Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Dopamine and Neuroinflammation in Schizophrenia – Interpreting the Findings from Translocator Protein (18kDa) PET Imaging
Authors Iliopoulou SM ![]() , Tsartsalis S, Kaiser S, Millet P, Tournier BB
, Tsartsalis S, Kaiser S, Millet P, Tournier BB
Received 13 August 2021
Accepted for publication 9 October 2021
Published 18 November 2021 Volume 2021:17 Pages 3345—3357
DOI https://doi.org/10.2147/NDT.S334027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Sotiria Maria Iliopoulou,1 Stergios Tsartsalis,2 Stefan Kaiser,1,2 Philippe Millet,1,2 Benjamin B Tournier1,2
1Adult Psychiatry Division, Department of Psychiatry, Geneva University Hospitals (HUG), Geneva, 1225, Switzerland; 2Faculty of Medicine, University of Geneva, Geneva, 1204, Switzerland
Correspondence: Sotiria Maria Iliopoulou Rue de Lausanne 20bis, 1225 Chêne Bourg, Geneva, Switzerland
Tel +41766897182
Fax +41223054599
Email [email protected]
Abstract: Schizophrenia is a complex disease whose pathophysiology is not yet fully understood. In addition to the long prevailing dopaminergic hypothesis, the evidence suggests that neuroinflammation plays a role in the pathophysiology of the disease. Recent studies using positron emission tomography (PET) that target a 18kDa translocator protein (TSPO) in activated microglial cells in an attempt to measure neuroinflammation in patients have shown a decrease or a lack of an increase in TSPO binding. Many biological and methodological considerations have been formulated to explain these findings. Although dopamine has been described as an immunomodulatory molecule, its potential role in neuroinflammation has not been explored in the aforementioned studies. In this review, we discuss the interactions between dopamine and neuroinflammation in psychotic states. Dopamine may inhibit neuroinflammation in activated microglia. Proinflammatory molecules released from microglia may decrease dopaminergic transmission. This could potentially explain why the levels of neuroinflammation in the brain of patients with schizophrenia seem to be unchanged or decreased compared to those in healthy subjects. However, most data are indirect and are derived from animal studies or from studies performed outside the field of schizophrenia. Further studies are needed to combine TSPO and dopamine imaging to study the association between microglial activation and dopamine system function.
Keywords: microglia, inflammation, biomarker, neuroimaging, PET, translocator protein
Introduction
Schizophrenia is a complex psychiatric disorder characterized by positive, negative and cognitive symptoms. The etiology of the disease is not clearly understood. Disturbances in several neurotransmitter pathways, various inflammatory mechanisms, environmental risk factors and genetic components have been suggested to be among the underlying mechanisms.1
The dopaminergic hypothesis has been the most prevailing theory about the pathophysiology of the illness. It associates symptoms of schizophrenia with frontal hypodopaminergia and striatal hyperdopaminergia.2 In addition to its function as a neurotransmitter, dopamine also acts on dopamine receptors on human and rodent microglia, as well as on peripheral immune cells.3–5 Interestingly, dopamine seems to modulate microglial activation state, their motility and their phagocytic activity, possibly exercising a recently reviewed immunomodulatory role in several affections.6,7
An increasing body of evidence suggests that inflammatory processes in the central nervous system (CNS) contribute to the development of schizophrenia, mainly via proinflammatory cytokines, glial and peripheral immune cells.8 Firstly, elevated levels of proinflammatory cytokines have been found in the serum and cerebrospinal fluid of patients with schizophrenia.9,10 Although penetration into the CNS of peripheral pro-inflammatory molecules is limited because of the blood-brain barrier, there is evidence of a certain level of permeability between the CNS and the periphery.11 Secondly, postmortem studies have demonstrated elevated levels of pro-inflammatory markers and microglial density in the brains of patients with schizophrenia, but the results have been heterogeneous.12–14 Supporting the hypothesis of glial activation, a transcriptional profiling study recently demonstrated the over-expression of genes linked to the inflammatory response in the cortex, hippocampus and striatum in schizophrenia.15 Thirdly, a meta-analysis of clinical trials showed that anti-inflammatory drugs may alleviate psychotic symptoms and cognitive deficits, although the sample sizes in the original studies were small.16
In recent years, the study of neuroinflammation in schizophrenia has become possible with the use of positron emission tomography (PET) and the development of radiotracers that target the 18 kDa translocator protein (TSPO). TSPO is overexpressed in activated microglial cells and astrocytes, and it has been suggested that it reflects inflammation in the brain.17–19 However, recent in vivo studies have shown decreased or similar levels of TSPO in patients with first-episode or chronic schizophrenia as compared to controls.19
In this review, we explore the role of dopamine on microglial activation and TSPO binding in schizophrenia, after reviewing the results of in vivo PET randomized controlled trials using second-generation TSPO radioligands as a biomarker of microglial activation. We thus examine whether a dopamine-induced microglial modulation could be a possible explanation for the decreased/unaltered TSPO levels shown in these studies, in addition to their methodological and biological limitations that have been discussed so far.
Search Strategy
Concerning the review of the in vivo findings on TSPO binding in schizophrenia, the PubMed database was searched with the following keywords:
((Positron Emission Tomography OR PET) AND (schizophrenia OR schizophreniform OR psychosis)) AND (microglia* OR microglia* activation OR TSPO OR Translocator protein OR peripheral benzodiazepine receptor)
up to 30 September 2021. The research yielded 112 results. We identified three meta-analysis and thirteen in vivo imaging studies which are discussed below.
Concerning the review of the interactions between neuroinflammation and the dopaminergic system, PubMed was searched using the following keywords: (dopamine[Text Word] OR “dopamine synthesis” OR BH4 OR tetrahydrobiopterin OR “dopamine transporter” OR VMAT OR “dopamine receptor”) AND (microglia[Text Word] OR cytokines[Text Word] OR pro inflammatory[Text Word] OR neuroinflammation OR TSPO) which yielded 2878 results (Microglia AND TSPO) with 464 results (Dopamine AND TSPO AND Microglia) with 12 results and (schizophrenia OR schizophreniform OR psychosis) AND (imaging OR PET OR SPECT) AND (dopamine OR receptor) with 2598 results. Sixty-four studies were included to represent the heterogeneous results of every query. Thirty-seven out of 64 were selected among the results of the search algorithms, and 27 out of 64 were used as support/evidence of the pathways or theories mentioned in this review. Given the scarcity of human in vivo studies in schizophrenia patients, we also mentioned representative findings from in vitro and animal studies as well as studies out of the field of schizophrenia research.
TSPO Binding Alterations in Patients with Schizophrenia
Overview of the Meta-Analytic Findings
The three recent meta-analysis of the literature identified thirteen in vivo imaging PET TSPO studies with approximately 200 subjects suffering from early or chronic schizophrenia-spectrum disorders.20–22
In the study of Marques et al, two separate meta-analysis were conducted depending on the measure used to quantify TSPO tracing in total grey matter.22 All but one study used the first-generation radioligand [11C]-PK11195, which has a high nonspecific binding and lipophilicity, characteristics that limit the accuracy of its quantification.23 Several parameters are used to measure the density of radiotracer receptors and quantify the tracer distribution, such as the binding potential (BP), distribution volume ratio (DVR) and total volume of distribution (VT). A significant elevation of TSPO binding potential (BP) in patients was found across all studies (g = 0.31, Confidence Intervals CI: 0.02–0.6) with moderate heterogeneity (I2 = 58%).24–28 A sensitivity analysis restricted to the five studies using PK11195 found a similar effect size of 0.35 (CI: 0.01–0.7). The only study using the second-generation ligand [11C]-DAA1106 found no difference in patients versus controls but was not identified as an outlier in the funnel plot.29 No difference in results was observed when adjusting for publication bias.
The authors found no difference in TSPO VT between patients and controls.30–35 The majority of the studies in this meta-analysis used second-generation radioligands, which presents considerably less nonspecific binding than [11C]-PK11195 and has an increased signal-to-noise ratio. This ligand thus has an increased potential to detect more subtle alterations in TSPO binding. Heterogeneity was found to be moderate to high (I2 = 53%).
