Back to Journals » Journal of Pain Research » Volume 19
Dorsal Root Ganglion as a Hub for Peripheral Sensitization: A Hierarchical Regulation Model and Translational Progress
Authors Cui A ![]() , Meng X
, Meng X ![]() , Wang Z, Wu Q
, Wang Z, Wu Q ![]() , Li M
, Li M ![]()
Received 5 February 2026
Accepted for publication 1 July 2026
Published 10 July 2026 Volume 2026:19 601365
DOI https://doi.org/10.2147/JPR.S601365
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alaa Abd-Elsayed
Aoyun Cui,1 Xianyu Meng,2 Zhen Wang,1 Qingyuan Wu,1 Mengdi Li3
1Department of Orthopedics, The First Clinical Medical College, Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, People’s Republic of China; 2Department of Orthopedics Ward II, The First Affiliated Hospital, Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, People’s Republic of China; 3Department of Rehabilitation, Qilu Hospital of Shandong University Dezhou Hospital, Dezhou, Shandong, People’s Republic of China
Correspondence: Xianyu Meng, Department of Orthopedics Ward II, The First Affiliated Hospital, Heilongjiang University of Chinese Medicine, No. 26, Heping Road, Xiangfang District, Harbin, Heilongjiang, 150040, People’s Republic of China, Email [email protected]
Abstract: The high attrition rate of analgesic candidates in clinical trials stems largely from a reductionist reliance on rodent models and a neglect of the spatiotemporal complexity of nociception. Positioning the dorsal root ganglion (DRG) as the core computational hub for peripheral sensitization, this review proposes a “Hierarchical Regulation Model” to integrate fragmented mechanisms into a systemic framework. We stratify peripheral sensitization into three interlocking tiers: an acute tier driven by ion channel functional remodeling (eg, Nav1.7/1.8, Transient Receptor Potential (TRP) family); an intermediate tier orchestrated by neuroinflammation and glia-immune networks (eg, Satellite glial cell (SGC) activation, B-cell infiltration); and a long-term tier anchored by epigenetic reprogramming (eg, histone modifications, DNA methylation) that constitutes the “molecular memory” of pain. Crucially, this model reveals that pain chronification arises from the “pathological homeostasis” formed by cross-hierarchical feedback loops. Importantly, we extend this framework to pathology-specific trajectories, highlighting “acute layer jumping” in chemotherapy-induced peripheral neuropathy and “early metabolic solidification” in diabetic peripheral neuropathy. Finally, anchored by the recent breakthrough approval of the selective Nav1.8 inhibitor Suzetrigine (VX-548), we validate the “Human-First” drug development paradigm. We conclude that overcoming translational barriers requires shifting from “one-size-fits-all” approaches to precision strategies stratified by pathological tier, disease stage, and biological sex.
Keywords: dorsal root ganglion, peripheral sensitization, neuropathic pain chronification, epigenetics, therapeutic translation
Introduction
Chronic pain is estimated to affect 20% of the adult population worldwide, resulting in a significant socioeconomic burden.1,2 Current pain relief medicines (predominantly opioids, NSAIDs, anticonvulsants) have several drawbacks in clinical use. Progress continues to be made in basic research, but successful translation to new clinical therapeutics is disconcertingly rare. This may in large part be due to a historical emphasis on reductionist rather than network-oriented views of mechanistic knowledge.
Moreover, another major reason for translational failure is the vast difference between human and rodent dorsal root ganglia (DRG).3 Cross-species transcriptomic studies show that while the neuronal subtypes per se are broadly similar across species, the broader systemic picture differs in terms of which functional neuropeptides and ion channels are expressed by sensory neurons. For example, studies show that there is differential expression of TRP channels between species. Further, the Calcitonin gene-related peptide(CGRP)/P2X3 receptor (P2X3R) distribution pattern used to distinguish peptidergic from non-peptidergic nociceptors in mice is not present in humans—human DRGs contain a considerable population of neurons which co-express CGRP and P2X3R.4 This basic difference leads to the potential that any therapeutic aimed at rodent-defined neuronal populations could also have off target effects, or not work in humans. New advancements in single-cell transcriptomics, spatial omics, and electrophysiological methods allow for the direct study of human DRGs, revolutionizing the field of pain research.5
Structurally, the DRG provides the requisite anatomical infrastructure for this systemic orchestration. Instead of serving as a simple relay station, as commonly portrayed, DRGs represent complex neuro-glial-immune ecosystems.6,7 Within this compact microcosm, peripheral sensitization is not a consequence of discrete neuronal activity, but emerges from intercellular crosstalk of different nociceptor subtypes and the interaction of those neurons with SGCs and a panoply of immunological cells that infiltrate that closed microenvironment. This intricate cytoarchitecture functions as a computational hub for signal amplification and integration, thereby providing the physical basis for the hierarchical regulation model proposed herein (Figure 1).
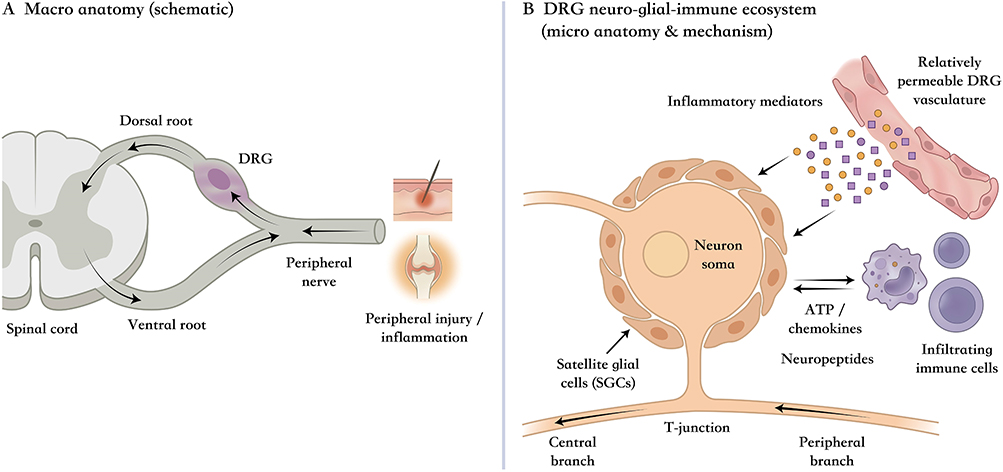 |
Figure 1 The DRG as an integration hub for peripheral sensitization across anatomical scales. (A) Overview of the sensory pathway emphasizing the DRG’s strategic position between peripheral input and central transmission, where injury-related signals can be consolidated before propagating centrally. (B) Local cellular ecosystem highlighting how neuron–glia–immune interactions, facilitated by the DRG’s permissive vascular interface, create a compartment that supports amplification, cross-talk, and persistence of sensitization—providing the anatomical rationale for a DRG-centered hierarchical regulation framework. |
Earlier conceptual frameworks distinguished early post-translational from later transcription-dependent pain plasticity and established central sensitization as a major mechanism of persistent pain hypersensitivity.8,9 Building on these foundations, we propose a DRG-centered Hierarchical Regulation Model that shifts the focus to peripheral sensitization and adds three elements: a tiered organization of acute ion channel remodeling, intermediate neuro-glial-immune communication, and long-term epigenetic consolidation; cross-hierarchical feedback rather than a purely linear temporal sequence; and pathology-specific trajectories across distinct pain states. Accordingly, this review introduces the model, examines its acute, intermediate, and long-term regulatory tiers, evaluates its pathology-specific manifestations, and discusses its translational implications.
The Hierarchical Regulation Model of Peripheral Sensitization in the DRG: Conceptual Framework and Temporal Dimensions
Based on analyses of literature from the last three years, we propose a “Hierarchical Regulation Model” of peripheral sensitization in the DRG. The key principle underlying our model is that pain chronification is not a progression along the timeline of one singular mechanism, but rather a cumulative manifestation of regulatory processes acting over timescales that overlap and layer upon each other.
Acute regulatory tier (seconds-to-minutes scale) focuses on functional changes to ion channels: reduced activation thresholds of sodium channels (Nav1.7/1.8/1.9), sensitization of TRP channels (TRPV1/TRPA1/TRPM8), downregulation of potassium channels (KCNQ, KCNN1)—all largely effected by post-translational modifications. Intermediate regulatory tier (hours-to-days scale) is primarily concerned with neuroinflammation and intercellular communication: activation of chemokine networks (CCL2/CCR2, CXCL12/CXCR4), functional alteration of SGCs (eg, Kir4.1 downregulated), infiltration of immune cells. Long-term regulatory tier (days-to-months scale) involves epigenetic remodeling, in which continued regulation of target genes by histone-modifying enzymes (G9a, PRMT5, HDAC2) constitutes the “molecular memory” of pain.
The core theoretical value of our model resides in explicating the “pathological homeostasis” arising from cross-hierarchical interplays. Pain chronification does not flow in one direction, but rather emerges as a self-sustaining interlocked feedback loop to generate pathology (Figure 2): while ion channel remodeling in the acute phase represents the final effector of electrophysiological disturbances, the instigator for this phenomenon often lies within the fluidic interplay between neuroinflammatory networks and glial cells in the intermediate tier. Of particular concern is that the fleeting extracellular signals do not terminate at the plasma membrane. Signals enter into the nucleus and generate epigenetic remodeling within the long-term tier. This web of molecular perturbations constitutes a “memory anchor”, solidifying the temporary signals of injury into a persistent pathological state by locking the expression of the relevant genes. The “signaling-transcription-function” loop between the three tiers is what underpins the stolid recalcitrance of chronification.
