")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Dose comparison of conivaptan (Vaprisol®) in patients with euvolemic or hypervolemic hyponatremia – efficacy, safety, and pharmacokinetics
Authors Palmer B, Rock A, Woodward E
Received 28 August 2015
Accepted for publication 1 December 2015
Published 18 January 2016 Volume 2016:10 Pages 339—351
DOI https://doi.org/10.2147/DDDT.S95326
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Biff F Palmer,1 Amy D Rock,2 Emily J Woodward2
1Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, 2Department of Research and Development, Cumberland Pharmaceuticals Inc., Nashville, TN, USA
Purpose: This study aimed to evaluate the efficacy, safety, and pharmacokinetics of 20 and 40 mg/day conivaptan (Vaprisol®) in patients with hypervolemic or euvolemic hyponatremia.
Methods: Hyponatremic patients – serum sodium (sNa) ≤130 mEq/L – received either 20 or 40 mg/day of conivaptan for 4 days, following an initial 20 mg loading dose. Efficacy was evaluated by the magnitude and extent of change in sNa. Safety was evaluated by the incidence of adverse events, changes in vital signs and laboratory parameters, rate of sNa correction, and frequency of infusion-site reactions. Pharmacokinetic parameters were also measured.
Results: A total of 37 patients received 20 mg/day and 214 patients received 40 mg/day conivaptan. Baseline-adjusted sNa-area under the concentration–time curve increased by an average of 753.8±499.9 mEq·hr/L (20 mg/day) and 689.2±417.3 mEq·hr/L (40 mg/day) over the course of the 4-day treatment period. The majority of patients in both treatment groups achieved a 4 mEq/L increase in sNa over baseline in ~24 hours (82.5%). Average increase in sNa after 4 days was ~10 mEq/L, varying with dosage level and baseline volume status. Treatment success (normal sNa or increase of ≥6 mEq/L) was attained by 70.3% of patients in the 20 mg/day group and 72.0% in the 40 mg/day group.
Conclusion: Both 20 and 40 mg/day doses of conivaptan are efficacious in increasing sNa over 4 days of treatment with no observed increase in the frequency of adverse events or specific infusion-site reactions using the higher dose. The pharmacokinetic parameters of both doses were similar to what has been reported previously, exhibiting greater-than-dose-proportional plasma concentrations.
Keywords: critical care, electrolyte, SIADH, V2 receptor antagonist, thiazide, osmotic demyelination syndrome
Introduction
Hyponatremia generally is defined as the condition where serum sodium (sNa) concentration is below the lower limit of normal, ie, <135 mEq/L. When thus defined, the incidence of hyponatremia may be as high as 30% in hospitalized patients.1 However, even levels below 138 mEq/L have been significantly associated with an increased risk of mortality,2 and in general, risk of mortality increases with decreasing levels of sNa.3,4 The presence of asymptomatic hyponatremia has been associated with an increased risk of perioperative complications and gait instability (leading to falls and fractures) as well as a negative economic impact due to an increase in the rate of admission to the intensive care unit, prolonged hospital stays, and increased hospital costs.5
The choice of treatment for hyponatremia should be based on the underlying causative etiology and volume status of the patient. Historically, treatment options such as fluid restriction, infusions of isotonic or hypertonic saline, loop diuretics, and the use of demeclocycline or urea have been associated with varying levels of success. Vaptans are a class of drugs that have more recently become available for treating euvolemic and hypervolemic hyponatremia. Vaptans (eg, conivaptan and tolvaptan) represent a targeted approach to the treatment of hyponatremia by inhibiting the interaction of arginine vasopressin (AVP) with the V2 receptor. Mechanistically, AVP binds to V2 receptors present on renal collecting ducts causing translocation of aquaporin-2 channels to the cell’s luminal surface, ultimately resulting in water retention and hyponatremia. Elevation of endogenous AVP (eg, SIADH – syndrome of inappropriate antidiuretic hormone secretion) or administration of exogenous vasopressin receptor agonists is responsible for the majority of cases of hyponatremia,6 and endogenous AVP production can be stimulated by many pathophysiological pathways. Thus, vaptans are a logical treatment option for most cases of hyponatremia – the exceptions include pseudohyponatremia (elevated plasma lipids or proteins), hypertonic hyponatremia (elevated effective plasma solutes), and hypovolemic hypotonic hyponatremia.5 There are two approved vaptans in the USA – conivaptan (Vaprisol®; Cumberland Pharmaceuticals Inc, Nashville, TN, USA) and tolvaptan (Samsca; Otsuka Pharmaceutical Co, Ltd, Tokyo, Japan).7,8 Conivaptan is supplied as a solution for intravenous (IV) infusion whereas tolvaptan is supplied as an oral tablet. Both compounds antagonize V1A and V2 receptors, although conivaptan is essentially nonselective in contrast to tolvaptan which has a much higher affinity for the V2 receptor.9,10 Conivaptan is used in the hospital setting as an IV preparation. Although tolvaptan is an oral formulation and commonly used in an outpatient setting, it must be initiated or reinitiated in the hospital under the supervision of a physician to determine the appropriate dosage and monitor for adverse effects.
