")
Back to Journals » OncoTargets and Therapy » Volume 11
Down-regulation of CXCL 11 inhibits colorectal cancer cell growth and epithelial-mesenchymal transition
Authors Gao YJ, Liu DL, Li S, Yuan GF, Li L, Zhu HY, Cao GY
Received 11 March 2018
Accepted for publication 22 July 2018
Published 23 October 2018 Volume 2018:11 Pages 7333—7343
DOI https://doi.org/10.2147/OTT.S167872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Yu Jie Gao,1 De Lin Liu,2 Sheng Li,2 Gao Feng Yuan,1 Li Li,1 Hong Yan Zhu,3 Guan Yi Cao3
1Department of Medical Oncology, Suqian First Hospital, Suqian, Jiangsu, China; 2Department of Medical Oncology, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, Jiangsu, China; 3Department of General Surgery, Suqian First Hospital, Suqian, Jiangsu, China
Background: The poor prognosis of colorectal cancer (CRC) largely results from local invasion and tumor metastases. Epithelial-mesenchymal transition (EMT) is a key step in the progression of solid tumors and plays a vital role in tumor metastasis. Recent studies demonstrate that C-X-C motif chemokine 11 (CXCL11) is involved in various cancers’ progression. However, its biological activity in CRC needs deeper exploration.
Methods: The level of CXCL11 in CRC tissues and cell lines was determined using the quantitative real-time PCR (qRT-PCR) assay. The MTT, colony formation, wound healing and Transwell invasion assays were applied to assess the role of CXCL11 in CRC cell growth, migration and invasion, in vitro, respectively. A xenograft model was constructed to analyze the function of CXCL11 in CRC cell growth in vivo.
Results: CXCL11 was over-expressed in CRC tissues and cell lines. Repression of CXCL11 significantly inhibited CRC cell migration, invasion and EMT in vitro. In addition, down-regulation of CXCL11 reduced CRC cell growth and metastasis in vivo. Finally, we revealed that repression of CXCL11 inhibited the metastatic ability of CRC cell in a N-cadherin dependent manner.
Conclusion: In summary, this study explicates the oncogenic activities of CXCL11 in CRC cell growth and metastasis.
Keywords: colon cancer, CXCL11, migration, invasion, EMT
Introduction
Colorectal cancer (CRC), which is the most common digestive cancer, is one of the leading causes for cancer deaths in the world.1 Nowadays, in spite of recent advances in treatment of CRC, including chemotherapy and radiation therapy, the prognosis of patients with CRC remains relatively poor.2 Advanced CRC is associated with a higher mortality rate, and incidence of metastases to distant organs. During tumor metastasis, epithelial mesenchymal transmission (EMT) is considered a vital process during which epithelial cells lose cellular polarity and cell-cell adhesion, and are transformed into separable and aggressive mesenchymal cells.3 EMT is controlled by numerous growth factors and dissimilar factors, including Wnt, TGF-β, Notch and tyrosine kinase receptor.4 The activation and initiation of these signaling pathways result in transcriptional suppression of numerous target genes which are involved in the loss functional activity of E-cadherin, which is a renowned marker of EMT.5 The loss of E-cadherin could lead to the release of β-catenin in cytosol, and subsequent translocation to the cell nucleus, which could trigger LEF/TCF activation and increase the expression of Snail, Twist and Slug which leads to the EMT.6
Chemokines are small, secreted proteins that are best known for their roles in mediating immune cell trafficking and lymphoid tissue development.7,8 Apart from guiding the migration of immune cells, the interaction of chemokines and their receptors also modulate the cellular activities of target cells.9 Consistent with other CXC cytokines, CXCL11 can mediate inflammatory responses, leukocyte trafficking, adaptive resistance, hematopoiesis, cancer cell transfer and angiogenesis.10 CXCL11 elicits its effects by binding to the G-protein-coupled (chemokine) receptors which are grouped into four subgroups including; CC, CXC, CX3C, and C and stimulating numerous down-stream signaling pathways which are involved in tumorigenicity, growth, apoptosis, angiogenesis, invasion, and metastasis of cancer cells.11 Besides the activities in tumor survival, metastasis, and angiogenesis, CXCL11 association with the poor response to therapy has also been demonstrated. A previous investigation has proved that radiation treatment could have a significant effect on the expression of CXCL11, thus demonstrating the role of CXCL11 during administration of chemotherapy and radiotherapy.12 Hence, CXCL11 appears to be an attractive molecular target and monoclonal antibodies against chemokine receptors have been used to inhibit the growth and colony formation of malignant tumors in preclinical studies.13 However, the expression level of CXCL11 and the activity of CXCL11 in the metastasis of CRC cancer cell are still unidentified. This study aimed to investigate the expression levels and elucidate on the oncogenic activities of CXCL11 in CRC.
Materials and methods
Cell lines and tissues
Non-tumorigenic human colonic epithelial cell (HCoEpiC) and CRC cell lines (SW480, SW116, HT-29 and COLO-205) were purchased from GuangZhou Jennio Biotech (GuangZhou, GuangDong, Chian). These cell lines were cultured in medium containing with 10% FBS and maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37°C. Thirty-six cases of human CRC tissues and paired surrounding normal tissues were clinically diagnosed at the Suqian First Hospital between 2016–2017. Written informed consent for participation in the study was obtained from all patients and the approval from the Institutional Research Ethics Committee was obtained. The study conforms with the Code of Ethics of the World Medical Association (Declaration of Helsinki) printed in the British Medical Journal (18 July 1964). The detailed clinical pathological parameters including age, metastasis and tumor size are summarized in Table S1.
siRNA and transfection
siRNA oligonucleotides specific for CXCL11 (Thermo Fisher Scientific, Waltham, MA, USA) were synthesized: siRNA 5′-CUGGUUACCAUCGGAGUUU-3′ (sense) and 5′-AAACUCCGAUGGUAACCAG-3′ (antisense). Cells were transfected with CXCL11 siRNA or non-specific siRNA (0.15 μg/well for 96 well culture plates and 2 μg/well for 6 well culture plates) using the Lipofectamine 2000 reagent (Thermo Fisher Scientific) for 48 hours. Next, proliferation, migration and invasion assays were performed on the cells. A N-cadherin expression construct was generated by subcloning PCR-amplied full-length human N-cadherin cDNA into the pMSCV retrovirus plasmid (synthesized by GenePharma, Shanghai, China). In co-transfection, cells in 6 well culture plates were co-transfected with CXCL11 siRNA (6 μg/well) and N-cadherin over-expression plasmids (6 μg/well in total) for 48 hours. Forty-eight hours post-transfection, the cells were assessed for proliferation, migration and invasion assay.
