")
Back to Journals » International Journal of Nanomedicine » Volume 19
Donepezil-Loaded Nanocarriers for the Treatment of Alzheimer’s Disease: Superior Efficacy of Extracellular Vesicles Over Polymeric Nanoparticles
Authors Oliveira Silva R , Counil H, Rabanel JM , Haddad M, Zaouter C, Ben Khedher MR, Patten SA, Ramassamy C
Received 24 November 2023
Accepted for publication 7 January 2024
Published 1 February 2024 Volume 2024:19 Pages 1077—1096
DOI https://doi.org/10.2147/IJN.S449227
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Misra
Rummenigge Oliveira Silva,1 Hermine Counil,1 Jean-Michel Rabanel,2 Mohamed Haddad,1 Charlotte Zaouter,1 Mohamed Raâfet Ben Khedher,1,3 Shunmoogum A Patten,1 Charles Ramassamy1
1Centre Armand-Frappier Santé Biotechnologie, Institut National de la Recherche Scientifique, Laval, Québec, Canada; 2Faculté de Pharmacie, Université de Montréal, Montréal, Québec, Canada; 3Higher Institute of Biotechnology of Beja, University of Jendouba, Beja, Tunisia
Correspondence: Charles Ramassamy, INRS, Centre Armand-Frappier Santé Biotechnologie, 531 Boul des Prairies, Laval, QC, H7V 1B7, Canada, Tel +450-687 50 10, Email [email protected]
Introduction: Drug delivery across the blood-brain barrier (BBB) is challenging and therefore severely restricts neurodegenerative diseases therapy such as Alzheimer’s disease (AD). Donepezil (DNZ) is an acetylcholinesterase (AChE) inhibitor largely prescribed to AD patients, but its use is limited due to peripheral adverse events. Nanodelivery strategies with the polymer Poly (lactic acid)-poly(ethylene glycol)-based nanoparticles (NPs-PLA-PEG) and the extracellular vesicles (EVs) were developed with the aim to improve the ability of DNZ to cross the BBB, its brain targeting and efficacy.
Methods: EVs were isolated from human plasma and PLA-PEG NPs were synthesized by nanoprecipitation. The toxicity, brain targeting capacity and cholinergic activities of the formulations were evaluated both in vitro and in vivo.
Results: EVs and NPs-PLA-PEG were designed to be similar in size and charge, efficiently encapsulated DNZ and allowed sustained drug release. In vitro study showed that both formulations EVs-DNZ and NPs-PLA-PEG-DNZ were highly internalized by the endothelial cells bEnd.3. These cells cultured on the Transwell® model were used to analyze the transcytosis of both formulations after validation of the presence of tight junctions, the transendothelial electrical resistance (TEER) values and the permeability of the Dextran-FITC. In vivo study showed that both formulations were not toxic to zebrafish larvae (Danio rerio). However, hyperactivity was evidenced in the NPs-PLA-PEG-DNZ and free DNZ groups but not the EVs-DNZ formulations. Biodistribution analysis in zebrafish larvae showed that EVs were present in the brain parenchyma, while NPs-PLA-PEG remained mainly in the bloodstream.
Conclusion: The EVs-DNZ formulation was more efficient to inhibit the AChE enzyme activity in the zebrafish larvae head. Thus, the bioinspired delivery system (EVs) is a promising alternative strategy for brain-targeted delivery by substantially improving the activity of DNZ for the treatment of AD.
Keywords: Alzheimer’s disease, donepezil, extracellular vesicular, PLA-PEG, bEnd.3 cells, zebrafish
Graphical Abstract:
Introduction
Alzheimer’s disease (AD) is a progressive neuropathological disease. In 2021, the World Alzheimer Report estimated that more than 55 million people were living with AD worldwide, a number that is expected to increase to 78 million by 2030 if no alternative treatment is presented.1
To date, there is still no effective treatment to reverse or slow the progression of AD. So far, the approved treatment of AD by the United States Food and Drug Administration (FDA) is primarily based on the inhibition of the acetylcholine esterase (AChE), the main enzyme that degrades the neurotransmitter acetylcholine in the brain2–4 and the monoclonal antibodies (Aducanumab, Lecanemab, Gantenerumab) targeting amyloid-β which are new bets to reduce the progression of the disease.5 However, until recently, very limited clinical efficacy based on the anti-amyloid-β immunotherapy have provided beneficial effects in AD.6 Among the AChE inhibitors, donepezil (DNZ) is a piperidine-based, non-competitive and reversible inhibitor of AChE, and the most potent approved drug for the therapeutic management of mild to moderate AD.7,8 Differing to what was previously thought, recent publications have shown that DNZ could also provide a neuroprotective effect that can help to slow down the progression of the disease in addition to symptomatic treatment.9,10 However, DNZ exhibits extensive first-pass metabolism resulting in a low brain bioavailability.11 This leads to the need for higher doses to achieve and maintain therapeutic effect in the brain.12 As a result, patients experience more peripheral adverse events that are mainly associated with cholinergic functions (nausea, diarrhea, dizziness, and insomnia) causing a low compliance to treatment.13–15
Nano delivery strategies are widely studied to improve drug distribution by associating lower side effects, targeting, reducing rapid liver metabolism, and better therapeutic efficacy.16,17 Nanoparticles (NPs) are drug encapsulation vehicles with ultra-small dimensions (10–1000 nm), high surface area, and functionalizable structure.18,19 The ability of these systems to cross cell membranes, reduce drug degradation can be achieved by optimizing the size, surface charge, shape, chemical composition, and surface properties.20 NPs were developed with the biocompatible and biodegradable aliphatic homopolymer PLA, which is approved by the FDA and the European regulatory authorities for a wide range of biomedical and pharmaceutical applications.21 We and others have demonstrated that NPs-PLA are able to project sustained release, surface changes that make it possible to target organs or cells, and in addition to higher protection of drugs against degradation.22–24 When NPs-PLA are functionalized through PEGylation (adding PEG to the surface of NPs), this process has been shown to be a powerful way to prolong the circulating half-life of drug-loaded nanocarriers, enabling drug transport across the BBB, and being able to improve the accumulation and diffusion of NPs in the brain parenchyma.24–28
Although synthetic drug delivery systems are currently the most investigated clinically, biological or bioinspired nanocarriers are increasingly considered as an alternative because they have numerous advantages over existing synthetic systems.29 An example of this innovation for drug delivery purposes are EVs. EVs are nanometer-sized vesicles released naturally by almost all cell types in the body and are classified into exosomes, microvesicles, and apoptotic bodies depending on their size.30 EVs are composed of a complex composition of bilayers containing different lipids, proteins, carbohydrates, DNA, RNAs, and surface-associated molecules.31 The ability to transport and deliver biomolecules over short or long distances makes them attractive for drug delivery purposes.32,33 EVs are constantly released into circulation by all cell types in the body, making human peripheral blood a safe source and potential therapeutic tool.34,35 When it comes to pharmacokinetics, native EVs formed in the body may have longer circulation half-lives and/or site-specific targeting mechanisms that undergo less hepatic clearance.36 Higher biocompatibility, less immunogenicity, and the possibility of being obtained from the patient’s own cells (autologous) or through blood/plasma (autologous/alogenous) are some of its positive features.37 This approach is in line with personalized medicine, which aims to tailor clinical treatments to the specific characteristics of each patient’s disease. EVs have gained more attention as drug delivery systems to the brain particularly because some studies showed that they can cross the BBB from the systemic circulation to the brain and vice-versa.38,39
Therefore, this work aims to compare the efficacy of DNZ-loaded synthetic (NPs-PLA-PEG) and bioinspired (EVs) nano delivery systems to cross the BBB and to release the drug. To achieve this goal, DNZ was encapsulated in EVs isolated from healthy human plasma and in NPs-PLA-PEG. Subsequently, the delivery systems were side-by-side evaluated for cellular internalization, toxicity, ability to cross biological barriers, biodistribution, and inhibition of the AChE enzyme activity and on in vivo model of zebrafish larvae.
Materials and Methods
EVs Isolation
Peripheral blood was collected from healthy donors between 18 and 60 years of age and gave written informed consent. In brief, blood was collected into EDTA tubes and plasma was prepared by centrifugation at 3000 × g for 15 min at 4°C. Plasma was aliquoted and stored in tubes at −20°C until further analysis. All methods were carried out by relevant guidelines and regulations and all experimental protocols were approved by the Ethical Committee of the Institut National de la Recherche Scientifique (INRS-CER 20-582).
Samples were defrosted and EVs enrichment was achieved by precipitation according to the manufacturer’s protocol with some modifications (ExoQuick - Invitrogen™ by Life Technologies Inc., Carlsbad, CA, USA). In brief, 60 μL of ExoQuick was added to a 300μL plasma sample and incubated for 30 min at 4°C. After incubation, the precipitate was spun down (10.000 x g, 5 min at 4°C), and the resulting pellets were washed, filtrated, and resuspended in PBS. The EVs fraction was stored at −20°C until further use.
Total Protein Determination
The EVs fraction were initially extracted using RIPA buffer (Sigma-Aldrich, St Louis, USA) with a cocktail of protease inhibitors (Sigma-Aldrich) in the proportion of 1 to 4 EVs then protein contents were measured using the BCA protein assay kit (Thermo Scientific Pierce, Rockford, IL, USA). The plate was incubated for 30 min at 37°C, before being analyzed with a spectrophotometer at 562nm in a microplate reader (SynergyTM HT, BioTek Instruments, Inc. Winooski, VT, USA).
Donepezil (DNZ)-Free Base Preparation
To ensure good encapsulation efficiency, the drug DNZ-HCL was converted to its base form. The DNZ-HCL (Sigma-Aldrich, St Louis, USA) was dissolved in distilled water and treated with 1% sodium hydroxide. The precipitate was dissolved in methanol (Sigma-Aldrich, St Louis, USA) and the dissolved base was then dried at room temperature.
Preparation of EVs-DNZ
To prepare DNZ-loaded EVs (DNZ-EVs), DNZ (1000 μg) was sonicated in Milli-Q water (250 μL) for 5 min to increase its water solubility and then added to 1 mg/mL EVs in PBS. The mixture was sonicated three times using a QSonica Q125 sonicator (Newtown, CT) with a 1/8′′ diameter probe operating at 20% amplitude and 6 cycles of 4 s on/2 s off, with 2 min intervals on ice between cycles. To recover the EVs, the sonicated solution was incubated at 37 °C for 60 min. EVs were washed in PBS using an ultrafiltration device (Amicon Ultra-2 centrifugal filters, MWCO 100k, Merck Millipore, Darmstadt, Germany) to remove unloaded DNZ. DNZ-EVs were kept at −20 °C until further use.
