")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Dose–response of an extrafine dry powder inhaler formulation of glycopyrronium bromide: randomized, double-blind, placebo-controlled, dose-ranging study (GlycoNEXT)
Authors Beeh KM , Emirova A , Prunier H, Santoro D, Nandeuil MA
Received 17 March 2018
Accepted for publication 26 April 2018
Published 25 May 2018 Volume 2018:13 Pages 1701—1711
DOI https://doi.org/10.2147/COPD.S168493
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Kai M Beeh,1 Aida Emirova,2 Hélène Prunier,2 Debora Santoro,3 Marie Anna Nandeuil2
1Insaf Respiratory Research Institute, Wiesbaden, Germany; 2Global Clinical Development, Chiesi S.A.S, Bois-Colombes, France; 3Global Clinical Development, Chiesi Farmaceutici SpA, Parma, Italy
Introduction: An extrafine formulation of the long-acting muscarinic antagonist, glycopyrronium bromide (GB), has been developed for delivery via the NEXThaler dry powder inhaler (DPI). This study assessed the bronchodilator efficacy and safety of different doses of this formulation in patients with COPD to identify the optimal dose for further development.
Patients and methods: This was a multicenter, randomized, double-blind, placebo-controlled, incomplete block, three-way crossover study, including three 28-day treatment periods, each separated by a 21-day washout period. Eligible patients had a diagnosis of COPD and post-bronchodilator forced expiratory volume in 1 s (FEV1) 40%–70% predicted. Treatments administered were GB 6.25, 12.5, 25 and 50 µg or matched placebo; all were given twice daily (BID) via DPI, with spirometry assessed on Days 1 and 28 of each treatment period. The primary end point was FEV1 area under the curve from 0 to 12 h (AUC0–12 h) on Day 28.
Results: A total of 202 patients were randomized (61% male, mean age 62.6 years), with 178 (88%) completing all the three treatment periods. For the primary end point, all the four GB doses were superior to placebo (p<0.001) with mean differences (95% CI) of 114 (74, 154), 125 (85, 166), 143 (104, 183) and 187 (147, 228) mL for GB 6.25, 12.5, 25 and 50 µg BID, respectively. All four GB doses were also statistically superior to placebo for all secondary efficacy end points, showing clear dose–response relationships for most of the endpoints. Accordingly, GB 25 µg BID met the criteria for the minimally acceptable dose. Adverse events were reported by 15.5, 16.2, 10.9 and 14.3% of patients receiving GB 6.25, 12.5, 25 and 50 µg BID, respectively, and 14.8% receiving placebo.
Conclusion: This study supports the selection of GB 25 µg BID as the minimal effective dose for patients with COPD when delivered with this extrafine DPI formulation.
Keywords: pulmonary disease, chronic obstructive, muscarinic antagonists, pulmonary function tests, dose–response relationship, drug, metered-dose inhalers
Introduction
A number of formulations of the long-acting muscarinic antagonist (LAMA), glycopyrronium bromide (GB), are available as a component of the management of COPD. This includes an extrafine formulation (ie, containing particles with mass median aerodynamic diameter of <2 μm), which has been approved in COPD for delivery via a pressurized metered-dose inhaler (pMDI) as a triple-therapy combination with the long-acting β2-agonist (LABA), formoterol fumarate (FF), and the inhaled corticosteroid (ICS), beclometasone dipropionate (BDP).1 The GB dose used in this pMDI formulation is 25 μg twice daily (BID), selected on the basis of a number of studies, including a single-dose, dose-escalation comparison with placebo, a 7-day repeat-dose comparison with placebo and a 7-day repeat-dose study in which the addition of GB to BDP/FF was compared with BDP/FF alone.2,3
An extrafine dry powder inhaler (DPI) formulation of GB has now been developed for delivery via the NEXThaler device (Chiesi Farmaceutici SpA, Parma, Italy). In comparison to standard pMDIs, DPIs have the advantage of not requiring actuation to be coordinated with inhalation.4,5 Furthermore, the NEXThaler is a reservoir-based multi-dose DPI, avoiding the need for insertion of capsules as is the case with single-dose DPIs. It incorporates a breath actuation mechanism that helps to ensure that aerosolization and delivery are independent of the inspiratory flow rate.6
The aim of the current study was to assess the bronchodilator efficacy and safety of different doses of this DPI formulation in patients with COPD to identify the optimal dose for further development.
Patients and methods
Trial design
This was a multicenter, randomized, double-blind, placebo-controlled, incomplete block, three-way crossover study. Following a prescreening visit, patients’ eligibility to participate was evaluated at a screening visit. After a 14-day run-in period, eligible patients were randomly assigned to one of 10 sequences, each comprising three treatment periods. The three treatment periods were 28 days in duration, separated by a 21-day washout period. On Days 1 and 28 of each treatment period, serial forced vital capacity (FVC) maneuvers were performed at pre-dose and at 15, 30 and 45 min, and 1, 2, 4, 6, 8, 10, 11.5 and 12 h post-dose, with slow vital capacity maneuvers (for the collection of inspiratory capacity [IC] data) performed at pre-dose, prior to the FVC maneuvers. Adverse events (AEs) and serious adverse events (SAEs) were monitored throughout the study, and vital signs (systolic and diastolic blood pressure) and electrocardiogram (ECG) data were collected pre-dose on Day 1 and post-dose on Day 28.
All patients received an inhaled short-acting β2-agonist (SABA)/short-acting muscarinic antagonist (SAMA) fixed combination (ipratropium bromide/fenoterol hydrobromide 20/50 μg pMDI) as rescue medication for “on-demand” use over the entire study period, with a minimum of 8 h between the use of rescue medication and the spirometric measurements. The following medication was prohibited from the indicated time prior to the screening visit and for the duration of the study: SABAs (6 h), SAMAs (8 h), SABA/SAMA combinations (8 h), LABAs (12 h; 72 h for ultra-LABAs), LAMAs (72 h), leukotriene modifiers (7 days), oral xanthine derivatives (7 days), phosphodiesterase-4 inhibitors (4 weeks), and depot corticosteroids (2 months).