Cell proliferation assay
Cell viability was determined using the TT assaye (Promega, Madison, WI, USA). Briefly, cells were seeded in 96-well cell culture plates. After incubation for an indicated time period (24 h, 48 h, 72 h or 96 h), 20 μL of MTT reagent were added to each well and incubation was continued for additional 4 hours. Then, 200 μL DMSO was added to each well. The absorbance was measured at 450 nm using a Synergy™ HT Multi-Mode Microplate Reader (Bio-Tek, Winooski, VT, USA). Cell viability was calculated as a percent of viable cells compared with control cells.
Western blotting
Cells were lysed, and total protein was subsequently separated by 8% SDS-PAGE, and then transferred onto PDVF membranes (Millipore, Bedford, MA, USA). Next, the membranes were incubated with antibodies (E-cadherin, N-cadherin and fibronectin) followed by incubation with horseradish peroxidase-conjugated IgGs (1:10,000, Bioworld Biotechnology, Shanghai, China). Bands were detected using an ECL kit (Millipore, Braunschweig, Germany).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) and the first-strand cDNA was synthesized using a SuperScript II Reverse Transcriptase kit (Thermo Fisher Scientific). The quantitative PCR analysis was carried out using the ABI PRISM 7000 Sequence Detection System (Thermo Fisher Scientic). The primers used for PCR of E-cadherin, N-cadherin, fibronectin and CXCL11 were as follows: E-cadherin: (forward primer) 5′-CGAGAGCTACACGTTCACGGT-3′ and (reverse primer) 5′-GGGTGTCGAGGGAAAAATAGG-3′. N-cadherin: (forward primer) 5′-AGCTCCATTCCGACTTAGACA-3′ and (reverse primer) 5′-CAGCCTGAGCACGAAGAGTG-3′; fibronectin: (forward primer) 5′-TCTGTGCCTCCTATCTATGTGC-3′ and (reverse primer) 5′-GAGGGACCACGACAACTCTTC-3′. CXCL11: (forward primer) 5′-GACGCTGTCTTTGCATAGGC-3′ and (reverse primer) 5′-GGATTTAGGCATCGTTGTCCTTT-3′. GAPDH: (forward primer) 5′-CTGGGCTACACTGAGCACC-3′ and (reverse primer) 5′-AAGTGGTCGTTGAGGGCAATG-3′. The cDNA amplification and relative expression values were obtained from three independent experiments and the comparative cycle threshold (Ct) method was applied to quantify the expression levels through 2(−ΔΔCt) method.
Immunofluorescence
Cells were cultured on coverslips and were fixed in 4% paraformaldehyde for 10 minutes at room temperature. Next, cells were washed with PBS for three times, blocked with 10% goat serum for 30 minutes and incubated with the antibodies (E-cadherin, N-cadherin and fibronectin) overnight. Then cells were incubated with the FITC-conjugated goat anti-rabbit IgG (Bioworld Biotechnology, Shanghai, China) at the temperature of 37°C for 30 minutes. The nuclei were then stained with DAPI (Bioworld). The images were photographed using the ZEN 2011 imaging software system on a Zeiss invert microscope (CarlZeiss, Hallbergnoos, Germany).
Wound-healing assay
SW480 cells were seeded into six-well plates for overnight. Cells were treated with 10 μg/mL Mitomycin C (BP25312, Fisher Scientific, Hampton, NH, USA) in culture media for 2 hours to eliminate proliferation. Next, cells were washed with PBS and an artificial uniform wound was created. Cells were then washed with serum-free medium. Images of cells wound closure were photographed at timepoints of 0 hours and 24 hours.14
Cell invasion
Cells were pre-treated with 10 μg/mL mitomycin C for 2 hours. Next, cells were detached using the trypsin digestion method and 1 × 105 SW480 cells were added to the upper chamber of a Transwell filter (8-μm pores, Costar, Cambridge, MA, USA), and DMEM medium with 20% FBS was added to the lower chamber. After 24 hours, the non-invaded cells were scratched off from the filter and the cells which had invaded the lower side of the upper chamber were fixed and stained with 1% crystal violet. Invasive cells were counted in five randomly selected fields on a counting chamber and under an inverted microscope (CarlZeiss, Hallbergnoos, Germany).15
CRC xenograft model
SW480 cells (5 × 106) were re-suspended in 100 μL PBS and then subcutaneously injected into BALB/c nude mice. The length and width of tumors was measured using calipers once every three days. The tumor volume was calculated using the following formula: length × width2 × 0.5. After 25 days, the mice were sacrificed, and all the tumors were collected and weighed. The animal experiments were approved by Medical Laboratory Animal Welfare and Ethics Committee of SuQian First Hospital and the experiments were conducted in accordance with Institutional Guidelines and the Guide for the Care and Use of Laboratory Animals (NIH publication no 85–23, revised 1996). The tumors were then fixed and applied for immunohistochemical staining with Ki67 antibody.
In vivo metastasis
SW480 cells with transfected siCXCL11 were inoculated into BALB/c mice via the lateral tail vein at a concentration of 1 × 106 cells. Mice were sacrificed 2 weeks after inoculation and all organs were examined for the presence of macroscopic metastases. The lung tissues were to the stained with H&E for visual identification of metastasis.16
Statistical analysis
Histological examination was performed by two pathologists. SPSS version 17.0 (IBM, New York City, NY, USA) was adopted for statistic assessment. Significant differences were assessed using either the two-tailed Student’s t-test or one-way ANOVA followed by post hoc Dunnett’s test. A P-value <0.05 was identified as being statistically significant.