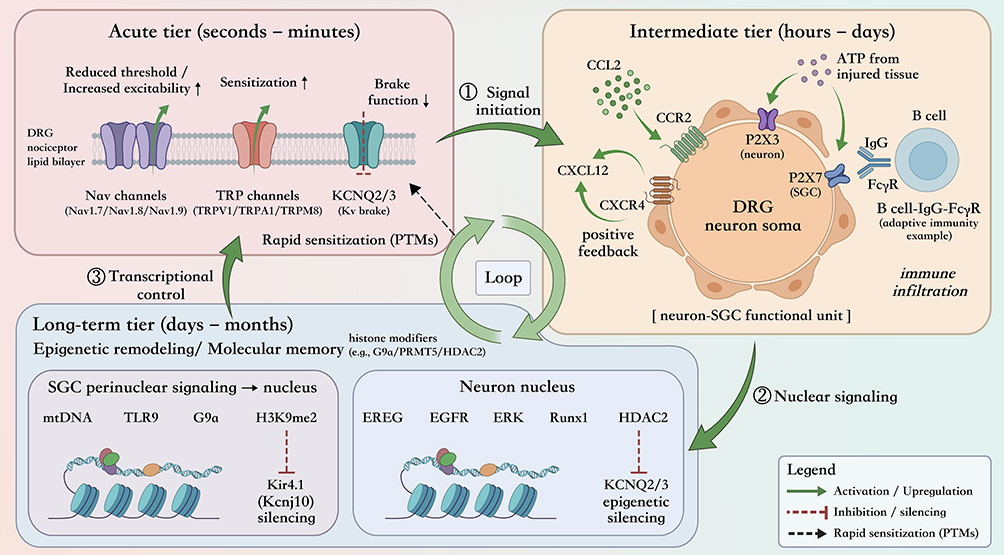 |
Figure 2 Conceptual architecture of the “Hierarchical Regulation Model” and the self-sustaining signaling–transcription–function loop in the DRG. Schematic summary of three overlapping regulatory tiers that operate on distinct timescales—acute (seconds–minutes), intermediate (hours–days), and long-term (days–months)—and collectively drive pain chronification. The acute tier emphasizes rapid functional remodeling of excitability “effectors” (ion-channel function) largely through post-translational mechanisms; the intermediate tier captures neuroinflammatory and intercellular communication programs (eg, chemokine/purinergic signaling and immune engagement) that amplify and maintain the sensitizing milieu; and the long-term tier illustrates how transient extracellular signals are converted into durable transcriptional reprogramming via epigenetic remodeling, forming a “memory anchor”. Cross-tier coupling generates a closed feedback architecture (“pathological homeostasis”), whereby intermediate-tier signaling promotes nuclear remodeling and transcriptional control that subsequently stabilizes (and re-feeds) acute electrophysiological dysfunction. In the schematic labels, upward arrows (↑) indicate increased excitability, sensitization, or functional output; downward arrows (↓) indicate reduced function, including reduced potassium-channel braking function; and right arrows (→) indicate directional signaling flow or causal progression between regulatory events. Solid green arrows represent activation or upregulation, red blunt-ended lines indicate inhibition or silencing, and black dashed arrows indicate rapid PTM-associated sensitization. Abbreviation: PTM, post-translational modification. |
Acute Regulatory Tier: Functional Remodeling of Ion Channels
The acute tier of the Hierarchical Regulation Model is the first point of entry of pain sensitisation. We will examine the functional re-modelling of the ion channels in this part, which molecularly execute the re-modelling. This process begins with the first transduction of chemical and mechanical stimuli by “multimodal sensors” in the form of TRP family and Acid-Sensing Ion Channels (ASICs) channels followed by the “amplification effect” of voltage-gated sodium channels (Nav) and simultaneous failure of potassium channels (Kv) “braking mechanisms”, all of which together trigger the rapid collapse of electrophysiological homeostasis. This transient burst of electrical signaling not only generates the acute pain experience but also provides the necessary driving energy to trigger subsequent medium-term and long-term pathological cascades.
Core Execution and Amplification: Voltage-Gated Sodium Channels
Nav1.7, 1.8, and 1.9 are the terminal executors of nociception, validated by strong genetic evidence linking them to congenital pain disorders.10 Functionally, Nav1.7 initiates action potentials, Nav1.8 sustains repetitive firing, and Nav1.9 regulates the resting membrane potential.
Stewart et al2 validated the electrophysiological effects of the selective Nav1.8 inhibitor, suzetrigine, in human DRG neurons, confirming that the drug significantly inhibits the firing frequency of action potentials in nociceptive neurons without significant impact on the cardiac isoform Nav1.5 and central isoforms Nav1.1/1.2.11 Notably, this study was conducted entirely using human DRG neurons rather than relying on traditional rodent models—this “human-first” drug development pathway establishes a new paradigm in the field of pain research. A comparative study by Zhang et al on Nav1.9 biophysics in human and rat DRG neurons revealed that despite the conservation of persistence and low activation thresholds, significant inter-species differences exist. Specifically, human Nav1.9 displays a higher inactivation threshold, slower deactivation, and—unlike the enhancement seen in rodents—is inhibited by inflammatory mediators.12 This fundamental divergence raises a critical concern: preclinical screens relying on rodent Nav1.9 gain-of-function models may have yielded false leads, necessitating a re-evaluation of data derived from non-human platforms.
Unlike Nav1.8, the clinical translation of Nav1.7 inhibitors has been blocked by traps of target selectivity and functional compensation. To deal with this, the ongoing research of Nav1.7 has extended beyond simple pore block. Novel small molecules such as QLS-2783 and indole 3-hydroxy derivatives13 have shown powerful effects by selectively stabilizing the channel in the inactivated state. However, Nav1.7 is also dynamically reshaped by pathological environments. Liu et al14 revealed that the SARS-CoV-2 nucleocapsid protein directly interacts with Nav1.7 to enhance sodium currents, driving “COVID-pain” by essentially hijacking the acute electrophysiological machinery. Beyond the channel pore, functional output is amplified by interacting partners. Feng et al15 reported a “supra-additive” effect of concurrent Nav1.7 and sodium-calcium exchanger NCX1 inhibition to pain, where it is suggested that ionic imbalance can be maintained by this synergy. Lastly, the membrane density of these acutely acting executors is orchestrated by upstream mechanisms—the long non-coding RNA (lncRNA) SNHG5 recruits CDK9 to the SCN9A promoter to increase Nav1.7 expression. This “SNHG5-CDK9-Nav1.7” axis replenishes the functional channel pool required to sustain high-frequency firing and neuroinflammation-induced apoptosis,16 illustrating how transcriptional events directly feed into and perpetuate the acute tier of hyperexcitability.
Multimodal Signal Transduction: TRP Family and ASIC Channels
The activation of Nav does not occur in a vacuum. It happens after the upstream occurrence of the initial transduction of thermal, mechanical and chemical stimuli by molecular sensors. The TRP family and ASICs form a “multimodal sensory frontline” and produce generator potentials that are required to activate Nav for the generation of action potentials. Among this superfamily, TRPV1 is defined as the prototypic polymodal receptor for capsaicin, heat and acid. However, rodent models of this have little translatability owing to inter-species heterogeneity. Cross-species comparative analyses show that TRPV1 is more broadly expressed in human DRGs than in mice, and across many neuronal subtypes including C-LTMRs and the cold-sensing receptors.17 This divergence holds critical implications for interpreting preclinical data and designing TRPV1-targeted therapeutics for human applications.
The functionality of TRPV1 is not fixed but is stringently reshaped by dynamic regulatory networks ranging from epigenetic modifications to neuro-immune-endocrine interactions. Yeh et al showed that histone methylation modifications induce TRPV1 upregulation directly at the transcriptional level in paclitaxel-induced Chemotherapy-Induced Peripheral Neuropathy (CIPN),18 acting as an upstream epigenetic switch. Following translation, channel activity is modulated by the inflammatory microenvironment; Li et al19 demonstrated that the ApoA-I binding protein regulates TRPV1 channel activity by interfering with TLR4-lipid raft complex formation. Meanwhile, as Franco-Enzástiga et al20 exemplified in human DRG neurons that Type I interferons can potentiate TRPV1 sensitization, presenting a pathway to pain in autoimmune diseases. The addition of variation in sex hormones presents another axis to this regulation. Artero-Morales et al21 demonstrated that estrogen can upregulate TRPV1 expression and function in the direct endocrine manipulation of ion channels via ERα/ERβ. Candler Paige et al22 found that the prolactin receptor (Prlr) mediates hyperalgesic priming in female, but not in male, Nav1.8+ nociceptors. Direct endocrine-regulation of channel function may compel some female patients to seek therapies aligned to hormonal cycles.
Beyond TRPV1, other TRP isoforms mediate specific senses and disease states. Broad et al23 described Phase I results for the TRPA1 antagonist LY3526318. Designed to be particularly potent toward human TRPA1, this novel compound proved analgesic in several models and human-safe, scoring a major goal in the ongoing effort to translate TRP targeting to the clinic. Mechanistically, Lee et al24 found that DJ-1/PARK7 regulates CIPN through TRPA1. Other family members also contribute to specific sensory deficits: Du et al25 confirmed that IL-33/ST2 signaling mediates cold allodynia via TRPM8, and Gao et al26 identified Copine-6 as a TRPM3 chaperone protein that regulates its sensitivity to thermal stimuli.
Distinct from the TRP family, ASICs function as ligand-gated cation channels specializing in detecting tissue acidification. ASIC3 is highly expressed in DRG nociceptive neurons and responds to mild acidification within the physiological pH range (pH 6.6–7.0), generating both transient and sustained current components.27 Recent evidences suggest that ASIC3 functions extend beyond simple acid sensing to act as an integrated receptor for the inflammatory milieu through non-canonical mechanisms. Marra et al28 demonstrated that lysophosphatidylcholine (LPC) and arachidonic acid (AA) in human inflammatory exudates can persistently activate ASIC3 at physiological pH (non-acidic conditions), inducing DRG neuron depolarization and increased C-fiber firing—an effect reversible by the ASIC3 blocker APETx2. This functional complexity is mirrored by structural and expression diversity. ASIC3 and P2X3 receptors have been shown to co-localize within DRG neurons to form functional protein complexes,29 where ASIC3 activation inhibits P2X3 currents through a Ca2+-dependent mechanism that allows crosstalk between acidic and purinergic signaling. In disease conditions, ASIC3 mRNA and protein expression levels are all increased in DRGs from rats with bone cancer pain, and exhibited greater co-localization in IB4-positive small-diameter neurons.30 Pharmacological blockade of peripheral ASIC3 via the specific toxin APETx2 decreases migraine-associated pain behavior such as trigeminal nociceptive sensitization and periorbital mechanical sensitivity.31 Analogous to TRPV1, inter-species investigations by Papalampropoulou-Tsiridou et al32 revealed that human DRGs appear to express ASIC1 and ASIC3 on a greater range of neuronal subpopulations than rodents. This highlights that even ASICs are active drug targets, their clinical translation is hamstrung by a bottleneck in terms of both molecular selectivity and species-specific expression profiles.