Treatments options such as fluid restriction, saline, diuretics, and vaptans are associated with caveats and potential risks. A recent registry study of 3,087 hyponatremic patients demonstrated that fluid restriction was the most commonly used treatment approach (35%) and resulted in a median increase of 2 mEq/L during the first 24 hours of therapy.11 Fluid restriction is hampered by low patient compliance and, as suggested by the registry study, is the least effective treatment option available. Isotonic saline was used in 15% of the cases and produced a median 3 mEq/L increase from baseline. Isotonic or normal saline is typically used for volume repletion in hypovolemic individuals and may not be appropriate for individuals who are hypervolemic. Hypertonic saline and tolvaptan were used in a minority of cases (2% and 5%, respectively) but were associated with more robust increases in the levels of sNa (5 and 4 mEq/L, respectively). The incidence of conivaptan use was not reported.
The risk of overly rapid sNa correction leading to osmotic demyelination syndrome (ODS) is a concern with any hyponatremia therapy. In acute hyponatremia, the risk of ODS increases with the presence of very low levels of sNa (<120 mEq/L), due to the osmotic compensatory mechanisms present in the brain. In chronic hyponatremia, the risk of ODS is increased when hypokalemia, alcoholism, malnutrition, or advanced liver disease are present.5 In the earlier-referenced registry study, overly rapid correction (ie, ≥12 mEq/L in any 24-hour period) was noted in 7.9% of patients regardless of the therapy followed.11 Interestingly, even patients who did not receive any therapy for hyponatremia experienced overly rapid correction at a rate of 1.4%. The relative risk (RR) of overly rapid correction was highest when hypertonic saline was used (RR 12.01, observed in 17.1% of patients who received hypertonic saline), a result which highlights the common perception that using hypertonic saline to treat hyponatremia is problematic and not easily standardized.12–14 In addition, 10.8% of patients receiving tolvaptan experienced overly rapid correction. Though overly rapid correction was observed, not a single case of ODS was reported in the registry. One of the aims of the present study was to monitor the relative rate of correction in sNa in patients receiving either 20 or 40 mg doses of conivaptan and to assess the safety and treatment success of both doses.
Conivaptan (Vaprisol®), which was approved by FDA in 2005, was the first marketed drug in this class. It is currently available as a premixed IV infusion that can be administered peripherally or centrally. The FDA-approved therapeutic regimen involves initiation of treatment with a 20 mg bolus loading dose given over 30 minutes followed by a continuous infusion of 20 mg/day for 4 days until the target level of sNa is achieved.7 A modified regimen consisting of twice daily, 30-minute infusions has been used in clinical trials and was found to be equally efficacious to the continuous infusion regimen.15 Based on the effect of the drug on sNa levels, the dose may be increased to 40 mg/day after the first 24 hours of continuous infusion. To increase the dose, two 20 mg/100 mL doses are given consecutively during the same 24-hour period. Since conivaptan infusion can be associated with vascular irritation, an additional aim of this study was to assess the frequency and types of infusion-site reactions (ISRs) experienced by patients receiving 20 or 40 mg/day doses of conivaptan as a continuous infusion.
Methods
Study design
This study was reviewed and approved by the Institutional Review Board or Independent Ethics Committee of record at each participating clinical center and was conducted in accordance with the ethical principles which have as their foundation the Declaration of Helsinki including the guidelines for Good Clinical Practice. Written informed consent was obtained from all the patients or their legally authorized representative prior to their enrollment in the study, and a Health Insurance Portability and Accountability Act authorization was also obtained from all patients enrolled in the USA.
This open-label, multicenter study evaluated conivaptan doses of either 20 or 40 mg/day administered as a continuous IV infusion for 4 days following an initial 20 mg loading dose given as an IV bolus. The protocol mandated that the site of infusion be a large vein in the arm and be rotated every 24 hours. Peripheral catheters and infusion pumps were used, and particular attention was given to the emergence of ISRs as part of the evaluation of safety.
During the screening period, baseline assessments were performed, which included medical and medication history, demographics, physical examination, vital signs, volume status, urinalysis, serum clinical chemistry, and hematology parameters. Baseline sNa was confirmed by two independent measurements taken at least 4 hours apart. In addition to continuous IV conivaptan infusion, subjects underwent the following measurements during the 4-day treatment period: sNa level was determined at regular intervals through day 4, serum clinical chemistry on days 2 and 4, hematology and urinalysis on day 4, vital signs and volume status on days 2 through 4, physical examination and a review for adverse events (AEs) once per day, and an electrocardiogram (ECG) was conducted on days 2 through 4. Sodium, calorie, and fluid intake remained consistent with what each patient received during the screening period. Subjects were evaluated twice during the posttreatment period on days 11 and 34. On day 11, subjects underwent a physical exam, vital signs and volume status were recorded, and a sample for sNa was taken. On day 34, subjects underwent a physical examination, vital signs and volume status were recorded, and samples for serum chemistry and hematology were collected. Pharmacokinetic (PK) assessments conducted during the study are outlined in the following section.
