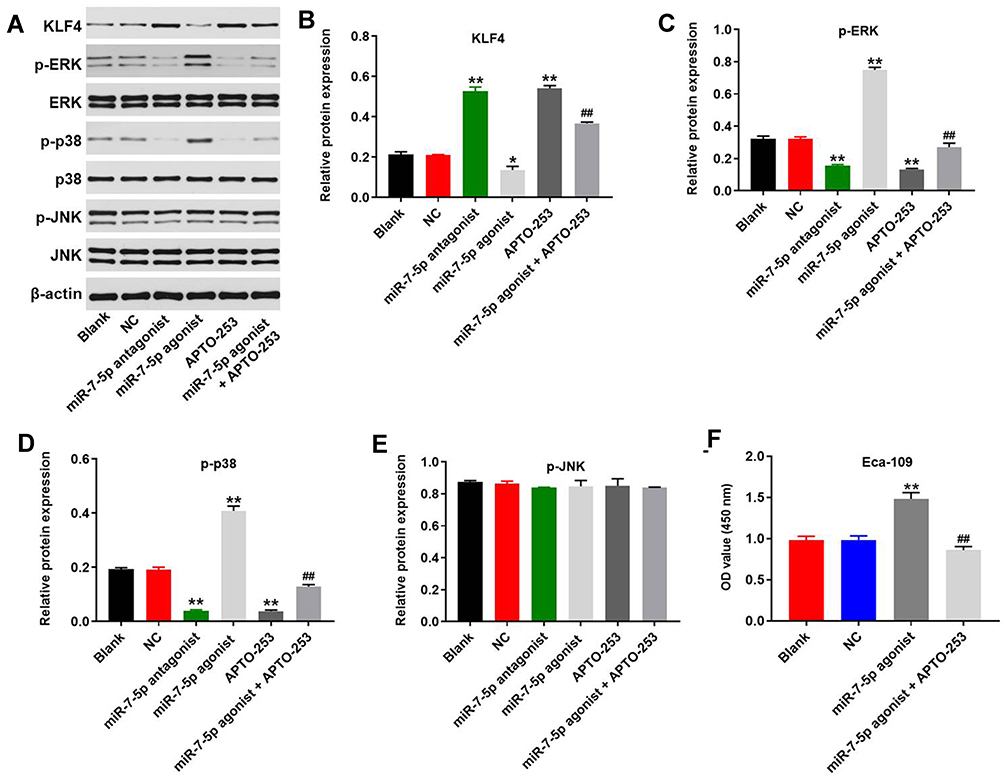 |
Figure 7 MiR-7-5p promoted the progression of EC via activation of MAPK signaling. (A) The protein expression of KLF4, p38, p-ERK, ERK, JNK, p-JNK and p-p38 in Eca-109 cells was detected by Western blot. (B) The relative expression of KLF4 was quantified via normalizing to β-actin. (C) The relative expression of p-ERK was quantified via normalizing to β-actin. (D) The relative expression of p-p38 was quantified via normalizing to β-actin. (E) The relative expression of p-JNK was quantified via normalizing to β-actin. Eca-109 cells were treated with NC, miR-7-5p agonist or miR-7-5p agonist+ APTO-253. (F) After 72 h of incubation, the OD value of Eca-109 cells was tested by CCK-8 assay. *P < 0.05, **P < 0.01 compared to control. ##P < 0.01 compared to miR-7-5p agonist. |
miR-7-5p Antagonist Significantly Attenuated the Tumor Growth of EC in vivo
Finally, to explore the effect of miR-7-5p on EC in vivo, xenograft mice model was established. As indicated in Figure 8A and B, the tumor size in mice was obviously decreased by the downregulation of miR-7-5p. Similarly, miR-7-5p antagonist notably reduced the tumor weight of mice (Figure 8C). Moreover, miR-7-5p antagonist obviously suppressed the expression of p-p38 and p-ERK and increased the expression of KLF4 in tumor tissues of mice (Figure 8D-G). Altogether, the downregulation of miR-7-5p significantly attenuated the tumor growth of EC in vivo.