Results
CXCL11 is overexpressed in CRC
To determine the level of CXCL11 in CRC tissues, the expression of CXCL11 was analyzed in CRC tissues and adjacent non-cancer tissues by the qRT-PCR assay. As shown in Figure 1A, the level of CXCL11 was distinctly higher in CRC tissues than in the surrounding normal tissues. CXCL11 protein levels were then determined using immunohistochemistry in CRC tissue. CXCL11 was highly expressed in CRC compared with the control tissue (Figure 1B). Expression of CXCL11 in the CRC cell lines (SW480, SW116, HT-29 and COLO-205) and the normal human colonic epithelial cell line (HCoEpiC) was investigated further by Western blot. As shown in Figure 1C, CXCL11 was over-expressed in all CRC cell lines, and significantly elevated in SW480, as compared with the level of CXCL11 in HCoEpiC. Oncomine analysis of neoplastic vs normal tissue revealed that CXCL11 was significantly overexpressed in CRC in different datasets (Figure 1D).17–19 To evaluate the clinical relevance of CXCL11, we investigated the levels of CXCL11 from clinical specimens that collected in The Human Protein Atlas (www.proteinatlas.org). CXCL11 was strongly expressed in CRC tissues, and moderate expression in normal tissues (Figure 1E). All these data suggest that CXCL11 is over-expressed in CRC tissue and cell lines.
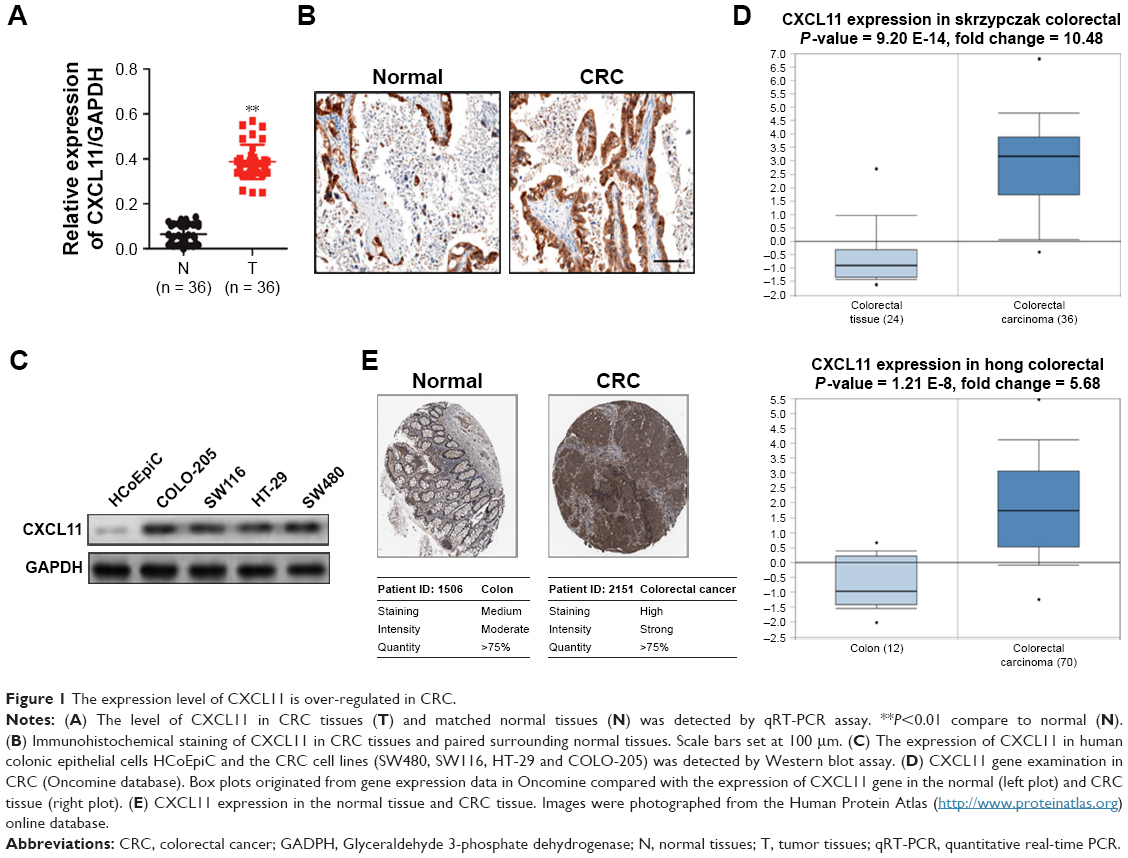 | Figure 1 The expression level of CXCL11 is over-regulated in CRC. |
CXCL11 knocked-down inhibits SW480 cell proliferation and colony formation
To investigate the role of CXCL11 in CRC, SW480 cells were transfected with CXCL11 siRNA to suppress the expression of CXCL11. As shown in Figure 2A and B, there was lower expression of CXCL11 in the SW480 cells than those transfected with siCXCL11. Since CXCL11 is known to promote cell proliferation, the effects of silencing CXCL11 on the proliferation of CRC cells was determined. As shown in Figure 2C, CXCL11 knocked-down remarkably inhibited SW480 cells proliferation compared with the control cells. The anchorage-independent growth capacity of tumor cells is considered essential for cancer progression. Colony formation experiments in SW480 cells were adopted to assess whether CXCL11 knockdown affected colony forming capacity of SW480 cell. As shown in Figure 2D, silencing CXCL11 dramatically reduced the colony formation ability of SW480 cells. These results suggest a pivotal role of CXCL11 in the growth of CRC cell in vitro.