All patients provided written informed consent prior to any study-related procedure. The study was approved by the independent ethics committees at each institution (Supplementary materials) and was performed according to the principles of the Declaration of Helsinki and the International Conference on Harmonization notes for guidance on Good Clinical Practice (ICH/CPMP/135/95). The study is registered in ClinicalTrials.gov (NCT02680197). There were no substantial protocol amendments.
Participants
The key inclusion criteria were as follows: male or female patients aged ≥40 years, with a diagnosis of COPD, post-bronchodilator forced expiratory volume in 1 s (FEV1)≥40% and ≤70% of the predicted normal value (measured 30–45 min after the administration of 80 μg ipratropium via pMDI) and a post-bronchodilator FEV1/FVC<0.7. All eligible patients were to have a change in FEV1 from pre- to post-bronchodilator value of at least 5% at the screening visit and were to be receiving a LABA and/or a LAMA (with or without an ICS) for at least 4 weeks prior to screening. All patients receiving ICSs were switched to the equivalent daily dose of fluticasone propionate DPI after they provided informed consent. The ICS regimen was then maintained for the entire run-in period and for the remainder of the study. Current and ex-smokers were eligible.
The main exclusion criteria were as follows: diagnosis of asthma or other respiratory disorders (other than COPD) which, in the investigator’s opinion, may have interfered with data interpretation; a COPD exacerbation or a lower respiratory tract infection within 8 weeks prior to screening; and a history of at least two exacerbations within the 12 months prior to screening. Full inclusion and exclusion criteria are listed in the “Supplementary materials” section.
Interventions
Treatments administered were GB 6.25, 12.5, 25 and 50 μg BID (total daily doses of 12.5, 25, 50 and 100 μg) or matched placebo, all via DPI. In each treatment period, patients received one of the five treatments, such that over the three periods each patient received three different treatments. Patients were assigned to treatment sequences through an interactive response technology system using an incomplete balanced block randomization scheme prepared by the contract research organization statistician. Patients, investigators and their staff, monitors and the sponsor’s clinical team were all blinded to treatment.
Outcomes
The objective of this study was to identify the optimal dose of GB DPI to be further developed for the treatment of patients with COPD. The primary efficacy variable was FEV1 area under the curve from 0 to 12 h (AUC0–12 h) normalized by time on Day 28. Secondary efficacy variables included the following: FEV1 AUC0–12 h on Day 1; trough and peak FEV1 and FVC on Days 1 and 28 (trough values were assessed from the mean of the measurements at 11.5 and 12 h post morning dose; peak values used data collected up to 4 h post-dose); morning pre-dose FEV1 on Day 28 (calculated from the mean of measurements at 45 and 10 min pre-dose); and change from baseline in pre-dose (morning) IC on Day 28. An additional post hoc analysis was performed on the percentage of patients with an increase from baseline in morning pre-dose FEV1 of at least 100 mL (ie, responders) on Day 28. Safety and tolerability of the study treatments were assessed in terms of treatment-emergent AEs and SAEs, vital signs (systolic and diastolic blood pressure) and ECG data.
Sample size and statistical analyses
For the primary end point, a total of 140 evaluable patients (14 per sequence) ensured a power of 94% to detect a mean difference of 120 mL (which was prespecified as the minimal important difference) between each dose of GB and placebo at a two-sided significance level of 0.0125 (Bonferroni adjustment), assuming a within-patient SD of 160 mL. Estimating a non-evaluable rate of 20%, it was planned to randomize approximately 180 patients. Since there were four primary comparisons (each GB dose vs placebo), the single-step Dunnett procedure was used to control the family-wise type I error rate at 0.05 (two-sided). This ensured the required power for each test, since the single-step Dunnett procedure is uniformly more powerful than the Bonferroni procedure.
The primary end point was analyzed using an analysis of covariance (ANCOVA) model including treatment, period and patient as fixed effects and baseline FEV1 as a covariate. The adjusted mean differences between each GB dose and placebo were calculated with Dunnett’s simultaneous 95% CIs and p-values. At each dose level, superiority of GB over placebo was demonstrated if the lower limit of the simultaneous CI for the mean difference was higher than 0. The secondary efficacy end points were analyzed using a similar ANCOVA model to the primary end point, with no adjustment for multiplicity. For the FEV1 responder analysis, the odds ratio was calculated using a conditional logistic regression model with treatment and period as fixed effects, baseline FEV1 as covariate and patient as stratum.
The primary end point was analyzed in both the intention-to-treat (ITT; primary analysis) and per-protocol (PP) populations. Secondary and post hoc efficacy variables were analyzed in the ITT population only. The ITT population was defined as all randomized patients who received at least one dose of study medication and with available efficacy data (primary or secondary efficacy variables) in at least two treatment periods. The PP population was defined as all patients from the ITT population without any major protocol deviations (ie, wrong inclusions, poor compliance and non-permitted medications). The safety population comprised all randomized patients who received at least one dose of study medication.
Results
Participants
The study was conducted between February 2016 and February 2017. Overall, 262 patients were screened, with 202 randomized in 21 study centers across four countries (Czech Republic, Germany, Hungary, Romania; Table 1). A total of 178 patients (88.1%) completed all the three treatment periods; 14 discontinued due to AEs, eight withdrew consent, one was lost to follow up and one discontinued due to other reasons.
 | Table 1 Baseline demographics and disease characteristics (safety population) |
Outcomes
Primary end point
In the analysis of data from the ITT population, all GB doses were statistically superior to placebo, with the three highest doses exceeding the prespecified 120 mL margin (Figure 1). There was a clear dose–response relationship for this end point, with GB 50 μg BID superior to the other GB doses. The results for the PP analysis were consistent with the ITT population, with a clear dose–response relationship, and all doses being statistically superior to placebo.