Plavén-Sigray et al conducted an individual participant data meta-analysis of second-generation radioligand studies using VT in three regions of interest (frontal and temporal lobe and hippocampus) as the outcome measure.20 These results were recently extended to include two additional datasets.21 Using Bayesian model comparison, a reduction in VT in all three regions in patients versus controls was found (estimated standardized difference in VT in patients versus controls: −0.42 (CI: −0.74, −0.08) in the frontal cortex, −0.38 (CI: −0.73, −0.04) in the temporal cortex and −0.52 (CI: −0.84, −0.21) in the hippocampus). Heterogeneity was low with an I2 found less than 15% in all analyses.
Overall, both Plavén-Sigray et al and Marques et al do not observe an increase in VT in patients with schizophrenia compared to controls. While Plavén-Sigray et al found decreased levels of TSPO in patients versus controls, Marques et al did not observe a significant difference between groups. Some possible explanations for the different results between the two studies have been mentioned by Plavén-Sigray et al: the authors included one more in vivo study showing lower levels in VT without regional specificity between 13 patients suffering from a first psychotic episode versus 15 controls.36 They also excluded two patients out of fourteen in the study of Bloomfield et al, due to lack of controls with the same TSPO genotype, and they did not include the second-generation study by Takano et al that did not control for TSPO genotype for this very reason.20 This study was included in the meta-analysis by Marques et al.22 Finally, they identified an overlap of fourteen control subjects in Kenk et al and Hafizi et al, assigning these subjects to Hafizi et al data set, as to best match the patient group.20 Methodologically, Plaven-Sigray et al estimated the effect sizes using individual patient data, which permitted them to control for potential confounding effects, such as age, sex, or medication, rather than using aggregate data as did Marques et al.37
Limitations of TSPO Measurement Accuracy
Possible confounders such as age, gender and duration of illness have been recently discussed by De Picker et al.38 Gender and duration of illness were not found to account for differences in TSPO in patients versus controls in the meta-analysis by Plavén-Sigray et al as mentioned above. Most studies found no difference in TSPO levels in patients suffering from recent-onset or chronic schizophrenia versus those in healthy controls.29–33,35,39 The meta-analysis of Plavén-Sigray et al found no age-by-group interaction effect on VT in the examined regions.21 However, a recent retrospective multicenter study found positive correlations between age and TSPO VT in the frontal and temporal cortex of 140 healthy subjects with a positive correlation between VT and age in all regions in male subjects.40 The same study found a significant negative correlation of BMI with tracer binding but came with several limitations such as heterogeneity in demographic participants’ data and the unknown smoking status of several subjects.40
As far as smoking is concerned, two trials controlled for smoking, and no correlation was found.28,32 Given the scarcity of data directly examining the effect of smoking on neuroinflammation in vivo, further research is needed to make robust conclusions.
As far as antipsychotics are concerned, preclinical evidence suggests that antipsychotic medication may alter neuroinflammatory reactions by attenuating microglial activity.41–43 The meta-analysis by Plavén-Sigray et al showed no changes in radioligand binding in medicated subjects versus controls.20 The inverse effects have been observed in an in vivo rodent study after chronic antipsychotic treatment with haloperidol and olanzapine at high doses.44
The parameters used to measure the density of radiotracer receptors have each their own strengths and weaknesses, and the selection of a specific measure may alter the results of TSPO studies. For example, the systematic review of Marques et al reported a significant elevation in tracer binding in trials using BP as the outcome measure; this was not the case with VT.22 The methodological differences of TSPO quantification have been explored in the literature, but their description is out of the scope of this study.45
As far as radioligands are concerned, Conen et al published a large dataset using PK11195, showing no difference in BP TSPO levels in the anterior cingulate gyrus between recent onset or chronic schizophrenia patients versus their age-matched controls, likely reflecting that this methodology is not sensitive to detect subtle differences in TSPO.46 This lack of reduction may be dependent not only on the tracer properties but also on the poor reliability of the outcome measures used, as noted in the previous paragraph.47,48 Second-generation radioligands are considered to display inter- and within-subject variability.49 Interindividual variations in binding affinity have been found to be associated with a single nucleotide polymorphism (rs6971) in TSPO, resulting in the existence of apparent “low-affinity binders”, “high-affinity binders” and “mixed-affinity binders” when second-generation radioligand are used. Low-affinity binders present no measurable TSPO radiotracer binding and are currently routinely excluded from PET studies.50,51 While the majority of trials account for this polymorphism, one out of the eight second-generation tracer trials did not, suggesting that the results may underestimate tracer binding if low-affinity binders are included in the study.29 For this very reason, this study was not included in the meta-analysis of Plavén-Sigray et al; hence, this limitation does not apply for this meta-analysis. The effect of genotype rs6971 in the TSPO gene was studied in two separate sub-analysis of the five studies using the VT method that accounted for genotyping in the meta-analysis of Marques et al.22,31–35 The authors showed significant group differences in mixed-affinity binders (effect size −0,56, CI −1,08, −0.04), but not in high-affinity binder group.
Biological Explanations for Decreased TSPO Levels in Schizophrenia
It has been implied that alterations in TSPO binding may not be directly correlated with glial cell activation markers.52 An infection-mediated schizophrenia mouse model study by Notter et al demonstrated reduced TSPO binding in the prefrontal cortex of symptomatic female mice and a lack of association between microglial morphology and TSPO expression.53 Another in vitro animal study found that TSPO ligands inhibit toll-like receptor (TRL)-activated microglia and astrocytes.54 Furthermore, studies concerning TSPO expression in M1 microglia have been inconclusive. In in vitro rodent microglia, mRNA TSPO expression was increased after pro-inflammatory stimulation, but only a slight increase on protein levels was observed.55 Similar findings stem from a recent study.56 On the other hand, an in vitro study showed that TSPO expression did not increase in primary human microglia upon pro-inflammatory activation contrary to rodent microglia.57 TSPO expression decreased during anti-inflammatory M2 microglial polarization in a mice model of brain hypoxia.58
Moreover, reduced TSPO levels in schizophrenia patients could reflect mitochondrial dysfunction in this patient group, including a decrease in the number of mitochondria or the alternation of the number of TSPO binding sites.59–61
Indirect data outside the field of schizophrenia research have shown that systemic proinflammatory signals may reduce TSPO levels in the brain, suggesting the existence of compensatory mechanisms to attenuate neuroinflammation or processes unrelated to the activation of glial cells.62 Increased levels of peripheral proinflammatory cytokines have been shown to exist along with the downregulation of TSPO in the prefrontal cortex in a schizophrenia mouse model, and this occurred without an elevation in local cytokine expression, which might be explained by the anti-inflammatory role of TSPO.53,63,64 Animal and human models have suggested that TSPO immunoreactivity is also found in vascular endothelial cells, which may play a role in modifying TSPO levels in the brain, as proposed in a schizophrenia animal model.53,65
Recent studies on Alzheimer’s disease suggest that not every microglial subtype expresses TSPO.66–68 Some studies failed to show the upregulation in TSPO mRNA in human activated microglia ex vivo, but the results were heterogeneous and not specific to schizophrenia.69,70
The aforementioned considerations strengthen the hypothesis that the conceptualization of TSPO alterations as indicators of neuroinflammatory status is not straightforward.