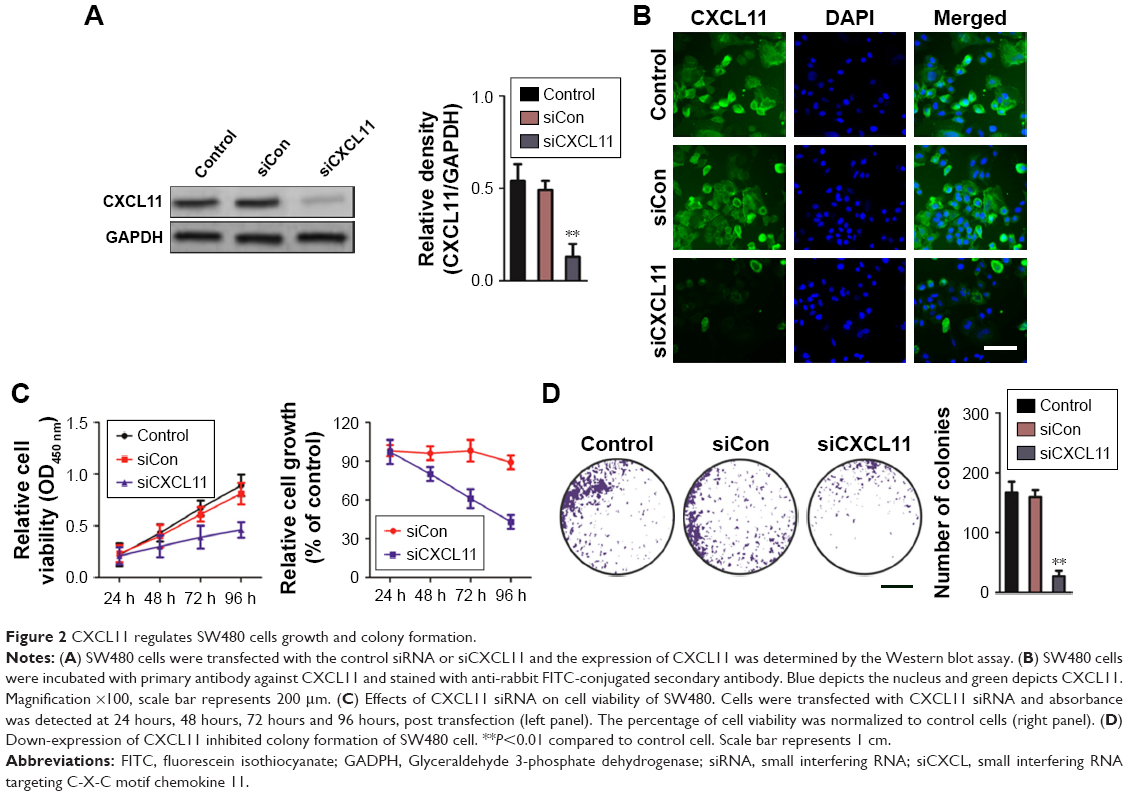 | Figure 2 CXCL11 regulates SW480 cells growth and colony formation. |
CXCL11 knocked-down inhibits migration and invasion of SW480 cell
In order to assess the effects of CXCL11 on CRC cell migration and invasion, CXCL11 was depleted in SW480 cells, and the function of CXCL11 on the migration ability of SW480 cells was determined by a wound healing assay. As presented in Figure 3A, the cell migration was largely inhibited in CXCL11-downexpressing SW480 cells. Consistently, the invasion of SW480 cells was dramatically decreased as compared to the parental cells (Figure 3B). Taken together, these data demonstrate that CXCL11 knock-down inhibits the aggressiveness of CRC SW480 cells.
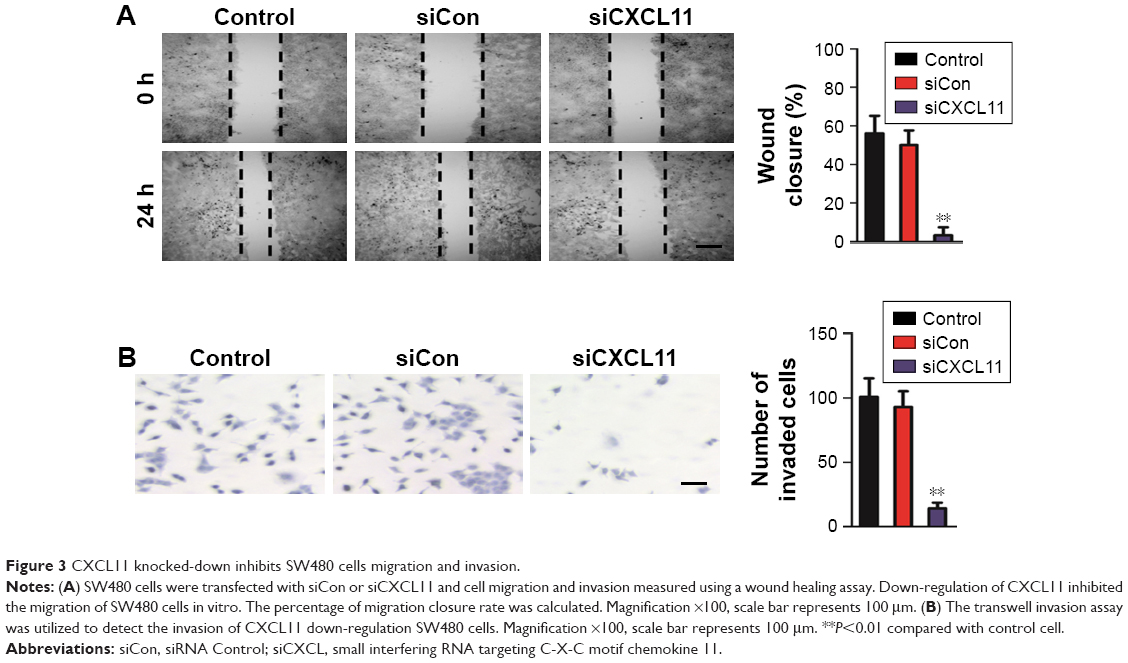 | Figure 3 CXCL11 knocked-down inhibits SW480 cells migration and invasion. |
CXCL11 knocked-down suppresses EMT of SW480 cells
Core elements of EMT contain the decrease of cell-cell adherence via the transcriptional suppression and delocalization of cadherins. To explore the role of CXCL11 in CRC cell EMT, the epithelial or mesenchymal marker in CXCL11 down-expressing SW480 cell were measured by immunofluorescence and western blotting assays. As shown in Figure 4A, a marked reduction of N-cadherin and fibronectin expression was found in CXCL11-downexpressing SW480 cells compared with that of the control cells, whereas down-expression of CXCL11 in SW480 cells resulted in up-regulation of epithelial marker, E-cadherin compared to control cell. Consistently, western blotting analysis showed a significant reduction in the expression of N-cadherin and fibronectin in CXCL11-depleted SW480 cells as compared with that of the control cell, whereas down-regulation of CXCL11 in SW480 cell increased the level of E-cadherin (Figure 4B). Further examination of transcription of EMT-associated markers by qRT-PCR analysis revealed that depletion of CXCL11 in SW480 cells increased the mRNA level of the E-cadherin gene and decreased the mRNA level of N-cadherin and fibronectin (Figure 4C). These results indicate that down-expression of CXCL11 inhibits mesenchymal phenotypes of CRC cells, which indicates that CXCL11 plays an essential role in the EMT process.