Potassium Channel Dysregulation and Excitability Balance
The maintenance of chronic pain involves not only an excitatory drive, but also the failure of the inhibitory “pull”—the functional downregulation of potassium channels. This “brake failure” allows neurons prone to repetitive firing, and can also involve upstream transcriptional reprogramming (a type of hierarchical cascade). Zhang et al33 observed that in bone cancer pain, the EREG/EGFR-ERK-Runx1 axis silences KCNQ2/KCNQ3 expression via HDAC2. Another example of silencing provided by Wang et al6 identified ESRRG-controlled KCNN1 downregulation as contributing to neuropathic pain. Outside of purely transcriptional mechanisms, Habib et al34 identified CASPR2 autoantibodies leading to hyperexcitability through Kv current reduction, establishing links between autoimmune pathologies and dysregulation of ion channels.
HCN pacemaker currents constitute a “stuck accelerator” while braking fails, breaking the balance of excitability. In this regard, Lei et al7 showed that activation of the cannabinoid CB1 receptor can inhibit HCN channel function to restore homeostasis. The “push-pull” imbalance (upregulation of sodium/HCN forces at the same time as a downregulation of potassium forces) underlies neuronal hyperexcitability and suggests a single common molecular basis for it.
Crucially, a successful deployment of these stabilizing strategies relies on the quality of preclinical human physiology models. Addressing this translational gap, authors such as Zurek et al35 benchmarked the electrophysiological characteristics of human DRG against human induced pluripotent stem cell-derived sensory neurons (hiPSC-SNs). They found differences in fundamental excitability properties, confirmed sexual dimorphism and also highlighted how human-specific ion channel kinetics cannot be accounted for by a rodent surrogate. These studies establish the context for developing high-fidelity human cellular models, and that when therapeutics aimed at restoring the ionic brake are developed and subsequently clinically translated, they must be rightly calibrated for the specific electrophysiological architecture of the human DRG.
Intermediate Regulatory Tier: Neuroinflammation and Intercellular Communication
Inflammatory Mediator Networks: Cytokines, Neurotrophic Factors, and Chemokines
The inflammatory cascade initiated through tissue injury represents the key “intermediate tier” of this sensitization model, coordinating pathways that convert peripheral injury into sustained hyperexcitability. Following peripheral tissue injury, pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) activated in the DRG initiate canonical signaling pathways including NF-κB, p38 MAPK, and JAK/STAT. These converging signals serve to functionally modulate as well as increase transcription of key ion channels such as Nav1.7 and TRPV1, driving inflammatory pain sensitization.36–38 Of fundamental importance is that this network is functionally redundant. Hernandez et al39 established a defining axiom of this network architecture: they demonstrated that only the combined downregulation of IL-6st, TNFR1 and IL-1R1 silences hyperalgesia via CRISPR epigenome editing, while independent blockade fails. This is the key difference from the way in which neuroinflammation can operate unlike systemic immunity—making single-cytokine antagonists ineffective in the chronic setting through functional redundancy.
Nerve growth factor (NGF) acts as a pivotal molecular hub linking inflammation to neural plasticity. Via the TrkA receptor, NGF activates the PI3K/Akt and MAPK/ERK signaling cascades, effectively translating extracellular inflammatory signals into intracellular excitability changes and upregulating TRPV1 expression.40 O-Sullivan et al41 used sensory neuron-specific TrkA conditional knockout mice to demonstrate that TrkA deletion markedly improved osteoarthritis-induced mechanical hyperalgesia and significantly reduced spinal glial activation. While Phase III clinical trials of the anti-NGF monoclonal antibody tanezumab confirmed its efficacy in osteoarthritis and cancer pain,42,43 safety concerns about rapidly progressive osteoarthritis have constituted a major regulatory hurdle. Furthermore, Sharma et al44 demonstrated that proneurotrophin-3 (proNT3) drives CIPN by inducing CCL2 upregulation via TrkC, revealing a distinct form of crosstalk where neurotrophic signaling actively recruits chemokine networks to drive pathology.
Chemokines coordinate the inflammatory microenvironment and initiate structural remodeling. The CCL2/CCR2 axis serves as a central pathway for DRG neuroinflammation. Midavaine et al45 developed the CCR2-targeting pepducin PP101, which effectively blocks T-cell infiltration and produces analgesia in bone cancer and CCI models. Zhang et al46 found that the RNA-binding protein SYNCRIP promotes neuropathic pain by stabilizing CCR2 mRNA, which highlights the critical role of post-transcriptional regulation in maintaining chemokine levels. Leng et al47 uncovered evidence about chronification, suggested that plasma CXCL12 induces a self-propagating positive feedback loop with CXCR4, which might serve as a molecular switch tipping the balance in promoting chronic pain. In the end, prolonged sensitization is a crash of active resolution programs. Yang et al48 showed that α7nAChR signaling restores cholinergic anti-inflammatory pathways to suppress HMGB1 release. It is the failure of resolution mechanisms that induces pathology, not inflammatory surges.
Satellite Glial Cell Activation and Purinergic Signaling
SGCs closely ensheath the somata of DRG neurons, forming an operable structure that is dynamically involved in peripheral sensitization through tight regulation of ionic homeostasis and gliotransmitter secretion. The key of homeostatic function is the Kir4.1 potassium channel. Luo et al49 provided an explanation of SGC dysfunction with a mechanistic basis, showing that the downstream signaling of an activated surface TLR9 receptor by paclitaxel acts not only to block the pore of the channel, but also to recruit the methyltransferase G9a, effecting epigenetic silencing of Kcnj10 via H3K9me2. Thus, the loss of K+ buffering capacity is not ephemeral. It generates a persistent potassium-rich microenvironment that effectively lowers the activation threshold of the neuron and perpetuates hyperexcitability dynamics long past chemotherapy.
Purinergic signaling provides a basis for the communication between neuron and glia. ATP acts as a ubiquitous “danger signal” released from damaged tissues, signaling two different pathological paths. On the side of the neuron, it activates P2X3 receptors, confined to nociceptors and high sensitivity.50–54 In models of bone cancer pain, their expression is upregulated around 50%, directly driving nociceptive gain.55 P2X3 does not function alone. Stephan et al29 confirmed that P2X3 forms a physiologically relevant protein complex with the acid-sensing ion channel ASIC3, enabling nociceptors to integrate acidic (tissue acidosis) and purinergic signals together, ensuring efficient transduction of metabolic stress to neuronal firing.
On the glial side, ATP drives constant remodeling via P2X7 receptors. Receptor activation does not exhibit the rapid gating of the neuronal P2X3 in this case, but rather signals through to other pathways that modify neuronal phenotype. As covered by Wang H et al,56 SGCs that downregulate P2X7 possess neurons with lower levels of TRPV1 expression. In this case, glial purinergic signaling operates trans-cellularly to “tune” ion channel expression: glial and neuronal compartments connected.
Finally, SGCs have the potential to modulate neurotransmission in an active capacity through the controlled release of soluble factors. Li et al57 demonstrated that SGCs secrete diazepam-binding inhibitor (DBI) which acts as a negative modulator of neuronal GABAA receptors, thereby selectively gating mechanosensory transmission. In pathological communication, vesicular transport is implicated. Zhao et al58 discovered that SGCs in an oxaliplatin exposed environment release exosomes with pro-nociceptive cargo to neurons, delivering a “sensitization signal” to them. Zhang et al59 demonstrated conclusion on the metabolic pathology side. The research showed that diabetes activates NR2A-Wnt-TLR2 signaling in SGCs. “Glial Metabolic Inflammation” is suggested to underlie diabetic neuropathy, whereby hyperglycemic stress is biologically transduced into an immunologic barricade of neuronal inhibition.
Neuro-Immune Interaction and Microenvironmental Remodeling
Upstream injury-interface mechanisms, including Schwann-cell-mediated neuroinflammatory signaling, retrograde axonal transport of lesion-derived cues, and altered blood–nerve/blood–DRG barrier permeability, may further shape the extent and timing of immune access to the DRG, although a detailed discussion of these processes lies beyond the central scope of this review. Immune cell recruitment is a critical step in the process of chronic pain, involving the infiltration of many branches of both adaptive and innate immunity to rewire the DRG environment. Unlike the common bias towards either T cells or the microglia, Lacagnina et al60 demonstrated that B cells act as instigators of neuropathic pain through the release of IgG and FcγR signaling. Their data reveal that FcγR knockout completely abolishes mechanical hyperalgesia and neuronal hyperexcitability, implicating humoral immunity in the existing pain paradigm. This hint provides potential treatment strategies for small cohorts of patients (especially those resistant to conventional treatments) specifically with B-cell therapies, or techniques aimed at clearing IgG such as plasmapheresis. Likewise, Martin et al61 discovered that anti-CV2/CRMP5 autoantibodies potentiate DRG neuronal hyperexcitability directly, establishing a thorough molecular basis of pain in autoimmune neuropathy.
About innate immunity, macrophages act as elaborate sensors, connecting global physiological status to local sensation. Konnova et al62 showed that modulation of macrophage potassium channels affect DRG neurons sensitisation. Correlating global pathology to local pain, Chivers et al63 discovered that chronic intermittent hypoxia drives M1 polarization of macrophages in the DRG. This explains a key mechanism through which pain sensitivity in patients with obstructive sleep apnea is worsened by systemic comorbidities, affecting peripheral sensitization through immune bridges.