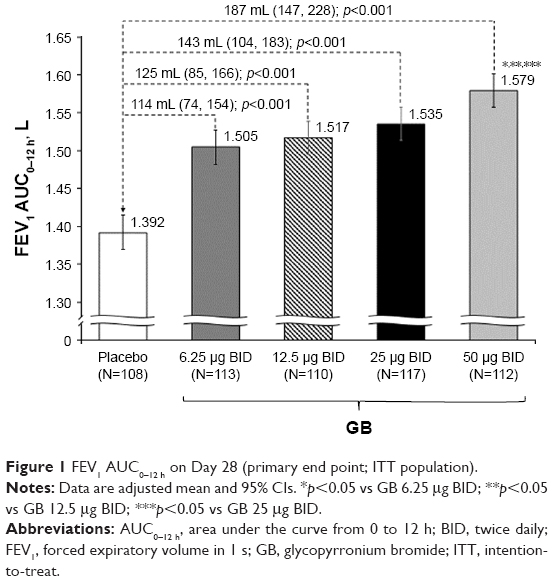 | Figure 1 FEV1 AUC0–12 h on Day 28 (primary end point; ITT population). |
Secondary end points
In the analysis of FEV1 AUC0–12 h on Day 1, although all the four GB doses were statistically superior to placebo, only the 25 and 50 μg BID doses exceeded the prespecified 120 mL margin, with both doses being superior to the two lower GB doses (Figure 2). Similarly, for trough FEV1 on Days 1 and 28, all the four GB doses were statistically superior to placebo, with a clear dose–response relationship (Figure 3A and B), and GB 25 and 50 μg BID statistically superior to the two lower doses on Day 1 (Figure 3A) and to GB 6.25 μg BID on Day 28 (Figure 3B). Trough FVC results were consistent with trough FEV1, with all GB doses statistically superior to placebo on both days (p≤0.001), and a clear dose–response relationship on Day 28, with GB 25 and 50 μg BID providing the greatest efficacy on Day 1. For peak FEV1 on Days 1 and 28, all GB doses were superior to placebo (Figure 4A and B). GB 25 and 50 μg BID were superior to the two lower doses on Day 1 (Figure 4A), with GB 50 μg BID superior to the other GB doses on Day 28 (Figure 4B). Peak FVC results were consistent with peak FEV1, with all GB doses superior to placebo on both Day 1 and Day 28 (p<0.001), and a clear dose–response relationship. For this end point, GB 25 and 50 μg BID were superior to the 6.25 and 12.5 μg doses on Day 1 (p<0.01), with GB 50 μg BID superior to all the three lower doses on Day 28 (p<0.05).
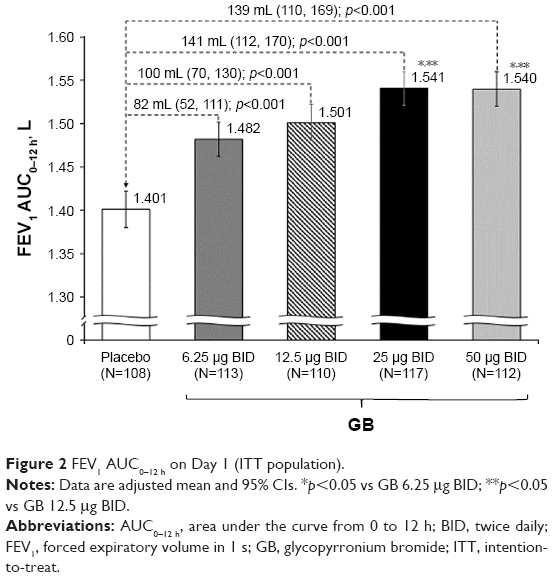 | Figure 2 FEV1 AUC0–12 h on Day 1 (ITT population). |