PKH26-Labeled EVs
100μg of EVs were resuspended in 250 μL of Diluent C (Sigma) and then mixed with 250 μL of 8 μM of PKH26 (Sigma) in Diluent C. The dye mixture and EVs were incubated for 10 minutes protected from light with gentle mixing by pipetting every minute. Excess dye was removed with 50μL of exosome-depleted fetal bovine serum (Sigma-Aldrich). Then, PKH26-labeled EVs were diluted in 1mL of PBS and transferred to Amicon Ultra-2 centrifugal filters 100 kDa MWCO filters (Merck Millipore, Darmstadt, Germany) and centrifuged at 3000×g for 15 min at 4°C. This step was repeated twice then the sample was diluted to the necessary concentration for the experiments.
NPs-PLA-PEG Synthesis
PEG2000-b-PLA450 diblock copolymers were obtained with Poly(ethylene glycol) (PEG, MW 2000) and poly(DL-lactic acid) by ring-opening polymerization, and NPs produced as previously described.24 Polymer (25 mg of PLA-PEG) and DNZ base (5 mg) were dissolved in 4 mL of acetone (Sigma-Aldrich, St Louis, USA). The organic phase was then added dropwise to the aqueous phase under stirring at 1000 rpm. Finally, the above mixture was added dropwise at an injection speed of 1 mL/min (Harvard Apparatus 22, Harvard, USA) to 10 mL of MilliQ water. The organic solvent was removed by using a rotary evaporator IKA RV 10 Control (IKA-Werke GmbH & Co. KG) at 40°C and 400 m bar pressure for 1 hour. NPs were then transferred to an ultrafiltration device (Amicon Ultra centrifugal filters, MWCO 100k) and washed 3 times with PBS to remove the unentrapped drug.
NPs-PLA-PEG Labeling with Cy5
Fluorescent NPs (PLA-PEG-Cy5) were also produced with the same nanoprecipitation method described above with the addition of the Cy5 dye. To remove the free dye, NPs were dialyzed for 2 days in MilliQ water using cellulose dialysis (12–15 kD cut-off, Sigma-Aldrich, ON Canada). The dialysis medium was changed 2–3 times a day. To confirm the absence of free dye in the formulation, samples were ultracentrifuged 3 times with PBS using an ultrafiltration device (Amicon Ultra centrifugal filters, MWCO 100k) at 10,000×g for 15 min at 4°C. The filtrate from the last wash was used as a control. NPs-PLA-PEG-Cy5 were kept at 4 °C until further use.
Nanoparticle Tracking Analysis (NTA)
The particle size and concentration of DNZ-EVs or NPs-PLA-PEG-Cy5 formulations were measured using NTA NS300 (Nanosight, Malvern Panalytical. UK). The system is equipped with a 488 nm laser and running with NTA 2.3 analytical software package. Samples were diluted in Milli-Q water to a range of 1×109-1.5 × 1011 particles/mL and analyzed with the camera level set at 14 and detection threshold set at 7. For each sample, three consecutive 60-sec videos were recorded at room temperature while the sample was injected with a syringe pump at the speed 100.
Polydispersity Index (PDI) and Surface Charge
The NPs were dispersed in MilliQ water after adequate dilution (1:100, v/v) for PDI and Zeta potential analysis using the Malvern Zetasizer 3000 (Malvern Instruments, UK). PDI was measured to determine the dispersity of the particle size distribution and zeta potential was studied to determine the surface charge of the NPs. All the measurements were carried out in triplicate at 25 °C.
Transmission Electron Microscopy (TEM)
The morphology of EVs and NPs-PLA-PEG were investigated by TEM. In brief, samples were fixed with 2% paraformaldehyde (PFA), added (10 µL/grid) to Formvar-carbon coated grid, and adsorbed for 5 min. Samples grids were then incubated with 2% uranyl-acetate (EVs) or one drop of 3% (w/v) phosphotungstic acid (NPs-PLA-PEG) for 1 min for negative staining. Excess stain was soaked off by a filter paper and the samples were examined in a Hitachi HT 7100 (Hitachi, Tokyo, Japan) electron microscope at 75 kV at 17000X–40000X magnification.
Capillary Western Blot Analysis (Jess)
EVs-specific proteins were analyzed using the capillary Western blot Jess system (ProteinSimple, CA, USA) according to the manufacturer’s protocol. Briefly, samples were diluted to a concentration of 1 mg/mL, denatured at 95 °C for 5 min, and processed using the 12–230 kDa Jess Separation Module with different antibodies: anti-TSG101 antibody (Novus Biologicals LLC, Cat. NB200-112, 1/10), anti-CD63 antibody (R&D Systems; Cat. MAB50482, 1:20), anti-calnexin antibody (Novus Biologicals LLC; Cat. NB100-1965, 1:25). The fluorescence detection was quantified with streptavidin-HRP (ProteinSimple; Cat. 042–414). After protein migration (25 minutes at 375 V) they were incubated for 30 minutes with the primary antibodies, followed by a washing step and a 30-minute incubation with the secondary antibodies. Finally, the Compass Simple Western software (version 5.0.1, ProteinSimple) was used to capture the digital image of the capillary immunodetection.
Encapsulation Efficiency (EE %) and Drug Loading (DL%)
The EE% and DL% of DNZ in the nanoformulations were determined using a Multiskan SkyHigh Microplate Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Briefly, DNZ (1 mg) was dissolved in Acetonitrile/Methanol/Water (50/40/10%) and diluted to different concentrations. The absorbance values at 267nm was plotted to the standard curve of DNZ to get the concentration. The linearity range of the calibration curve was within 0.5–25 μg/mL with a correlation coefficient of 0.9988. Ultrafiltration was used to determine the EE. For this, samples were placed in an ultrafiltration device (Amicon Ultra centrifugal filters, MWCO 100k) and centrifuged at 10.000 rpm for 15 min at 4°C to remove the free DNZ then the filtrated was analyzed at 267 nm. The EE% and DL% were calculated using the following equation:


In vitro Release Profiles
The equivalent to 300 μg/mL of DNZ nanoformulations were transferred to a dialysis membrane immersed in a vial containing 10 mL fresh PBS (pH 7.4) maintained at 37°C, with continuous magnetic stirring (100 rpm) for 24h. At different time intervals, a small volume of the release medium was removed and replaced by the same volume of PBS. The DNZ content in the release medium was quantified in triplicate using the same method to determine the encapsulation efficiency (see above).
Stability Assay
To study the formulations stability and their potential to withstand atmospheric/environmental changes, the EVs samples were stored at –20°C and NPs-PLA-PE at 4°C. Samples were withdrawn at day 1, 30, and 60-day time intervals and analyzed for mean particle size, zeta potential, PDI, and drug content using the previously mentioned methods. Each study was performed in triplicate.
Cells Culture
bEnd.3 cell line was purchased from ATCC (ATCC CRL-2299TM), and cultured in Dulbecco’s Modified Eagle’s Media (DMEM)(Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) and antibiotics (100 U/mL penicillin and 100 g/mL streptomycin). Human neuroblastoma cell line (SK-N-SH) was cultured in Eagle’s minimal essential medium (EMEM) supplemented with 10% FBS, 1% sodium pyruvate (1 mM), and 1% (v/v) of both penicillin and streptomycin. Cells were grown in an incubator with optimal culture conditions of 37 °C and 5% CO2, and the medium was routinely replaced every 2–3 days.
Cytotoxicity (Resazurin Assay)
The potential cytotoxic of all formulations was performed with the Resazurin reduction assay. For this, bEnd.3 and SK-N-SH were harvested using trypsin/0.25% EDTA. Cells were plated with 100 µL/well at the density 2 × 104 cells/mL in 96-well plates (Corning Incorporated, Corning, NY, USA). After 48 h of proliferation, the medium was removed and replaced with fresh complete medium containing the free DNZ or DNZ-loaded formulations with an equivalent of DNZ at 0.1, 1, 10, and 30 μg/mL for 24 h. Cell viability was determined with the excitation at 530 nm and emission wavelength at 590 nm with the SynergyTM HT plate reader (BioTek Instruments, Inc. Winooski, VT, USA). Cell viability obtained with the PBS-treated cells was considered as 100%.
AChE Inhibition Assay
The activity of AChE was determined spectrophotometrically in a 96-well microplates by the Ellman’s method with slight modifications.40 The inhibitory effect of 10 μg/mL of free DNZ, DNZ-loaded NPs-PLA-PEG or EVs was determined on SK-N-SH neuronal cells in a 6-well plate. For this, after 24 hours of treatment, cells were washed 3 times with PBS (pH 7.4), total proteins were extracted followed by the addition of a cocktail of protease and phosphatase inhibitors (Roche® Basel, Switzerland) and protein was quantified by the BCA assay (Thermo Scientific Pierce, Rockford, IL, USA).
The reaction solution was prepared by mixing 15 mL of Sodium phosphate monobasic buffer at 10 mM, with the pH 7.5–8.0 (Sigma-Aldrich), 200 μL of the enzymatic substrate acetylthiocholine iodide at 75 mM (Sigma-Aldrich), and 500 μL of 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB) at 10 mM in sodium phosphate monobasic. The Ellman’s assay was initiated by adding the reaction solution to 10 μg of the cell lysates in a 96-well plate for a total volume adjusted to 200 μL. After 10 min of incubation, the absorbance was measured at 412 nm every minute for up to 30 min using a Multiskan SkyHigh Microplate Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The 30 min time point was chosen because it was in the linear phase of the assay. The percent of inhibition of the enzyme activity was calculated by the equation given below:

Cells Internalization of Nanoformulations
bEnd.3 and SK-N-SH cells were seeded in a 24-well plate (Sarstedt, Montreal, Canada) at a density of 1×105 cells/cm2. After reaching 90% confluence, 1.98×1010 particle/cm2 of PKH26-labeled EVs and Cy5-labeled NPs-PLA-PEG were added to the cell culture medium. 24 h after, cells were washed twice with ice-cold PBS then trypsinized (Trypsin/0.25% EDTA) at 37 °C for 5 min. Cell suspensions were transferred to 5 mL RIA tubes (Falcon, Corning, USA) and centrifuged at 4000 rpm for 4 min at RT (Sorvall Legend, ThermoScientific). After media removal, cells were washed twice with PBS at pH 7.4 and finally resuspended in 0.5% PFA/PBS. Cells were kept at rest at 4 °C for 1 h before analysis. The uptake of PKH26-labeled EVs and Cy5-labeled NPs-PLA-PEG was analyzed on a BD LSR Fortessa cytometer (Becton-Dickinson, Boston, MA, USA) with a minimum of 10,000 events recorded/sample. The fluorescence of NPs-PLA-PEG-Cy5 was followed at λ Exc. 633 nm/λ Em. 647 nm (Alexa 647) and EVs-PKH26 in λ Exc.551 nm/λ Em. 567nm (PE). Analysis was performed using FlowJo V10.5 software (FlowJo LLC, Ashland, OR, USA) with an FSC-A and SSC-A to exclude debris and dead cells followed by FSC-H versus FSC-A to exclude doublets. Finally, fluorescent positive cells were blocked in the appropriate fluorescent channels: FSC-A x PE-H for PKH26 and FSC-A x Alexa 647 for Cy5.