Reciprocal Interactions Between Neuroinflammation and the Dopaminergic System
Overview
In terms of the biological interpretation of the findings on PET-TSPO binding, the complex interaction between dopamine and neuroinflammation has received little attention so far. In vitro, microglia can express five dopamine receptors (D1 to D5) depending on the species.4,71 Different microglia populations may express different dopamine receptors and exhibit different responses to dopamine according to their activation phenotype.72,73 For example, M1 or pro-inflammatory microglia produce proinflammatory cytokines (such as tumor necrosis factor alpha TNF‐α, interleukin (IL)‐1β and IL‐12) and high levels of inducible nitric oxide (iNOS) in response to dopamine. On the other hand, M2 or anti-inflammatory microglia express anti-inflammatory cytokines (such as IL-10 and Transforming growth factor beta TGFb).74
Influence of Dopamine on Neuroinflammation
In vitro evidence suggests that dopamine decreases lipopolysaccharide (LPS)-induced nitric oxide (NO) release, but not TNF-alpha and IL-6, via D1-like (D1DR) and D2-like dopamine receptors (D2DR).71,75,76 It has been shown that in LPS-treated microglia, the stimulation of D1DR and D2DR inhibits the activity of the local renin-angiotensin system in the brain, leading to the suppression of the pro-inflammatory AT1/NADPH-oxidase/superoxide axis, a major intermediate in oxidative stress pathways.77 A possible mechanism proposed is the downregulation of the extracellular signal–regulated kinase (ERK) 1/2 and p38 mitogen-activated protein kinase (p38MAPK) (Figure 1).73,78,79 It has also been reported that D1/2DR in microglial cells inhibits the NLRP3 inflammasome and interleukin-1 beta in an in vitro mouse model of intracerebral hemorrhage.76 Indirectly, astrocytic D2DR has also been shown to inhibit microglial pro-inflammatory activation through the production of αB-crystallin in astrocytes.80
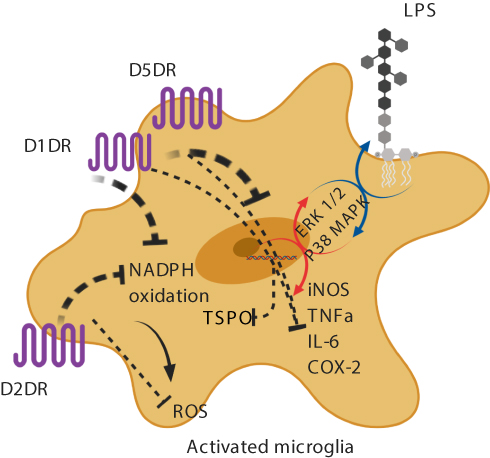 |
Figure 1 Activation of dopamine receptors on microglia and microglial inhibition. Abbreviations: LPS, lipopolysaccharide; D1DR, D1 dopamine receptor; D2DR, D2 dopamine receptor; D5DR, D5 dopamine receptor NADPH, nicotinamide adenine dinucleotide phosphate; ERK, extracellular signal–regulated kinase; p38 MAPK, p38 mitogen-activated protein kinase; iNOS, inducible nitric oxide synthase; TNFa, tumor necrosis factor alpha; IL-6, interleukin-6; COX-2, cyclooxygenase-2; ROS, reactive oxygen species; TSPO, translocator protein; dotted line, inhibition; circular arrow, uninhibited pathway; colorful arrow, uninhibited pathway. Image created with biorender.com. Notes: In LPS-treated microglia, it has been shown that dopamine inhibits the AT1/NADPHoxidase/superoxide axis and the synthesis of NO and pro-inflammatory cytokines. Dopamine-induced downregulation of ERK 1/2 and p38MAPK activity has been suggested as a potential mechanism. Created with BioRender.com. |
Findings outside of the field of schizophrenia research are in line with the aforementioned data. In Parkinson’s disease, animal studies have shown that acetyl-L-carnitine-induced upregulation of D1 receptors, attenuating microglial activation in the prefrontal cortex and hippocampus.81 Genetic deficiency of the D3 receptor has been found to inhibit neuroinflammatory pathways in a murine model of Parkinson’s disease (PD).82,83 De novo synthesized D2 receptors on microglia inhibit astrogliosis and neuroinflammation after ischemic stroke.84 Conversely, an increase of dopamine release in the striatum after cocaine injections in mice temporarily increased TNF-a mRNA and protein levels in microglia by acting on their D2 receptors in an in vivo study about addiction.85
Overall, high levels of dopamine may inhibit microglial activation by acting on their D1 and D2 dopaminergic receptors.
Microglial Modulation by Dopamine and PET-TSPO Findings
Dopamine synthesis and release has been shown to be increased in the striatum of patients with schizophrenia. Therefore, the dopamine-mediated attenuation of pro-inflammatory microglial activity could account at least in part for the absence of statistically significant differences in TSPO binding in the striatum of patients versus controls.29,33,86 The lack of increase in TSPO binding in the thalamus of patients, where an excess innervation of dopaminergic terminals was initially suspected in post-mortem findings, could also be in line with dopamine-induced microglial inhibition.29,35,46,87
In contrast, in the prefrontal cortex and other extra-striatal areas decreased dopamine release has been suggested.88 Concerning the frontal cortex, Kenk et al did not show an increase in TSPO binding, while Plaven-Sigray et al found decreased TSPO binding.88,89 As far as other extra-striatal regions are concerned, the meta-analysis of Plaven-Sigray showed a decrease in TSPO binding in temporal cortex and hippocampus where dopamine synthesis and release has been shown to be decreased.89 Controversially, preliminary findings of a recent cross-sectional in vivo study found a TSPO overexpression in the hippocampus in a subset of participants, which included antipsychotic-free schizophrenia patients and subjects at clinical high risk of psychosis.90 However, authors did not provide data about dopamine release or D1/2DR in the hippocampus per se but suggest another potential mechanism explaining the potential neuroinflammation in this region.90
Outside of the field of schizophrenia research, the reduction of microglial activation by dopamine has been explored in PD and Alzheimer’s disease (AD). Firstly, in vivo imaging data in PD have shown that TSPO binding may be increased or unchanged in nigrostriatal and cortical regions of patients where lower levels of dopamine have been found.91,92 Secondly, in rodent models of Alzheimer’s disease (AD), the dopaminergic degeneration of the ventral tegmental area (VTA) was associated with reduced dopamine levels in the hippocampus where microglial TSPO binding has been found to be increased.93,94 However, we did not find any in vivo trials in patients measuring regionally both TSPO and dopamine levels, and the evidence for an association is only indirect so far.
On a receptor level, increased or unaltered levels in striatal D2/3DR radioligand binding in drug-free and drug-naïve patients, respectively, has been observed, whereas findings about extrastriatal binding of D2/3DR have been mixed.95,96 However, associating existent data concerning D2/D3 receptors in schizophrenia and regional specific microglial modulation by these receptors is not feasible. Firstly, it has not been specified if and up to which extent alternations in dopamine receptor-binding concern microglia. Furthermore, DRD2 exists in an active and inactive state and the increase of the first is not necessarily associated with an increase in total receptor binding in patients.97 Finally, it has been suggested that antipsychotics may increase active DRD2 in the striatum.98 In sum, we cannot exclude that possible alterations on DR levels in several brain regions in schizophrenia may concern microglia, but if and how these changes interfere with dopamine-induced microglial inhibition is unclear.
Whether or not the dopamine-induced microglial inhibition is followed by a decrease or a lack of an increase in TSPO binding in patients suffering from schizophrenia versus controls remains to be established. Rodent studies have shown that inhibiting microglial activation with minocycline decreases gliosis and TSPO binding.99 Decreased microglial activation seemed to be the case after 12 weeks of administration of oral minocycline in patients suffering from brain injury.100 A recent study showed unaltered TSPO binding in patients suffering from chronic major depressive disorder after administration of oral minocycline, which may be a disease-specific effect according to the authors.101
In sum, further studies are warranted to measure both TSPO and dopamine release and specify whether a causal and regionally specific link exists between them in schizophrenia.
Influence of TSPO on Microglial Activation in Animal and Human Studies
While trying to elucidate dopamine and microglial interactions, it should be taken into consideration that TSPO itself may modulate microglial activation.
Interestingly, in vivo and in vitro findings in animal models of neurodegenerating diseases showed that TSPO ligands significantly reduced microgliosis and dopamine depletion with neuroprotecting effects.102–104 Contradictory in vitro data on rodent microglia exist about the action of TSPO ligands on microglial activation concerning both their pro – and their anti-inflammatory polarization. TSPO ligands may reduce M1 polarization or increase the production of reactive oxygen species and pro-inflammatory cytokines from microglia or contribute to M2 polarization.58,103,105–107 Those differences may be associated with the different ligands and mitochondrial inflammatory induction methods that have been used. In a human microglial in vitro study, two TSPO ligands stimulated the release of anti-inflammatory cytokines, and TSPO knockdown microglia released more pro-inflammatory cytokines in comparison with wildtype microglia after mitochondrial activation.55 It is currently unknown if TSPO further inhibits pro-inflammatory microglial activation along with dopamine in schizophrenia.
Influence of (Neuro)inflammation and TSPO on Dopamine
It would be simplistic to consider that dopamine-mediated microglial modulation is unidirectional. Interestingly, the role of inflammation on mesolimbic dopamine modulation and motivational drive has been recently reviewed.108 However, no study, to our knowledge, has examined the association – if any – between inflammatory cytokines and regional dopamine alterations and symptoms in schizophrenia. Three potential mechanisms by which pro-inflammatory cytokines interfere with dopaminergic pathways are discussed below.