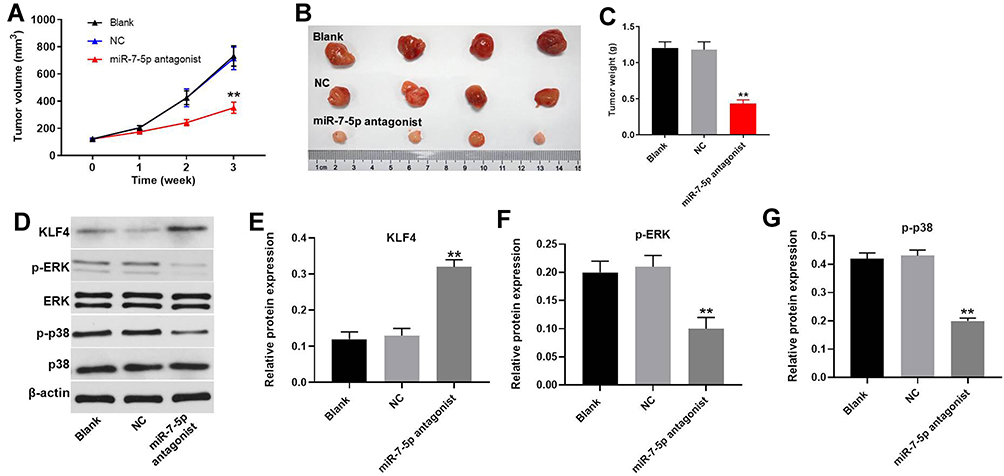 |
Figure 8 Downregulation of miR-7-5p significantly attenuated the tumor growth of EC in vivo. Eca-109 cells were transplanted subcutaneously in each mouse. Then, mice were treated with NC or miR-7-5p antagonist twice a week. (A) Tumor volumes of mice were measured weekly. (B) At the end of the study, tumor tissues of mice were collected and pictured. (C) Tumor weights in each group of mice were calculated. (D) The protein expressions of p38, p-p38, ERK, p-ERK and KLF4 in tumor tissues of mice were detected by Western blot. (E) The relative protein expression of KLF4 was quantified via normalizing to β-actin. (F) The relative protein expression of p-ERK was quantified via normalizing to β-actin. (G) The relative protein expression of p-p38 was quantified via normalizing to β-actin. **P < 0.01 compared to control. |
Discussion
MiRNAs have been confirmed to play important roles in the occurrence of EC.19–21 In this research, we found that the downregulation of miR-7-5p could notably suppress the growth of EC cells. Our study firstly explored the role of miR-7-5p in EC. Tong et al indicated that miR-7-5p could enhance the cell growth and metastasis in gastric cancer.22 Based on our finding and this previous report, miR-7-5p could as a key biomarker during the tumorigenesis. However, Zhong et al demonstrated that miR-7-5p was downregulated in nasopharyngeal carcinoma cells, and miR-7-5p directly targeted E2F transcription factor 3 (E2F3).23 E2F3 has been confirmed to promote cancer cell growth,24 while KLF4 has been verified to be downregulated in EC.25 Thus, this discrepancy might result from the different functions between E2F3 and KLF4.
Kruppel-like factor 4 (KLF4) is an important factor that mediates genes participated in cell growth, differentiation, and metastasis.26,27 Additionally, overexpression of KLF4 has been studied in multiple malignant tumors and has been found to regulate various biological functions, including cell proliferation, survival and metastasis.11,28,29 We have indicated that KLF4 was a downstream target of miR-7-5p. It has been previously confirmed that miR-9 increased the cell apoptosis in ovarian cancer by directly targeting KLF4.30 Our finding was consistent with this previous research. However, KLF4 has been reported that it could induce the metastasis of colorectal cancer,31 while we found KLF4 was negatively correlated with the tumorigenesis of EC. Since our findings were similar to the previous data that KLF4 was downregulated in EC, different functions of KLF4 in different tumor type might result in this discrepancy.
P38 can mediate many biological processes.32 It has been regarded that p38 can activate MAPK to promote the cancer development and progression.33–35 In addition, previous studies have confirmed the association between KLF4 and MAPK/p38 signaling.36,37 Our study further verified the relation between KLF4 and MAPK/p38, suggesting that KLF4 could at as a key modulator on MAPK/p38 signaling. Frankly speaking, this research only focused on MAPK/p38 signaling so far. Since it has been reported that PI3K/Akt signaling could play a key role during the development of EC,38 we will verify the function of miR-7-5p on this pathway.
In conclusion, miR-7-5p antagonist could inhibit the tumorigenesis of EC via the mediation of KLF4/MAPK signaling pathway. Therefore, miR-7-5p could serve as a potential target for the treatment of EC.
Disclosure
The authors declared no competing interests in this study.