Establishment of the BBB Model and in vitro Transcytosis of Nanoformulations
For the transcytosis experiment, the bEnd.3 monolayer cell line were cultured on the PET Transwell® filters with 1 μm pore diameter and 0.3 cm2 surface area (Corning Costar, Cambridge, MA) as previously described.41 Cells were seeded at a density of 5×104 cells/insert, the media in the apical and basal compartment was 0.5 mL each and was changed every other day during the 7–10 days of culture. Cells were allowed to build tight junction (TJ) for at least 10 days.42 The bEnd.3 monolayer cell integrity was validated by measuring the TEER, the permeability of the fluorescein dextran isothiocyanate, and finally the immunofluorescence of tight junction proteins as previously described.41 Details are described in the Supplementary Data Section.
On the basolateral side, SK-N-SH cells were seeded on the bottom of the basal compartment at a density of 5×104 cells/insert to examine the internalization of nanoformulations released by the bEnd.3 cells.
For the transcytosis assay, labeled formulations (1.2 × 1010 particles) were added to the apical compartment for 24h at 37°C. Media on the basolateral side were collected and evaluated by nanoflow cytometry (NanoAnalyzer U30 instrument, NanoFCM Inc., Nottingham, UK). The nFCM flow nanoanalyzer was used to calculate the number of uncaptured/suspended NPs and their labeling following the manufacturer’s instructions. Two single photon count avalanche photodiodes (APDs) were used for the simultaneous detection of side scatter (SSC) and single particle fluorescence. The fluorescence of NPs-PLA-PEG-Cy5 was monitored in the bandpass filter (680/40) and EVs-PKH26 in the bandpass filter (580/40). Measurements were made for 1 minute at a sampling pressure of 1.0 kPa with a particle count between 3000 to 10,000/min. The instrument was calibrated for concentration and size using a standard of 250 nm silica nanoparticles of known concentration and a cocktail of 4-modal silica nanospheres (NanoFCM Inc., S16M-Exo), respectively. Data processing was performed using the nFCM Professional Suite v1.8 software. Subsequently, SK-N-SH cells on the basolateral side were washed with PBS, trypsinized, harvested, and the uptake was quantitatively measured by flow cytometry (LSRFortessaTM BD Biosciences) using the same parameters to determine the cellular internalization.
Zebrafish Maintenance
Wild-type and Tg (flk1:EGFP) adult zebrafish (Danio rerio) were maintained at 28 °C at a light/dark cycle of 12/12 h, staged, and maintained according to standard procedures.43 The zebrafish flk1 promoter is able to drive the expression of green fluorescent protein (EGFP) which is expressed in all blood vessels in embryos and larvae making it a good model for biodistribution studies.44,45 Zebrafish were bred and the embryos were raised in embryo media (E3 medium) prepared from 5 mM NaCl, 0.17 mM KCl, 0.4 mM CaCI2, and 0.15 mM MgSO4 in 1 L of MilliQ water. Eggs containing dead or poor-quality embryos were removed. Embryos were raised in E3 medium in a dark incubator at 28°C for subsequent experiments. Until 24 hours postfertilization (hpf), E3 medium was supplemented with 0.3 mg of methylene blue/liter to prevent fungal growth. For imaging studies, pigment formation was blocked by adding 0.003% phenylthiourea (PTU) dissolved in egg water at 24 hpf. All experiments were performed in line with the guidelines of the Canadian Council for Animal Care (CCAC) and the Institut National de la Recherche Scientifique (INRS) animal ethics committee (#2005-01).
Acute Toxicity Test in the Zebrafish Embryo
Fertilized eggs from wild-type zebrafish were harvested and collected under a microscope, and dead/unfertilized embryos were removed to avoid contamination of the test solution. These eggs were incubated in E3 medium in a 6-well plate at a density of 10 eggs/well in 3 mL of the medium at 28°C and all experiments were performed in triplicate.
Acute toxicity of free DNZ toxicity was assessed (see Supplementary Data) by exposing the embryos to DNZ (1, 10, 30, 50, and 100 μg/mL) diluted in fish water. Survival, hatching time and malformation were recorded for 3 days.
The toxicity of the nanoformulations composed by the free DNZ, EVs-DNZ, NPs-PLA-PEG-DNZ groups at 10 μg/mL, and the control group was evaluated in 3 days. The exposure solutions were refreshed every 24 h to avoid aggregation of the NPs. Toxicity was assessed based on survival rate, hatching time, and gross morphological changes (viable larvae without deformations) at each time point: 24, 48, and 72 hpf using a stereomicroscope (Leica S6, Germany). Deformities included pericardial edema, yolk sac edema, tail deformities, and spinal curvature.
Larvae Behavior Test
After continuous exposure to the aforementioned treatments for 144 hpf (6 days), the zebrafish larvae were transferred to a 96-well plate (one fish per well). Subsequently, the plate was placed inside the DanioVision™ observation chamber (Noldus Information Technology, Leesburg, VA) for 30 minutes in the dark at 28.5°C. Then the light source inside the DanioVision observation chamber was turned on for 2 hours and the total distance traveled by each larva was tracked during dark and light periods by EthoVision® XT video tracking software (Noldus Information Technology, Leesburg, VA).
Intravenous Injection into Zebrafish Embryos
Zebrafish embryos 48 hpf were anesthetized with (0.02%) tricaine (Sigma) and positioned on a 35mm glass bottom Petri dishes (MatTek Corporation, MA, USA) coated with 1% low-melting agarose (UltraPure™LMP Agarose, Invitrogen, Life Technologies, Canada). The formulations were loaded into borosilicate glass capillary needles (Harvard Apparatus) and then 4 nL were microinjected into the duct of Cuvier. The injections were performed using a Pneumatic nanoinjector (FemtoJet 4i, Eppendorf, USA) and a manual micromanipulator MN-153 (Narishige, Zeiss Canada Ltd., ON Canada).
Drug Treatments and in vivo Determination of Acetylcholinesterase Activity in the Head
Briefly, 48 hpf zebrafish embryos were anesthetized and 4 nL (0.4 ng of DNZ) of the nanoformulations (free DNZ, EVs-DNZ, and NPs-PLA-PEG-DNZ at 100 μg/mL) were microinjected. After 24h, larvae from each group were anesthetized in ice-cold PBS (2–4 °C) and sacrificed by decapitation after cessation of opercular movements. Then they were homogenized using a portable motorized mini homogenizer (Cole Parmer, Vernon Hills, IL, USA). Homogenates from each group were centrifuged at 10,000 g for 10 min at 4°C and the supernatant was collected. AChE activity was determined for each exposure group in the larval homogenate supernatant following the same protocol previously described for cells.40 The experiments were conducted using samples containing a pool of 7 larval brains and all enzymatic assays were performed in at least three different experiments, and each was performed in triplicate.
Image Acquisition (ZF Larvae Biodistribution)
Tg (flk1:EGFP) zebrafish embryos were cultured under standard conditions at 28°C in E3 medium containing PTU. Injections of 4nL of the formulations (NPs-PLA-PEG-Cy5 and EVs-PKH26) into zebrafish embryos (48 hpf) were performed as described above. Such injection represents the amount of 7.92×105 NPs. For image acquisition, zebrafish larvae were kept under anesthesia and embedded in low-melting agarose. Image of successfully injected zebrafish embryos was performed at 24h post-injection (hpi) using a Zeiss LSM780 system confocal microscope (Zeiss, Germany) equipped with a Plan-Apochromat 10x/0.45 objective (whole embryo) and 20x/0.8 objective (brain). Optical filters providing excitation/emission at 569nm/631nm, 636nm/737nm, and 488nm/510nm were used for the detection of PKH26, Cy5, and EGFP, respectively. Acquisitions were performed using Zen® 2011 software (Zeiss, Germany), while image analysis was processed using the Fiji Image J software.46
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software, version 9.5.1 (GraphPad Software, San Diego, CA, USA), and results with a P value <0.05 were considered statistically significant. Each experiment was repeated at least three times. All data were presented as the mean ± standard error of the mean (SEM). Differences between two groups were compared using a two-tailed Student’s t-test, and more than two groups were compared using a two-way or one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test.
Results
EVs and NPs Characterizations
TEM and NTA showed the typical “cup-shaped” morphology of EVs with a mean diameter of 126 ± 0.38 nm (Figure 1A and B). Western blotting showed the presence of EV specific marker proteins (CD63 and TSG101) and the absence calnexin supporting the absence of contaminating intracellular particles during the EVs preparation steps (Figure 1C). The average protein content determined by the BCA assay kit was 2.5 ± 0.21µg/µL.