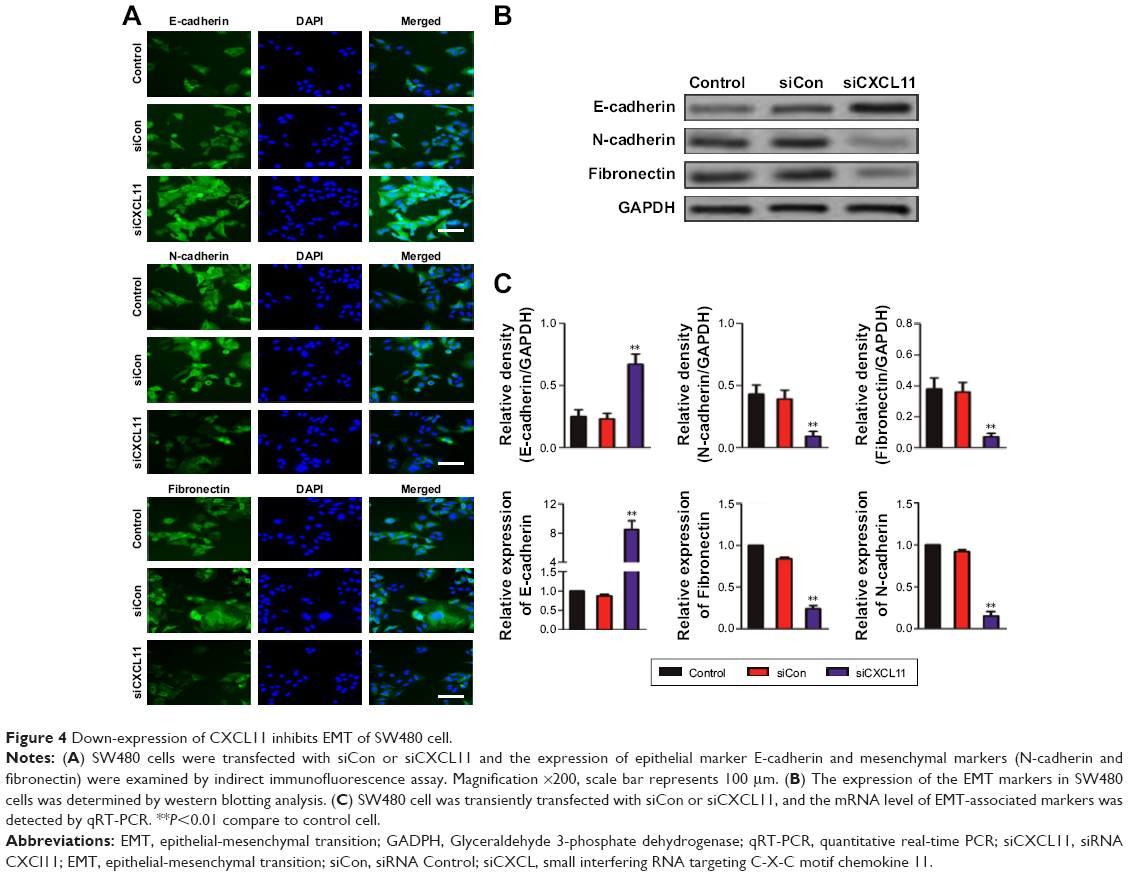 | Figure 4 Down-expression of CXCL11 inhibits EMT of SW480 cell. |
CXCL11 knocked-down suppresses SW480 cell growth and metastasis in vivo
To explore the role of CXCL11 on CRC cell growth in vivo, the parental cells or siCXCL11 SW480 cells were injected subcutaneously into nude mice. As shown in Figure 5A, tumors derived from siCXCL11 SW480 cells grew much slower than those originating from the parental cells. As expected, Ki-67, a proliferation marker of tumor cells was greatly reduced in tumors that formed by siCXCL11 SW480 cells (Figure 5B). To further explore the function of CXCL11 in CRC metastasis in vivo, control or CXCL11 siRNA transfected SW480 cells were injected into BALB/c nude mice via the tail vein. The results indicated that the number of lung metastasis foci was significantly decreased in mice that injected with siCXCL11 SW480 cells in comparison with those injected with control SW480 cells (Figure 5C). The results showed that the down-regulation of CXCL11 greatly suppressed metastasis of CRC cells in vivo.
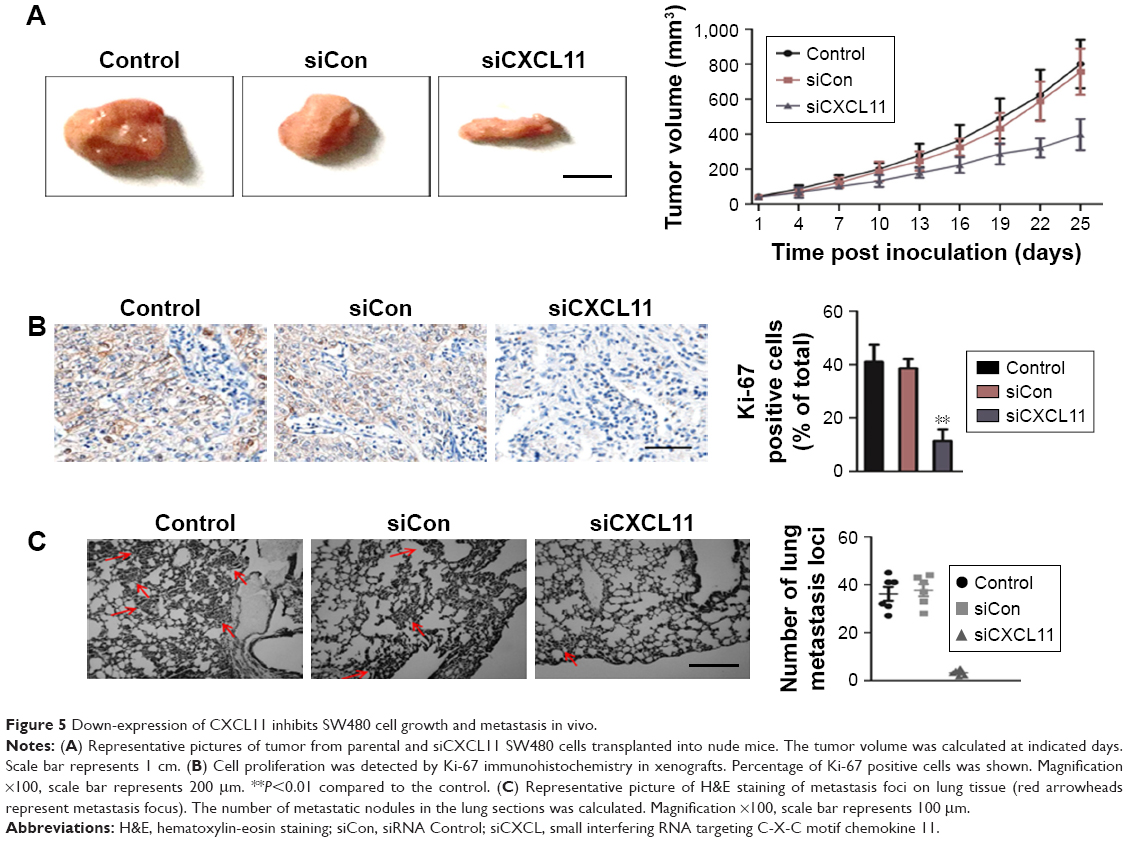 | Figure 5 Down-expression of CXCL11 inhibits SW480 cell growth and metastasis in vivo. |
N-cadherin is involved in the biological behaviors of CXCL11
To confirm the role of CXCL11 in CRC cell proliferation, colony-formation as well as metastasis, gain of function assay was conducted. Firstly, SW480 cells were transfected with vector containing CXCL11 (Figure 6A). Consistent with previous results that CXCL11 acted as an oncogene in CRC, over-expression of CXCL11 significantly increased proliferation, colony formation and invasion of SW480 cells (Figure 6B–D). In order to explore the role of N-cadherin or E-cadherin in CXCL11-medtiated CRC cell migration and invasion, the relationship of N-cadherin, fibronectin or E-cadherin and CXCL11 in CRC tissues