Patients
This study was conducted at 28 centers in the USA, Israel, and South Africa. Eligible patients (N=250, planned for enrollment) were either male or female and at least 18 years of age with a diagnosis of hyponatremia. Hyponatremia was defined as two consecutive sNa measurements no greater than 130 mEq/L taken at least 4 hours, but no more than 24 hours, apart, and no earlier than 24 hours prior to the first dose of study drug. History of hyponatremia was characterized for each patient and included five variables: the amount of time since the earliest known occurrence of hyponatremia (months), the amount of time since the current episode began (days), the subject’s current volume status, the primary underlying cause of hyponatremia, and the presence of congestive heart failure (CHF). The patient’s volume status had to be either euvolemic (absence of pitting edema or ascites) or hypervolemic (edematous), and hypovolemic (volume depleted or dehydrated) patients were excluded. The other exclusion criteria included blood glucose levels above 275 mg/dL; being a nursing or pregnant female – as confirmed by a pregnancy test during screening; uncontrolled hyper- or hypotension; uncontrolled arrhythmias; untreated thyroid or adrenal disease; elevated alanine aminotransferase (ALT) or aspartate aminotransferase (AST, elevated is defined as >5× upper limit of normal) or depressed albumin (defined as <1.5 g/dL); evidence of renal insufficiency in the form of estimated glomerular filtration rate <20 mL/min2 or known urinary outflow obstruction; and a white blood cell (WBC) count <3,000 cells/μL.
Test agent
Conivaptan hydrochloride injection (Vaprisol®; Cumberland Pharmaceuticals Inc., Nashville, TN, USA) was manufactured by Yamanouchi Pharmaceutical Company, Ltd (Tokyo, Japan). The initial loading dose of 20 mg was diluted in 100 mL of 5% dextrose in water (D5W) and given over a period of 30 minutes. All subjects received the same loading dose. For continuous infusions, 20 or 40 mg of the drug was diluted in 250 mL of D5W and infused over 24 hours. Treatment allocation was chronological, that is, all patients enrolled under the original protocol or amendment one received a dosage of 40 mg/day, and those enrolled under amendment two received 20 mg/day.
Efficacy assessments
The primary efficacy measure in this study was baseline-adjusted change in the area under the concentration–time curve (AUC) for sNa over the 4-day treatment period. Additional parameters that were measured to determine the efficacy of the drug included: the time from the first dose until a ≥4 mEq/L increase in sNa relative to baseline was observed, the total time during the treatment period a patient had sNa levels ≥4 mEq/L higher than baseline, the overall change in sNa at the end of the treatment period relative to baseline, and the number of patients who had either a ≥6 mEq/L increase in sNa from baseline or a normal sNa level (ie, ≥135 mEq/L) at any time during the study.
Safety assessments
Safety was evaluated by changes in physical exam; vital signs; body weight; volume status; clinical laboratory parameters (sNa, blood urea nitrogen [BUN], serum creatinine [sCr], ALT, AST, alkaline phosphatase, bilirubin, WBC count, neutrophil count, and coagulation tests); urinalysis results; 12-lead ECG; the emergence of ISRs; and relative frequencies of AEs as reported by patients receiving 20 mg/day vs those receiving 40 mg/day.
ISRs observed during the treatment period were of particular importance; the infusion site was changed every 24 hours, and a specially designed data collection tool was used to uniformly assess ISRs. Data regarding the type and severity of ISR, the outcome, whether a dose was interrupted due to ISR, and the time of onset and duration of the ISR were collected.
Patients were observed and queried for AEs at the end of each treatment day as well as on days 11 and 34. Treatment-emergent AEs (TEAEs) were defined as any events that occurred after the first dose of study drug, and posttreatment AEs (PTAEs) were defined as those that occurred at any time after discontinuation of study drug through day 34. Serious AEs (SAEs) were recorded through the 15 days after the posttreatment period (ie, day 34). SAEs are those that result in death, hospitalization, disability, or birth defect; are considered life-threatening; or are likely to result in one of the above outcomes without medical intervention. Clinically significant laboratory abnormalities were verified by repeat testing and reported as AEs. Special attention was given to blood dyscrasias (eg, leukopenia, WBC <1,500/μL, or neutrophils <500/μL) and severe volume loss resulting in declining renal function (eg, BUN increased by >40 mg/mL or SCr increased by >1.5 mg/dL).
Pharmacokinetics
Blood samples were obtained from all patients for the evaluation of PK parameters by means of venipuncture or cannula from the arm opposite that used for treatment at the following time points of the study: day 1, hour 0 (pre-dose) and hour 0.5 (end of loading dose); day 3, hour 24; day 4, hour 24; day 11 ±1 day. Twenty-seven subjects from two clinical sites provided PK samples at additional time points: day 1, hours 1, 4, and 24; day 2, hour 24; and day 4, hours 1, 2, 7, 12, and 24. These patients constituted the “PK-rich” analysis set. For all subjects, if an infusion was prematurely terminated, a sample for the evaluation of PK parameters was taken at that time.
Over the course of the study, 1,433 plasma concentration measurements were made using a validated liquid chromatography-mass spectrometry method in compliance with Good Laboratory Practices. Concentrations below the lower limit of quantitation were set to zero for the purposes of descriptive statistics but were excluded from the determination of terminal half-life. The range of linearity for the assay was 0.50–2,000.0 ng/mL; the average correlation coefficient was 0.9992; precision ranged from 3.5% to 5.4%; and accuracy ranged from −1.8% to 5.1%. Noncompartmental methods were used to calculate PK parameters using WinNonlin version 4.0.1 or later (Pharsight, Princeton, NJ, USA).