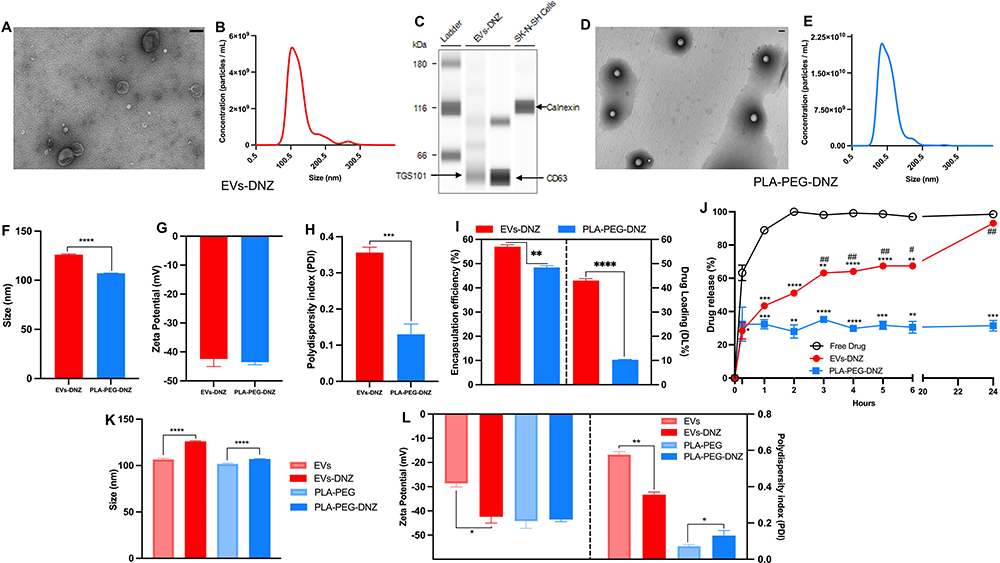 |
Figure 1 Characterization of EVs-DNZ and synthetic NPs-PLA-PEG-DNZ. (A and D) A representative TEM image of EVs-DNZ and NPs-PLA-PEG-DNZ. Scale bar, 100 nm. (B, E, and F) Size distribution of EVs-DNZ and NPs-PLA-PEG-DNZ measured by NTA. (C) Western blot analysis of EVs isolated from human plasma and cell lysates to confirm the expression of the EVs marker proteins CD63, TSG101, and Calnexin (as negative control). (G) Zeta potential, (H) PDI, and (I) encapsulation efficiency and drug loading of EVs-DNZ and NPs-PLA-PEG-DNZ. Data are shown as mean ± SEM. n = 3 per group, unpaired t-test. (J) In vitro drug release study of free DNZ, EVs-DNZ, and NPs-PLA-PEG-DNZ. Data are presented as mean ± SEM (n = 3 per group) with *p < 0.05, **p < 0.01, ***p< 0.001, and ****p < 0.0001 vs free DNZ; and #p < 0.05, ##p < 0.01 vs NPs-PLA-PEG-DNZ. (K and L) Effect of drug encapsulation on size, zeta potential, and PDI. Data are presented as mean ± SEM (n=3). |
On the other hand, NPs-PLA-PEG-DNZ, obtained by nanoprecipitation, had spherical morphology and an average size of 107 ± 0.15 nm (Figure 1D and E). The zeta potential of EVs-DNZ and NPs-PLA-PEG-DNZ was similar with −42.43 ± 1.52 mV and −43.56 ± 0.50 mV, respectively (Figure 1F and G). However, the size distribution was more uniform for NPs-PLA-PEG-DNZ (PDI: 0.13 ± 0.016) as compared to EVs-DNZ (PDI: 0.36 ± 0.01) (Figure 1H).
Some changes after drug encapsulation in the systems were observed. The NTA analysis showed that after drug loading, the size of EVs-DNZ and NPs-PLA-PEG-DNZ was statistically increased compared to the blank EVs and NPs-PLA-PEG (Figures 1K and S1A). In addition, DLS analysis showed that drug loading caused a decrease in the absolute value of the zeta potential of EVs-DNZ and a reduction in PDI (Figure 1L).
Increases in EVs and NPs size and size distribution indirectly reflect the successful loading of DNZ, while the negative zeta potential implies that EVs-DNZ were likely more stable and more resistant to aggregation as observed in the stability study (Figure S1A). The negative zeta potential may contribute to the greater stability of the EVs-DNZ formulations during storage at −20°C as no significant changes in particle size and PDI were observed after thawing (Figure S1A and B).
Regarding NPs-PLA-PEG, drug loading did result in an increase of the PDI but no change in the absolute value of the zeta potential was recorded (Figure 1K and L). Zeta potential values lower than −30 mV recorded for the NPs-PLA-PEG-DNZ formulations during the stability study also contributed to maintain a monodisperse particle population of the formulations and low particle size (Figure S1C).
DNZ encapsulation efficiencies were 57.6 ± 1.45% and 48.2 ± 0.92% in EVs-DNZ and NPs-PLA-PEG-DNZ, respectively. Drug loading values were 43.0±0.81 for EVs-DNZ and 10.3±0.14 for NPs-PLA-PEG-DNZ (Figure 1I). Even after 2 months of storage, drug encapsulation values in the system remained above 38% (Figure S1D). Considering that donepezil free base is less soluble in water (33 g/L) and the physicochemical properties of EVs, the drug may be retained in parts in the lipid bilayer and in its interior/core.47,48 In relation to PLA-PEG, encapsulation occurs by trapping the drug within the polymeric matrix after the drug and polymer are dissolved in an organic solvent and then in contact with an aqueous solution (nanoprecipitation method).49 The in vitro release of DNZ from the formulations using the dialysis bag method was compared to the diffusion of a free DNZ solution (Figure 1J). EVs-DNZ and NPs-PLA-PEG-DNZ showed a slower and more sustained drug release than free DNZ as expected. In the first hour, 43.4 ± 1.98% and 32.3 ± 2.81% of the encapsulated drug were released from the EVs and NPs-PLA-PEG respectively, compared to more than 88.9 ± 1.50% of the free DNZ. When evaluated over a 24h period, the DNZ release profile of EVs displayed an initial burst release followed by a slower release phase until release reached almost 100% (93.1 ± 1.25%). For NPs-PLA-PEG, the amount of DNZ released in 24 h remained close to 32% indicating that the synthetic NPs had a slower release profile in vitro compared to EVs and free DNZ (Figure 1J).
Effects of Formulation on Cell Viability and AChE Activity in vitro
Resazurin-based assays were used to analyze the toxicity of bEnd.3 and SK-N-SH cells induced by different concentrations of DNZ and to determine whether its encapsulation could reduce free DNZ-associated in vitro toxicity (Figure 2A). bEnd.3 is the most widely endothelial cells used to study the permeability of the tight junctions which are representative of the BBB.50 Free DNZ and NPs-PLA-PEG-DNZ were not toxic to bEnd.3 cells while, they induced, respectively, 57.2 ± 8.58% and 73.4± 6.56% reduction of the SK-N-SH cells viability at 30 mg/mL (Figure 2A). Interestingly, the EVs-DNZ formulation was not toxic to both cell lines (Figure 2A). No toxicity was observed in cells treated with the blank formulations (Figure S2). Based on these results, 10 µg/mL of DNZ was selected for the rest of the study (encapsulated or not).
 |
Figure 2 Evaluation of cell viability and AChE activity in neuronal cells. (A) bEnd.3 and SK-N-SH cells viability after treatment with different concentrations of free DNZ, EVs-DNZ, and NPs-PLA-PEG-DNZ for 24h as determined by resazurin assay. Data presented are mean ± SEM (n = 6), **p < 0.01 vs control without treatment. (B) Effect of formulations on AChE inhibition in neuronal cells (SK-N-SH). Inhibition of the enzyme activity in percentage of the control group without treatment with n = 3 per group. ****p < 0.0001 vs control and ####p < 0.0001 vs PLA-PEG. |
For the inhibition of the enzyme activity, SK-N-SH cells were treated with 10 µg/mL of free DNZ, or with either both formulations EVs-DNZ or NPs-PLA-PEG-DNZ containing an equivalent of 10 µg/mL of DNZ. After 24 hours of treatment, free DNZ and EVs-DNZ significantly inhibited the AChE activity by 34.34 ± 2.02% and 34.30 ± 1.06%, respectively. Interestingly, the inhibitory effect observed with free DNZ and EVs-DNZ was two fold higher than with the NPs-PLA-PEG-DNZ formulation (15.51 ± 1.13%) (Figure 2B). These results could be due to lower release of DNZ by the formulation NPs-PLA-PEG-DNZ or higher uptake of free DNZ and EVs-DNZ by neuronal cells which could confirm better biocompatibility of the EVs-DNZ formulation.
Internalization of EVs and NPs-PLA-PEG
For the uptake studies, the nanoformulations were labeled with the fluorescent dyes PKH26 (EVs-PKH26) and Cy5 (PLA-PEG-Cy5) (Figure 3A). PKH26 has aliphatic tails that are anchored in the lipid bilayer of the plasma membrane of EVs.51 Cy5 has proved to be a good platform for rapid assessment of the biodistribution of NPs in vivo in many studies.24,52 The fluorescence intensity of EVs-PKH26 and PLA-PEG-Cy5 was confirmed by the nanoscale flow cytometer (nFCM) specially designed for small particles detection (Figure 3B). Then, the uptake of EVs-PKH26 and PLA-PEG-Cy5 by SK-N-SH and bEnd.3 cells was analyzed by FACS. After 24h incubation of bEnd.3 cells with EVs-PKH26 and NPs-PLA-PEG-Cy5, 92.57 ± 0.17% and 92.83 ± 0.32% of the cells were, respectively, fluorescent (Figure 3C and D). For SK-N-SH cells, 99.03 ± 0.37% and 99.22 ± 0.20% were fluorescent when incubated with EVs-PKH26 and NPs-PLA-PEG-Cy5, respectively (Figure 3E and F). Overall, the uptake efficacy by both cell types was similar for both nanoformulations.
 |
Figure 3 Uptake of EVs and NPs-PLA-PEG by bEnd.3 and SK-N-SH cells. (A) Schematic representation of staining of EVs and NPs-PLA-PEG by PKH26 and Cy5, respectively. Created with BioRender.com. (B) Representative of nFCM fluorescence intensity resulting from staining EVs with PKH26 (red) or NPs-PLA-PEG with Cy5 (blue). Black signal resulted from EVs and NPs-PLA-PEG without fluorescence labelling (control). Flow cytometric analysis of EVs-PKH26 or NPs-PLA-PEG-Cy5 uptake by bEnd.3 (C and D) and SK-N-SH (E and F) cells after 24h of incubation. Controls represent cells that were not incubated with any formulation (untreated). The filtrate from the last wash after labeling and the unlabeled nanoformulations (EVs and NPs-PLA-PEG) were used to determine the background. The percentages of PKH26 and Cy5 positive cells are indicated inside the gates. |
In vitro BBB Transcytosis Study
After 8 days of culture, the integrity of the tight junctions (TJ) of the bEnd.3 cell monolayers was validated with the TEER recording values of 29 ± 0.3 Ω × cm2, the permeability of 6.7×10−6 ± 0.31 cm s−1 to Dex-FITC, and the expression of TJ proteins ZO-1 and claudin-5 proteins (Figure S3) confirming the restricted paracellular transport53 (Figure 4A). The permeability of the fluorescent NPs through the bEnd.3 monolayer cells and their uptake by SK-N-SH cells seeded at the bottom of the basal compartment (the “brain side” of the Transwell®) were quantified by flow cytometry (Figure 4B and C).