Interestingly, this interaction is influenced by sexual dimorphism, an intriguing variable which has often been forgotten. The classic study of Sorge et al64 showed that males depend on the microglia/macrophage system (P2X4R-BDNF) to drive hyperalgesia while females depend on adaptive immune cells (possibly T lymphocytes). However, this is not simply a feature of rodent models. Transcriptomic data from human DRGs published by Ray et al65 confirmed this divergence: pain-related genes in males are enriched in neural plasticity and pro-inflammatory cytokines pathways, while females specifically engage unique immunomodulatory pathways (eg, interferon signaling). This mechanistic divergence dictates that clinical immunomodulatory trials must ensure rigorous sex stratification to avoid obscuring efficacy.
Finally, the intermediate tier of signaling extends beyond chemical signaling to that of physical remodeling. A recent paper by Xie et al66 revealed a newly discovered “neuro-vascular-mechanical” mechanism for spontaneous pain. Following nerve injury, vascular density increases due to inflammation-induced angiogenesis, and proliferation of pericytes allows pulsations of this vasculature to “mechanically fire” synchronous “bursting” activity in nearby sensory neurons via Piezo2 ion channels. This concept combines vascular remodeling and mechanotransduction to provide an explanation for the paroxysmal stabbing pain that patients report, and suggests that targeting angiogenic activity or Piezo2 modulation is likely to have distinctive analgesic properties.
Long-Term Regulatory Tier: Epigenetic Remodeling and Pain Memory
If the post-translational modifications via phosphorylation of ion channels are the “transient response” of pain, then epigenetic remodeling is the engraving of the “molecular memory” of pain. This layer reveals why chronic pain persists after the primary injury has mended. Unlike mutations that can alter DNA itself, epigenetic modifications are “reprogramming” of gene expression programs over the long haul by networks of histone modifications, DNA methylation and RNA modification, which transform transient noxious signals into persistent pathological states, creating a bridge across acute signaling and chronic memory.
Histone Modification: “Reading and Writing” of Chromatin States and Gene Switches
Histone-modifying enzymes are tasked with sealing the deal, converting transient signaling to a durable form of transcriptional reprogramming. These epigenetic “writers” inscribe the sensitization events we have previously touched on—upregulated TRP channels, collapse of potassium buffering—into the chromatin.
Mechanistically, this remodeling reflects a combined “push-pull” dysregulation of the electrophysiological imbalance described. On the excitatory side, PRMT5 catalyzes the symmetric dimethylation of Histone H3 (H3R2me2s) at the TRPV1 promoter to recruit the WDR5 complex and deposit active H3K4me3 marks, chemically converting transient stress signals to stable nociceptor hyperexcitability.18 Concurrently, the inhibitory tone is “cut” by G9a-mediated chromatin repression. In a foundational study, Laumet et al showed that nerve injury increases H3K9me2 enrichment at the promoters of multiple K⁺ channel genes in DRG neurons, leading to their coordinated transcriptional silencing during the acute-to-chronic pain transition. G9a inhibition or sensory neuron-specific deletion restored K⁺ channel expression and attenuated neuropathic pain, establishing this pathway as a causal epigenetic mechanism of persistent hyperexcitability.67 A complementary glial mechanism was later identified by Luo et al, who showed that TLR9-G9a signaling silences Kcnj10/Kir4.1 in SGCs through H3K9me2, thereby impairing K⁺ buffering and further amplifying neuronal excitability.49 Additionally, HDAC2 is the signal integrator for the microenvironment; EREG-EGFR activates it and it’s recruited by MeCP2 in a corepressor complex to silence KCNQ2/3 specifically.33 These enzymes do not merely modify histones but fundamentally rewrite the transcriptional logic of the DRG to sustain a pathological equilibrium.
DNA Methylation: The Most Persistent “Molecular Imprint”
While histone modifications are reversible through biological processes, DNA methylation is much more stable and long-lasting, thus it is considered as a “foundation” of epigenetic memory and provided strong evidence to the persistence of chronic pain.
A foundational study by Zhao et al demonstrated that peripheral nerve injury induces OCT1-dependent upregulation of DNMT3a in injured DRG neurons, which then binds the Kcna2 promoter and increases its DNA methylation, leading to transcriptional repression of Kcna2/Kv1.2. Blocking DNMT3a upregulation restored Kcna2 expression and attenuated neuropathic pain behaviors, whereas DNMT3a overexpression in naïve DRGs reduced Kv currents, increased neuronal excitability, triggered spinal dorsal horn sensitization, and produced neuropathic pain-like symptoms. These findings established DNMT3a-mediated Kcna2 silencing as a causal DNA methylation mechanism through which transient nerve injury is converted into a persistent pro-excitatory transcriptional state in primary afferent neurons.68
However, Chen et al69 using whole-genome bisulfite sequencing presented insight into the fact that these deep epigenetic marks are not strictly set in stone. They showed exercise preconditioning to be analgesic by the selective reversal of the hypermethylation of Ryr1 (calcium modulating) and Xirp2 (cytoskeletal remodeling). This is particularly clinically relevant as it ties body activity to epigenetic plasticity of the DRG, giving a solid epigenetic underpinning to the non-pharmaceutical treatment (eg, physical therapy) of pain.
The Epitranscriptomic Bridge: Locking the Chromatin Remodeling Machinery
Even though m6A RNA modification takes place largely after transcription, an increasing amount of research points towards these modifications having an essential role of connecting time-dependent cellular signal changes to changes in the transcriptional environment, such as chromatin remodeling.
Importantly, m6A can also promote pain memory indirectly by regulating the stability of epigenetic enzymes themselves. As shown by Li et al,70 nerve injury induces the upregulation of FTO (m6A eraser) in the DRG FTO selectively removes the m6A modification on the mRNA of Ehmt2 (encoding G9a) in DRG neurons, preventing YTHDF2-mediated degradation of this mRNA, ultimately stabilizing the accumulation of G9a protein. This “FTO-m6A-G9a” axis highlights the hierarchical transformation that occurs whereby RNA modifications provide “logistics support” for the continuous supply of necessary histone methyltransferases required for histone methylation/maintaining chromatin silencing.
This epitranscriptomic instability appears to be a conserved feature across the somatosensory axis. In the setting of inflammatory pain, Pan et al71 showed that the m6A writer METTL3 is downregulated in the spinal cord, resulting in TET1-mediated central sensitization. Although the central terminal is in focus in this work, it highlights a “whole body” collapse of m6A homeostasis, raising the possibility that methyltransferase targeted approaches might more durably restore epigenetic stability to both ends of the somatosensory pathway.
Post-Transcriptional Network Stabilization: Coordinated Maintenance of Pathological Homeostasis
In contrast to the simple on/off functions of single genes, non-coding RNAs (miRNAs, circRNAs) preserve the “pathological homeostasis” of sensitized neurons through the construction of elaborate networks of regulators. The most archetypical result is the “coordinated disinhibition” effect.
The miR-183 cluster (miR-96/182/183) is enriched in DRG nociceptive neurons. Tao et al72 reported that injury-mediated downregulation of miR-183 disrupts an anti-inflammatory axis where loss of inhibition on TGFα signaling enables a cascade within the CCL2/CCR2 axis that engenders simultaneous upregulation of pro-nociceptive effectors Nav1.7, Nav1.8, and TRPV1. This release of an entire excitatory module results in a sensitized state that is impervious to compensatory action that targets single ion channels.
Non-coding RNAs further increase the dimensionality of these networks by assuming non-canonical signaling functions to promote pathological feedback loops. Zhang et al73 discovered that miR-21 functions multifacetedly not only as a post transcriptional repressor, but also as a ligand for the Toll-like receptor 8 (TLR8) in sensory neurons. This secretion-based mechanism means that hyperexcitability can be relayed between cells in a paracrine fashion. Following nerve injury, upregulated miR-21 is secreted into endosomes, where it binds and activates TLR8 to fuel a pERK-mediated inflammatory cascade exacerbating neuronal excitability. Herein lies the “vicious cycle” where the non-coding RNA is functioning as a signaling agonist, entrapping the neuron in a self-perpetuating hyperexcitable state that is independent of the initial injury. Taken together these processes that range from the general to the specific, from coordinated unshackling of ion channels to novel RNA-receptor pairs form a new pathological homeostasis. To this end, a targeted approach to breaking into these non-coding RNA networks offers a “system reset” approach to chipping away at the large molecular edifice of “treatment resistant” pain.
Reversibility of Epigenetic Modifications and the Therapeutic Window
The translational potential of epigenetic therapies is severely constrained by biological complexity, in which “reversibility” is tightly bound by biological context.
A major factor is the timing of intervention, which differs between isoforms. Broad Class I HDAC inhibitors have a narrow therapeutic window restricted to pre-injury “induction phase”,74 but Westlund et al showed that selective inhibition of Class IIa HDAC5 is effective up to three weeks following injury.75 This hints at a molecular “handover” as different isoforms dictate the formation and maintenance of pain memory. Conversely, DNA methylation remodeling has considerable longevity and a tendency towards consolidation,76 indicating a deeply entrenched epigenetic change in chronic pain. This raises a concern that drugs that only target early stage mechanisms might fail to reverse stable methylation states.
Superimposed over these time constraints is the potent effect of biological sex. ATAC-seq profiling of the human DRG reveals a surprising architecture—there is much more chromatin accessibility on the X chromosome in female neurons than in males, and deeper accessibility in GABAergic and interferon gene clusters.77 The finding suggested that memory for pain in females is utilising much wider networks of genomic real estate and molecular programmes. A successful translation process needs to embrace stratified precision medicine and harness techniques that are specifically tailored to the stage of disease, and to the sex-determined epigenetic profile of the patient.
Validation and Variation of the Hierarchical Model in Specific Pathologies
While the “Acute-Intermediate-Long-term” hierarchical regulation model provides a generic framework for peripheral sensitization, the specifics of the spatiotemporal dynamics have significant differences in pathology-specific ways. Understanding the “acceleration”, “jumping” or “stagnation” in sub-models is important to specify the applications for stratified therapies (Figure 3).