was detected by qRT-PCR assay. As show in Figure S1, there was a significant correlation the expression of-cadherin and CXCL11 (P<0.01) whereas there was no significant correlation between CXCL11 and E-cadherin (P>0.05). The expression of N-cadherin is closely associated with acquisition of EMT phenotype and with enhanced invasion in cancer cells. Hence, we assumed that N-cadherin potentially plays a role in CXCL11 regulated CRC cells invasion. To confirm N-cadherin was the functional mediator of CXCL11-induced cell phonotype change, we ectopically expressed N-cadherin in CXCL11 knocked-down SW480 cells. Re-expression of N-cadherin was detected using Western blotting (Figure 6E). As expected, transfection of N-cadherin in CXCL11 knocked-down SW480 cells profoundly rescued the cell growth and colony formation compared with siCXCL11 transfection alone (Figure 6F and G). And cell invasion suppressed by CXCL11 was significantly neutralized by the re-expression of N-cadherin (Figure 6H). These findings show that N-cadherin partially rescued the down-expression of CXCL11 induced inhibition of proliferation, colony formation and invasion in CRC cell, which suggest that N-cadherin is a functional mediator of CXCL11 in CRC.
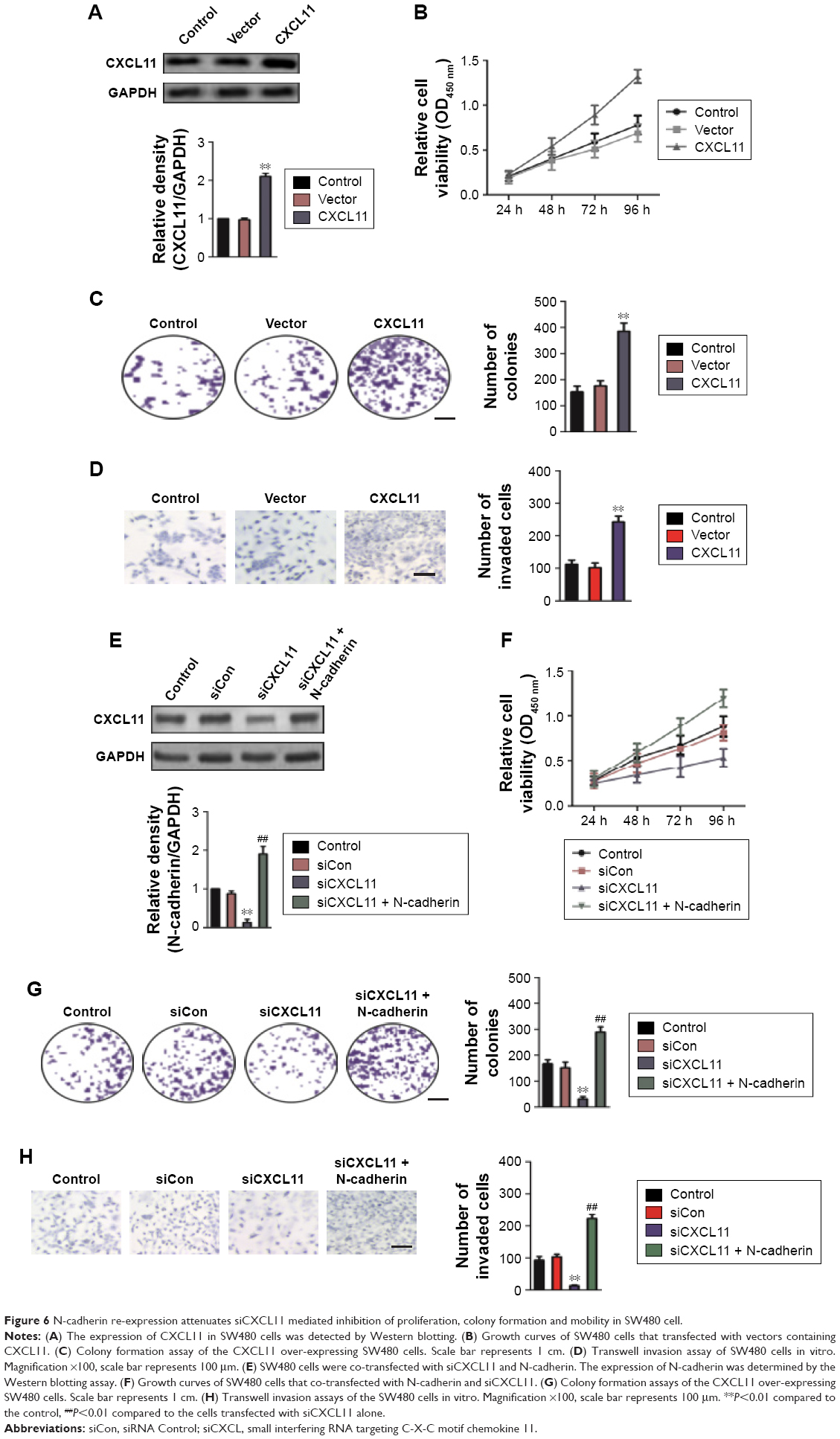 | Figure 6 N-cadherin re-expression attenuates siCXCL11 mediated inhibition of proliferation, colony formation and mobility in SW480 cell. |
Discussion
CRC remains a medical challenge owing to aggressive metastasis and the minimal effectual treatment schemes available.20 EMT plays an essential role in cancer cell invasion and metastasis, which is triggered by a varied set of stimuli that could lead to steady reprogramming of epithelial cells to mesenchymal cells.21 We identified that CXCL11 is overexpressed in CRC cell lines and revealed the essential role of CXCL11 in CRC cell growth and metastasis.