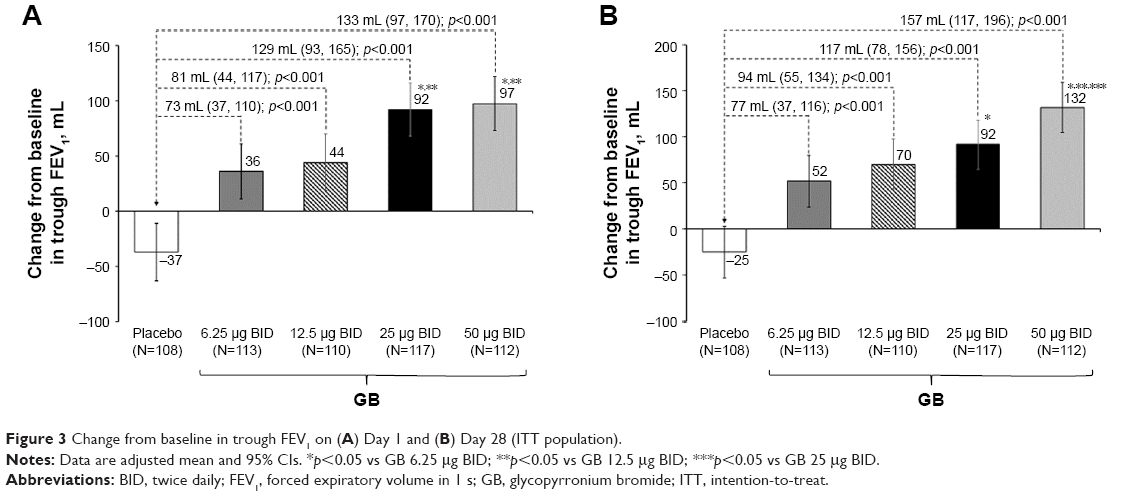 | Figure 3 Change from baseline in trough FEV1 on (A) Day 1 and (B) Day 28 (ITT population). |
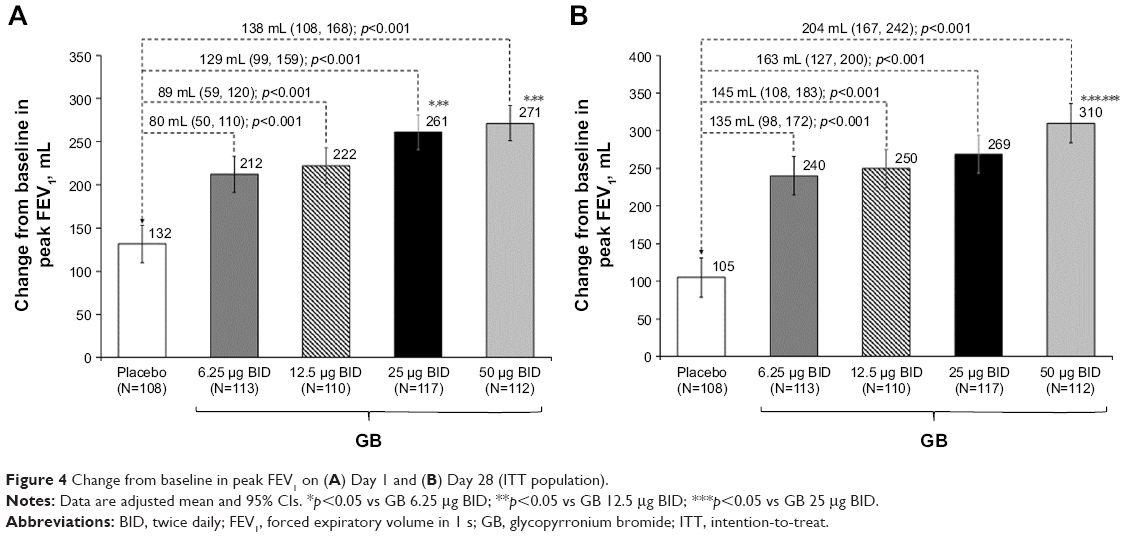 | Figure 4 Change from baseline in peak FEV1 on (A) Day 1 and (B) Day 28 (ITT population). |
In the evaluation of morning pre-dose FEV1 on Day 28, all GB doses were superior to placebo, with GB 50 μg BID superior to the other doses (Figure 5). Patients were significantly more likely to be FEV1 responders (ie, change from baseline in morning pre-dose FEV1 ≥100 mL) when receiving any of the GB doses than placebo (p<0.001), with the percentage of responders increasing with increasing GB dose (Table S1). In the analysis of pre-dose (morning) IC on Day 28, all GB doses were statistically superior to placebo (Figure S1). The dose–response relationship was less clear for this end point, with the 50 μg BID dose being superior to the 12.5 μg BID dose.
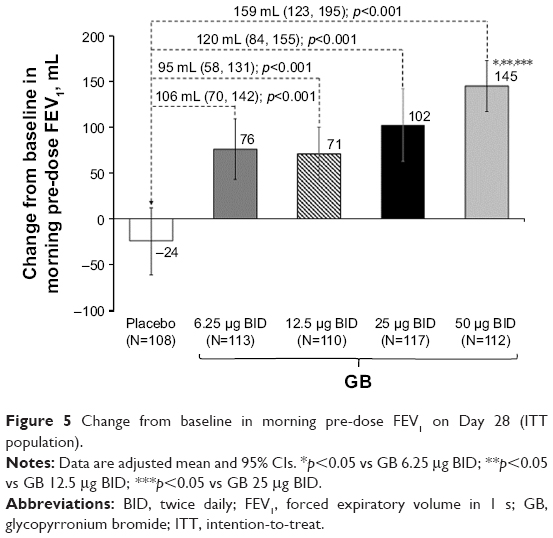 | Figure 5 Change from baseline in morning pre-dose FEV1 on Day 28 (ITT population). |
Safety
Overall, a similar proportion of patients experienced AEs with all five treatments (Table 2). There were few AEs considered related to treatment, and only one was considered severe, an episode of oropharyngeal candidiasis in a patient receiving GB 25 μg BID. There were no treatment-related SAEs and no AEs leading to death. There were no clinically relevant changes from baseline in mean systolic or diastolic blood pressure or heart rate and no evidence of a GB dose effect. No cardiac disorders were reported during treatment with GB.