Statistical analyses
Efficacy, safety, and PK data were presented descriptively and sample size was based on clinical judgment and previous findings. The analysis population included all the enrolled subjects who received the medication under study, unless noted otherwise in the “Results” section. Data were summarized by treatment group and volume status. Continuous variables were summarized descriptively using number (N), mean, SD, median, minimum (min), and maximum (max). Categorical data were displayed as percentages or frequencies. The final visit assessment for any parameter was defined as the last measurement taken during the treatment period. Medications (historical, concomitant, or restricted) were summarized according to the Anatomical Therapeutic Chemical (ATC) drug class and the World Health Organization Drug Dictionary (WHO-DD) reference. AEs were classified based on the Medical Dictionary for Regulatory Activities (MedDRA) version 6.0. SAS version 6.12 (SAS Institute Inc, Cary, NC, USA) was used for all analyses.
The primary efficacy parameter was change from baseline in sNa over the duration of the 4-day treatment period as measured by AUC and corrected for baseline sNa. Baseline sNa was defined as the average of all the three pretreatment measures (two values taken at the time of screening plus one taken at hour 0, pre-dose). Patients without all three values were excluded from the primary efficacy analysis. AUC was computed from the ten available sNa measures using linear interpolation for missing internal values and the integral average method for missing terminal values.
Results
Disposition, demographics, and baseline characteristics
A total of 251 patients were enrolled in the study. All patients received the initial 20 mg loading dose followed by either 20 mg/day (N=37, 14.7%) or 40 mg/day (N=214, 85.3%) conivaptan (Figure 1). The majority of patients completed the study – including follow-up visits and receiving all 4 days of treatment (80.9%). Reasons for discontinuation in both groups were similarly represented and included: withdrawal of consent, AEs, administrative (lack of venous access, improved sNa requiring no further treatment, prohibited concomitant medication), and death. Enrollees were predominantly female (65.7%), white and non-Hispanic (88.8%) with an average age of 72.2±19.9 years. The treatment groups did not differ appreciably in terms of any demographic parameter (Table 1).
 | Figure 1 Disposition of patients. |
 | Table 1 Demographic and baseline characteristics of patients per treatment group |
Other baseline characteristics were also comparable between the treatment groups. The majority of patients were euvolemic (72.5%) and did not have a history of congestive heart failure (non-CHF, 71.7%). There were slightly more hypervolemic patients in the 20 mg/day group (37.8%) than in the 40 mg/day group (25.7%). The predominant cause of hyponatremia in both the groups was SIADH (43.4%), and the overall distribution of etiologies was similar between the treatment groups, with an exception being the “unknown” category which was almost twice as frequently listed as the cause of hyponatremia for members of the 40 mg/day group as compared to the 20 mg/day group. In the 20 mg/day group, four of the seven listed as “other” were due to diuretic treatment, and in the 40 mg/day group, 31 of the 52 “other” cases included diuretic use either alone or with another etiology. The duration of the current episode of hyponatremia was similar between treatment groups.
Drug exposure and treatment compliance
The mean total loading dose was 19.1±3.34 mg for the 20 mg group (N=37) and was similar for the 40 mg group (19.7±2.07 mg, N=214). The mean total infused dose over the treatment period was 68.2±20.02 mg for the 20 mg/day group and 133.2±43.16 mg for the 40 mg/day group. Overall compliance and mean total infusion time (average =83 hours, for all treated patients) were similar between the treatment groups.
Efficacy
Baseline-adjusted sNa-AUC during treatment
All treated patients were included in the primary efficacy analysis. The mean baseline sNa-AUC was similar between treatment groups, both overall and when segregated by volume status (Table 2). Numerical differences were seen in the mean treatment period baseline-adjusted sNa-AUC between patients receiving 20 and 40 mg/day conivaptan. The directionality of these differences changed relative to segregation by volume status (hypervolemic: 40 mg AUC >20 mg AUC vs euvolemic: 20 mg AUC > 40 mg AUC).
 | Table 2 Baseline sNa-AUC and baseline-adjusted sNa-AUC during the treatment period by treatment group and volume status |
Time to first confirmed ≥4 mEq/L increase in sNa relative to baseline
The majority of patients (82.5%) obtained at least a 4 mEq/L increase in sNa by ~24 hours after initiation of the study drug (Table 3). Hypervolemic patients receiving 20 mg/day conivaptan took a longer amount of time to reach a 4 mEq/L increase in sNa (median =58.5 hours), whereas euvolemic patients receiving 20 mg/day conivaptan demonstrated the shortest time to increase sNa levels (median =12.0 hours).
 | Table 3 Time (hours) to first confirmed ≥4 mEq/L increase in sNa relative to baseline |
Cumulative time sNa was increased by at least 4 mEq/L relative to baseline
The total amount of time that patients demonstrated at least a 4 mEq/L increase in sNa was, on average, 60 hours (Table 4). Hypervolemic patients receiving 20 mg/day conivaptan had the shortest duration of effect (42.9 hours), whereas euvolemic patients receiving 20 mg/day demonstrated the longest average duration of effect (71.4 hours). Maximum duration of effect was similar for all groups (~93 hours).