 |
Figure 4 Transcytosis efficacy of nanoformulations through the bEnd.3 cells and their uptake by SK-N-SH cells. (A) Schematic representation of the experimental design with the bEnd.3 cells monolayer cultured in the upper chamber (apical) of the insert Transwell® with 1 μm membrane pores and SK-N-SH cells seeded on the bottom of the basal compartment (the “brain side” of the Transwell®). Fluorescent formulations were added in the apical chamber at the equivalent concentration of 1.2 × 1010 particles and incubated for 24 h. Created with BioRender.com. (B) Flow cytometry plots indicating the percentage of nanoformulations (EVs-PKH26 and NPs-PLA-PEG-Cy5) taken-up by SK-N-SH after 24 h of incubation; the percentages of PKH26-positive or Cy5-positive cells are indicated inside the gates. (C) bar graph representing the quantification of the EVs-PKH26 (in red) and NPs-PLA-PEG-Cy5 (in blue) uptake and control cells (in black) were without nanoformulation. Data presented are mean ± SEM (n = 6). ***p < 0.001 vs control and ####p < 0.0001 vs control and EVs-PKH26. (D) Bivariate dot plots of PE (PKH26) and Cy5 fluorescence versus SSC of the nanoformulations (EVs-PKH26 and NPs-PLA-PEG-Cy5) present in the culture medium of the basal compartment of the Transwell®). (E) Quantification of nanoformulations in suspension in the basal medium after 24h of incubation. Data shown are mean ± SEM (n = 5), ****p < 0.0001 vs control and EVs-PKH26. (F) Representative fluorescence images showing the uptake of EVs-PKH26 and NPs-PLA-PEG-Cy5 by bEnd.3 cells (monolayer grown on the insert filter) after 24h incubation. EVs-PKH26 and NPs-PLA-PEG-Cy5 appear in red; core, blue; and claudin-5 (in green). Scale bar, 30 μm. Orthogonal views of 3D image stacks confirm the uptake of EVs-PKH26 (G) and NPs-PLA-PEG-Cy5 (H) in bEnd.3 cells. Scale bar, 60 μm. |
The images showed that most of the bEnd.3 monolayer cells in the Transwell® were strongly fluorescent after incubation with the labeled nanoformulations corroborating the high uptake shown by the flow cytometry (Figure 3C and D). Live confocal microscopy also confirmed that both EVs-PKH26 and NPs-PLA-PEG-Cy5 were not merely bound to the cells membrane but were internalized by bEnd.3 cells (Figure 4F, G and H).
The transcytosis of NPs-PLA-PEG-Cy5 and EVs-PKH26 across the monolayer was 65.9 ± 4.27% and 13.7 ± 2.08%, respectively. No interaction between the NPs-PLA-PEG with the Transwell® filter was observed as they crossed the pores of the cell-free Transwell®. However, around 30% of the EVs added to the apical compartment interact with the filter (data not shown). NPs were quantified in the culture medium of the basal compartment by nanoscale flow cytometry (nFCM). After 24 h, 3.31 ± 0.56% of NPs-PLA-PEG-Cy5 while only 0.17 ± 0.02% of EVs-PKH26 remained in suspension in the basal medium suggesting a greater fraction of EVs-PKH26 is associated to SK-N-SH cells (Figure 4D and E).
Acute Toxicity of DNZ and Nanoformulations Exposed to Larvae to Embryos Zebrafish
Zebrafish larvae were exposed to different concentrations of the free drug DNZ (1, 10, 30, 50, and 100 μg/mL) and the survival rate, hatch rate, and gross morphological changes were examined at different time point (Figure 5A). The highest concentration without causing toxicity (malformation and embryonic mortality) until 72h was 10μg/mL (Figures 5B and S4A–D). Thus, nanoformulations with an equivalent concentration of 10μg/mL of DNZ was selected for the in vivo experiments.
 |
Figure 5 Acute toxicity in the zebrafish embryo and larval behavior test. (A) Timeline of the experiments. Created with BioRender.com. (B) Mortality (%) and (C) hatching (%) of zebrafish embryos from the control group and after exposure to 10 μg/mL DNZ, EVs-DNZ, and NPs-PLA-PEG-DNZ containing an equivalent of 10 μg/mL of DNZ during 72 h. (D) Representative morphology of zebrafish embryos exposed to the nanoformulations. (E) The locomotor activity of zebrafish larvae on the sixth day after treatment with the free drug DNZ, or both nanoformulations. Tests were performed from 3 replicate trials using 10 embryos at each dose. Values are expressed as mean ± SEM, *p < 0.05 vs control. One-way ANOVA followed by Tukey as a post hoc comparison. |
However, exposure to DNZ-loaded EVs anticipated the hatching time of the embryos (Figure 5C) as compared to NPs-PLA-PEG-DNZ, and free dugs (*p < 0.05). No obvious morphological changes (viable larvae without deformations) or growth retardation were observed for any group until 72 hpf exposure to nanoformulations containing the equivalent of 10μg/L of DNZ (Figure 5D). Behavioral test on the sixth day after DNZ or NPs-PLA-PEG-DNZ treatment revealed that the locomotor activity, in relation to the total distance traveled, was higher than in EVs-DNZ treated zebrafish or controls (Figure 5E). Higher locomotor activity is mainly due to the peripheral inhibition of AChE induced by DNZ or NPs-PLA-PEG-DNZ while EVs-DNZ did not induce peripheral side effects.
Image Acquisition (Biodistribution) and Acetylcholinesterase Activity in the Larvae Head
Our objectives were to increase the delivery of DNZ into the brain through the nanoformulations. For this, NPs-PLA-PEG-Cy5 or EVS-PKH26 systems were injected intravenously (Cuvier’s duct) into the transgenic Tg (flk1:EGFP) larvae 48 hpf (Figure 6A). There was evident accumulation of both particles along the trunk and tail 24 h after injection, suggesting an interaction with cells in this region. Sinusoidal endothelial (or scavenger) cells and macrophages located in the tail region of the embryonic zebrafish are known to be an important cell type in the binding, absorption and elimination of nanoparticles (Figure 6B and D).54 Interestingly, both nanoformulations were found in the brain with higher levels of EVs-PKH26 in cerebral blood vessels and in the brain parenchyma (Figure 6C and E). These results demonstrate higher brain accessibility of EVs-PKH26 as compared to NPs-PLA-PEG-Cy5 following an intravenous injection.
 |
Figure 6 In vivo biodistribution of nanoformulations in zebrafish embryos and acetylcholinesterase activity in the head. (A) Scheme showing the injection site (i.v.) in embryonic zebrafish (48 hpf) and imaging period. NPs-PLA-PEG-Cy5 and EVs-PKH26 were microinjected into the Tg (flk1:EGFP) zebrafish larvae at a dose equivalent to 7.92×105 NPs (4nL). Vascular endothelial cells are in green and labeled nanoformulations are in red. The nanoformulations were screened for in vivo distribution after 24h in the lateral position. Created with BioRender.com. (B and C) Whole embryo (10× magnification) and head-level (20× magnification) views of EVs-PKH26 biodistribution. EVs-PKH26 remains associated with the dorsal region of the embryo. Bright red dots were observed in the brain region of the larvae, corresponding to EVs-PKH26. The gray dots correspond to the presence of nanoformulations in the respective regions. (D and E) Whole embryos and head-level views of biodistribution. NPs-PLA-PEG-Cy5 predominantly circulated freely and was distributed throughout the vasculature of the embryo. High levels of NPs-PLA-PEG-Cy5 were confined to the vasculature but low level in the brain parenchyma (gray). Scale bars: 200µm (whole embryo) and 100µm (head). (F) Schematic representation of the injection and AChE activity analysis. Created with BioRender.com. (G) Effect of nanoformulations on the inhibition of the AChE activity in the zebrafish brain. The percentage of inhibition values was calculated in comparison to the activity of the control group without treatment. Data presented are mean ± SEM (n=3), six fish per sample. ***p <0.001, ****p < 0.0001 vs control, ###p < 0.001, ####p < 0.0001 vs DNZ, and $p < 0.05 vs NPs-PLA-PEG. One-way ANOVA followed by Tukey’s post hoc test. |
AChE inhibition was measured in the head of embryos following an intravenous injection of nanoformulations at 48 hpf (Figure 6F). All treatments led to the inhibition of the AChE activity in the zebrafish head after 24 h. More interestingly, the inhibition of the enzyme activity was 1.36 higher with the EVs-DNZ nanoformulation reaching 38.62% ± 9.75%, as compared to the NPs-PLA-PEG-DNZ nanoformulation (28.42% ± 2.0%) and free DNZ (14.43% ± 9.63%) as compared to control larvae without treatment (0.55% ± 0.32%) (Figure 6G).
Discussion
Treatment of many brain diseases remains challenging due to the impermeability of the endothelial cells that makes up the BBB, limiting the entry into the brain of about 98% of drugs.55,56 Among strategies to overcome the BBB, one of the most promising is the use of nanocarriers.57 We and others have demonstrated that synthetic NPs-PLA-PEG can cross the BBB, synthetic NPs-PLA-PEG and natural-occurring EVs are potential nanocarriers for drug delivery to the CNS.24,39,58–60 The use of smart carriers in recent decades has expanded into the field of targeted drug delivery. However, little is known about the advantages of EV-based drug delivery over the synthetic carriers.61 To date, a direct comparison between EVs and synthetic NPs (PLA-PEG) for drug delivery to the brain has not been performed. This dialogue can help us to better understand EVs as delivery systems and facilitate their medical application using the knowledge gained during the last decade by synthetic NPs.
As a first objective, the complete physicochemical characterization of nanometric delivery systems is critical because its determine the interactions with the biological medium and cell surfaces. As for the hydrodynamic size, they showed the same size range and zeta potential, an increase in size after the encapsulation step, which can be attributed to the accommodation of the drug in the structure of the systems.62 Sonication (the process used in EVs) could also be another factor as it is expected that the energy generated in the process could lead to vesicle membrane remodeling or fusion resulting in a larger vesicle. This could also have impacted the surface charge of EVs-DNZ due to changes in the proteins and/or lipids of the membrane exposed to the surface of the vesicles.63,64 The zeta potential for NPs-PLA-PEG showed an overall negative charge without modification after drug loading. Low values of zeta potential (−40 mV) for both nanoformulations indicate reduced aggregation by the electrostatic repulsion of particles helping to increase physical stability. This is an essential requirement for efficient delivery of the drugs.65,66 Furthermore, NPs with a negative surface charge is more efficient for drug delivery across the BBB.67 These findings demonstrate that after drug loading, both nanoformulations were designed to be within the size range of 100 nm to 300 nm which is considered to be favorable for the transport of drugs across the BBB.68
The second objective was to assess biological safety and absence of cytotoxicity which are important parameters for biological applications and drug delivery systems. As previously observed, we also found that DNZ was toxic to neuronal cells from 30 μg/mL.69,70 Interestingly, EVs-DNZ nanoformulation was not toxic and even protect against DNZ induced-neuronal and bEnd.3 cells toxicity until 30 μg/mL but the same protective effect was not observed with the NPs-PLA-PEG-DNZ nanoformulation. One hypothesis for a lower cytotoxicity of EVs-DNZ would be the sustained release of the drug while the free drug diffuses rapidly and unrestrictedly into the cells.71 Moreover, a previous study showed that blood-derived-EVs from healthy donors, which included from multiple cells and with a complex composition, have a neuroprotective effect in addition to having low immunogenicity and low cytotoxicity, can also improve cell survival and/or reduce cell death.72–75 In addition, the in vivo efficacy of EVs-DNZ to inhibit the AChE activity is higher than DNZ and NPs-PLA-PEG-DNZ nanoformulation.