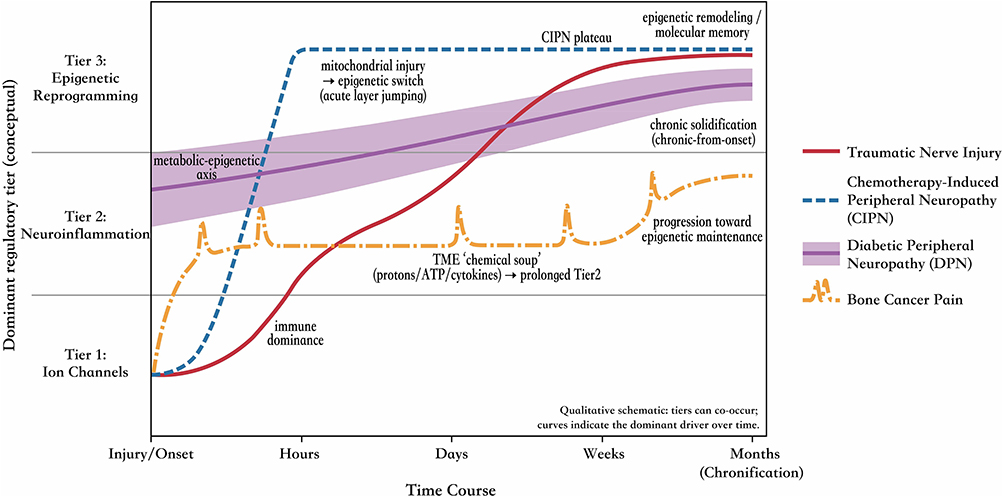 |
Figure 3 Pathology-specific trajectories of the hierarchical regulation model across time. Qualitative schematic illustrating how the dominant regulatory driver shifts over the course from onset to chronification, while acknowledging that tiers can co-occur. Distinct curves depict representative pain etiologies with different spatiotemporal “kinetics” of tier engagement—capturing acceleration, stagnation, or layer jumping rather than a uniform sequence. Traumatic nerve injury follows a canonical multi-step cascade, progressing from early electrophysiological dysfunction to a prominent neuroimmune phase and ultimately consolidating into epigenetic remodeling, with intermediate-tier immune dominance shaping the clinical phenotype. Chemotherapy-induced peripheral neuropathy (CIPN) exemplifies mitochondrial-driven “acute layer jumping”, in which direct organelle injury rapidly engages metabolic/epigenetic programs and compresses (or bypasses) the expected early window dominated by excitability changes. Diabetic peripheral neuropathy (DPN) shows “chronic-from-onset” solidification, where persistent systemic metabolic stress produces early, diffuse long-term-tier activation without a discrete acute trigger point. Bone cancer pain features a prolonged intermediate tier driven by tumor–nerve interface signals (a sustained “chemical soup” and growth-factor signaling), with downstream recruitment of durable transcriptional/epigenetic reprogramming. Symbol explanation: right arrows (→) indicate proposed directional mechanistic progression between pathological events and regulatory-tier engagement. |
CIPN: Mitochondrial-Driven “Layer Jumping”
CIPN’s pathological progression differs greatly from other types of traumatic nerve injury. Unlike the electrophysiological initiation followed by inflammation in trauma, this disorder has a significant feature called “Acute Layer Jumping”.
The principal targets of these chemotherapeutics (eg, paclitaxel, oxaliplatin) have a significant effect on intracellular mitochondria. It has been demonstrated that platinum drugs can directly form adducts with mtDNA resulting in energy failure, whereas paclitaxel induces opening of the mitochondrial permeability transition pore (mPTP).78,79 This direct organelle damage implies that CIPN mechanisms effectively bypass the purely electrophysiological induction phase, directly triggering metabolic remodeling and epigenetic changes characteristic of the long-term tier.
Luo et al49 supported this idiosyncratic trajectory. They found that activation of the TLR9-G9a pathway in SGCs is not dependent on neuronal electrical activity but rather is triggered by the direct release of mtDNA, causing transcriptional silencing of Kir4.1 at a relatively early timepoint. This sheds light on why “classical” sodium channel blockers are often futile at treating CIPN since the therapeutic window must be shifted forward (upstream). Strategies such as using lansoprazole as OCT2 transport inhibitor80 inherently adopt a “prophylactic” or “immediate-early” paradigm and achieve blockade against mitochondrial toxicity before “layer jumping” to epigenetic silence becomes permanent.
Traumatic Neuropathic Pain: Full Hierarchical Cascade with Immune Dominance
Traumatic nerve injury (eg, SNL/CCI models) exemplifies the standard mode of hierarchical control, featuring a transparent “three-step” cascade: axonal breakage leads to a more rapid accumulation of Nav channels (acute tier), after triggering Wallerian degeneration, inciting massive immune infiltration (intermediate tier), then evolves to enduring remodeling of the chromatin (long-term tier).
However, a defining phenotypic feature of this pathology is the excessive weighting and importance given to the intermediate layer and the adaptive immune response. Following traumatic rupture of the blood-nerve barrier (BNB), the B cell-IgG-FcγR signaling axis uncovered by Lacagnina et al60 showed that after a physical nerve injury, the humoral immune response does not just act as an amplifier of the environment but as a driver of sensitization in itself. The assumption of autoantibodies as paving the way for this deleterious role is also amplified by mechanistic parallels exhibited by Martin et al61 in paraneoplastic syndromes, whereby anti-CV2/CRMP5 autoantibodies facilitated sensory neuron hyperexcitability. These findings underscore an “Immune-Dominant” profile and suggest that immunomodulatory interventions targeting the intermediate tier (eg, antibody clearance or B-cell depletion) possess superior translational potential for this specific patient cohort compared to other pain etiologies.
Diabetic Peripheral Neuropathy (DPN): Metabolic-Epigenetic “Chronic Solidification”
DPN represents another extreme of the pathological spectrum: “insidious onset and early solidification”. Due to the absence of a distinct temporal point of mechanical or chemical insult, the systemic persistence of a hyperglycemic environment results in DPN lacking a clear “acute trigger point”. Instead, the model in DPN manifests as the early and diffuse activation of the long-term tier.
Driven by systemic metabolic stress, signaling axes such as the NR2A-Wnt-TLR2 pathway in SGCs remain in a state of chronic activation.59 This leads to extensive remodeling of the neuronal gene expression profile via epigenetic mechanisms before the emergence of overt pain behaviors. For instance, Ascl1/Lhx6-dependent GABAergic neuronal dysfunction has been confirmed not as an acute event, but rather as a collapse of transcriptional programs resulting from prolonged metabolic toxicity.81
This “chronic-from-onset” characteristic implies that for DPN patients, searching for a traditional “acute-phase intervention window” (eg, blocking sodium channels) may be of limited utility. This characteristic dictates a shift in clinical trial design: DPN cohorts must be strictly stratified by metabolic history rather than pain intensity alone. The therapeutic focus should be directed toward erasing these solidified “metabolic memories”—for example, by targeting Sirtuin activators or HDAC inhibitors to reverse epigenetic imprints—strategies that address the fundamental transcriptional collapse that renders standard sodium channel blockers ineffective.
Bone Cancer Pain: A Unique “Tumor-Nerve Interaction Layer”
Bone cancer pain introduces an exogenous biological variable into the hierarchical model—the tumor microenvironment (TME)—which results in a profoundly elongated and complicated intermediate tier.
Unfurling to the interconnected lore of the TME, the alterations create an unbearable “chemical soup” consisting of protons and ATP, tonically activating the ASIC3-P2X3 complexes described above (Satellite Glial Cell Activation and Purinergic Signaling). Beyond these acute activators, growth factors secreted by the tumor (EREG, for example) directly hijack the neuronal transcriptional machinery. Developed by Zhang ZX et al,33 the EREG/EGFR-ERK-Runx1-HDAC2-KCNQ axis exemplifies how pathological EREG dysregulation directly underpins epigenetic silencing in the long-term tier via signaling molecules in the mid-tier.
This brings us to another caveat—you cannot treat bone cancer pain by simply repairing the aberrant DRG as a functioning unit. Strategy dictates total severing of this tumor-specific signaling interface with the nerve (eg, EGFR, Runx1) to stop the pathologic source of its aberrant input contributing endlessly to the epigenetic remodeling of the nociceptor. Severing this conduit might stop a malignant feed-forward loop backwards in which neuropeptide released by the neurons (substance P, CGRP) also purportedly promotes tumor growth in the microenvironment.
Translational Progress and Future Directions
Establishment of the “Human-First” Paradigm
Suzetrigine (VX-548) is the most consequential translational success in the pain field of the last several years—the first approved acute analgesic based on a new peripheral target in decades.82 Suzetrigine’s development forged a path for the newly minted “Human-First” approach: the target (loss-of-function mutations in SCN10A) comes from human genetic evidence, and efficacy evaluation occurs on human DRG neurons from organ donors.2,82 Importantly, the drug has high selectivity for Nav1.8 and its distinct profile compared to earlier rodent-active compounds, the development strategy fundamentally emphasized human tissue data, proving that “You don’t have to cure mice to cure people sometimes”.
This paradigm is being extended to other molecular targets. A TRPA1 antagonist LY3526318 has completed Phase I trials,23 and recent humanized anti-P2X4 antibodies1 and CCR2 pepducins45 hold potential as therapeutics for “Intermediate Tier” mechanisms. In addition to designing new candidate drugs, repositioning existing agents based on similar mechanism and pathway repositioning is another accelerated method of drug discovery. To support the predictiveness of our pathology-specific models, lansoprazole has been demonstrated effective prophylactically against CIPN by inhibiting OCT2-mediated accumulation and addressing the “layer jumping” mechanism (Sec 6.1).80 Baricitinib83 and Stattic84 aim for the JAK/STAT signaling hub (Sec 4.1), highlighting how definition of the “Intermediate Tier” facilitates repurposing of existing immunomodulators for neuropathic pain.