 | Table 4 Cumulative time (hours) sNa was increased by at least ≥4 mEq/L relative to baseline |
Rate of sNa increase during treatment with conivaptan
Mean sNa markedly increased over the first 6 hours of treatment regardless of dosage and baseline volume status (overall population: 4.3±3.7 mEq/L with 20 mg/day and 3.9±4.5 mEq/L with 40 mg/day) and continued to rise steadily over the first 24 hours of treatment (Figure 2). Euvolemic patients receiving either dose plateaued at ~132 mEq/L after 24 hours of treatment and maintained that level through treatment day 4. Patients who were hypervolemic at baseline and received 20 mg/day conivaptan demonstrated the slowest rate of increase in sNa, but reached the 132 mEq/L level around treatment day 4.
 | Figure 2 Rate of sNa correction in hypervolemic and euvolemic patients over 4 days of treatment with either 20 or 40 mg/day conivaptan. |
Change in sNa from baseline through study day 4
By the end of treatment, patients receiving 20 mg/day conivaptan experienced an average rise in sNa (9.4±5.3 mEq/L) (Figure 3). Euvolemic patients receiving 20 mg/day demonstrated the greatest average rise in sNa (10.8±5.22 mEq/L) and this was comparable to euvolemic patients receiving 40 mg/day (9.2±5.24 mEq/L). Hypervolemic patients also demonstrated substantial increases in sNa (20 mg/day: 7.1±4.8 and 40 mg/day: 7.6±5.8). On average, patients attained near normal sNa values by the end of treatment (~132 mEq/L, regardless of treatment and baseline volemic status).
 | Figure 3 Mean serum sodium (sNa) values following 4 days of treatment with conivaptan 20 mg/day (A) or 40 mg/day (B). |
Number of patients with at least a 6 mEq/L increase in sNa relative to baseline
The majority of patients treated with 20 mg/day conivaptan obtained either a normal sNa measurement (≥135 mEq/L) or an increase of ≥6 mEq/L (70.3%) and similar results were observed for patients receiving 40 mg/day (72.0%). A similar proportion of euvolemic patients treated with either 20 or 40 mg/day conivaptan attained this treatment goal (82.6% vs 74.8%, respectively). In comparison, slightly more hypervolemic patients treated with 40 mg/day (63.6%) attained either a 6 mEq/L increase or a normal sNa measurement than did hypervolemic patients receiving 20 mg/day (50.0%).
Mean sNa at follow-up on days 11 and 34
Patients returned to the clinical center on study days 11 and 34 and their overall condition and sNa levels were assessed (Table 5). By study day 34, both hypervolemic and euvolemic patients who had been treated with either dose of conivaptan exhibited approximately normal sNa values (average, 132–135 mEq/L).
 | Table 5 Serum sodium (mEq/L) at follow-up relative to baseline |
Safety
Adverse events and safety laboratory parameters
Overall, 97.6% of patients experienced at least one AE, and this rate was similar regardless of the dose received (94.6%, 20 mg/day vs 98.1%, 40 mg/day). The majority of AEs were considered mild (89.6%) or moderate (70.1%), and only 7.6% of patients discontinued the study drug due to an AE. The proportion of patients experiencing an AE, distribution of severities, frequency of treatment-related events, and discontinuation of the study drug due to AEs were similar regardless of the dosage received.
The most common AEs experienced by either treatment group were infusion-site phlebitis and ISR; these were similarly represented regardless of the dosage level (Table 6) and were generally considered to be related to the study drug by the investigator. The majority of individual AEs were similarly represented in both treatment groups. Gastrointestinal disorders were more commonly represented in the 40 mg/day treatment group (39.3%, 40 mg/day vs 13.5%, 20 mg/day) as were musculoskeletal and connective tissue disorders (7.9%, 40 mg/day vs 0%, 20 mg/day) and AEs affecting the renal system (22.0%, 40 mg/day vs 16.2%, 20 mg/day). Alternatively, vascular AEs were experienced by slightly more patients in the 20 mg/day group (37.8%, 20 mg/day vs 24.3%, 40 mg/day). All other body systems were similarly represented in terms of the relative proportion of AEs experienced the by patients.
 | Table 6 All adverse events occurring in at least 5% of patients in either treatment group |
The mean change from baseline for all hematology parameters, leukocyte counts, and coagulation parameters were not clinically meaningful. In addition, few hematological AEs were reported. Anemia was experienced by 19 patients, thrombocytopenia by three patients, and febrile neutropenia by one patient. Similarly mean change from baseline for clinical chemistry parameters of relevance – BUN, sCr, AST, and ALT – were not clinically meaningful. Urinalysis results were similar between the treatment groups and mean change from baseline in urinalysis parameters were not clinically meaningful. Baseline vital signs were similar between the treatment groups as were the mean change from baseline values for all vital signs measured. Blood pressure abnormalities such as hypertension (9.2% overall), hypotension (6.0% overall), and orthostatic hypotension (8.0% overall) were noted as AEs and were similarly represented in both treatment groups. Finally, ECG ST segment depression was noted for two patients receiving 20 mg/day conivaptan. However, a separate placebo-controlled trial has previously demonstrated that conivaptan does not affect cardiac repolarization or conduction.7
Withdrawals due to overly rapid correction
Eight patients experienced a ≥12 mEq/L increase in sNa in under 24 hours, which led to the discontinuation of study treatment per protocol. An additional two patients were discontinued from treatment due to the rate of increase in sNa, but they did not strictly meet the above definition (patient identifier 11205 and 11404, Table 7). One patient was included in the 20 mg/day group and the other nine received 40 mg/day. Euvolemia and hypervolemia were equally represented. Each patient’s baseline and final sNa values as well as the diagnosed cause of hyponatremia and potentially confounding medical history are listed in Table 7.