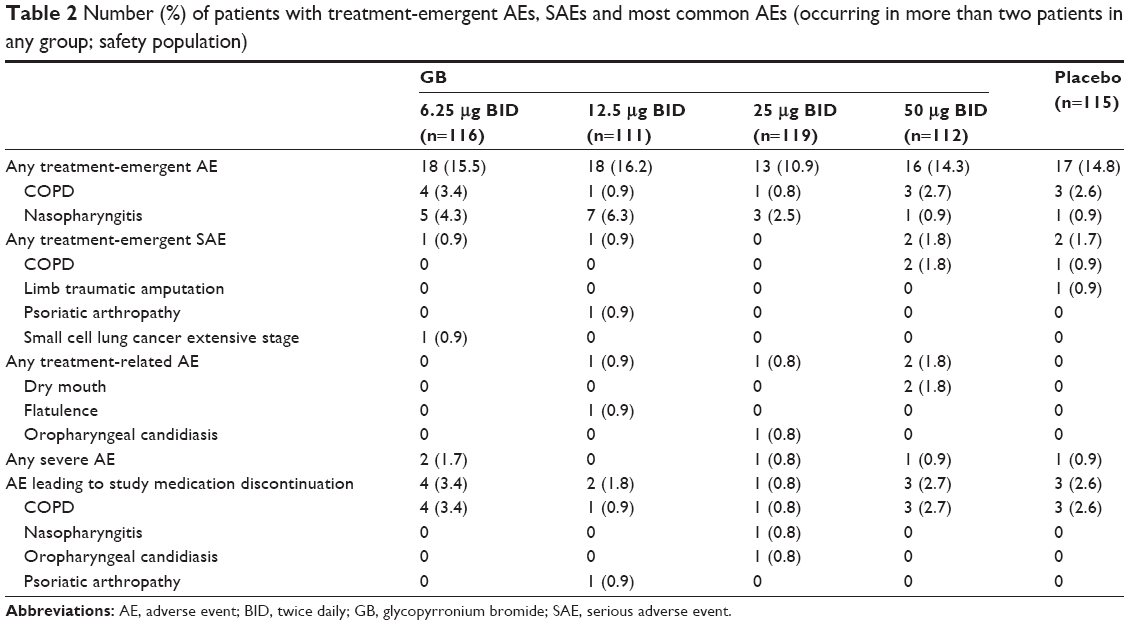 | Table 2 Number (%) of patients with treatment-emergent AEs, SAEs and most common AEs (occurring in more than two patients in any group; safety population) |
Discussion
In the efficacy analyses presented in this article, all the four GB doses were consistently and statistically superior to placebo, with clear dose–response relationships for most of the end points. The GB dose that consistently met criteria for the minimally acceptable dose was 25 μg BID. This was the lowest dose to exceed the prespecified 120 mL difference from placebo for FEV1 AUC0–12 h on both Day 1 and Day 28, peak FEV1 on both days, morning pre-dose FEV1 on Day 28 and trough FEV1 on Day 1 (missing this threshold by only 3 mL for trough FEV1 on Day 28). Furthermore, this dose was superior to the two lower doses for trough and peak FEV1 on Day 1 and to the lowest GB dose for trough FEV1 on Day 28. In addition, in the post hoc FEV1 responder analysis, GB 25 μg BID was the lowest dose to which >50% of patients responded.
Dose–response to bronchodilators in COPD is typically assessed using FEV1 analyses. However, given one of the classic features of COPD is hyperinflation, it is also useful to consider the effects of a bronchodilator on measures such as IC. In this study, all the four GB doses provided statistically significant improvements versus placebo in pre-dose IC. The greatest improvement in IC was observed with the two higher GB doses, which provided differences from placebo consistent with those seen previously with tiotropium and aclidinium.7,8 These data therefore provide further support to the selection of 25 μg BID as the minimal effective dose of this formulation of GB. Of note, GB 25 μg BID is also the minimal effective dose for the extrafine pMDI formulation, in terms of both comparisons with placebo and as an add-on to BDP/FF.2,3 In particular, the FEV1 AUC0–12 h treatment–placebo difference for GB 25 μg BID was 143 mL at Day 28 for the DPI formulation in the current study compared with 142 mL at Day 7 for the pMDI formulation.3 In the previous study, GB 25 μg BID via pMDI was statistically superior to tiotropium 18 μg once daily (OD) for pre-dose FEV1 after 7 days of dosing.3 Furthermore, the total daily dose of 50 μg is the approved dose for a different GB formulation that is administered OD (although a study of that formulation showed little difference in efficacy when the same daily dose was administered OD or BID).9
The current study recruited a broad COPD population, with wide FEV1 inclusion criteria, and so these results are likely to be representative. Patients were required to have some degree of reversibility to an SAMA, an approach that is consistent with previous dose-ranging studies.3,10,11 This perhaps limits the ability to generalize the findings slightly, as does the requirement for all patients to be receiving inhaled long-acting bronchodilator therapy. However, the reversibility requirement was only 5%; given a number of studies have shown that at a population level average reversibility of patients with COPD is at least 10%, this is unlikely to substantially limit the applicability of the population recruited and therefore of the overall findings.12,13 Indeed, nearly two-thirds of the population recruited into UPLIFT (the 4-year trial of tiotropium efficacy and safety) had a 15% improvement in FEV1 following the administration of ipratropium.14
We acknowledge that the efficacy of the 50 μg BID dose was consistently numerically greater than that of the 25 μg BID dose, indicating that the range of doses selected did not go sufficiently high to reach the plateau of the dose–response curve. Indeed, in a previous study with the pMDI formulation, a single 100 μg dose provided numerically higher efficacy to a single 50 μg dose.3 This illustrates one of the challenges of conducting dose-ranging studies – what level of efficacy is sufficient (assuming no safety signal is observed). When powering the study, we selected a difference from placebo (120 mL) for the primary end point that was consistent with other previous studies of bronchodilators in COPD.3,11 This value is also within the 100–140 mL range proposed by an American Thoracic Society/European Respiratory Society task force as the minimal important difference for FEV1.15 Importantly, the GB dose selection was not influenced by the safety findings, with the observed AE, vital signs and ECG results providing reassurance over the safety of this GB formulation. There were few serious or severe AEs (with no relationship between incidence and GB dose), and the percentage of patients experiencing AEs with GB 25 μg BID was lower than with placebo, with no cardiovascular safety signal. Overall, the safety profile of this formulation was consistent with that of the GB component in the BDP/FF/GB fixed-dose combination pMDI formulation, which has demonstrated a good overall safety profile in studies of up to 52 weeks in duration.16,17 Given the need of a broader armamentarium for treating COPD patients with different phenotypes/endotypes, the availability of this GB DPI formulation might facilitate the development of new combinations with different classes of inhaled anti-inflammatory agents, such as with novel phosphodiesterase-4 inhibitors.