Finally, we have also seen DRG stimulation (DRG-S) used as a macroscopic treatment, with the accurate trial demonstrating that DRG-S is superior to SCS for the treatment of CRPS.85 By acting directly on the excitability of the soma—the “Hub” of sensitization—DRG-S blocks nociceptive transmission without regarding to the molecular context of that transmission. The success of DRG-S lends clinical weight to the concept of the dorsal root ganglion, and specifically renders it a surgical target.
Multidimensional Analysis of Translational Barriers
Despite the breakthroughs brought by the “human-first” paradigm, the pain field still faces systemic obstacles ranging from target validation to clinical assessment.
Complexity at the Target Level
The cautionary tale of the Nav1.7 translational gap showed that despite having the most robust human genetic evidence, several very selective Nav1.7 inhibitors failed in Phase II/III trials. Yang et al86 noticed a systemic mismatch between the preclinical and clinical world, in which rodent models are often young male rodents with acute inflammation, yet trials are often in heterogeneous cohorts with chronic neuropathic pain. Mechanistic concerns were highlighted by Eagles et al87 around this “functional redundancy” barrier, whereby efficacy in vivo is blunted by the presence/upregulation of other sodium channel isoforms (such as Nav1.9). Stewart et al2 found that in human DRG, even in the presence of complete Nav1.8 block, Nav1.7 can still fire action potentials (and vice versa). Based on this redundancy, combinatorial strategies are more rational than monotherapies, surpassing the limitations of the “one-gene, one-drug” model.
Infrastructure vs. Species Barriers
While it is now well-accepted that transcriptomic differences exist between human and rodent DRGs,4,5,17 the dilemma lies in standardizing their use, in which major consortia like the PRECISION Human Pain Network under the NIH HEAL Initiative are currently working towards the systematic characterization of human pain tissues.82 By harmonizing the global recovery protocols of human tissues and developing matched datasets with hiPSC-SNs,35 the field is transitioning from merely acknowledging species differences to constructing a human-focused preclinical infrastructure that reduces translational attrition.
The Metrics Crisis: Subjective vs. Objective
Lastly, we have methodological roadblocks in therapeutic assessment. The classical tool of subjective metrics (VAS/NRS) turns out to be fraught with variability and placebo response rates reaching 30–40% that essentially shrink the window of detectable therapeutic and increase the Number Needed to Treat (NNT).88–90 In a fight against such noise, phenotypic objective stratification is key. Quantitative Sensory Testing (QST) is a partial answer to this;91 we have now come to the “golden time” of the fluid biomarks. Neurofilament Light Chain (NfL) in serum is a readout of structural neuronal damage;92 circulating miRNAs (miR-183-5p downregulation) provides hints about their epigenetic status, for example.93 Such biomarkers make the “mechanism based stratification” possible—if we are guaranteed that patients are enrolled based on matching biological drivers (active degeneration vs. epigenetic remodeling), then trials can sift out the phenotypic “noise” and significantly reduce the NNT of the novel therapeutics.
Limitations of the Review
Several limitations of this review warrant acknowledgment. First, regarding mechanistic coverage, we prioritized the three core tiers of ion channels, neuroinflammation, and epigenetics. Thus, while we addressed mitochondrial toxicity in the specific context of chemotherapy-induced neuropathy (CIPN: Mitochondrial-Driven “Layer Jumping”), a broader discussion of metabolic dynamics (such as autophagy regulation and the gut-brain-DRG axis) across other pain etiologies remains relatively limited. Second, despite emphasizing the importance of human DRG research, current studies on human tissues face challenges regarding sample representativeness: organ donors are primarily brain-dead patients whose physiological status may diverge from healthy populations. Furthermore, sample acquisition is constrained by ethical approval and tissue preservation conditions, hindering the expansion of research scales. Third, although sex-specific mechanisms are highlighted throughout the text (eg, in immune interaction and chromatin accessibility), a systematic, distinct integration of these variables warrants a dedicated future review. Finally, this review strictly prioritizes peripheral sensitization; readers are encouraged to consult specialized literature for comprehensive insights into central sensitization and peripheral-central gating mechanisms.
Conclusions
The evidence reviewed here supports a DRG-centered Hierarchical Regulation Model in which pain chronification emerges from the interaction of acute ion-channel remodeling, intermediate neuro-glial-immune communication, and long-term epigenetic consolidation. Its pathology-specific manifestations—including “layer jumping” in CIPN, immune-dominant progression after traumatic nerve injury, and early metabolic-epigenetic solidification in DPN—further argue against a uniform linear account of chronic pain. For clinicians, researchers, and ultimately patients, this framework provides a rationale for mechanism-informed disease stratification, more human-relevant target validation, and more precise therapeutic development.
The success of Suzetrigine marks the dawn of a new “human-first” era in analgesic drug development. As Copits et al82 noted, the next two to five years will represent a critical window for translating knowledge gained from human DRG research into highly targeted therapeutics. To move this agenda beyond a general call for precision medicine, several immediate steps are needed: prospective sex-stratified efficacy and safety analyses in DRG-related clinical trials, given the growing evidence for sex-dependent molecular and epigenomic programs in human DRGs;65,77 greater use of human DRG neurons or validated human sensory-neuron platforms as translational filters in analgesic candidate selection;2,35 and, in the longer term, progressive incorporation of such human-relevant pharmacological evidence into future regulatory evaluation frameworks for mechanism-based analgesics.
Acknowledgments
The authors appreciate the time and effort expended by the participants. We are also grateful to Professor Zhen Wang for support with his inspiration.
Funding
This work is financially and experimentally supported by the following provincial research project: Study on the Method and Mechanism of Functional Bundle Identification and Promoting Chemotactic Repair during Peripheral Nerve Transection Injury. Grant Number: Natural Science Foundation of Heilongjiang Province (PL2024H218).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zurek NA, Shilling MW, Demeter JB, et al. Humanized anti-P2X4 scFv reduces ATP-induced P2X4 currents and modulates excitability in human DRG neurons. Mol Pain. 2025;21:17448069251376200. doi:10.1177/17448069251376200
2. Stewart RG, Osorno T, Fujita A, et al. Modulation of human dorsal root ganglion neuron firing by the Nav1.8 inhibitor suzetrigine. Proc Natl Acad Sci U S A. 2025;122(22):e2503570122. doi:10.1073/pnas.2503570122
3. Su M, Ouyang X, Zhou P, et al. Inhibition of TTX-S Na(+) currents by a novel blocker QLS-278 for antinociception. J Pharmacol Exp Ther. 2025;392(2):100030. doi:10.1124/jpet.124.002273
4. Shiers S, Klein RM, Price TJ. Quantitative differences in neuronal subpopulations between mouse and human dorsal root ganglia demonstrated with RNAscope in situ hybridization. Pain. 2020;161(10):2410–17. doi:10.1097/j.pain.0000000000001973
5. O’Brien JA, Lesnak JB, Price TJ. Neuropharmacology and physiology of human dorsal root ganglion neurons responsible for nociception: state-of-the-art and future directions. Pain. 2025;166(11s):S8–s14. doi:10.1097/j.pain.0000000000003663
6. Wang H, Zuo W, Feng X, et al. ESRRG-controlled downregulation of KCNN1 in primary sensory neurons is required for neuropathic pain. JCI Insight. 2024;9(12). doi:10.1172/jci.insight.180085
7. Lei X, Chen M, Liu X, et al. Activation of cannabinoid CB1 receptors suppresses HCN channels function in dorsal root ganglion neurons of rats. Brain Res Bull. 2025;229:111425. doi:10.1016/j.brainresbull.2025.111425
8. Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci U S A. 1999;96(14):7723–7730. doi:10.1073/pnas.96.14.7723
9. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926. doi:10.1016/j.jpain.2009.06.012
10. Goodwin G, McMahon SB. The physiological function of different voltage-gated sodium channels in pain. Nat Rev Neurosci. 2021;22(5):263–274. doi:10.1038/s41583-021-00444-w
11. Osteen JD, Immani S, Tapley TL, et al. Pharmacology and mechanism of action of suzetrigine, a potent and selective Na(V)1.8 pain signal inhibitor for the treatment of moderate to severe pain. Pain Ther. 2025;14(2):655–674. doi:10.1007/s40122-024-00697-0