 | Table 7 Characteristics of subjects discontinued from treatment due to a rapid increase in sNa |
Four patients who were discontinued for overly rapid correction had baseline sNa of 120 mEq/L or less. Four subjects were diagnosed with CHF as the cause of their current episode of hyponatremia; two patients were diagnosed with SIADH alone. The remaining four patients had been treated with diuretics prior to their inclusion in this study, and this was considered the causative etiology of hyponatremia. Three of these four subjects had disothiazide listed as the causative agent, specifically. All ten of these patients had a history of hypertension (100%) and all but one (90%) had a history of some form of dyslipidemia. No instances of ODS were reported in the study, and all ten patients were alive at follow-up on day 34.
Infusion-site reactions
AEs occurring at the infusion site were equally represented in both treatment groups (70.3%, 20 mg/day; 78.0%, 40 mg/day) with the exception of erythema which was experienced by more patients in the 40 mg/day group (7.0%) (Table 8).
 | Table 8 All infusion-site reactions |
Most reactions began within the first 48 hours of treatment (56.7%, 20 mg/day; 71%, 40 mg/day) and the overwhelming majority of these reactions recovered with no sequelae (92.3%, 20 mg/day; 95.8%, 40 mg/day). However, a proportion of ISRs necessitated a dose interruption (30.8%, 20 mg/day; 27.5%, 40 mg/day), and a minority necessitated discontinuation of the study drug (3.8%, 20 mg/day; 4.2%, 40 mg/day), but these rates were similar between treatment groups.
Serious adverse events
SAEs were experienced by 30.7% of patients and the proportion of patients experiencing at least one SAE was similar regardless of treatment allocation (24.3%, 20 mg/day vs 31.8%, 40 mg/day, Table 9). Additionally, the majority of unique AEs were reported by one patient each; the events that occurred in at least three patients of either treatment group are listed in Table 9. Hyponatremia was the most common SAE and was similarly represented in both treatment groups. All SAEs of hyponatremia were recurrences during the follow-up period, and all but one were deemed unrelated to the study drug by the investigator.
 | Table 9 All serious adverse events occurring in ≥3 patients of either treatment group |
Withdrawals and deaths
Overall, 19 patients (7.6%) discontinued treatment with the study drug due to an AE. The proportion of discontinued patients was similar regardless of treatment allocation (5.4%, 20 mg/day vs 7.9%, 40 mg/day). The most common AE leading to withdrawal from study drug was infusion-site phlebitis (2.7%, 20 mg/day; 2.8%, 40 mg/day).
A total of 26 patients (10.4%) died during the study, and nine (34.6%) of these deaths occurred more than 30 days following treatment initiation. Of the 17 deaths that occurred within 30 days of the first dose of study drug, two were in the 20 mg/day group and deemed not related to the study medication (gastric malignancy and cerebrovascular accident). Fourteen of the 15 deaths in the 40 mg/day treatment group were deemed not related to study drug; the one event that was considered to be possibly related was a death due to myocardial infarction in a patient with a history of coronary artery bypass graft and ischemic cardiomyopathy. Other causes of death included multiorgan failure (N=3), sudden death (N=2), and the rest were unique occurrences.
Pharmacokinetics
Median plasma concentrations were highest following completion of the loading dose, at study hour 0.5, for all patients regardless of treatment allocation. By hour 72, median plasma concentrations of conivaptan were nearly dose proportional. By study day 10, virtually 100% of the drug had been cleared. Plasma concentrations were nearly identical for all patients, regardless of age group, for both dose levels tested, which may be due to an extensive overlap between the geriatric (>65 years) and PK populations (refer to sample size table, Figure 4).
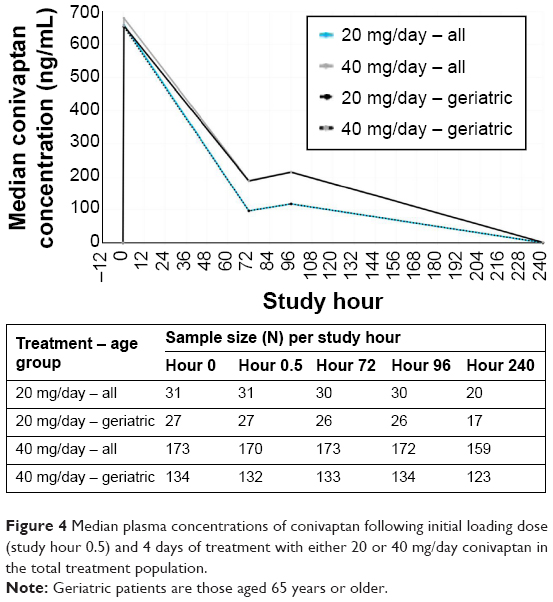 | Figure 4 Median plasma concentrations of conivaptan following initial loading dose (study hour 0.5) and 4 days of treatment with either 20 or 40 mg/day conivaptan in the total treatment population. |
A subset of patients (N = eight per treatment group, seven of whom were ≥65 years of age) provided additional samples for the analysis of PK parameters through study hour 120 (“PK-rich” group, Figure 5). The data obtained from this subpopulation parallels those obtained from the overall study population wherein plasma concentrations are highest following the initial loading dose and are subsequently maintained at a nearly constant level throughout the 96-hour treatment period (Figure 5). Plasma concentrations were less dose proportional than what was measured in the total study population; blood levels in the 40 mg/day group were more than twice those seen in the 20 mg/day group. Median AUC was 6,199.8 ng·hr/mL for the 20 mg/day regimen and 21,487.4 ng·hr/mL for the 40 mg/day group, indicating a greater-than-dose-proportional increase in exposure in the PK-rich population. Plasma levels of conivaptan dropped following the end of the treatment period as evidenced by the samples taken at hours 97, 98, and 103. For the 20 and 40 mg/day groups, median half-life was 5.3 and 8.1 hours, respectively, and median clearance was 16.1 and 8.7 L/hour, respectively.