As a third objective, the nanoformulations were investigated for cell internalization, and ability to cross an in vitro BBB model. We found that the uptake by both cell types was very high (greater than 90%) for the two nanoformulations after 24h of exposure. It has been reported that the uptake of EVs and NPs-PLA-PEG could occur very rapidly as they were identified within cells after one hour of exposure.24,76 Transcytosis rates after 24h of ~50% were also observed for NPs-PLA with PEG length similar to that used in our study (2000g×mol−1).28 Although PEG is considered a non-toxic compound, NPs-PLA-PEGylated can induce an acute inflammatory response and an increased rate of endocytosis in bEnd.3 cells within 24h.77 Considering that the transcytosis previously requires endocytosis, this process may explain the exocytosis of the NPs-PLA-PEG in the basal compartment. We have previously showed that the transcytosis of NPs-PLA-PEG through the bEnd.3 cells occurred mainly by the macropinocytosis pathway which possibly led to their exocytosis.24 Although to a lesser extent compared to synthetic nanoparticles, our results showed the important finding that EVs could also overcome the BBB Transwell® model likely by macropinocytosis along with the clathrin-dependent pathway78,79 Neuronal activity can efficiently stimulate the uptake of EVs from the circulation which could also explain the low level of EVs in the basal medium.80 Regarding the NPs-PLA-PEG-Cy5, we have previously demonstrated that these neuronal cells can release them over time (after 6 hours) after being taken-up.24 Overall, our data suggest that EVs-PKH26 can be rapidly uptake by neuronal cells while NPs-PLA-PEG-Cy5 achieved a balance between uptake and release.
As a fourth and final objective, the evaluation of in vivo safety and brain distribution in zebrafish larvae model is fundamental for pharmacological applications. Zebrafish are used as a screening platform to improve the effectiveness of treatments as it is a reliable intermediate model that bridges the gap between in vitro cellular testing and in vivo mammalian experimentation.81 We found that both nanoformulations were not toxic to zebrafish embryos. However, early hatching was observed in embryos exposed to EVs-DNZ without impacting mortality or development. This finding has been observed before and is associated with the high sequence identity of the human marker CD63 in Zebrafish, associated with the pre-polster tissue responsible for the origin of the hatching gland.82,83 Another study showed that when CD63 knockdown was performed in embryos, there was a failure in hatching or delay in hatching, evidencing that tetraspanin CD63 found in EVs-DNZ (Figure 1C) plays a role in the mechanism of hatching involving probably surface proteins.84 NPs-PLA-PEG-DNZ and free DNZ were found to increase locomotor activity in zebrafish while the EVs-DNZ group was not different from the control. Similar hyperactivity-induced by DNZ was reported in response to the acoustic stimulus.85 Zebrafish larvae have acetylcholinesterase with enzymatic activity like other vertebrates. Moreover, they possess neuronal-type nicotinic acetylcholine receptors in the peripheral nervous and central system, in addition to muscle-type receptors that mediate the neuromuscular transmission at the neuromuscular junctions.86,87 A rapid tissue distribution of free DNZ or peripheral uptake of the NPs-PLA-PEG-DNZ system may have caused the peripheral action of the drug, resulting in the observed hyperactivity in locomotion test (Figure 5F). This response may be associated with the peripheral cholinergic side effects often associated with acetylcholinesterase inhibitor drugs in AD patients.88 Considering that EVs-DNZ can deliver a higher drug concentration to the brain, less peripheral effect was observed compared to free drug and NPs-PLA-PEG-DNZ.
Zebrafish larvae is considered a valid in vivo model to assess the brain permeability of drugs due to the functionality and structure of the BBB which are homologous to the human.89,90 24 hours after the peripheral injection, both nanoformulations were observed around the plexus of the caudal vein region which is known to have large number of highly endocytic endothelial cells similar to those in mammals and a high tortuosity favoring particles uptake.91 Moreover, it has been reported that the negative surface charge of NPs-PLA-PEG, EVs, or even other negatively charged NPs activates their uptake by cells.54 Interestingly, both nanoformulations were able to cross the BBB with higher brain distribution of PKH26-tagged EVs as compared to NPs-PLA-PEG. Some studies have also demonstrated that EVs were also capable of delivering drugs into the brain of zebrafish. It seems that the complex composition of EVs can help to circumvent natural barriers, and the presence of the CD63 biomarker may be one of the reasons for the selectivity of EVs by the BBB, allowing them to cross via receptor-mediated endocytosis.92 In another study, the brain targeting of quercetin was higher when it is loaded in plasma-derived EVs after an intravenous administration. Furthermore, the nanoformulation quercetin-EVs promoted improvements in AD symptoms by inhibiting phosphorylated tau-mediated neurofibrillary tangles.93 Previous study reported that exosomes can widely diffuse once they have entered the brain including the hippocampus, and be taken up particularly by damaged neurons and astrocytes.94 On the other hand, the peripheral distribution of NPs-PLA-PEG was higher than their cerebral uptake. More importantly, in addition to the brain targeting properties, EV-DNZ were able to deliver their cargo DNZ more efficiently than NPs-PLA-PEG-DNZ and induced higher reduction of AChE enzyme activity than free DNZ or NPs-PLA-PEG-DNZ.
One of the main difference between synthetic and bioinspired NPs is the presence and the composition of protein corona, which may affect biodistribution and targeting ability.95,96 The present results suggest that the PEGylated NPs-PLA may triggered the interaction of complement proteins adsorbed on the surface of the NPs.97–99 In zebrafish many complement proteins such as C3 are transferred from mothers to eggs and can activate the opsonization process, facilitating phagocytosis and particle clearance.100,101 Thus, corona proteins may have been one of the factors explain different observations between in vitro and in vivo properties and between the polymeric and bioinspired NPs which interfered with their brain delivery and their therapeutic performance.
Regarding the investigation of the role of the BBB in the delivery of nutrients or drugs to the CNS, it was demonstrated that the in vitro model is an important tool to predict the transcytosis capacity of nanoformulations. However, as the BBB has a complex nature that is difficult to capture in a single in vitro model, hence the limitation that may have led to an overestimation (PLA-PEG) and underestimation (pEVs) of brain uptake when compared to the in vivo model (zebrafish).
Conclusion
In summary, bioinspired nanoformulations, compared to freely administered drugs and synthetic NPs, were superior in increasing DNZ drug bioavailability in the brain while maintaining safety. This study highlights the multifunctionality of EVs largely due to a complex biological structure that allows them to overcome some limitations found in synthetic NPs in addition to acting as efficient vehicles for drug delivery to the brain.
The head-to-head study design allowed us to demonstrate that DNZ-loaded EVs from healthy donors had numerous advantages over free drug therapies and synthetic NPs, evidencing their potential for the treatment of neurological disorders. First, EVs were more efficient in encapsulating the drug and showed less cellular toxicity. In vivo, both systems were biocompatible, with NPs-PLA-PEG showing better targeting efficiency than free drug, but less than EVs. Thus, DNZ-loaded EVs when injected systemically in an animal model had higher pharmacological response, lower peripheral side effect, and without toxicity. As perspective, it would be interesting to decorate the surface of EVs to enhance their permeability through the BBB and to further improve their brain therapeutic index.
Abbreviations
AChE, Acetylcholinesterase; AD, Alzheimer’s disease; BCA, Bicinchoninic acid; BBB, blood-brain barrier; CNS, Central Nervous System; DNZ, Donepezil; DMEM, Dulbecco’s Modified Eagle’s Media; DL%, Drug loading; EMEM, Eagle’s minimal essential medium; EDTA, Ethylenediaminetetraacetic acid; EE %, Encapsulation efficiency; FDA, United States Food and Drug Administration; hpf, Hours Post-Fertilization; INRS-CER, Ethical Committee of the Institut National de la Recherche Scientifique; EVs, Extracellular Vesicles; FBS, Fetal Bovine Serum; FACS, Fluorescence-activated cell sorting; nFCM, nanoscale flow cytometry; NPs nanoparticles; NTA, Nanoparticle tracking analysis; PET, Polyester; PTU, phenylthiourea; PDI, Polydispersity index; PLA-PEG, Poly (lactic acid)-poly(ethylene glycol); SEM, Standard Error of the Mean; TEER, Transendothelial Electrical Resistance; TJ, tight junctions; TEM, Transmission electron microscopy.
Acknowledgments
This study was supported by Research Chair Louise & André Charron on Alzheimer’s disease, the Armand-Frappier Foundation, NSERC (RGPIN-2018-06182) Government of Canada), Canadian Institute of Health Research, NSERC/CRSNG, and Canada Foundation for Innovation. The authors are grateful to Pr D. Girard for the use of the DLS Malvern Zetasizer; Pr Maritza Jaramillo for the use of the Microplate Spectrophotometer; Jessy Tremblay (INRS, Center Armand-Frappier) for help with experiments involving flow cytometry and confocal microscopy; Arnaldo Nakamura (INRS, Center Armand-Frappier) for help with the transmission electron microscope. Tg (flk1:EGFP) was a generous gift from Dr. Xiao-Yan Wen (Univ. of Toronto, Toronto, ON, Canada). Graphical abstract is created by the main author using BioRender.com.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gauthier S, Rosa-Neto P, Morais J, Webster C. World Alzheimer report 2021: journey through the diagnosis of dementia. Alzheimer’s Dis Int. 2021;2021:30.