Altered Dopamine Synthesis
To our knowledge, little is known of the effects of TSPO ligands on dopamine. In a PD mouse model, a TSPO agonist decreased dopamine turnover rates in MPTP-treated animals and dopamine levels became comparable with those measured in control animals.104
Besides TSPO, pro-inflammatory cytokines have been shown to activate the biosynthesis and the oxidative use of 5,6,7,8-tetrahydrobiopterin (BH4), a cofactor involved in the synthesis of dopamine, serotonin and nitric oxide.109 Furthermore, increased glutamate release under the influence of pro-inflammatory cytokines may increase the effects of oxidative stress on BH4 and dopamine synthesis according to in vitro studies.110–112
In patients with schizophrenia, a significant decrease in plasma BH4 was observed and was correlated with CSF levels.113 The hypothesis of a dopaminergic hypofunction due to deficiency of BH4 has thus been tested in animal studies. Yang and colleagues developed a strain of genetically modified mice that produced approximately 60% less BH4 in the brain compared to wildtype that had a significant reduction (over 90%) in the levels of dopamine in the caudate putamen and cortex and presented symptoms similar to those observed in phencyclidine or amphetamine-induced animal models of schizophrenia.114,115
Impaired Packaging and Release
The cytoplasmic dopamine concentration is mainly regulated by two transporters: the membranic dopamine transporter (DAT), which translocates dopamine from extracellular spaces into the extravesicular cytoplasmic compartment of neurons, and the vesicular monoamine transporter 2 (VMAT2) on synaptic vesicles, which mediates the packaging of dopamine into cytoplasmic vesicles.116,117
In rat enterochromaffin-like cells, IL-1 and TNF have been associated with decreased expression of VMAT2.118 In an animal study, the anti-inflammatory pituitary adenylate cyclase-activating polypeptide 38 was shown to increase VMAT2 expression and thus lead to reduced dopamine release from and/or increased reuptake into vesicles after METH administration, potentially resulting in elevated cytoplasmic dopamine levels, auto-oxidation and generation of reactive oxygen species.119,120 Reduced dopamine release in the striatum of rhesus monkeys after four weeks of peripheral interferon-α (INF-α) has been observed and associated with anhedonia-like symptoms.121
In patients with hepatitis C, peripheral administration of INF-a for four to six weeks was followed by increased dopamine uptake and decreased turnover in ventral striatum during winning trials when a gambling task was performed.122 A decreased activity in those regions was also demonstrated and associated to anhedonia, similarly to findings in schizophrenia suggesting a negative association between ventral striatal activation and apathy.122,123
However, no direct evidence concerning the interactions of neuro-inflammatory mechanisms and DAT/VMAT dysregulation or dopamine release in schizophrenia has been published to our knowledge.
Impaired Dopamine Receptor Signaling
Less is known about the effects of inflammation on dopamine receptor signaling. Decreased D2 receptor binding (but not DAT binding) was observed in the striatum of rhesus monkeys in the study of Felger et al.121 A recent study showed that interactions between the microglial complement C3 receptor and complement C3 protein are associated with immune activation and downregulation of D1 receptors in the nucleus accumbens in adolescent male rats.124
In sum, the in vitro and indirect data suggest that proinflammatory cytokines and the modulation of TSPO activity may alter dopamine synthesis, trigger oxidative stress in neurons by increasing intracellular dopamine levels and impair dopamine receptor function. However, these functions in schizophrenia have yet to be demonstrated, and it is not known up to which extent specific brain regions are concerned, whether the alterations in dopamine receptors number or activation status are concerned and how or if the dopamine receptors on microglia are influenced in order to conclude in an association between TSPO levels and neuro-inflammation based on imaging trials (Figure 2). For example, while a cytokine-induced decrease in dopamine release was confirmed in brain regions where reduced dopamine levels have been demonstrated, it is not known if an increase in microglial activation and in TSPO levels is present, and the latter has not been found increased in the frontal cortex in schizophrenia patients in in vivo PET-TSPO trials. Further research is warranted in order to measure dopamine receptors number and activation status in microglia which can be done with already described techniques, along with cytokine levels in specific brain regions in patients.125
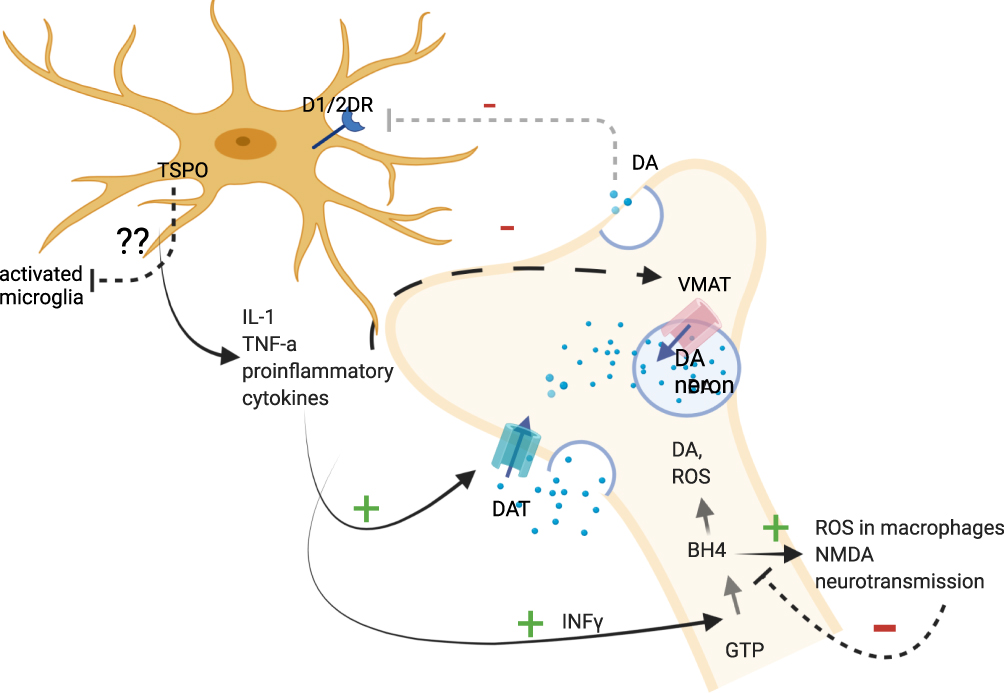 |
Figure 2 Pathways between proinflammatory cytokines, dopaminergic neurons and microglia. Abbreviations: GTP, guanosine triphosphate; INFγ, interferon gamma; BH4, tetrahydrobiopterin; DA, dopamine; VMAT, vesicular monoamine transporter; DAT, dopamine transporter; ROS, reactive oxygen species; IL-1, interleukin-1; TNF-a, tumor necrosis factor-a; DRD1,2, dopamine receptor 1,2; dotted line/arrow, inhibition; arrows, activated pathway; +, activation; -, inhibition; ??, unknown. Image created with biorender.com. Notes: Proinflammatory cytokines have been shown to activate the biosynthesis of BH4, which is involved in the synthesis of dopamine and stimulates the production of reactive oxygen species in macrophages and NMDA neurotransmission. IL-1 and TNF have been associated with decreased expression of VMAT2. Pro-inflammatory cytokines have been associated with increased expression of DAT, which results in the elevation of cytoplasmic DA levels in neurons. Decreases in dopamine release may result in decreased dopamine-induced microglial inhibition. The role of TSPO on activated microglia where it is expressed remains to be established, but some studies suggested that it promotes anti-inflammatory microglial polarization. Created with BioRender.com. |
Non-Dopaminergic Neurotransmitter Receptors on Microglia
Microglial cells also express glutamate receptors (recently reviewed by Spampinato et al), as well as other neurotransmitter receptors.126,127 In vitro, metabotropic glutamate receptor 5 (mGluR5) inhibits microglial activation with a reduction in nitric oxide, reactive oxygen species, and TNFa production, whereas mGluR2 but not mGluR3 receptors induce TNFa release and microglial activation.128–132 In rodent models of schizophrenia, mGlu2 and mGlu3 mRNA and protein levels have been found to be decreased in the frontal cortex, but this decrease was not necessarily microglial-specific.133 Post-mortem findings showed a reduction in mGlu5 receptor signaling in the dorsolateral prefrontal cortex of individuals with schizophrenia.134 However, this does not seem to be in line with the lack of increase in TSPO binding observed in this region of interest.33 Although glutamate and glutamine levels have been measured in several brain regions in patients, imaging limitations with glutamate proton magnetic resonance spectroscopy need to be taken into account as this method does not differentiate between intra- and extracellular compartments.135 Further research is warranted in order to specify how and where glutamate transmission modulates microglial activation as reflected by TSPO imaging, taking into consideration the methodological limitations of glutamate imaging and its interactions with N-methyl-D-aspartate receptors and dopaminergic neurons. The overview of the role of neurotransmitters other than dopamine on microglia is out of the scope of this study.