 | Figure 5 Median plasma concentrations of conivaptan following initial loading dose (study hour 0.5) and 4 days of treatment with either 20 or 40 mg/day conivaptan in the PK-rich sampling group (N=8 per group). |
Discussion
Assessing the relative safety, efficacy, and PK parameters of a 20 and 40 mg/day dose of conivaptan was the primary goal of this study. Conivaptan given at doses of 20 and 40 mg/day continuously for 4 days following a 20 mg bolus loading dose was well tolerated. Although a majority of patients experienced at least one AE during the study, the overall profile of AEs was consistent with that reported in previous studies of conivaptan and are not unexpected in this hospitalized population.7
Importantly, the frequency of ISRs in the 40 mg/day group (78.0%) was similar to that reported in patients receiving 20 mg/day (70.3%) and most events resolved with no residual effects. The similar frequency of ISRs between treatment groups suggests that the increased infusion rate required by the 40 mg/day dose and the resulting effective plasma concentration of conivaptan are no more irritating to the vasculature than what is seen as a result of the 20 mg/day dose.
One case of rhabdomyolysis was reported in this study by a subject allocated to the 40 mg/day treatment regimen who was concomitantly receiving CYP3A inhibitors. The event was moderate in severity, not deemed serious or related to study drug, and the patient recovered without sequelae. As noted in the package insert, conivaptan should not be given in concert with drugs that interact with CYP3A.7
The risk of overly rapid sNa correction culminating in ODS is a major concern when treating hyponatremia in patients who also have hypokalemia, alcoholism, malnutrition, or advanced liver disease. In this study, ten patients were discontinued from the study treatment because they showed a rapid rise in sNa, eight of these patients met the definition of “overly rapid” in the protocol (≥12 mEq/L increase in ≤24 hours). No additional measures to slow or reverse the sNa rise were noted in the case report forms, and all ten were alive at follow-up on day 34. Four of these patients also experienced an SAE, all of which were deemed unrelated to the study drug by the investigator and occurred ≥8 days after the discontinuation of treatment. None of these ten patients exhibited symptomology consistent with the development of ODS. It is important to comment on the medical history and cause of hyponatremia in these ten patients because these factors influence the choice of treatment and risk of ODS. Three of these patients (30%) had thiazide diuretic treatment recorded as the causative agent of their hyponatremia. Thiazide diuretics impair sodium reabsorption in the distal nephron and can therefore result in hyponatremia. The appropriate treatment for thiazide-induced hyponatremia (TIH) includes discontinuation of the thiazide and fluid restriction.16–19 These patients should not have been treated with a V2 receptor antagonist because they would be considered hypovolemic and increasing free water clearance would only be expected to precipitate an overly rapid rise in sNa. Furthermore, 90% of the patients who experienced overly rapid correction had some form of preexisting hyperlipidemia. When indirect potentiometry or flame photometry are used, high serum lipid levels (or protein) can interfere with the measurement of sNa, leading to a misdiagnosis of hyponatremia (ie, pseudohyponatremia) and a physiological state not well suited for treatment with a V2 receptor antagonist.20,21 Hyperlipidemia is also associated with the emergence of TIH.22 Cumulatively, these observations suggest that patients with a history of hyperlipidemia and recent thiazide use should not be treated with V2 receptor antagonists. A conservative estimate of the frequency of overly rapid correction in this study would be 3.98% and possibly as little as 1.99% if the patients not meeting the standard definition of overly rapid correction and those with TIH are excluded. Both rates are lower than what has been reported as a result of hypertonic saline (17.1%) and tolvaptan (10.8%) treatment in a recent registry study.11
The efficacy of conivaptan in correcting serious, persistent, and unresponsive hyponatremia was well established prior to the conduct of this study. Previous analyses indicate that the most serious complications of hyponatremia can be alleviated with as little as a 4–6 mEq/L increase in sNa.23,24 Thus, treatment success may be defined as a patient obtaining either a normal sNa value (135 mEq/L) or at least a 6 mEq/L increase from baseline. In this study, 70.3% of patients treated with 20 mg/day conivaptan and 72.0% of patients treated with 40 mg/day conivaptan attained this treatment goal.
The data presented here indicate that the 20 and 40 mg/day doses of conivaptan are similarly efficacious in euvolemic patients. However, the data from hypervolemic patients suggest a potential dose–response relationship. Hypervolemic patients receiving 20 mg/day lagged in comparison to those receiving 40 mg/day in terms of the rate of increase in sNa, the absolute increase in sNa, and the duration of effect. Fluid-overloaded patients potentially require a longer time to eliminate excess water and normalize their sNa levels in comparison to euvolemic patients. Regardless of mechanism, these data suggest that the choice of conivaptan treatment regimen (20 or 40 mg/day) should take into account the patient’s initial volume status.