12. Zhang X, Hartung JE, Gold MS. Persistent (Na v 1.9) sodium currents in human dorsal root ganglion neurons. Pain. 2025;166(2):448–459. doi:10.1097/j.pain.0000000000003394
13. Wang Y, Shu J, Yang H, et al. Nav1.7 modulator bearing a 3-hydroxyindole backbone holds the potential to reverse neuropathic pain. ACS Chem Neurosci. 2024;15(6):1063–1073. doi:10.1021/acschemneuro.3c00353
14. Liu JK, Zhou ZS, Wang SH, et al. SARS-CoV-2 N protein interacts with Na v 1.7 to promote pathological pain. Pain. 2025;166(11):e635–e651. doi:10.1097/j.pain.0000000000003675
15. Feng Y, Yan F, Chen D, et al. Synergistic Inhibition of Nav1.7 and NCX1: a novel strategy for treating cancer-induced bone pain by modulating pain sensitization and neuronal inflammation. CNS Neurosci Ther. 2025;31(4):e70389. doi:10.1111/cns.70389
16. Wang C, Chen R, Zhu X, Zhang X, Lian N. Long noncoding RNA small nucleolar RNA host gene 5 facilitates neuropathic pain in spinal nerve injury by promoting SCN9A expression via CDK9. Hum Cell. 2024;37(2):451–464. doi:10.1007/s13577-023-01019-w
17. Bhuiyan SA, Xu M, Yang L, et al. Harmonized cross-species cell atlases of trigeminal and dorsal root ganglia. Sci Adv. 2024;10(25):eadj9173. doi:10.1126/sciadv.adj9173
18. Yeh CM, Lai CY, Peng HY, et al. Protein arginine methyltransferase 5 contributes to paclitaxel-induced neuropathic pain by activating transient receptor potential vanilloid 1 epigenetic modification in dorsal root ganglion. Anesth Analg. 2024;138(5):1107–1119. doi:10.1213/ane.0000000000006595
19. Li Y, Uhelski ML, North RY, et al. ApoA-I binding protein (AIBP) regulates transient receptor potential vanilloid 1 (TRPV1) activity in rat dorsal root ganglion neurons by selective disruption of toll-like receptor 4 (TLR4)-lipid rafts. Brain Behav Immun. 2025;123:644–655. doi:10.1016/j.bbi.2024.10.017
20. Franco-Enzástiga Ú, Natarajan K, Espinosa F, Granja-Vazquez R, Mydugolam H, Price TJ. Type I IFNs enhance human dorsal root ganglion nociceptor excitability and induce TRPV1 sensitization. JCI Insight. 2025;10(19). doi:10.1172/jci.insight.194987
21. Artero-Morales M, González-Rodríguez S, Ferrer-Montiel A. TRP channels as potential targets for sex-related differences in migraine pain. Front Mol Biosci. 2018;5:73. doi:10.3389/fmolb.2018.00073
22. Paige C, Barba-Escobedo PA, Mecklenburg J, et al. Neuroendocrine mechanisms governing sex differences in hyperalgesic priming involve prolactin receptor sensory neuron signaling. J Neurosci. 2020;40(37):7080–7090. doi:10.1523/jneurosci.1499-20.2020
23. Broad LM, Suico JG, Turner PK, et al. Preclinical and clinical evaluation of a novel TRPA1 antagonist LY3526318. Pain. 2025;166(8):1893–1908. doi:10.1097/j.pain.0000000000003570
24. Lee SH, Tonello R, Lee K, et al. The Parkinson’s disease DJ-1/PARK7 gene controls peripheral neuronal excitability and painful neuropathy. Brain. 2025;148(5):1639–1651. doi:10.1093/brain/awae341
25. Du L, Zhu J, Liu S, et al. Transient receptor potential melastatin 8 contributes to the interleukin-33-mediated cold allodynia in a mouse model of neuropathic pain. Pain. 2025;166(2):347–359. doi:10.1097/j.pain.0000000000003346
26. Gao Y, Yan S, Zhang Z, et al. Copine-6 is a TRPM3 escort protein controlling the sensitivity of sensory neurons to noxious heat. EMBO J. 2025;44(15):4222–4251. doi:10.1038/s44318-025-00487-0
27. Deval E, Noël J, Gasull X, et al. Acid-sensing ion channels in postoperative pain. J Neurosci. 2011;31(16):6059–6066. doi:10.1523/jneurosci.5266-10.2011
28. Marra S, Ferru-Clément R, Breuil V, et al. Non-acidic activation of pain-related Acid-Sensing Ion Channel 3 by lipids. EMBO J. 2016;35(4):414–428. doi:10.15252/embj.201592335
29. Stephan G, Huang L, Tang Y, et al. The ASIC3/P2X3 cognate receptor is a pain-relevant and ligand-gated cationic channel. Nat Commun. 2018;9(1):1354. doi:10.1038/s41467-018-03728-5
30. Qiu F, Wei X, Zhang S, Yuan W, Mi W. Increased expression of acid-sensing ion channel 3 within dorsal root ganglia in a rat model of bone cancer pain. Neuroreport. 2014;25(12):887–893. doi:10.1097/wnr.0000000000000182
31. Holton CM, Strother LC, Dripps I, Pradhan AA, Goadsby PJ, Holland PR. Acid-sensing ion channel 3 blockade inhibits durovascular and nitric oxide-mediated trigeminal pain. Br J Pharmacol. 2020;177(11):2478–2486. doi:10.1111/bph.14990
32. Papalampropoulou-Tsiridou M, Shiers S, Wang F, Godin AG, Price TJ, De Koninck Y. Distribution of acid-sensing ion channel subunits in human sensory neurons contrasts with that in rodents. Brain Commun. 2022;4(6):fcac256. doi:10.1093/braincomms/fcac256
33. Zhang ZX, Tian Y, Li S, et al. Involvement of HDAC2-mediated kcnq2/kcnq3 genes transcription repression activated by EREG/EGFR-ERK-Runx1 signaling in bone cancer pain. Cell Commun Signal. 2024;22(1):416. doi:10.1186/s12964-024-01797-2
34. Habib M, Wiessler AL, Fischer P, et al. Neuropathic pain and distinct CASPR2 autoantibody IgG subclasses drive neuronal hyperexcitability. Neurol Neuroimmunol Neuroinflamm. 2025;12(4):e200423. doi:10.1212/nxi.0000000000200423
35. Zurek NA, Ehsanian R, Goins AE, et al. Electrophysiological analyses of human dorsal root ganglia and human induced pluripotent stem cell-derived sensory neurons from male and female donors. J Pain. 2024;25(6):104451. doi:10.1016/j.jpain.2023.12.008
36. Pinho-Ribeiro FA, Verri WA Jr, Chiu IM. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol. 2017;38(1):5–19. doi:10.1016/j.it.2016.10.001
37. de Macedo FHP, Aires RD, Fonseca EG, et al. TNF-α mediated upregulation of Na(V)1.7 currents in rat dorsal root ganglion neurons is independent of CRMP2 SUMOylation. Mol Brain. 2019;12(1):117. doi:10.1186/s13041-019-0538-0
38. Fang D, Kong LY, Cai J, et al. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: roles in the development of bone cancer pain in a rat model. Pain. 2015;156(6):1124–1144. doi:10.1097/j.pain.0000000000000158
39. Stover JD, Farhang N, Lawrence B, Bowles RD. Multiplex epigenome editing of dorsal root ganglion neuron receptors abolishes redundant interleukin 6, tumor necrosis factor alpha, and interleukin 1β signaling by the degenerative intervertebral disc. Hum Gene Ther. 2019;30(9):1147–1160. doi:10.1089/hum.2019.032
40. Denk F, Bennett DL, McMahon SB. Nerve growth factor and pain mechanisms. Annu Rev Neurosci. 2017;40:307–325. doi:10.1146/annurev-neuro-072116-031121
41. O-Sullivan I, Kc R, Singh G, et al. Sensory neuron-specific deletion of tropomyosin receptor kinase A (TrkA) in mice abolishes osteoarthritis (OA) pain via NGF/TrkA intervention of peripheral sensitization. Int J Mol Sci. 2022;23(20). doi:10.3390/ijms232012076
42. Schnitzer TJ, Marks JA. A systematic review of the efficacy and general safety of antibodies to NGF in the treatment of OA of the Hip or knee. Osteoarthritis Cartilage. 2015;23(Suppl 1):S8–17. doi:10.1016/j.joca.2014.10.003
43. Fallon M, Sopata M, Dragon E, et al. A randomized placebo-controlled trial of the anti-nerve growth factor antibody tanezumab in subjects with cancer pain due to bone metastasis. Oncologist. 2023;28(12):e1268–e1278. doi:10.1093/oncolo/oyad188
44. Sharma D, Feng X, Wang B, et al. Proneurotrophin-3 contributes to chemotherapy-induced neuropathic pain through TrkC-mediated CCL2 elevation in DRG neurons. EMBO Rep. 2025;26(24):6141–6158. doi:10.1038/s44319-025-00534-1
45. Midavaine É, Brouillette RL, Théberge E, et al. Discovery of a CCR2-targeting pepducin therapy for chronic pain. Pharmacol Res. 2024;205:107242. doi:10.1016/j.phrs.2024.107242
46. Zhang Y, Wang B, Feng X, et al. RNA-binding protein SYNCRIP contributes to neuropathic pain through stabilising CCR2 expression in primary sensory neurones. Br J Anaesth. 2024;133(5):1028–1041. doi:10.1016/j.bja.2024.07.024
47. Leng SZ, Fang MJ, Wang YM, et al. Elevated plasma CXCL12 leads to pain chronicity via positive feedback upregulation of CXCL12/CXCR4 axis in pain synapses. J Headache Pain. 2024;25(1):213. doi:10.1186/s10194-024-01917-w
48. Yang H, Morgan TS, Petruzzelli S, et al. Nociceptor α7nAChR activation blunts neuronal HMGB1 release and attenuates inflammation and nociceptive behavior. Mol Med. 2025;31(1):324. doi:10.1186/s10020-025-01387-z
49. Luo Q, Jiang J, Zhu Q, et al. TLR9 in satellite glial cells promotes paclitaxel-induced neuropathic pain by reducing Kir4.1 transcription through histone methylation activation. Brain Behav Immun. 2025;128:65–82. doi:10.1016/j.bbi.2025.03.037
50. Jayaraj ND, Bhattacharyya BJ, Belmadani AA, et al. Reducing CXCR4-mediated nociceptor hyperexcitability reverses painful diabetic neuropathy. J Clin Invest. 2018;128(6):2205–2225. doi:10.1172/jci92117
51. Chen Z, Zhang C, Song X, et al. BzATP activates satellite glial cells and increases the excitability of dorsal root ganglia neurons in vivo. Cells. 2022;11(15). doi:10.3390/cells11152280