Conclusion and Future Directions
The goal of this review was to explore the role of dopamine in the modulation of microglial activity, as reflected by TSPO levels in schizophrenia, according to the two most recent meta-analysis of in vivo human trials. Previously investigated confounding factors in these trials, biological and methodological interpretations seem unlikely to fully account for the decreased/unaltered TSPO findings in schizophrenia. We have proposed a new theoretical framework in which dopamine may inhibit microglial activation leaving TSPO levels unaltered or decreased in patients compared to controls. However, the interactions between dopamine and neuroinflammation are not uni-directional and the TSPO itself may exercise possible immunomodulatory effects as presented above. Furthermore, it remains to be elucidated which extent TSPO expression reflects a pro- or anti-inflammatory microglial activation, given the conflicting evidence discussed above and the fact that the spectrum of microglial phenotypes is actually thought to be wider and more complex than the simplified M1/2 classification.74
It is known that the dopamine levels and D2 availability differ across different brain regions of patients, but a regionally specific alteration of TSPO levels due to dopamine-induced microglial inhibition does not seem to be the case for the most part according to existing data.89,136–139 A possible explanation could be that PET TSPO studies have not tested for the receptor’ density in the same brain regions as PET DA studies.22 Dopamine release in schizophrenia is not fully mapped, and the dopamine receptor density measured in patients is not specific for microglia to our knowledge. Future research is needed to test our hypothesis, combining TSPO and dopamine imaging to explore the association between microglial activation and dopamine levels brain region by brain region, ideally quantifying microglial dopamine receptors at the same time. Shedding light on these mechanisms will both ameliorate our understanding of how the microglia respond to altered dopamine levels and if they are affected by dopamine receptors’ dysregulation in schizophrenia.
Acknowledgments
This work was supported by the Swiss National Science Foundation (no. 320030-184713).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Henriksen MG, Nordgaard J, Jansson LB. Genetics of Schizophrenia: overview of Methods, Findings and Limitations. Front Hum Neurosci. 2017;11:322. doi:10.3389/fnhum.2017.00322
2. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull. 2009;35(3):549–562. doi:10.1093/schbul/sbp006
3. Arreola R, Alvarez-Herrera S, Perez-Sanchez G, et al. Immunomodulatory Effects Mediated by Dopamine. J Immunol Res. 2016;2016:3160486. doi:10.1155/2016/3160486
4. Mastroeni D, Grover A, Leonard B, et al. Microglial responses to dopamine in a cell culture model of Parkinson’s disease. Neurobiol Aging. 2009;30(11):1805–1817. doi:10.1016/j.neurobiolaging.2008.01.001
5. Sarkar C, Basu B, Chakroborty D, Dasgupta PS, Basu S. The immunoregulatory role of dopamine: an update. Brain Behav Immun. 2010;24(4):525–528. doi:10.1016/j.bbi.2009.10.015
6. Feng Y, Lu Y. Immunomodulatory Effects of Dopamine in Inflammatory Diseases. Front Immunol. 2021;12:663102. doi:10.3389/fimmu.2021.663102
7. Albertini G, Etienne F, Roumier A. Regulation of microglia by neuromodulators: modulations in major and minor modes. Neurosci Lett. 2020;733:135000. doi:10.1016/j.neulet.2020.135000
8. Mongan D, Ramesar M, Focking M, Cannon M, Cotter D. Role of inflammation in the pathogenesis of schizophrenia: a review of the evidence, proposed mechanisms and implications for treatment. Early Interv Psychiatry. 2019;14(4):385.
9. Upthegrove R, Manzanares-Teson N, Barnes NM. Cytokine function in medication-naive first episode psychosis: a systematic review and meta-analysis. Schizophr Res. 2014;155(1–3):101–108. doi:10.1016/j.schres.2014.03.005
10. Dickerson F, Stallings C, Origoni A, et al. Inflammatory Markers in Recent Onset Psychosis and Chronic Schizophrenia. Schizophr Bull. 2016;42(1):134–141. doi:10.1093/schbul/sbv108
11. Mueller NBK. The mild encephalitis concept for psychiatric disorders revisited in the light of current psychoneuroimmunological findings. Neurol Psychiatry Brain Res. 2013;19:87–101. doi:10.1016/j.npbr.2013.04.004
12. Trepanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP. Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry. 2016;21(8):1009–1026. doi:10.1038/mp.2016.90
13. van Kesteren CF, Gremmels H, de Witte LD, et al. Immune involvement in the pathogenesis of schizophrenia: a meta-analysis on postmortem brain studies. Transl Psychiatry. 2017;7(3):e1075. doi:10.1038/tp.2017.4
14. Murphy CE, Lawther AJ, Webster MJ, et al. Nuclear factor kappa B activation appears weaker in schizophrenia patients with high brain cytokines than in non-schizophrenic controls with high brain cytokines. J Neuroinflammation. 2020;17(1):215. doi:10.1186/s12974-020-01890-6
15. Lanz TA, Reinhart V, Sheehan MJ, et al. Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: a comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Transl Psychiatry. 2019;9(1):151. doi:10.1038/s41398-019-0492-8
16. Hong J, Bang M. Anti-inflammatory Strategies for Schizophrenia: a Review of Evidence for Therapeutic Applications and Drug Repurposing. Clin Psychopharmacol Neurosci. 2020;18(1):10–24. doi:10.9758/cpn.2020.18.1.10
17. Rupprecht R, Papadopoulos V, Rammes G, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9(12):971–988. doi:10.1038/nrd3295
18. Guilarte TR. TSPO in diverse CNS pathologies and psychiatric disease: a critical review and a way forward. Pharmacol Ther. 2019;194:44–58. doi:10.1016/j.pharmthera.2018.09.003
19. Tournier BB, Tsartsalis S, Ceyzériat K, Garibotto V, Millet P. In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells. 2020;9(9):1941. doi:10.3390/cells9091941
20. Plaven-Sigray P, Matheson GJ, Collste K, et al. Positron Emission Tomography Studies of the Glial Cell Marker Translocator Protein in Patients With Psychosis: a Meta-analysis Using Individual Participant Data. Biol Psychiatry. 2018;84(6):433–442. doi:10.1016/j.biopsych.2018.02.1171
21. Plavén-Sigray P, Matheson GJ, Coughlin JM, et al.Meta-analysis of the Glial Marker TSPO in Psychosis Revisited: reconciling Inconclusive Findings of Patient-Control Differences. Biol Psychiatry. 2020;(3):e5–e8. doi:10.1016/j.biopsych.2020.05.028
22. Marques TR, Ashok AH, Pillinger T, et al. Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. Psychol Med. 2019;49(13):2186–2196. doi:10.1017/S0033291718003057
23. Fujita M, Imaizumi M, Zoghbi SS, et al. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. NeuroImage. 2008;40(1):43–52. doi:10.1016/j.neuroimage.2007.11.011
24. Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nuclear Med. 2009;50(11):1801–1807. doi:10.2967/jnumed.109.066647
25. van Berckel BN, Bossong MG, Boellaard R, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64(9):820–822. doi:10.1016/j.biopsych.2008.04.025
26. Holmes SE, Hinz R, Drake RJ, et al. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: a [(11)C](R)-PK11195 positron emission tomography study. Mol Psychiatry. 2016;21(12):1672–1679. doi:10.1038/mp.2016.180
27. Di Biase MA, Zalesky A, O’Keefe G, et al. PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Transl Psychiatry. 2017;7(8):e1225. doi:10.1038/tp.2017.193