Conclusion
In this study, over 70% of patients treated with either 20 or 40 mg/day conivaptan reached the treatment goal of either a normal sNa measurement or an increase of at least 6 mEq/L from baseline. Seemingly small increases such as these are associated with a reduction in the most serious complications of hyponatremia. Both doses were well tolerated and demonstrated an AE profile typical of patients hospitalized for hyponatremia. The 40 mg/day dose of conivaptan was no more irritating to the vasculature than what was seen with the 20 mg/day dose. Finally, the rate of overly rapid correction in this study, 3.9%, was less than what has been reported elsewhere for other hyponatremia treatments. Conivaptan given at the doses of 20 and 40 mg/day continuously for 4 days following a 20 mg bolus loading dose is a safe and effective treatment for hyponatremia.
Disclosure
Biff F Palmer, MD, has received speaker’s fees from Cumberland Pharmaceuticals Inc. Emily J Woodward, PhD, and Amy D Rock, PhD, are employees of Cumberland Pharmaceuticals Inc., Nashville, TN, USA. The authors report no other conflicts of interest in this work.
References
Hawkins RC. Age and gender as risk factors for hyponatremia and hypernatremia. Clin Chim Acta. 2003;337(1–2):169–172. | ||
Wald R, Jaber BL, Price LL, Upadhyay A, Madias NE. Impact of hospital-associated hyponatremia on selected outcomes. Arch Intern Med. 2010;170(3):294–302. | ||
Terzian C, Frye EB, Piotrowski ZH. Admission hyponatremia in the elderly: factors influencing prognosis. J Gen Intern Med. 1994;9(2):89–91. | ||
Bennani SL, Abouqal R, Zeggwagh AA, et al. [Incidence, causes and prognostic factors of hyponatremia in intensive care]. Rev Med Interne. 2003;24(4):224–229. | ||
Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1–S42. | ||
Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Suppl 1):S36–S42. | ||
VAPRISOL (conivaptan hydrochloride) Injection, for intravenous use. Nashville, TN: Cumberland Pharmaceuticals Inc.; 2014. | ||
SAMSCA (tolvaptan) tablets for oral use. Tokyo, Japan: Otsuka Pharmaceutical Co., Ltd.; 2014. | ||
Tahara A, Saito M, Sugimoto T, et al. Pharmacological characterization of YM087, a potent, nonpeptide human vasopressin V1A and V2 receptor antagonist. Naunyn Schmiedebergs Arch Pharmacol. 1998;357(1):63–69. | ||
Yamamura Y, Nakamura S, Itoh S, et al. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J Pharmacol Exp Ther. 1998;287(3):860–867. | ||
Greenberg A, Verbalis JG, Amin AN, et al. Current treatment practice and outcomes. Report of the hyponatremia registry. Kidney Int. 2015;88(1):167–177. | ||
Diringer MN, Zazulia AR. Hyponatremia in neurologic patients: consequences and approaches to treatment. Neurologist. 2006;12(3):117–126. | ||
Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol. 2007;2(6):1110–1117. | ||
Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Eur J Endocrinol. 2014;170(3):G1–G47. | ||
Koren MJ, Hamad A, Klasen S, Abeyratne A, McNutt BE, Kalra S. Efficacy and safety of 30-minute infusions of conivaptan in euvolemic and hypervolemic hyponatremia. Am J Health Syst Pharm. 2011;68(9): 818–827. | ||
Barber J, McKeever TM, McDowell SE, et al. A systematic review and meta-analysis of thiazide-induced hyponatraemia: time to reconsider electrolyte monitoring regimens after thiazide initiation? Br J Clin Pharmacol. 2015;79(4):566–577. | ||
Diaconu CC, Balaceanu A, Bartos D. Diuretics, first-line antihypertensive agents: are they always safe in the elderly? Rom J Intern Med. 2014;52(2):87–90. | ||
Frenkel NJ, Vogt L, De Rooij SE, et al. Thiazide-induced hyponatraemia is associated with increased water intake and impaired urea-mediated water excretion at low plasma antidiuretic hormone and urine aquaporin-2. J Hypertens. 2015;33(3):627–633. | ||
Sardar GK, Eilbert WP. Severe hyponatremia associated with thiazide diuretic use. J Emerg Med. 2015;48(3):305–309. | ||
Hussain I, Ahmad Z, Garg A. Extreme hypercholesterolemia presenting with pseudohyponatremia – a case report and review of the literature. J Clin Lipidol. 2015;9(2):260–264. | ||
Aw TC, Kiechle FL. Pseudohyponatremia. Am J Emerg Med. 1985; 3(3):236–239. | ||
Rastogi D, Pelter MA, Deamer RL. Evaluations of hospitalizations associated with thiazide-associated hyponatremia. J Clin Hypertens (Greenwich). 2012;14(3):158–164. | ||
Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282–299. | ||
Aylwin S, Burst V, Peri A, Runkle I, Thatcher N. Dos and don’ts in the management of hyponatremia. Curr Med Res Opin. 2015;14:26. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.