Conclusion
Overall, this study supports the selection of extrafine GB 25 μg BID as the minimal effective dose for patients with COPD when delivered via the NEXThaler.
Data sharing statement
The data from this study are available on request, following submission of a valid research protocol to the corresponding author.
Acknowledgments
The authors would like to thank the investigators and patients at the investigative sites for their support of this study. Writing support was provided by David Young of Young Medical Communications and Consulting Ltd. This support was funded by Chiesi Farmaceutici SpA. This study was funded by Chiesi Farmaceutici SpA. Employees of the sponsor were involved in study design, oversaw the conduct and analysis of the study and (as authors) took part in the decision to submit the manuscript.
Disclosure
KMB declares that no personal payments were received from any pharmaceutical entity in the past 5 years. KMB is a full-time employee of Insaf Respiratory Research Institute. The institution has received compensation for services on advisory boards or consulting for Ablynx, Almirall, AstraZeneca, Berlin Chemie, Boehringer, Chiesi, Cytos, Mundipharma, Novartis, Pohl Boskamp and Zentiva. The institution has received compensation for speaker activities in scientific meetings supported by Almirall, AstraZeneca, Berlin Chemie, Boehringer, Cytos, ERT, GSK, Novartis, Pfizer, Pohl Boskamp and Takeda. The institution has further received compensation for design and performance of clinical trials from Almirall, Altana/Nycomed, AstraZeneca, Boehringer, Cytos, GSK, Infinity, Medapharma, MSD, Mundipharma, Novartis, Parexel, Pearl Therapeutics, Pfizer, Revotar, Teva, Sterna, and Zentiva. AE, HP, DS and MAN are employees of Chiesi, the study sponsor. The authors report no other conflicts of interest in this work.
References
Chiesi Limited. Trimbow 87 Micrograms/5 Micrograms/9 Micrograms Pressurised Inhalation, Solution: Summary of Product Characteristics. Manchester, UK 2017. | ||
Singh D, Schröder-Babo W, Cohuet G, et al; TRIDENT Study Investigators. The bronchodilator effects of extrafine glycopyrronium added to combination treatment with beclometasone dipropionate plus formoterol in COPD: a randomised crossover study (the TRIDENT study). Respir Med. 2016;114:84–90. | ||
Singh D, Scuri M, Collarini S, et al. Bronchodilator efficacy of extrafine glycopyrronium bromide: the Glyco 2 study. Int J Chron Obstruct Pulmon Dis. 2017;12:2001–2014. | ||
Rootmensen GN, van Keimpema ARJ, Jansen HM, de Haan RJ. Predictors of incorrect inhalation technique in patients with asthma or COPD: a study using a validated videotaped scoring method. J Aerosol Med Pulm Drug Deliv. 2010;23(5):323–328. | ||
Virchow JC, Crompton GK, Dal Negro R, et al. Importance of inhaler devices in the management of airway disease. Respir Med. 2008;102(1):10–19. | ||
Farkas Á, Lewis D, Church T, et al. Experimental and computational study of the effect of breath-actuated mechanism built in the NEXThaler® dry powder inhaler. Int J Pharm. 2017;533(1):225–235. | ||
O’Donnell DEE, Flüge T, Gerken F, et al. Effects of tiotropium on lung hyperinflation, dyspnoea and exercise tolerance in COPD. Eur Respir J. 2004;23(6):832–840. | ||
Beeh KM, Watz H, Puente-Maestu L, et al. Aclidinium improves exercise endurance, dyspnea, lung hyperinflation, and physical activity in patients with COPD: a randomized, placebo-controlled, crossover trial. BMC Pulm Med. 2014;14(1):209. | ||
Arievich H, Overend T, Renard D, et al. A novel model-based approach for dose determination of glycopyrronium bromide in COPD. BMC Pulm Med. 2012;12(1):74. | ||
Leaker BR, Barnes PJ, Jones CR, Tutuncu A, Singh D. Efficacy and safety of nebulized glycopyrrolate for administration using a high efficiency nebulizer in patients with chronic obstructive pulmonary disease. Br J Clin Pharmacol. 2015;79(3):492–500. | ||
Singh D, Ravi A, Reid F, Buck H, O’Connor G, Down G. Bronchodilator effects, pharmacokinetics and safety of PSX1002-GB, a novel glycopyrronium bromide formulation, in COPD patients; a randomised crossover study. Pulm Pharmacol Ther. 2016;37:9–14. | ||
Agusti A, Calverley PMA, Celli B, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res. 2010;11:122. | ||
Vestbo J, Anderson JA, Brook RD, et al. Fluticasone furoate and vilanterol and survival in chronic obstructive pulmonary disease with heightened cardiovascular risk (SUMMIT): a double-blind randomised controlled trial. Lancet. 2016;387(10030):1817–1826. | ||
Tashkin DP, Celli B, Decramer M, et al. Bronchodilator responsiveness in patients with COPD. Eur Respir J. 2008;31(4):742–750. | ||
Cazzola M, MacNee W, Martinez FJ, et al; American Thoracic Society, European Respiratory Society Task Force on Outcomes of COPD. Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J. 2008;31(2):416–469. | ||
Vestbo J, Papi A, Corradi M, et al. Single inhaler extrafine triple therapy versus long-acting muscarinic antagonist therapy for chronic obstructive pulmonary disease (TRINITY): a double-blind, parallel group, randomised controlled trial. Lancet. 2017;389(10082):1919–1929. | ||
Singh D, Papi A, Corradi M, et al. Single inhaler triple therapy versus inhaled corticosteroid plus long-acting β2-agonist therapy for chronic obstructive pulmonary disease (TRILOGY): a double-blind, parallel group, randomised controlled trial. Lancet. 2016;388(10048):963–973. |
Supplementary materials
Methods
Independent ethics committees
- Czech Republic: Etická komise, Fakultní nemocnice Královské Vinohrady (FNKV), Praha.