28. van der Doef TF, de Witte LD, Sutterland AL, et al. In vivo (R)-[(11)C]PK11195 PET imaging of 18kDa translocator protein in recent onset psychosis. NPJ Schizophrenia. 2016;2:16031. doi:10.1038/npjschz.2016.31
29. Takano A, Arakawa R, Ito H, et al. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int j Neuropsychopharmacol. 2010;13(7):943–950. doi:10.1017/S1461145710000313
30. Bloomfield PS, Selvaraj S, Veronese M, et al. Microglial Activity in People at Ultra High Risk of Psychosis and in Schizophrenia: an [(11)C]PBR28 PET Brain Imaging Study. Am J Psychiatry. 2016;173(1):44–52. doi:10.1176/appi.ajp.2015.14101358
31. Hafizi S, Tseng HH, Rao N, et al. Imaging Microglial Activation in Untreated First-Episode Psychosis: a PET Study With [(18)F]FEPPA. Am J Psychiatry. 2017;174(2):118–124. doi:10.1176/appi.ajp.2016.16020171
32. Coughlin JM, Wang Y, Ambinder EB, et al. In vivo markers of inflammatory response in recent-onset schizophrenia: a combined study using [(11)C]DPA-713 PET and analysis of CSF and plasma. Transl Psychiatry. 2016;6:e777. doi:10.1038/tp.2016.40
33. Kenk M, Selvanathan T, Rao N, et al. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull. 2015;41(1):85–93. doi:10.1093/schbul/sbu157
34. Collste K, Plaven-Sigray P, Fatouros-Bergman H, et al. Lower levels of the glial cell marker TSPO in drug-naive first-episode psychosis patients as measured using PET and [(11)C]PBR28. Mol Psychiatry. 2017;22(6):850–856. doi:10.1038/mp.2016.247
35. Ottoy J, De Picker L, Verhaeghe J, et al. (18)F-PBR111 PET Imaging in Healthy Controls and Schizophrenia: test-Retest Reproducibility and Quantification of Neuroinflammation. J Nuclear Med. 2018;59(8):1267–1274. doi:10.2967/jnumed.117.203315
36. Laurikainen H, Vuorela A, Toivonen A, et al. Elevated serum chemokine CCL22 levels in first-episode psychosis: associations with symptoms, peripheral immune state and in vivo brain glial cell function. Transl Psychiatry. 2020;10(1):94. doi:10.1038/s41398-020-0776-z
37. Tierney JF, Fisher DJ, Burdett S, Stewart LA, Parmar MKB. Comparison of aggregate and individual participant data approaches to meta-analysis of randomised trials: an observational study. PLoS Med. 2020;17(1):e1003019. doi:10.1371/journal.pmed.1003019
38. De Picker L, Morrens M. Perspective: solving the Heterogeneity Conundrum of TSPO PET Imaging in Psychosis. Front Psychiatry. 2020;11:362. doi:10.3389/fpsyt.2020.00362
39. De Picker L, Ottoy J, Verhaeghe J, et al. State-associated changes in longitudinal [(18)F]-PBR111 TSPO PET imaging of psychosis patients: evidence for the accelerated ageing hypothesis? Brain Behav Immun. 2019;77:46–54. doi:10.1016/j.bbi.2018.11.318
40. Tuisku J, Plavén-Sigray P, Gaiser EC, et al. Effects of age, BMI and sex on the glial cell marker TSPO - a multicentre [(11)C]PBR28 HRRT PET study. Eur J Nucl Med Mol Imaging. 2019;46(11):2329–2338. doi:10.1007/s00259-019-04403-7
41. Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42–48. doi:10.1016/j.pnpbp.2007.06.031
42. Kato TA, Monji A, Mizoguchi Y, et al. Anti-Inflammatory properties of antipsychotics via microglia modulations: are antipsychotics a ‘fire extinguisher’ in the brain of schizophrenia? Mini Rev Med Chem. 2011;11(7):565–574. doi:10.2174/138955711795906941
43. Zhu F, Zheng Y, Ding YQ, et al. Minocycline and risperidone prevent microglia activation and rescue behavioral deficits induced by neonatal intrahippocampal injection of lipopolysaccharide in rats. PLoS One. 2014;9(4):e93966. doi:10.1371/journal.pone.0093966
44. Cotel MC, Lenartowicz EM, Natesan S, et al. Microglial activation in the rat brain following chronic antipsychotic treatment at clinically relevant doses. Eur Neuropsychopharmacol. 2015;25(11):2098–2107. doi:10.1016/j.euroneuro.2015.08.004
45. Turkheimer FE, Rizzo G, Bloomfield PS, et al. The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans. 2015;43(4):586–592. doi:10.1042/BST20150058
46. Conen S, Gregory CJ, Hinz R, et al. Neuroinflammation as measured by positron emission tomography in patients with recent onset and established schizophrenia: implications for immune pathogenesis. Mol Psychiatry. 2020. doi:10.1038/s41380-020-0829-y
47. Plavén-Sigray P, Matheson GJ, Cselényi Z, Jucaite A, Farde L, Cervenka S. Test-retest reliability and convergent validity of (R)-[(11)C]PK11195 outcome measures without arterial input function. EJNMMI Res. 2018;8(1):102. doi:10.1186/s13550-018-0455-8
48. Plavén-Sigray P, Cervenka S. Meta-analytic studies of the glial cell marker TSPO in psychosis - a question of apples and pears?: a commentary on ‘Neuroinflammation in schizophrenia: metaanalysis of in-vivo microglial imaging’ by Marques et al. - ERRATUM. Psychol Med. 2019;49(9):1583–1584. doi:10.1017/S0033291719000746
49. Guo Q, Owen DR, Rabiner EA, Turkheimer FE, Gunn RN. Identifying improved TSPO PET imaging probes through biomathematics: the impact of multiple TSPO binding sites in vivo. NeuroImage. 2012;60(2):902–910. doi:10.1016/j.neuroimage.2011.12.078
50. Owen DR, Howell OW, Tang SP, et al. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cerebral Blood Flow Metab. 2010;30(9):1608–1618. doi:10.1038/jcbfm.2010.63
51. Owen DR, Yeo AJ, Gunn RN, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cerebral Blood Flow Metab. 2012;32(1):1–5. doi:10.1038/jcbfm.2011.147
52. Sneeboer MAM, van der Doef T, Litjens M, et al. Microglial activation in schizophrenia: is translocator 18kDa protein (TSPO) the right marker? Schizophr Res. 2020;215:167–172. doi:10.1093/schbul/13.4.589
53. Notter T, Coughlin JM, Gschwind T, et al. Translational evaluation of translocator protein as a marker of neuroinflammation in schizophrenia. Mol Psychiatry. 2018;23(2):323–334. doi:10.1038/mp.2016.248
54. Lee JW, Nam H, Yu SW. Systematic Analysis of Translocator Protein 18 kDa (TSPO) Ligands on Toll-like Receptors-mediated Pro-inflammatory Responses in Microglia and Astrocytes. Exp Neurobiol. 2016;25(5):262–268. doi:10.5607/en.2016.25.5.262
55. Pozzo ED, Tremolanti C, Costa B, et al. Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation. Int J Mol Sci. 2019;20(18):4467. doi:10.3390/ijms20184467
56. Pannell M, Economopoulos V, Wilson TC, et al. Imaging of translocator protein upregulation is selective for pro-inflammatory polarized astrocytes and microglia. Glia. 2020;68(2):280–297. doi:10.1002/glia.23716
57. Owen DR, Narayan N, Wells L, et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J Cerebral Blood Flow Metab. 2017;37(8):2679–2690. doi:10.1177/0271678X17710182
58. Zhou D, Ji L, Chen Y. TSPO Modulates IL-4-Induced Microglia/Macrophage M2 Polarization via PPAR-γ Pathway. J Mol Neurosci. 2020;70(4):542–549. doi:10.1007/s12031-019-01454-1
59. Manji H, Kato T, Di Prospero NA, et al. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13(5):293–307. doi:10.1038/nrn3229
60. Roberts RC. Postmortem studies on mitochondria in schizophrenia. Schizophr Res. 2017;187:17–25. doi:10.1016/j.schres.2017.01.056
61. Somerville SM, Conley RR, Roberts RC. Mitochondria in the striatum of subjects with schizophrenia. World j Biol Psychiatry. 2011;12(1):48–56. doi:10.3109/15622975.2010.505662
62. Forsberg A, Cervenka S, Jonsson Fagerlund M, et al. The immune response of the human brain to abdominal surgery. Ann Neurol. 2017;81(4):572–582. doi:10.1002/ana.24909
63. Zhang H, Ma L, Yin YL, et al. Over-expression of TSPO in the hippocampal CA1 area alleviates cognitive dysfunction caused by lipopolysaccharide in mice. Brain Res. 2016;1646:402–409. doi:10.1016/j.brainres.2016.06.001