References
1. Njei B, McCarty TR, Birk JW. Trends in esophageal cancer survival in United States adults from 1973 to 2009: a SEER database analysis. J Gastroenterol Hepatol. 2016;31(6):1141–1146. doi:10.1111/jgh.13289
2. Zheng R, Liu Y, Zhang X, Zhao P, Deng Q. miRNA-200c enhances radiosensitivity of esophageal cancer by cell cycle arrest and targeting P21. Biomed Pharmacother. 2017;90:517–523. doi:10.1016/j.biopha.2017.04.006
3. Xu J, Bai Y, Xu N, et al. Tislelizumab plus chemotherapy as first-line treatment for advanced esophageal squamous cell carcinoma and gastric/gastroesophageal junction adenocarcinoma. Clin Cancer Res. 2020. doi:10.1158/1078-0432.CCR-19-3561
4. Huang FL, Yu SJ. Esophageal cancer: risk factors, genetic association, and treatment. Asian J Surg. 2018;41(3):210–215. doi:10.1016/j.asjsur.2016.10.005
5. Bollschweiler E, Holscher AH. Prognostic relevance of tumor response after neoadjuvant therapy for patients with esophageal cancer. Ann Transl Med. 2019;7(Suppl 6):S228. doi:10.21037/atm.2019.08.36
6. Hasakova K, Reis R, Vician M, Zeman M, Herichova I. Expression of miR-34a-5p is up-regulated in human colorectal cancer and correlates with survival and clock gene PER2 expression. PLoS One. 2019;14(10):e0224396. doi:10.1371/journal.pone.0224396
7. Mohammadrezakhani H, Baradaran B, Shanehbandi D, et al. Overexpression and clinicopathological correlation of long noncoding RNA TMPO-AS1 in colorectal cancer patients. J Gastrointest Cancer. 2019.
8. Li J, Jin B, Wang T, et al. Serum microRNA expression profiling identifies serum biomarkers for HCV-related hepatocellular carcinoma. Cancer Biomark. 2019;26(4):501–512. doi:10.3233/CBM-181970
9. Akula SM, Abrams SL, Steelman LS, et al. RAS/RAF/MEK/ERK, PI3K/PTEN/AKT/mTORC1 and TP53 pathways and regulatory miRs as therapeutic targets in hepatocellular carcinoma. Expert Opin Ther Targets. 2019;1–15.
10. Feng Z, Zhang X, Li L, et al. Tumor-associated macrophage-derived exosomal microRNA-155-5p stimulates intracranial aneurysm formation and macrophage infiltration. Clin Sci (Lond). 2019;133(22):2265–2282. doi:10.1042/CS20190680
11. Wang W, Fu S, Lin X, et al. miR-92b-3p functions as a key gene in esophageal squamous cell cancer as determined by co-expression analysis. Onco Targets Ther. 2019;12:8339–8353. doi:10.2147/OTT.S220823
12. Yu T, Bai W, Su Y. et al. Enhanced expression of lncRNA ZXF1 promotes cisplatin resistance in lung cancer cell via MAPK axis. Exp Mol Pathol;2020. 104484. doi:10.1016/j.yexmp.2020.104484
13. Qi XT, Li YL, Zhang YQ, et al. KLF4 functions as an oncogene in promoting cancer stem cell-like characteristics in osteosarcoma cells. Acta Pharmacol Sin. 2019;40(4):546–555. doi:10.1038/s41401-018-0050-6
14. Rippa E, La Monica G, Allocca R, et al. Overexpression of gastrokine 1 in gastric cancer cells induces Fas-mediated apoptosis. J Cell Physiol. 2011;226(10):2571–2578. doi:10.1002/jcp.22601
15. Zheng L, Zhao Z, Rong L, Xue L, Song Y. RASSF6-TRIM16 axis promotes cell proliferation, migration and invasion in esophageal squamous cell carcinoma. J Genet Genomics. 2019;46(10):477–488. doi:10.1016/j.jgg.2019.10.004
16. Wu Z, Cai L, Lu J, et al. MicroRNA-93 mediates cabergoline-resistance by targeting ATG7 in prolactinoma. J Endocrinol. 2018.
17. Zhang X, Yang Z, Heng Y, Miao C. MicroRNA 181 exerts an inhibitory role during renal fibrosis by targeting early growth response factor1 and attenuating the expression of profibrotic markers. Mol Med Rep. 2019;19(4):3305–3313. doi:10.3892/mmr.2019.9964
18. Lu RH, Xiao ZQ, Zhou JD, et al. MiR-199a-5p represses the stemness of cutaneous squamous cell carcinoma stem cells by targeting Sirt1 and CD44ICD cleavage signaling. Cell Cycle. 2019;1–14.
19. Lin Y, Lin Z, Fang Z, et al. Plasma MicroRNA-34a as a potential biomarker for early diagnosis of esophageal cancer. Clin Lab. 2019;65:11. doi:10.7754/Clin.Lab.2019.190340
20. Sanchez-Sendra B, Garcia-Gimenez JL, Gonzalez-Munoz JF, et al. Circulating miRNA expression analysis reveals new potential biomarkers for human cutaneous melanoma staging. J Eur Acad Dermatol Venereol. 2019.