- Germany: Ethics commission at the Hesse Regional Medical Council, Landesärztekammer Hessen, Frankfurt; Ethikkommissionen (EK I und EK II) bei der Ärztekammer Schleswig-Holstein, 23795 Bad Segeberg; Ethikkommission an der medizinischen Fakultät der Rheinischen Friedrich-Wilhelms Universität Bonn, Bonn; Landesärztekammer Brandenburg, Hauptgeschäftsstelle Ethikkommission, Cottbus; Ethik-Kommission der Albert-Ludwigs-Universität Freiburg, Freiburg.
- Hungary: Ethics Committee for Clinical Pharmacology (ECCP), Budapest.
- Romania: National Bioethics Committee for Medicines and Medical Devices, Antonescu, Bucharest.
Inclusion criteria
Patients with COPD airflow obstruction had to meet all of the following inclusion criteria to be eligible for enrollment into the study:
- Male or female patients aged ≥40 years at screening visit, with signed informed consent obtained prior to initiation of any study-related procedure.
- Patients with a diagnosis of COPD (according to the Global Initiative for Chronic Obstructive Lung Disease guidelines 2015) at least 12 months before screening.
- Current smoker or ex-smoker with a smoking history of at least 10 pack-years defined as: 1 pack-year = ([number of cigarettes smoked per day] × [number of years of smoking])/20; if applicable, smoking cessation therapy had to be completed 3 months prior to screening or smoking cessation had to be accomplished at least 3 months prior to screening. The smoking status was not permitted to change between screening and the last study visit.
- A post-bronchodilator forced expiratory volume in 1 s FEV1≥40% and ≤70% of the predicted normal value (measured 30–45 min after the administration of 80 μg ipratropium via pressurized metered-dose inhaler [pMDI]); a post-bronchodilator FEV1/forced vital capacity (FVC)<0.7; and a change in FEV1 from the pre-bronchodilator value (reversibility) of at least 5% at screening (if the criterion was not met at screening, the test could be rescheduled once before randomization).
- Patients receiving any of the following for at least 4 weeks prior to screening: long-acting muscarinic antagonist (LAMA); long-acting β2-agonist (LABA); LAMA + LABA; inhaled corticosteroid (ICS) + LABA; ICS + LAMA. Patients with an FEV1<50% of the predicted normal value and a history of one exacerbation within the past 12 months had to be treated with ICS + LABA or ICS + LAMA before screening.
- Ability and cooperative attitude to understand and perform required outcome measurements of the protocol (eg, spirometry maneuvers) and ability to understand the risks involved. Ability to be trained to use the dry powder inhaler (DPI) inhalers.
All inclusion criteria were checked at screening. Criteria 3 and 6 were rechecked at randomization (visit [V]2).
Exclusion criteria
Patients with any of the following criteria were not enrolled:
- Diagnosis of asthma or other respiratory disorders (other than COPD) which may have interfered with data interpretation according to the investigator’s opinion.
- Patients who had a COPD exacerbation or a lower respiratory tract infection within 8 weeks prior to screening, or during the run-in period, that resulted in the use of an antibiotic, or oral or parenteral corticosteroids, or hospitalization.
- Patients with a history of ≥2 exacerbations within the last 12 months prior to screening (frequent exacerbators).
- Patients treated with oral/parenteral β2-agonists or nebulized bronchodilators or phosphodiesterase (PDE) inhibitors or who received LABA/LAMA/ICS treatment therapy in the 4 weeks prior to screening and during the run-in period.
- Patients on an ICS treatment that had been initiated, or with the effective dose that had been changed, within 4 weeks prior to screening or during the run-in period (patients on a stable dose of ICS for at least 4 weeks prior to screening were allowed).
- Patients requiring long-term (at least 12 h daily) oxygen therapy for chronic hypoxemia.
- Patients with known respiratory disorders other than COPD including but not limited to α-1 antitrypsin deficiency, active tuberculosis, lung cancer, bronchiectasis, sarcoidosis, lung fibrosis, pulmonary hypertension and interstitial lung disease.
- Patients with medical diagnosis of narrow-angle glaucoma, prostatic hypertrophy or bladder neck obstruction that, in the opinion of the investigator, would have prevented use of anticholinergic agents.
- Patients who had unstable concurrent disease: eg, uncontrolled hyperthyroidism, uncontrolled diabetes mellitus or other endocrine disease, significant hepatic impairment, significant renal impairment, history of cerebrovascular disease, uncontrolled gastrointestinal disease (eg, active peptic ulcer), neurological disease, uncontrolled hematological disease, uncontrolled autoimmune disorders or any other disease or other condition which might have placed the patient at undue risk or potentially compromised the results or interpretation of the study according to the investigator’s opinion.
- Patients who had concomitant disease of poor prognosis (eg, cancer).
- Patients who had clinically significant cardiovascular conditions, including unstable ischemic heart disease, New York Heart Association (NYHA) class III/IV left ventricular failure, acute ischemic heart disease within 1 year prior to screening, history of sustained and non-sustained cardiac arrhythmias diagnosed in the past 6 months (sustained meant lasting >30 s and/or ending only with external action, and/or leading to hemodynamic collapse; non-sustained meant >3 beats in <30 s, and/or ending spontaneously, and/or asymptomatic), clinically significant impulse conduction blocks.
- Patients with known atrial fibrillation (AF):
- Paroxysmal AF.
- Persistent: AF episode lasting >7 days or requiring termination by cardioversion, either with drugs or by direct current cardioversion (DCC) within 6 months prior to screening.
- Long standing persistent: continuous AF diagnosed for <6 months and/or without a rhythm control strategy.