2. S. Schneider L. A critical review of cholinesterase inhibitors as a treatment modality in Alzheimer’s disease. Dial Clin Neurosci. 2000;2(2):111–128. doi:10.31887/DCNS.2000.2.2/lschneider
3. Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Research Support, Non-U.S. Gov’t. Review. Brain. 2018;141(7):1917–1933. doi:10.1093/brain/awy132
4. Massoud F, Gauthier S. Update on the pharmacological treatment of Alzheimer’s disease. Curr Neuropharmacol. 2010;8(1):69–80. doi:10.2174/157015910790909520
5. Perneczky R, Jessen F, Grimmer T, et al. Anti-amyloid antibody therapies in Alzheimer’s disease. Brain. 2023;146(3):842–849. doi:10.1093/brain/awad005
6. Richard E, den Brok MG, van Gool WA. Bayes analysis supports null hypothesis of anti‐amyloid beta therapy in Alzheimer’s disease. Alzheimer’s Dementia. 2021;17(6):1051–1055. doi:10.1002/alz.12379
7. Wilkinson DG. The pharmacology of donepezil: a new treatment for Alzheimer’s disease. Expert Opinion Pharmacother. 1999;1(1):121–135. doi:10.1517/14656566.1.1.121
8. Eissa KI, Kamel MM, Mohamed LW, Kassab AE. Development of new Alzheimer’s disease drug candidates using donepezil as a key model. Arch Pharm. 2022;356(1):e2200398. doi:10.1002/ardp.202200398
9. Kim SH, Kandiah N, Hsu JL, Suthisisang C, Udommongkol C, Dash A. Beyond symptomatic effects: potential of donepezil as a neuroprotective agent and disease modifier in Alzheimer’s disease. Br. J. Pharmacol. 2017;174(23):4224–4232. doi:10.1111/bph.14030
10. Noh MY, Koh SH, Kim SM, Maurice T, Ku SK, Kim SH. Neuroprotective effects of donepezil against A β42‐induced neuronal toxicity are mediated through not only enhancing PP 2 A activity but also regulating GSK‐3β and n AChR s activity. J Neurochem. 2013;127(4):562–574. doi:10.1111/jnc.12319
11. Asiri YA, Mostafa GA. Donepezil. In: Profiles of Drug Substances, Excipients and Related Methodology. Elsevier; 2010:117–150.
12. Shin CY, Kim H-S, Cha K-H, et al. The effects of donepezil, an acetylcholinesterase inhibitor, on impaired learning and memory in rodents. Biomolecules Ther. 2018;26(3):274. doi:10.4062/biomolther.2017.189
13. Marcantonio ER, Palihnich K, Appleton P, Davis RB. Pilot randomized trial of donepezil hydrochloride for delirium after Hip fracture. J Am Geriatr Soc. 2011;59:S282–S288.
14. Wilkinson D, Doody R, Helme R, et al. Donepezil in vascular dementia: a randomized, placebo-controlled study. Neurology. 2003;61(4):479–486. doi:10.1212/01.WNL.0000078943.50032.FC
15. Carrasco MM, Agüera L, Gil P, Moríñigo A, Leon T. Safety and effectiveness of donepezil on behavioral symptoms in patients with Alzheimer disease. Alzheimer Dis Associated Disord. 2011;25(4):333–340. doi:10.1097/WAD.0b013e318212ab7a
16. Chamundeeswari M, Jeslin J, Verma ML. Nanocarriers for drug delivery applications. Environ Chem Lett. 2019;17(2):849–865. doi:10.1007/s10311-018-00841-1
17. Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine. 2011;6(4):715–728. doi:10.2217/nnm.11.19
18. Niemeyer CM. Nanoparticles, proteins, and nucleic acids: biotechnology meets materials science. Angew Chem Int Ed. 2001;40(22):4128–4158. doi:10.1002/1521-3773(20011119)40:22<4128::AID-ANIE4128>3.0.CO;2-S
19. Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8(2):147–166. doi:10.1016/j.nano.2011.05.016
20. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature Biotechnol. 2015;33(9):941–951. doi:10.1038/nbt.3330
21. Li G, Zhao M, Xu F, et al. Synthesis and biological application of polylactic acid. Review. Molecules. 2020;25(21):5023. doi:10.3390/molecules25215023
22. Rabanel JM, Faivre J, Paka GD, Ramassamy C, Hildgen P, Banquy X. Effect of polymer architecture on curcumin encapsulation and release from PEGylated polymer nanoparticles: toward a drug delivery nano-platform to the CNS. Comparative Study. Research support, Non-U.S. Gov’t. Eur J Pharm Biopharm. 2015;96:409–420. doi:10.1016/j.ejpb.2015.09.004
23. Dong Y, Feng SS. Nanoparticles of poly (D, L‐lactide)/methoxy poly (ethylene glycol)‐poly (D, L‐lactide) blends for controlled release of paclitaxel. J Biomed Mater Res Part A. 2006;78(1):12–19. doi:10.1002/jbm.a.30684
24. Rabanel J-M, Piec P-A, Landri S, Patten SA, Ramassamy C. Transport of PEGylated-PLA nanoparticles across a blood brain barrier model, entry into neuronal cells and in vivo brain bioavailability. J Control Release. 2020;328:679–695. doi:10.1016/j.jconrel.2020.09.042
25. Huang F-YJ, Chen W-J, Lee W-Y, S-T L, Lee T-W, J-M L. In vitro and in vivo evaluation of lactoferrin-conjugated liposomes as a novel carrier to improve the brain delivery. Int J Mol Sci. 2013;14(2):2862–2874. doi:10.3390/ijms14022862
26. Loureiro JA, Gomes B, Coelho MA, Carmo Pereira M, Rocha S. Targeting nanoparticles across the blood–brain barrier with monoclonal antibodies. Nanomedicine. 2014;9(5):709–722. doi:10.2217/nnm.14.27
27. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Delivery Rev. 2016;99(Pt A):28–51. doi:10.1016/j.addr.2015.09.012
28. Tehrani SF, Bernard-Patrzynski F, Puscas I, Leclair G, Hildgen P, Roullin VG. Length of surface PEG modulates nanocarrier transcytosis across brain vascular endothelial cells. Nanomedicine. 2019;16:185–194. doi:10.1016/j.nano.2018.11.016
29. Andaloussi SE, Lakhal S, Mäger I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Delivery Rev. 2013;65(3):391–397. doi:10.1016/j.addr.2012.08.008
30. Doyle LM, Wang MZ. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Research support, Non-U.S. Gov’t Review. Cells. 2019;8(7):727. doi:10.3390/cells8070727
31. Pegtel DM, Gould SJ. Exosomes. Annu. Rev. Biochem. 2019;88(1):487–514. doi:10.1146/annurev-biochem-013118-111902
32. Lai RC, Yeo RW, Tan KH, Lim SK. Exosomes for drug delivery - a novel application for the mesenchymal stem cell. Review. Biotechnol. Adv. 2013;31(5):543–551. doi:10.1016/j.biotechadv.2012.08.008
33. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Research support, Non-U.S. Gov’t. Review. Nat Rev Mol Cell Biol. 2018;19(4):213–228. doi:10.1038/nrm.2017.125
34. Caby M-P, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17(7):879–887. doi:10.1093/intimm/dxh267
35. Kang JY, Park H, Kim H, et al. Human peripheral blood‑derived exosomes for microRNA delivery. IntJ Mol Med. 2019;43(6):2319–2328. doi:10.3892/ijmm.2019.4150
36. Van den Boorn JG, Schlee M, Coch C, Hartmann G. SiRNA delivery with exosome nanoparticles. Nature Biotechnol. 2011;29(4):325–326. doi:10.1038/nbt.1830
37. Piffoux M, Nicolás-Boluda A, Mulens-Arias V, et al. Extracellular vesicles for personalized medicine: the input of physically triggered production, loading and theranostic properties. Adv Drug Delivery Rev. 2019;138:247–258. doi:10.1016/j.addr.2018.12.009
38. Amiri A, Bagherifar R, Ansari Dezfouli E, Kiaie SH, Jafari R, Ramezani R. Exosomes as bio-inspired nanocarriers for RNA delivery: preparation and applications. J Transl Med. 2022;20(1):1–16. doi:10.1186/s12967-022-03325-7
39. Qu M, Lin Q, Huang L, et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J Control Release. 2018;287:156–166. doi:10.1016/j.jconrel.2018.08.035
40. Ellman GL, Courtney KD, Andres JV, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7(2):88–95. doi:10.1016/0006-2952(61)90145-9
41. Rabanel J-M, Faivre J, Zaouter C, Patten SA, Banquy X, Ramassamy C. Nanoparticle shell structural cues drive in vitro transport properties, tissue distribution and brain accessibility in zebrafish. Biomaterials. 2021;277:121085. doi:10.1016/j.biomaterials.2021.121085
42. Koto T, Takubo K, Ishida S, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol. 2007;170(4):1389–1397. doi:10.2353/ajpath.2007.060693
43. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Research Support, U.S. Gov’t, P.H.S. Dev. Dyn. 1995;203(3):253–310. doi:10.1002/aja.1002030302
44. Cheng J, Gu Y-J, Wang Y, Cheng SH, Wong W-T. Nanotherapeutics in angiogenesis: synthesis and in vivo assessment of drug efficacy and biocompatibility in zebrafish embryos. Int j Nanomed. 2011;2007–2021. doi:10.2147/IJN.S20145
45. Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002;248(2):307–318. doi:10.1006/dbio.2002.0711
46. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9(7):676–682. doi:10.1038/nmeth.2019
47. Sutthapitaksakul L, Dass CR, Sriamornsak P. Donepezil—An updated review of challenges in dosage form design. J Drug Delivery Sci Technol. 2021;63:102549. doi:10.1016/j.jddst.2021.102549
48. van der Meel R, Fens MH, Vader P, Van Solinge WW, Eniola-Adefeso O, Schiffelers RM. Extracellular vesicles as drug delivery systems: lessons from the liposome field. J Control Release. 2014;195:72–85. doi:10.1016/j.jconrel.2014.07.049
49. Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int J Pharm. 2008;364(2):298–327. doi:10.1016/j.ijpharm.2008.04.042
50. Jayaraj RL, Tamilselvam K, Manivasagam T, Elangovan N. Neuroprotective effect of CNB-001, a novel pyrazole derivative of curcumin on biochemical and apoptotic markers against rotenone-induced SK-N-SH cellular model of Parkinson’s disease. J Mol Neurosci. 2013;51(3):863–870. doi:10.1007/s12031-013-0075-8
51. Chuo ST-Y, Chien JC-Y, Lai CP-K. Imaging extracellular vesicles: current and emerging methods. J Biomed Sci. 2018;25(1):1–10. doi:10.1186/s12929-018-0494-5
52. Tong R, Coyle VJ, Tang L, Barger AM, Fan TM, Cheng J. Polylactide nanoparticles containing stably incorporated cyanine dyes for in vitro and in vivo imaging applications. Microsc Res Tech. 2010;73(9):901–909. doi:10.1002/jemt.20824
53. Zhu L, Li R, Jiao S, et al. Blood-brain barrier permeable chitosan oligosaccharides interfere with β-Amyloid aggregation and alleviate β-amyloid protein mediated neurotoxicity and neuroinflammation in a dose-and degree of polymerization-dependent manner. Mar Drugs. 2020;18(10):488. doi:10.3390/md18100488