64. Daugherty DJ, Selvaraj V, Chechneva OV, Liu XB, Pleasure DE, Deng W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol Med. 2013;5(6):891–903. doi:10.1002/emmm.201202124
65. Cosenza-Nashat M, Zhao ML, Suh HS, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35(3):306–328. doi:10.1111/j.1365-2990.2008.01006.x
66. Keren-Shaul H, Spinrad A, Weiner A, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169(7):1276–1290.e1217. doi:10.1016/j.cell.2017.05.018
67. Friedman BA, Srinivasan K, Ayalon G, et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018;22(3):832–847. doi:10.1016/j.celrep.2017.12.066
68. Rangaraju S, Dammer EB, Raza SA, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):24. doi:10.1186/s13024-018-0254-8
69. Narayan N, Mandhair H, Smyth E, et al. The macrophage marker translocator protein (TSPO) is down-regulated on pro-inflammatory ‘M1ʹ human macrophages. PLoS One. 2017;12(10):e0185767. doi:10.1371/journal.pone.0185767
70. Beaino W, Janssen B, Kooij G, et al. Purinergic receptors P2Y12R and P2X7R: potential targets for PET imaging of microglia phenotypes in multiple sclerosis. J Neuroinflammation. 2017;14(1):259. doi:10.1186/s12974-017-1034-z
71. Farber K, Pannasch U, Kettenmann H. Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol Cell Neurosci. 2005;29(1):128–138. doi:10.1016/j.mcn.2005.01.003
72. McKenna F, McLaughlin PJ, Lewis BJ, et al. Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J Neuroimmunol. 2002;132(1–2):34–40. doi:10.1016/S0165-5728(02)00280-1
73. Fan Y, Chen Z, Pathak JL, Carneiro AMD, Chung CY. Differential Regulation of Adhesion and Phagocytosis of Resting and Activated Microglia by Dopamine. Front Cell Neurosci. 2018;12:309. doi:10.3389/fncel.2018.00309
74. Jurga AM, Paleczna M, Kuter KZ. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front Cell Neurosci. 2020;14:198. doi:10.3389/fncel.2020.00198
75. Chang JY, Liu LZ. Catecholamines inhibit microglial nitric oxide production. Brain Res Bull. 2000;52(6):525–530. doi:10.1016/S0361-9230(00)00291-4
76. Wang T, Nowrangi D, Yu L, et al. Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflammation. 2018;15(1):2. doi:10.1186/s12974-017-1039-7
77. Dominguez-Meijide A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL. Dopamine modulates astroglial and microglial activity via glial renin-angiotensin system in cultures. Brain Behav Immun. 2017;62:277–290. doi:10.1016/j.bbi.2017.02.013
78. Borroto-Escuela DO, Tarakanov AO, Bechter K, Fuxe K. IL1R2, CCR2, and CXCR4 May Form Heteroreceptor Complexes with NMDAR and D2R: relevance for Schizophrenia. Front Psychiatry. 2017;8:24. doi:10.3389/fpsyt.2017.00024
79. Sun J, Nan G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: a potential therapeutic target (Review). Int J Mol Med. 2017;39(6):1338–1346. doi:10.3892/ijmm.2017.2962
80. Shao W, Zhang SZ, Tang M, et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature. 2013;494(7435):90–94. doi:10.1038/nature11748
81. Singh S, Mishra A, Srivastava N, Shukla R, Shukla S. Acetyl-L-Carnitine via Upegulating Dopamine D1 Receptor and Attenuating Microglial Activation Prevents Neuronal Loss and Improves Memory Functions in Parkinsonian Rats. Mol Neurobiol. 2018;55(1):583–602. doi:10.1007/s12035-016-0293-5
82. Chen Y, Ni YY, Liu J, et al. Dopamine receptor 3 might be an essential molecule in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. BMC Neurosci. 2013;14:76. doi:10.1186/1471-2202-14-76
83. Gonzalez H, Contreras F, Prado C, et al. Dopamine receptor D3 expressed on CD4+ T cells favors neurodegeneration of dopaminergic neurons during Parkinson’s disease. J Immunol. 2013;190(10):5048–5056. doi:10.4049/jimmunol.1203121
84. Huck JH, Freyer D, Bottcher C, et al. De novo expression of dopamine D2 receptors on microglia after stroke. J Cerebral Blood Flow Metab. 2015;35(11):1804–1811.
85. Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D. Microglial TNF-α Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron. 2016;90(3):483–491. doi:10.1016/j.neuron.2016.03.030
86. Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK. Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry Suppl. 2007;51:s13–18. doi:10.1192/bjp.191.51.s13
87. Oke AF, Adams RN. Elevated thalamic dopamine: possible link to sensory dysfunctions in schizophrenia. Schizophr Bull. 1987;13(4):589–604.
88. Rao N, Northoff G, Tagore A, et al. Impaired Prefrontal Cortical Dopamine Release in Schizophrenia During a Cognitive Task: a [11C]FLB 457 Positron Emission Tomography Study. Schizophr Bull. 2019;45(3):670–679. doi:10.1093/schbul/sby076
89. Slifstein M, van de Giessen E, Van Snellenberg J, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry. 2015;72(4):316–324.
90. Schifani C, Hafizi S, Tseng HH, et al. Preliminary data indicating a connection between stress-induced prefrontal dopamine release and hippocampal TSPO expression in the psychosis spectrum. Schizophr Res. 2019;213:80–86. doi:10.1016/j.schres.2018.10.008
91. Gröger A, Kolb R, Schäfer R, Klose U. Dopamine reduction in the substantia nigra of Parkinson’s disease patients confirmed by in vivo magnetic resonance spectroscopic imaging. PLoS One. 2014;9(1):e84081. doi:10.1371/journal.pone.0084081
92. Belloli S, Morari M, Murtaj V, Valtorta S, Moresco RM, Gilardi MC. Translation Imaging in Parkinson’s Disease: focus on Neuroinflammation. Front Aging Neurosci. 2020;12:152. doi:10.3389/fnagi.2020.00152
93. Tournier BB, Tsartsalis S, Rigaud D, et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol Dis. 2019;121:95–105. doi:10.1016/j.nbd.2018.09.022
94. Nobili A, Latagliata EC, Viscomi MT, et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat Commun. 2017;8:14727. doi:10.1038/ncomms14727
95. Yang YK, Yu L, Yeh TL, Chiu NT, Chen PS, Lee IH. Associated alterations of striatal dopamine D2/D3 receptor and transporter binding in drug-naive patients with schizophrenia: a dual-isotope SPECT study. Am J Psychiatry. 2004;161(8):1496–1498. doi:10.1176/appi.ajp.161.8.1496
96. Kegeles LS, Abi-Dargham A, Frankle WG, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67(3):231–239. doi:10.1001/archgenpsychiatry.2010.10
97. Kubota M, Nagashima T, Takano H, et al. Affinity States of Striatal Dopamine D2 Receptors in Antipsychotic-Free Patients with Schizophrenia. Int j Neuropsychopharmacol. 2017;20(11):928–935. doi:10.1093/ijnp/pyx063
98. Chouinard G, Samaha AN, Chouinard VA, et al. Antipsychotic-Induced Dopamine Supersensitivity Psychosis: pharmacology, Criteria, and Therapy. Psychother Psychosom. 2017;86(4):189–219. doi:10.1159/000477313
99. Wolf BJ, Brackhan M, Bascuñana P, et al. TSPO PET Identifies Different Anti-inflammatory Minocycline Treatment Response in Two Rodent Models of Epileptogenesis. Neurotherapeutics. 2020;17(3):1228–1238. doi:10.1007/s13311-020-00834-5
100. Scott G, Zetterberg H, Jolly A, et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain. 2018;141(2):459–471. doi:10.1093/brain/awx339
101. Attwells S, Setiawan E, Rusjan PM, et al. A double-blind placebo-controlled trial of minocycline on translocator protein distribution volume in treatment-resistant major depressive disorder. Transl Psychiatry. 2021;11(1):334. doi:10.1038/s41398-021-01450-3