52. Kubo A, Iwata K, Shinoda M, Shinozuka H, Taguchi T, Mizumura K. Purinergic receptor- and metabotropic glutamate receptor 5-mediated interactions between satellite glial cells and neurons in rat trigeminal ganglia. Glia. 2026;74(2):e70126. doi:10.1002/glia.70126
53. Chen Z, Huang Q, Song X, et al. Purinergic signaling between neurons and satellite glial cells of mouse dorsal root ganglia modulates neuronal excitability in vivo. Pain. 2022;163(8):1636–1647. doi:10.1097/j.pain.0000000000002556
54. Belgi A, Burnley JV, MacRaild CA, et al. Alkyne-bridged α-conotoxin Vc1.1 potently reverses mechanical allodynia in neuropathic pain models. J Med Chem. 2021;64(6):3222–3233. doi:10.1021/acs.jmedchem.0c02151
55. Wu JX, Xu MY, Miao XR, et al. Functional up-regulation of P2X3 receptors in dorsal root ganglion in a rat model of bone cancer pain. Eur J Pain. 2012;16(10):1378–1388. doi:10.1002/j.1532-2149.2012.00149.x
56. Wang H, Chen L, Xing J, Shi X, Xu C. Reduction of TRPV1 expression on neurons due to downregulation of P2X7R in neonatal rat dorsal root ganglion satellite glial cells under co-culture conditions. Biol Cell. 2024;116(10):e2400021. doi:10.1111/boc.202400021
57. Li X, Prudente AS, Prato V, et al. Peripheral gating of mechanosensation by glial diazepam binding inhibitor. J Clin Invest. 2024;134(16). doi:10.1172/jci176227
58. Zhao L, Liu S, Zhang X, et al. Satellite glial cell-secreted exosomes after in-vitro oxaliplatin treatment presents a pro-nociceptive effect for dorsal root ganglion neurons and induce mechanical hypersensitivity in naïve mice. Mol Cell Neurosci. 2023;126:103881. doi:10.1016/j.mcn.2023.103881
59. Zhang YY, Zhu DX, Wang MY, et al. Activation of NR2A-Wnt-TLR2 signaling axis in satellite glial cells of the dorsal root ganglion contributes to neuropathic pain induced by nerve injury in diabetic mice. Mol Neurobiol. 2025;62(6):8013–8037. doi:10.1007/s12035-025-04754-3
60. Lacagnina MJ, Willcox KF, Boukelmoune N, et al. B cells drive neuropathic pain-related behaviors in mice through IgG-Fc gamma receptor signaling. Sci Transl Med. 2024;16(766):eadj1277. doi:10.1126/scitranslmed.adj1277
61. Martin L, Stratton HJ, Salih LY, et al. Anti-CV2/CRMP5 autoantibodies as drivers of sensory neuron excitability and pain in rats. Nat Commun. 2025;16(1):7311. doi:10.1038/s41467-025-62380-y
62. Konnova EA, Deftu AF, Chu Sin Chung P, Kirschmann G, Decosterd I, Suter MR. Potassium channel modulation in macrophages sensitizes dorsal root ganglion neurons after nerve injury. Glia. 2024;72(4):677–691. doi:10.1002/glia.24496
63. Chivers SB, Andrade MA, McLay CL, et al. Chronic intermittent hypoxia drives M1 macrophage polarization in dorsal root ganglia. Brain Behav Immun. 2025;129:442–452. doi:10.1016/j.bbi.2025.06.028
64. Sorge RE, Mapplebeck JC, Rosen S, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18(8):1081–1083. doi:10.1038/nn.4053
65. Ray PR, Shiers S, Caruso JP, et al. RNA profiling of human dorsal root ganglia reveals sex differences in mechanisms promoting neuropathic pain. Brain. 2023;146(2):749–766. doi:10.1093/brain/awac266
66. Xie W, Lückemeyer DD, Qualls KA, et al. Vascular motion in the dorsal root ganglion sensed by Piezo2 in sensory neurons triggers episodic pain. Neuron. 2025;113(11):1774–1788.e5. doi:10.1016/j.neuron.2025.03.006
67. Laumet G, Garriga J, Chen SR, et al. G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat Neurosci. 2015;18(12):1746–1755. doi:10.1038/nn.4165
68. Zhao JY, Liang L, Gu X, et al. DNA methyltransferase DNMT3a contributes to neuropathic pain by repressing Kcna2 in primary afferent neurons. Nat Commun. 2017;8:14712. doi:10.1038/ncomms14712
69. Chen B, Wang T, Zhu C, et al. Identification of potential intervention targets involved in prior exercise that attenuates peripheral neuropathic pain by integrating transcriptome and whole-genome bisulfite sequencing analyses. Mol Neurobiol. 2025;62(5):6562–6575. doi:10.1007/s12035-025-04696-w
70. Li Y, Guo X, Sun L, et al. N(6)-methyladenosine demethylase FTO contributes to neuropathic pain by stabilizing G9a expression in primary sensory neurons. Adv Sci. 2020;7(13):1902402. doi:10.1002/advs.201902402
71. Pan Z, Zhang Q, Liu X, et al. Methyltransferase-like 3 contributes to inflammatory pain by targeting TET1 in YTHDF2-dependent manner. Pain. 2021;162(7):1960–1976. doi:10.1097/j.pain.0000000000002218
72. Tao Z, Zhou Y, Zeng B, Yang X, Su M. MicroRNA-183 attenuates osteoarthritic pain by inhibiting the TGFα-mediated CCL2/CCR2 signalling axis. Bone Joint Res. 2021;10(8):548–557. doi:10.1302/2046-3758.108.Bjr-2019-0308.R2
73. Zhang ZJ, Guo JS, Li SS, et al. TLR8 and its endogenous ligand miR-21 contribute to neuropathic pain in murine DRG. J Exp Med. 2018;215(12):3019–3037. doi:10.1084/jem.20180800
74. Denk F, Huang W, Sidders B, et al. HDAC inhibitors attenuate the development of hypersensitivity in models of neuropathic pain. Pain. 2013;154(9):1668–1679. doi:10.1016/j.pain.2013.05.021
75. Westlund KN, Montera M, Goins AE, et al. Epigenetic HDAC5 inhibitor reverses craniofacial neuropathic pain in mice. J Pain. 2024;25(2):428–450. doi:10.1016/j.jpain.2023.09.015
76. Topham L, Gregoire S, Kang H, et al. The transition from acute to chronic pain: dynamic epigenetic reprogramming of the mouse prefrontal cortex up to 1 year after nerve injury. Pain. 2020;161(10):2394–2409. doi:10.1097/j.pain.0000000000001917
77. Franco-Enzástiga Ú, Inturi NN, Natarajan K, et al. Epigenomic landscape of the human dorsal root ganglion: sex differences and transcriptional regulation of nociceptive genes. Pain. 2025;166(3):614–630. doi:10.1097/j.pain.0000000000003508
78. Podratz JL, Knight AM, Ta LE, et al. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis. 2011;41(3):661–668. doi:10.1016/j.nbd.2010.11.017
79. Carré M, André N, Carles G, et al. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem. 2002;277(37):33664–33669. doi:10.1074/jbc.M203834200
80. Kobayashi A, Ikemura K, Ueno M, Wakai E, Yamane F, Okuda M. Lansoprazole identified as a prophylactic agent for oxaliplatin-induced peripheral neuropathy using integrated in silico, in vitro, and in vivo analyses. Biomed Pharmacother. 2025;189:118272. doi:10.1016/j.biopha.2025.118272
81. Hwang SM, Rahman MM, Go EJ, et al. Modulation of pain sensitivity by Ascl1- and Lhx6-dependent GABAergic neuronal function in streptozotocin diabetic mice. Mol Ther. 2025;33(2):786–804. doi:10.1016/j.ymthe.2024.12.039
82. Copits BA, Curatolo M, Dougherty PM, et al. Human pain neuroscience and the next generation of pain therapeutics. Neuron. 2025;113(9):1304–1306. doi:10.1016/j.neuron.2025.04.005
83. Simon N, Rudjito R, Moll L, et al. Characterisation of the antinociceptive effect of baricitinib in the collagen antibody-induced arthritis mouse model. Ann Rheum Dis. 2025;84(3):421–434. doi:10.1016/j.ard.2025.01.005
84. Li D, Pan S, Jiang W, Gao H. Subcutaneous administration of Stattic alleviates neuropathic pain by relieving inflammation in a mouse model of postherpetic neuralgia. Neurosci Lett. 2024;834:137831. doi:10.1016/j.neulet.2024.137831
85. Chapman KB, Sayed D, Lamer T, et al. Best practices for dorsal root ganglion stimulation for chronic pain: guidelines from the American Society of Pain and Neuroscience. J Pain Res. 2023;16:839–879. doi:10.2147/jpr.S364370
86. Yang J, Xie YF, Smith R, Ratté S, Prescott SA. Discordance between preclinical and clinical testing of Na V 1.7-selective inhibitors for pain. Pain. 2025;166(3):481–501. doi:10.1097/j.pain.0000000000003425
87. Eagles DA, Chow CY, King GF. Fifteen years of Na(V) 1.7 channels as an analgesic target: why has excellent in vitro pharmacology not translated into in vivo analgesic efficacy? Br J Pharmacol. 2022;179(14):3592–3611. doi:10.1111/bph.15327
88. Bjelkarøy MT, Benth J, Simonsen TB, et al. Measuring pain intensity in older adults. Can the visual analogue scale and the numeric rating scale be used interchangeably? Prog Neuropsychopharmacol Biol Psychiatry. 2024;130:110925. doi:10.1016/j.pnpbp.2023.110925
89. Colloca L. The placebo effect in pain therapies. Annu Rev Pharmacol Toxicol. 2019;59:191–211. doi:10.1146/annurev-pharmtox-010818-021542
90. Finnerup NB, Haroutounian S, Baron R, et al. Neuropathic pain clinical trials: factors associated with decreases in estimated drug efficacy. Pain. 2018;159(11):2339–2346. doi:10.1097/j.pain.0000000000001340
91. van Driel MEC, Huygen F, Rijsdijk M. Quantitative sensory testing: a practical guide and clinical applications. BJA Educ. 2024;24(9):326–334. doi:10.1016/j.bjae.2024.05.004
92. Andersen NE, Boehmerle W, Huehnchen P, Stage TB. Neurofilament light chain as a biomarker of chemotherapy-induced peripheral neuropathy. Trends Pharmacol Sci. 2024;45(10):872–879. doi:10.1016/j.tips.2024.08.001
93. Asadi G, Rezaei Varmaziar F, Karimi M, et al. Determination of the transcriptional level of long non-coding RNA NEAT-1, downstream target microRNAs, and genes targeted by microRNAs in diabetic neuropathy patients. Immunol Lett. 2021;232:20–26. doi:10.1016/j.imlet.2021.01.007
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.