54. Campbell F, Bos FL, Sieber S, et al. Directing nanoparticle biodistribution through evasion and exploitation of Stab2-dependent nanoparticle uptake. ACS nano. 2018;12(3):2138–2150. doi:10.1021/acsnano.7b06995
55. Beccaria K, Canney M, Bouchoux G, Puget S, Grill J, Carpentier A. Blood-brain barrier disruption with low-intensity pulsed ultrasound for the treatment of pediatric brain tumors: a review and perspectives. Neurosurg Focus. 2020;48(1):E10. doi:10.3171/2019.10.FOCUS19726
56. Almutairi M, Gong C, Xu YG, Chang Y, Shi H. Factors controlling permeability of the blood–brain barrier. Cell. Mol. Life Sci. 2016;73(1):57–77. doi:10.1007/s00018-015-2050-8
57. Rempe R, Cramer S, Qiao R, Galla H-J. Strategies to overcome the barrier: use of nanoparticles as carriers and modulators of barrier properties. Cell Tissue Res. 2014;355(3):717–726. doi:10.1007/s00441-014-1819-7
58. Tian T, Zhang H-X, C-P H, et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137–149. doi:10.1016/j.biomaterials.2017.10.012
59. Li AJ, Zheng YH, Liu GD, Liu WS, Cao PC, Bu ZF. Efficient delivery of docetaxel for the treatment of brain tumors by cyclic RGD-tagged polymeric micelles. Mol Med Rep. 2015;11(4):3078–3086. doi:10.3892/mmr.2014.3017
60. Zhou R, Zhu L, Zeng Z, et al. Targeted brain delivery of RVG29‐modified rifampicin‐loaded nanoparticles for Alzheimer’s disease treatment and diagnosis. Bioeng Transl Med 2022;7(3):e10395. doi:10.1002/btm2.10395
61. Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nature Nanotechnol. 2021;16(7):748–759. doi:10.1038/s41565-021-00931-2
62. Xu J, Chen Y, Jiang X, Gui Z, Zhang L. Development of hydrophilic drug encapsulation and controlled release using a modified nanoprecipitation method. Processes. 2019;7(6):331. doi:10.3390/pr7060331
63. Haney MJ, Klyachko NL, Zhao Y, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30. doi:10.1016/j.jconrel.2015.03.033
64. Al-Jipouri A, Almurisi SH, Al-Japairai K, Bakar LM, Doolaanea AA. Liposomes or Extracellular Vesicles: a Comprehensive Comparison of Both Lipid Bilayer Vesicles for Pulmonary Drug Delivery. Polymers. 2023;15(2):318. doi:10.3390/polym15020318
65. Singh R, Lillard JW. Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86(3):215–223. doi:10.1016/j.yexmp.2008.12.004
66. Heurtault B, Saulnier P, Pech B, Proust J-E, Benoit J-P. Physico-chemical stability of colloidal lipid particles. Biomaterials. 2003;24(23):4283–4300. doi:10.1016/S0142-9612(03)00331-4
67. Lockman PR, Koziara JM, Mumper RJ, Allen DD. Nanoparticle surface charges alter blood–brain barrier integrity and permeability. J Drug Targeting. 2004;12(9–10):635–641. doi:10.1080/10611860400015936
68. Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood–brain barrier by nanoparticles. J Control Release. 2012;161(2):264–273. doi:10.1016/j.jconrel.2011.08.017
69. Goldblum D, Gygax M, Böhnke M, Garweg JG. In vitro toxicity of rivastigmine and donepezil in cells of epithelial origin. Ophthal Res. 2002;34(2):97–103. doi:10.1159/000048336
70. Das JR, Tizabi Y. Additive protective effects of donepezil and nicotine against salsolinol-induced cytotoxicity in SH-SY5Y cells. Neurotox Res. 2009;16(3):194–204. doi:10.1007/s12640-009-9040-2
71. Kou L, Sun J, Zhai Y, He Z. The endocytosis and intracellular fate of nanomedicines: implication for rational design. Asian J Pharm Sci. 2013;8(1):1–10. doi:10.1016/j.ajps.2013.07.001
72. Yamamoto T, Kosaka N, Ochiya T. Latest advances in extracellular vesicles: from bench to bedside. Sci Technol Adv Mater. 2019;20(1):746–757. doi:10.1080/14686996.2019.1629835
73. Lee M, Ban -J-J, Yang S, Im W, Kim M. The exosome of adipose-derived stem cells reduces β-amyloid pathology and apoptosis of neuronal cells derived from the transgenic mouse model of Alzheimer’s disease. Brain Res. 2018;1691:87–93. doi:10.1016/j.brainres.2018.03.034
74. Bonafede R, Scambi I, Peroni D, et al. Exosome derived from murine adipose-derived stromal cells: neuroprotective effect on in vitro model of amyotrophic lateral sclerosis. Exp Cell Res 2016;340(1):150–158. doi:10.1016/j.yexcr.2015.12.009
75. Sun T, Ding Z-X, Luo X, Liu Q-S, Cheng Y, Hassanzadeh K. Blood exosomes have neuroprotective effects in a mouse model of Parkinson’s disease. Oxid Med Cell Longev. 2020;2020:1–14. doi:10.1155/2020/3807476
76. Feng D, Zhao WL, Ye YY, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11(5):675–687. doi:10.1111/j.1600-0854.2010.01041.x
77. Tehrani SF, Rabanel J-M, Legeay S, et al. Tailoring PEGylated nanoparticle surface modulates inflammatory response in vascular endothelial cells. Eur. J. Pharm. Biopharm. 2022;174:155–166. doi:10.1016/j.ejpb.2022.04.003
78. Morad G, Carman CV, Hagedorn EJ, et al. Tumor-derived extracellular vesicles breach the intact blood–brain barrier via transcytosis. ACS nano. 2019;13(12):13853–13865. doi:10.1021/acsnano.9b04397
79. Yuan D, Zhao Y, Banks WA, et al. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials. 2017;142:1–12. doi:10.1016/j.biomaterials.2017.07.011
80. Kur I-M, Prouvot P-H, Fu T, et al. Neuronal activity triggers uptake of hematopoietic extracellular vesicles in vivo. PLoS biol. 2020;18(3):e3000643. doi:10.1371/journal.pbio.3000643
81. Cascallar M, Alijas S, Pensado-López A, et al. What zebrafish and nanotechnology can offer for cancer treatments in the age of personalized medicine. Cancers. 2022;14(9):2238. doi:10.3390/cancers14092238
82. Matsuda A, Moirangthem A, Angom RS, et al. Safety of bovine milk derived extracellular vesicles used for delivery of RNA therapeutics in zebrafish and mice. J Appl Toxicol. 2020;40(5):706–718. doi:10.1002/jat.3938
83. Gritsman K, Talbot WS, Schier AF. Nodal signaling patterns the organizer. Development. 2000;127(5):921–932. doi:10.1242/dev.127.5.921
84. Trikić MZ, Monk P, Roehl H, Partridge LJ, Kanellopoulos J. Regulation of zebrafish hatching by tetraspanin cd63. PLoS One. 2011;6(5):e19683. doi:10.1371/journal.pone.0019683
85. Best JD, Berghmans S, Hunt JJ, et al. Non-associative learning in larval zebrafish. Neuropsychopharmacology. 2008;33(5):1206–1215. doi:10.1038/sj.npp.1301489
86. Svoboda KR, Vijayaraghavan S, Tanguay RL. Nicotinic receptors mediate changes in spinal motoneuron development and axonal pathfinding in embryonic zebrafish exposed to nicotine. J Neurosci. 2002;22(24):10731–10741. doi:10.1523/JNEUROSCI.22-24-10731.2002
87. Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19(2):99–113. doi:10.2307/2412448
88. Ibach B, Haen E. Acetylcholinesterase inhibition in Alzheimer’s Disease. Curr Pharm Des. 2004;10(3):231–251. doi:10.2174/1381612043386509
89. Li Y, Chen T, Miao X, et al. Zebrafish: a promising in vivo model for assessing the delivery of natural products, fluorescence dyes and drugs across the blood-brain barrier. Pharmacol Res. 2017;125:246–257. doi:10.1016/j.phrs.2017.08.017
90. Jeong J-Y, Kwon H-B, Ahn J-C, et al. Functional and developmental analysis of the blood–brain barrier in zebrafish. Brain Res Bull. 2008;75(5):619–628. doi:10.1016/j.brainresbull.2007.10.043
91. Lu W, Wan J, She Z, Jiang X. Brain delivery property and accelerated blood clearance of cationic albumin conjugated pegylated nanoparticle. J Control Release. 2007;118(1):38–53. doi:10.1016/j.jconrel.2006.11.015
92. Yang T, Martin P, Fogarty B, et al. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res. 2015;32(6):2003–2014. doi:10.1007/s11095-014-1593-y
93. Qi Y, Guo L, Jiang Y, Shi Y, Sui H, Zhao L. Brain delivery of quercetin-loaded exosomes improved cognitive function in AD mice by inhibiting phosphorylated tau-mediated neurofibrillary tangles. Drug Delivery. 2020;27(1):745–755. doi:10.1080/10717544.2020.1762262
94. Nakano M, Nagaishi K, Konari N, et al. Bone marrow-derived mesenchymal stem cells improve diabetes-induced cognitive impairment by exosome transfer into damaged neurons and astrocytes. Sci Rep. 2016;6(1):24805. doi:10.1038/srep24805
95. Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein Corona with possible implications for biological impacts. Proc Natl Acad Sci. 2008;105(38):14265–14270. doi:10.1073/pnas.0805135105
96. Moghimi SM, Hunter A, Andresen T. Factors controlling nanoparticle pharmacokinetics: an integrated analysis and perspective. Annu. Rev. Pharmacol. Toxicol. 2012;52(1):481–503. doi:10.1146/annurev-pharmtox-010611-134623
97. Grenier P, de Oliveira Viana IM, Lima EM, Bertrand N. Anti-polyethylene glycol antibodies alter the protein Corona deposited on nanoparticles and the physiological pathways regulating their fate in vivo. J Control Release. 2018;287:121–131. doi:10.1016/j.jconrel.2018.08.022
98. Saadati R, Dadashzadeh S, Abbasian Z, Soleimanjahi H. Accelerated blood clearance of PEGylated PLGA nanoparticles following repeated injections: effects of polymer dose, PEG coating, and encapsulated anticancer drug. Pharm Res. 2013;30(4):985–995. doi:10.1007/s11095-012-0934-y
99. Verhoef JJ, Carpenter JF, Anchordoquy TJ, Schellekens H. Potential induction of anti-PEG antibodies and complement activation toward PEGylated therapeutics. Drug Discovery Today. 2014;19(12):1945–1952. doi:10.1016/j.drudis.2014.08.015
100. Yang Q, Lai SK. Anti‐PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev. 2015;7(5):655–677. doi:10.1002/wnan.1339
101. Wang Z, Zhang S, Tong Z, Li L, Wang G, Fugmann SD. Maternal transfer and protective role of the alternative complement components in zebrafish Danio rerio. PLoS One. 2009;4(2):e4498. doi:10.1371/journal.pone.0004498
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.