102. Scholz R, Caramoy A, Bhuckory MB, et al. Targeting translocator protein (18 kDa) (TSPO) dampens pro-inflammatory microglia reactivity in the retina and protects from degeneration. J Neuroinflammation. 2015;12:201. doi:10.1186/s12974-015-0422-5
103. Choi J, Ifuku M, Noda M, Guilarte TR. Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia. 2011;59(2):219–230. doi:10.1002/glia.21091
104. Gong J, Szego ÉM, Leonov A, et al. Translocator Protein Ligand Protects against Neurodegeneration in the MPTP Mouse Model of Parkinsonism. J Neurosci. 2019;39(19):3752–3769. doi:10.1523/JNEUROSCI.2070-18.2019
105. Monga S, Denora N, Laquintana V, et al. The protective effect of the TSPO ligands 2,4-Di-Cl-MGV-1, CB86, and CB204 against LPS-induced M1 pro-inflammatory activation of microglia. Brain Behavior Immunity. 2020;5:100083. doi:10.1016/j.bbih.2020.100083
106. Azrad M, Zeineh N, Weizman A, Veenman L, Gavish M. The TSPO Ligands 2-Cl-MGV-1, MGV-1, and PK11195 Differentially Suppress the Inflammatory Response of BV-2 Microglial Cell to LPS. Int J Mol Sci. 2019;20:3. doi:10.3390/ijms20030594
107. Zhou J, Zhang X, Peng J, et al. TSPO ligand Ro5-4864 modulates microglia/macrophages polarization after subarachnoid hemorrhage in mice. Neurosci Lett. 2020;729:134977. doi:10.1016/j.neulet.2020.134977
108. Treadway MT, Cooper JA, Miller AH. Can’t or Won’t? Immunometabolic Constraints on Dopaminergic Drive. Trends Cogn Sci. 2019;23(5):435–448. doi:10.1016/j.tics.2019.03.003
109. Shi W, Meininger CJ, Haynes TE, Hatakeyama K, Wu G. Regulation of tetrahydrobiopterin synthesis and bioavailability in endothelial cells. Cell Biochem Biophys. 2004;41(3):415–434. doi:10.1385/CBB:41:3:415
110. Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflammation. 2012;9:275. doi:10.1186/1742-2094-9-275
111. Guillemin GJ. Quinolinic acid: neurotoxicity. FEBS J. 2012;279(8):1355. doi:10.1111/j.1742-4658.2012.08493.x
112. Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85(10):2059–2070. doi:10.1002/jnr.21325
113. Richardson MA, Read LL, Taylor Clelland CL, et al. Evidence for a tetrahydrobiopterin deficit in schizophrenia. Neuropsychobiology. 2005;52(4):190–201. doi:10.1159/000089002
114. Yang S, Lee YJ, Kim JM, et al. A murine model for human sepiapterin-reductase deficiency. Am J Hum Genet. 2006;78(4):575–587. doi:10.1086/501372
115. Choi YK, Tarazi FI. Alterations in dopamine and glutamate neurotransmission in tetrahydrobiopterin deficient spr-/- mice: relevance to schizophrenia. BMB Rep. 2010;43(9):593–598. doi:10.5483/BMBRep.2010.43.9.593
116. Giros B, El Mestikawy S, Godinot N, et al. Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Mol Pharmacol. 1992;42(3):383–390.
117. Lawal HO, Krantz DE. SLC18: vesicular neurotransmitter transporters for monoamines and acetylcholine. Mol Aspects Med. 2013;34(2–3):360–372. doi:10.1016/j.mam.2012.07.005
118. Kazumori H, Ishihara S, Rumi MA, Ortega-Cava CF, Kadowaki Y, Kinoshita Y. Transforming growth factor-alpha directly augments histidine decarboxylase and vesicular monoamine transporter 2 production in rat enterochromaffin-like cells. Am J Physiol Gastrointest Liver Physiol. 2004;286(3):G508–514. doi:10.1152/ajpgi.00269.2003
119. Felger JC, Miller AH. Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol. 2012;33(3):315–327. doi:10.1016/j.yfrne.2012.09.003
120. Guillot TS, Richardson JR, Wang MZ, et al. PACAP38 increases vesicular monoamine transporter 2 (VMAT2) expression and attenuates methamphetamine toxicity. Neuropeptides. 2008;42(4):423–434. doi:10.1016/j.npep.2008.04.003
121. Felger JC, Mun J, Kimmel HL, et al. Chronic interferon-alpha decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology. 2013;38(11):2179–2187. doi:10.1038/npp.2013.115
122. Capuron L, Pagnoni G, Drake DF, et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry. 2012;69(10):1044–1053. doi:10.1001/archgenpsychiatry.2011.2094
123. Bègue I, Kaiser S, Kirschner M. Pathophysiology of negative symptom dimensions of schizophrenia - Current developments and implications for treatment. Neurosci Biobehav Rev. 2020;116:74–88. doi:10.1016/j.neubiorev.2020.06.004
124. Kopec AM, Smith CJ, Ayre NR, Sweat SC, Bilbo SD. Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nat Commun. 2018;9(1):3769. doi:10.1038/s41467-018-06118-z
125. Tournier BB, Tsartsalis S, Ceyzériat K, et al. Fluorescence-activated cell sorting to reveal the cell origin of radioligand binding. J Cerebral Blood Flow Metab. 2020;40(6):1242–1255. doi:10.1177/0271678X19860408
126. Liu H, Leak RK, Hu X. Neurotransmitter receptors on microglia. Stroke Vascular Neurol. 2016;1(2):52–58. doi:10.1136/svn-2016-000012
127. Spampinato SF, Copani A, Nicoletti F, Sortino MA, Caraci F. Metabotropic Glutamate Receptors in Glial Cells: a New Potential Target for Neuroprotection? Front Mol Neurosci. 2018;11:414. doi:10.3389/fnmol.2018.00414
128. Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci. 2005;25(11):2952–2964. doi:10.1523/JNEUROSCI.4456-04.2005
129. Loane DJ, Stoica BA, Tchantchou F, et al. Novel mGluR5 positive allosteric modulator improves functional recovery, attenuates neurodegeneration, and alters microglial polarization after experimental traumatic brain injury. Neurotherapeutics. 2014;11(4):857–869. doi:10.1007/s13311-014-0298-6
130. Qiu JL, Zhu WL, Lu YJ, et al. The selective mGluR5 agonist CHPG attenuates SO2-induced oxidative stress and inflammation through TSG-6/NF-κB pathway in BV2 microglial cells. Neurochem Int. 2015;85–86:46–52. doi:10.1016/j.neuint.2015.04.007
131. Byrnes KR, Stoica B, Loane DJ, Riccio A, Davis MI, Faden AI. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia. 2009;57(5):550–560. doi:10.1002/glia.20783
132. Zinni M, Mairesse J, Pansiot J, et al. mGlu3 receptor regulates microglial cell reactivity in neonatal rats. J Neuroinflammation. 2021;18(1):13. doi:10.1186/s12974-020-02049-z
133. Matrisciano F, Tueting P, Maccari S, Nicoletti F, Guidotti A. Pharmacological activation of group-II metabotropic glutamate receptors corrects a schizophrenia-like phenotype induced by prenatal stress in mice. Neuropsychopharmacology. 2012;37(4):929–938. doi:10.1038/npp.2011.274
134. Wang H-Y, MacDonald ML, Borgmann-Winter KE, et al. mGluR5 hypofunction is integral to glutamatergic dysregulation in schizophrenia. Mol Psychiatry. 2020;25(4):750–760. doi:10.1038/s41380-018-0234-y
135. Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of Glutamate Alterations in Schizophrenia: a Meta-analysis of Proton Magnetic Resonance Spectroscopy Studies. JAMA Psychiatry. 2016;73(7):665–674. doi:10.1001/jamapsychiatry.2016.0442
136. Kegeles LS, Slifstein M, Xu X, et al. Striatal and extrastriatal dopamine D2/D3 receptors in schizophrenia evaluated with [18F]fallypride positron emission tomography. Biol Psychiatry. 2010;68(7):634–641. doi:10.1016/j.biopsych.2010.05.027
137. Lehrer DS, Christian BT, Mantil J, et al. Thalamic and prefrontal FDG uptake in never medicated patients with schizophrenia. Am J Psychiatry. 2005;162(5):931–938. doi:10.1176/appi.ajp.162.5.931
138. Talvik M, Nordström AL, Olsson H, Halldin C, Farde L. Decreased thalamic D2/D3 receptor binding in drug-naive patients with schizophrenia: a PET study with [11C]FLB 457. Int j Neuropsychopharmacol. 2003;6(4):361–370. doi:10.1017/S1461145703003699
139. Yasuno F, Suhara T, Okubo Y, et al. Low dopamine d(2) receptor binding in subregions of the thalamus in schizophrenia. Am J Psychiatry. 2004;161(6):1016–1022. doi:10.1176/appi.ajp.161.6.1016
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.