21. Sahraei M, Chaube B, Liu Y, et al. Suppressing miR-21 activity in tumor-associated macrophages promotes an antitumor immune response. J Clin Invest. 2019;129(12):5518–5536. doi:10.1172/JCI127125
22. Tong L, Shen S, Huang Q, et al. Proteasome-dependent degradation of Smad7 is critical for lung cancer metastasis. Cell Death Differ. 2019.
23. Zhong Q, Huang J, Wei J, Wu R. Circular RNA CDR1as sponges miR-7-5p to enhance E2F3 stability and promote the growth of nasopharyngeal carcinoma. Cancer Cell Int. 2019;19:252. doi:10.1186/s12935-019-0959-y
24. Chen H, Tan X, Ding Y. Knockdown SNHG20 suppresses nonsmall cell lung cancer development by repressing proliferation, migration and invasion, and inducing apoptosis by regulating miR-2467-3p/E2F3. Cancer Biother Radiopharm. 2020. doi:10.1089/cbr.2019.3430
25. Huang H, Wei L, Qin T, et al. Circular RNA ciRS-7 triggers the migration and invasion of esophageal squamous cell carcinoma via miR-7/KLF4 and NF-kappaB signals. Cancer Biol Ther. 2019;20(1):73–80. doi:10.1080/15384047.2018.1507254
26. Ji W, Shi H, Shen H, et al. Mechanism of KLF4 protection against acute liver injury via inhibition of apelin signaling. Oxid Med Cell Longev. 2019;2019:6140360. doi:10.1155/2019/6140360
27. Wang YD, Chen WD, Wang M, et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48(5):1632–1643. doi:10.1002/hep.22519
28. Khachigian LM. Transcription factors targeted by miRNAs regulating smooth muscle cell growth and intimal thickening after vascular injury. Int J Mol Sci. 2019;20:21. doi:10.3390/ijms20215445
29. Xue Y, Jia X, Li C, et al. DDX17 promotes hepatocellular carcinoma progression via inhibiting Klf4 transcriptional activity. Cell Death Dis. 2019;10(11):814. doi:10.1038/s41419-019-2044-9
30. Zhang L, Zhou Q, Qiu Q, et al. CircPLEKHM3 acts as a tumor suppressor through regulation of the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis in ovarian cancer. Mol Cancer. 2019;18(1):144. doi:10.1186/s12943-019-1080-5
31. Shao H, Dong D, Shao F. Long non-coding RNA TUG1-mediated down-regulation of KLF4 contributes to metastasis and the epithelial-to-mesenchymal transition of colorectal cancer by miR-153-1. Cancer Manag Res. 2019;11:8699–8710. doi:10.2147/CMAR.S208508
32. Silva Ferraz L, Torres da Costa R, Alves da Costa C, et al. Targeting mitochondria in melanoma: interplay between MAPK signaling pathway and mitochondrial dynamics. Biochem Pharmacol. 2020;178:114104. doi:10.1016/j.bcp.2020.114104
33. Bendell JC, Bischoff HG, Hwang J, et al. A Phase 1 dose-escalation study of checkpoint kinase 1 (CHK1) inhibitor prexasertib in combination with p38 mitogen-activated protein kinase (p38 MAPK) inhibitor ralimetinib in patients with advanced or metastatic cancer. Invest New Drugs. 2019.
34. Yen WC, Wu YH, Wu CC, et al. Impaired inflammasome activation and bacterial clearance in G6PD deficiency due to defective NOX/p38 MAPK/AP-1 redox signaling. Redox Biol. 2019;28:101363. doi:10.1016/j.redox.2019.101363
35. Wei T, Xiaojun X, Peilong C. Magnoflorine improves sensitivity to doxorubicin (DOX) of breast cancer cells via inducing apoptosis and autophagy through AKT/mTOR and p38 signaling pathways. Biomed Pharmacother. 2019;121:109139. doi:10.1016/j.biopha.2019.109139
36. Xu K, Ji M, Huang X, et al. Differential regulatory roles of MicroRNAs in porcine intramuscular and subcutaneous adipocytes. J Agric Food Chem. 2020;68(13):3954–3962. doi:10.1021/acs.jafc.9b08191
37. Kawano A, Ariyoshi W, Yoshioka Y, et al. Docosahexaenoic acid enhances M2 macrophage polarization via the p38 signaling pathway and autophagy. J Cell Biochem. 2019;120(8):12604–12617. doi:10.1002/jcb.28527
38. Shi N, Yu H, Chen T. Inhibition of esophageal cancer growth through the suppression of PI3K/AKT/mTOR signaling pathway. Onco Targets Ther. 2019;12:7637–7647. doi:10.2147/OTT.S205457
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.