Malignant CRC remains one of the dominant causes of cancer death due to its extreme metastatic ability.22 In recent decades, the incidence and death rate of CRC steadily increased and there are dedicated efforts to identify newer therapeutic strategies.23 In most circumstance, for patients with CRC, surgical resection remains the conventional therapy of choice. Few effective alternative therapies to target metastatic CRC, and the intricate molecular mechanisms underlying metastasis in CRC cells, have been developed. Consequently, the investigation of innovative cellular and molecular mechanisms is vital for the exploitation of novel therapeutic strategies. The alter-expression of genes which belongs to the chemokines family in tumors has been identified and it has been confirmed that CXC chemokines, and their receptors may be involved in tumor occurrence and metastasis.24 CXCL11 is secreted by various cells including; epithelial cells, neutrophils, endothelial cells, eosinophils, monocytes, stromal cells and cancer cells. The interactions between chemokines and their receptors are considered significant in the metastasis of various tumors.10 This research revealed that the expression of CXCL11 in CRC cell was much greater than that in the normal cells. The level of CXCL11 expression in CRC tumor tissue was also significantly higher than that in the normal colon tissue indicating that CXCL11 may act as an oncogene in CRC. CXCL11 has been shown to control tumor growth and metastatic behavior of tumor cells including; lung carcinoma and bladder carcinoma. In the present study, silencing CXCL11 could considerably inhibited the migration and invasion of CRC cells in vitro. The metastatic ability of CRC cells in vivo was also remarkably inhibited by down-regulation of CXCL11.
The incidence of EMT is associated with altered expression of quite a few molecular markers, such as E-cadherin, N-cadherin and fibronectin. This study also showed that the down-regulation of CXCL11 in CRC cells could increase the epithelial marker (E-cadherin) and decrease the expression of mesenchymal cell markers (N-cadherin and fibronectin). In sum, our investigations revealed that CXCL11 acted a crucial role in CRC metastasis and EMT. Silencing of CXCL11 in CRC cell could restrain EMT and cell migration, invasion in vitro and diminish the metastatic capacity of CRC cells in vivo.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhu J, Liu M, Liu Y, Zhang Y, Yang B, Zhang W. Zoledronic acid regulates autophagy and induces apoptosis in colon cancer cell line CT26. Biomed Res Int. 2017;2017:7203584–7203586. | ||
Arivazhagan R, Lee J, Bayarsaikhan D, et al. MicroRNA-340 inhibits the proliferation and promotes the apoptosis of colon cancer cells by modulating REV3L. Oncotarget. 2018;9(4):5155–5168. | ||
Peng Q, Lin K, Chang T, et al. Identification of genomic expression differences between right-sided and left-sided colon cancer based on bioinformatics analysis. Onco Targets Ther. 2018;11:609–618. | ||
Qin Y, Huo Z, Song X, Chen X, Tian X, Wang X. mir-106a regulates cell proliferation and apoptosis of colon cancer cells through targeting the PTEN/PI3K/AKT signaling pathway. Oncol Lett. 2018;15(3):3197–3201. | ||
Spalinger MR, Manzini R, Hering L, et al. PTPN2 regulates inflammasome activation and controls onset of intestinal inflammation and colon cancer. Cell Rep. 2018;22(7):1835–1848. | ||
Wang N, Zhang Y, Liang H. microRNA-598 inhibits cell proliferation and invasion of glioblastoma by directly targeting metastasis associated in colon cancer-1. Oncol Res. Epub 2018 Feb 14. | ||