- Permanent: AF diagnosed for at least 6 months with a resting ventricular rate ≥100/min not controlled with a rate control strategy (ie, selective β-blocker, calcium channel blocker, pacemaker placement, digoxin or ablation therapy).
- Patients who had clinically significant abnormal 12-lead electrocardiogram (ECG) that might have placed the patient at undue risk or potentially compromised the results or interpretation of the study according to the investigator’s opinion.
- Patients whose 12-lead ECG showed a QT interval corrected for heart rate (QTcF)>450 ms for males or QTcF>470 ms for females.
- Patients with clinically significant laboratory abnormalities indicating a significant or unstable concomitant disease that might have placed the patient at undue risk or potentially compromised the results or interpretation of the study according to the investigator’s opinion.
- Pregnant or lactating women and all women of childbearing potential, unless they were willing to use highly effective birth control methods, such as:
- Placement of an intrauterine device or intrauterine releasing system.
- Oral, intravaginal, transdermal combined estrogen and progestogen containing hormonal contraception or oral, injectable, implantable progestogen-only hormonal contraception.
- Bilateral tubal occlusion.
- Partner vasectomy (provided that the partner of the study participant was the sole sexual partner and that successful sterilization had been confirmed after surgery).
- Sexual abstinence defined as refraining from heterosexual intercourse during the entire period of risk associated with the study treatments.
For all women of childbearing potential, serum pregnancy tests were performed at V0, at V10 and at early termination (ET) visits. Urinary pregnancy tests were performed at V0 and on Day 1 of each treatment period (V2, V5 and V8); female patients of non-childbearing potential, defined as physiologically incapable of becoming pregnant (postmenopausal [no menses for 12 months without an alternative medical cause] or permanently sterilized by hysterectomy, bilateral salpingectomy or bilateral oophorectomy), were eligible. If indicated, as per investigator’s request, postmenopausal status may have been confirmed by follicle-stimulating hormone levels (according to local laboratory range) in women not using hormonal contraception or hormonal replacement therapy. - Known intolerance/hypersensitivity or any contraindication to treatment with M3-antagonists or any of the excipients contained in the formulations used in the study.
- Patients who had evidence of alcohol or drug abuse, not compliant with the study protocol or not compliant with the study treatments according to investigator’s judgment.
- Patients with major surgery within 3 months prior to screening or planned during the study, which may have affected patient’s compliance with study procedures.
- Patients who had participated in another clinical study with an investigational drug administered within 2 months prior to screening.
All exclusion criteria were checked at screening (V1). Criteria 2, 4, 5, 13 and 14 were rechecked at randomization (V2).
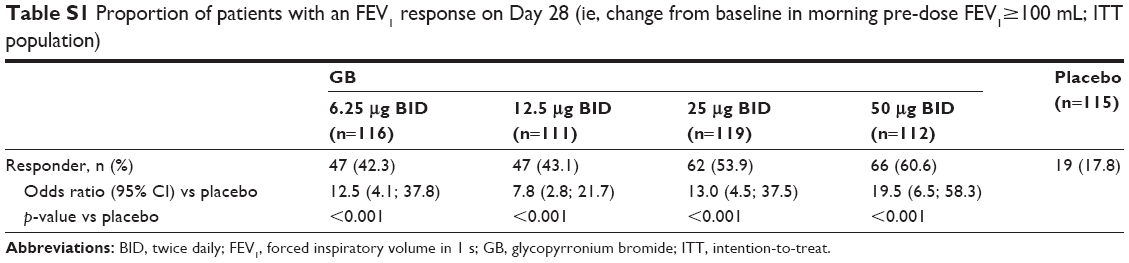 | Table S1 Proportion of patients with an FEV1 response on Day 28 (ie, change from baseline in morning pre-dose FEV1≥100 mL; ITT population) |
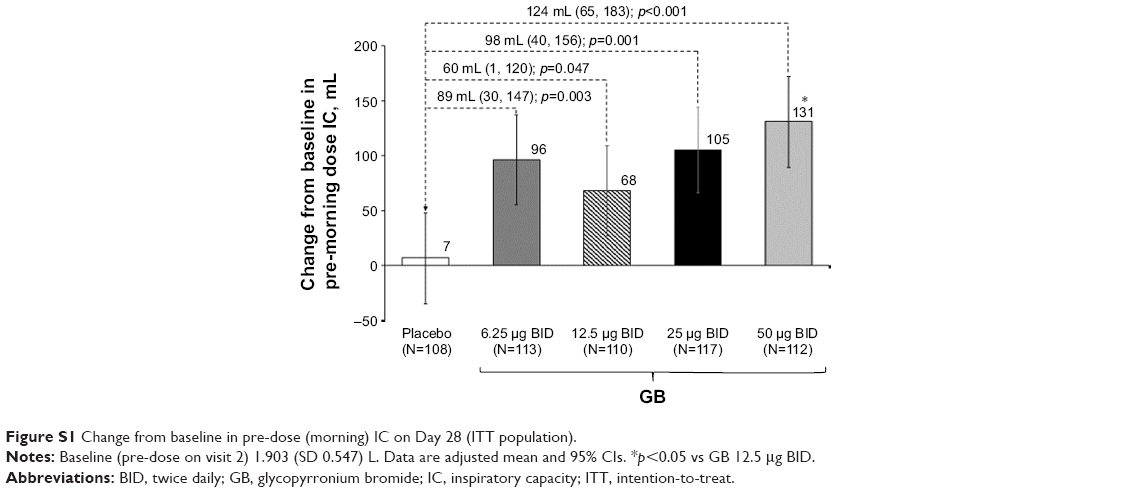 | Figure S1 Change from baseline in pre-dose (morning) IC on Day 28 (ITT population). |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.