Wu S, Singh S, Varney ML, Kindle S, Singh RK. Modulation of CXCL-8 expression in human melanoma cells regulates tumor growth, angiogenesis, invasion, and metastasis. Cancer Med. 2012;1(3):306–317. | ||
Mijatovic T, Mahieu T, Bruyère C, et al. UNBS5162, a novel naphthalimide that decreases CXCL chemokine expression in experimental prostate cancers. Neoplasia. 2008;10(6):573–586. | ||
Wang P, Yang X, Xu W, Li K, Chu Y, Xiong S. Integrating individual functional moieties of CXCL10 and CXCL11 into a novel chimeric chemokine leads to synergistic antitumor effects: a strategy for chemokine-based multi-target-directed cancer therapy. Cancer Immunol Immunother. 2010;59(11):1715–1726. | ||
Rupertus K, Sinistra J, Scheuer C, et al. Interaction of the chemokines I-TAC (CXCL11) and SDF-1 (CXCL12) in the regulation of tumor angiogenesis of colorectal cancer. Clin Exp Metastasis. 2014;31(4):447–459. | ||
Zeng YJ, Lai W, Wu H, et al. Neuroendocrine-like cells-derived CXCL10 and CXCL11 induce the infiltration of tumor-associated macrophage leading to the poor prognosis of colorectal cancer. Oncotarget. 2016;7(19):27394–27407. | ||
Guo G, Peng Y, Xiong B, et al. Involvement of chemokine CXCL11 in the development of morphine tolerance in rats with cancer-induced bone pain. J Neurochem. 2017;141(4):553–564. | ||
Koo YJ, Kim TJ, Min KJ, So KA, Jung US, Hong JH. CXCL11 mediates TWIST1-induced angiogenesis in epithelial ovarian cancer. Tumour Biol. 2017;39(5):1010428317706226. | ||
Chang H, Shi YH, Talaf TK, Lin C. Aquaporin-8 mediates human esophageal cancer Eca-109 cell migration via the EGFR-Erk1/2 pathway. Int J Clin Exp Pathol. 2014;7(11):7663–7671. | ||
Kojima S, Enokida H, Yoshino H, et al. The tumor-suppressive microRNA-143/145 cluster inhibits cell migration and invasion by targeting GOLM1 in prostate cancer. J Hum Genet. 2014;59(2):78–87. | ||
Ding C, Luo J, Li L, et al. Gab2 facilitates epithelial-to-mesenchymal transition via the MEK/ERK/MMP signaling in colorectal cancer. J Exp Clin Cancer Res. 2016;35(5):5. | ||
Ortega CE, Seidner Y, Dominguez I. Mining CK2 in cancer. PLoS One. 2014;9(12):e115609. | ||
Skrzypczak M, Goryca K, Rubel T, et al. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010;5(10):e13091. | ||
Hong Y, Downey T, Eu KW, Koh PK, Cheah PY. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin Exp Metastasis. 2010;27(2):83–90. | ||
Wang Y, Chai Z, Wang M, Jin Y, Yang A, Li M. COPB2 suppresses cell proliferation and induces cell cycle arrest in human colon cancer by regulating cell cycle-related proteins. Exp Ther Med. 2018;15(1):777–784. | ||
Wen YA, Xiong X, Zaytseva YY, et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018;9(3):265. | ||
Zhao P, Zhang Z. TNF-α promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol Lett. 2018;15(3):3820–3827. | ||
Zhong L, Huot J, Simard MJ. p38 activation induces production of miR-146a and miR-31 to repress E-selectin expression and inhibit transendothelial migration of colon cancer cells. Sci Rep. 2018;8(1):2334. | ||
Tokunaga R, Zhang W, Naseem M, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation – A target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47. |
Supplementary materials
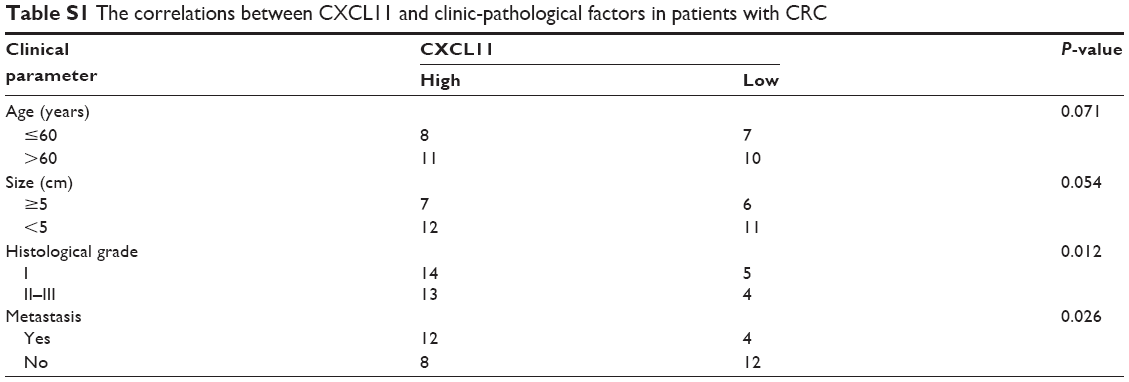 | Table S1 The correlations between CXCL11 and clinic-pathological factors in patients with CRC |
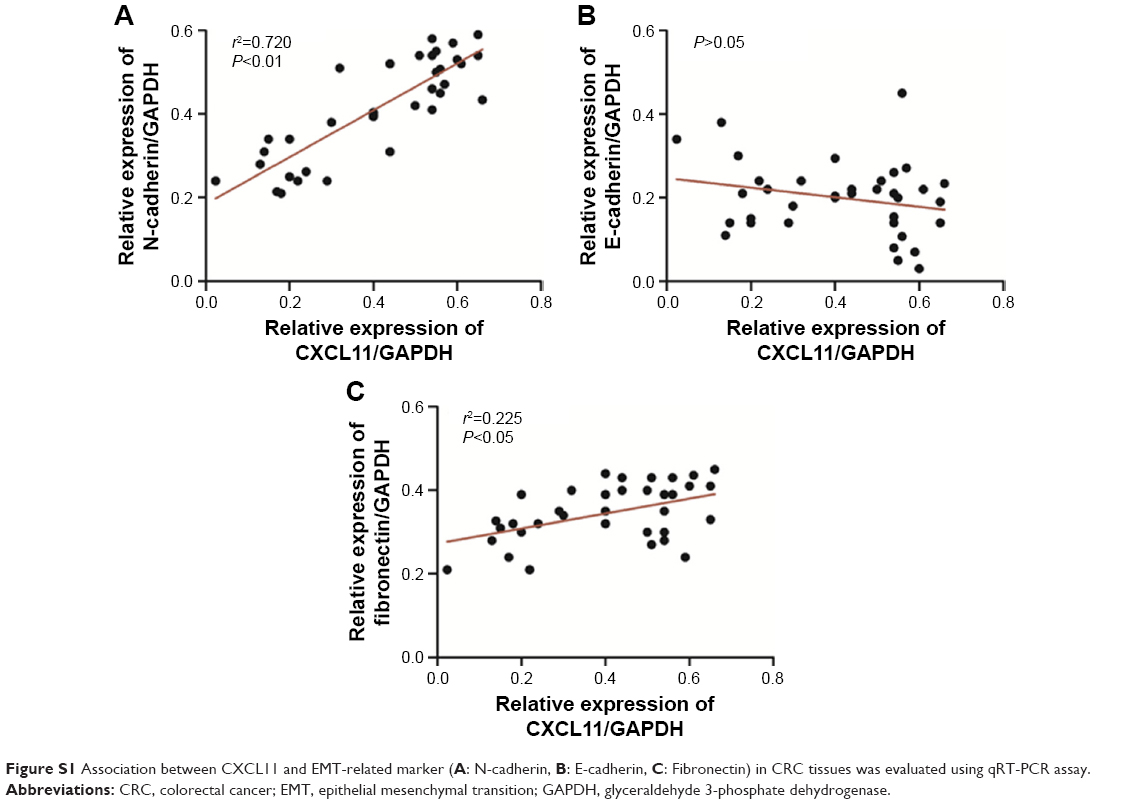 | Figure S1 Association between CXCL11 and EMT-related marker (A: N-cadherin, B: E-cadherin, C: Fibronectin) in CRC tissues was evaluated using qRT-PCR assay. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.